

Abstract
Babies born small are at an increased risk of developing myriad adult diseases. While growth restriction increases disease risk in all individuals, often a second hit is required to unmask ‘programmed’ impairments in physiology. Programmed disease outcomes are demonstrated more commonly in male offspring compared with females, with these sex‐specific outcomes partly attributed to different placenta‐regulated growth strategies of the male and female fetus. Pregnancy is known to be a major risk factor for unmasking a number of conditions and can be considered a ‘second hit’ for women who were born small. As such, female offspring often develop impairments of physiology for the first time during pregnancy that present as pregnancy complications. Numerous maternal stressors can further increase the risk of developing a maternal complication during pregnancy. Importantly, these maternal complications can have long‐term consequences for both the mother after pregnancy and the developing fetus. Conditions such as preeclampsia, gestational diabetes and hypertension as well as thyroid, liver and kidney diseases are all conditions that can complicate pregnancy and have long‐term consequences for maternal and offspring health. Babies born to mothers who develop these conditions are often at a greater risk of developing disease in adulthood. This has implications as a mechanism for transmission of disease across generations. In this review, we discuss the evidence surrounding long‐term intergenerational implications of being born small and/or experiencing stress during pregnancy on programming outcomes.
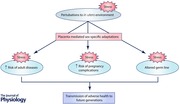
Introduction
The lifelong health of an individual is determined by the cumulative experiences he or she is exposed to throughout life. Often, the earlier an event occurs in an individual's life, the greater the impact it has on long‐term health outcomes. This is particularly true of fetal development, with numerous studies demonstrating that adult disease may be influenced by events that occurred in the womb (Gluckman et al. 2007). While some individuals who are ‘programmed’ to have an increased risk of disease will develop symptoms despite living a healthy lifestyle, others may develop disease only after a ‘second hit’ such as chronic exposure to factors including smoking, consumption of high‐salt or high‐fat diet, excessive consumption of alcohol and by living a sedentary lifestyle. An extremely common and severe ‘second hit’ for females, known to unmask a variety of conditions in adult life, is pregnancy. In fact, pregnancy is the greatest physiological ‘stress test’ that a woman can experience in her life. With large physiological adaptations occurring during normal pregnancies (Torgersen & Curran, 2006; Weissgerber & Wolfe, 2006), factors that negatively impact maternal physiology can have profound implications for the success of the pregnancy. Women who are programmed to have an increased risk of disease prior to pregnancy may develop disease for the first time during pregnancy. Common pregnancy complications include conditions such as preeclampsia, gestational diabetes and hypertension, as well as thyroid, liver and kidney diseases (Williams, 2003; Joshi et al. 2010; Steegers et al. 2010; Acharya et al. 2013). Women who develop these diseases during pregnancy may be at greater risk of disease long after the completion of the pregnancy. The fetus that developed in the womb may also have been exposed to suboptimal conditions and may be programmed to develop disease in later life. This potentially leads to the transgenerational transmission of disease.
This review will focus on the consequences of being born small due to uteroplacental insufficiency, the most common cause of growth restriction in Western society. We will discuss how females who were born small are at an increased risk of pregnancy complications and will consider how this may result in the transgenerational transmission of programmed disease. In addition, to understand the mechanisms involved, we will consider outcomes following experimentally induced uteroplacental insufficiency in a rat model of intrauterine growth restriction that induces offspring outcomes commonly observed following many different pregnancy perturbations.
Programming of adult diseases
The fetal origins of adult disease hypothesis was first described by David Barker, who proposed that disruptions to the intrauterine environment during fetal development have the potential to ‘program’ increased risks for developing disease during adulthood (Barker, 1994). This concept was put forward when Barker and his colleagues studied the birth and death records of 4654 men born in Hertfordshire and identified that low weight at birth and at 1 year of age was associated with increased mortality from ischaemic heart disease (Barker et al. 1989 a,b). Subsequent epidemiological and experimental studies have been able to further expand upon this observation and there is now strong evidence associating low birth weight with increased vulnerability to the development of a wide array of adult diseases (McMillen et al. 2005; Gluckman et al. 2007; Warner et al. 2010). Increased understanding that the early postnatal environment also has important consequences for later health has resulted in the field of developmental origins of health and disease (DOHaD).
Low birth weight is commonly used as a surrogate marker of intrauterine growth restriction (Moritz et al. 2009). It is defined as having a birth weight below the 10th percentile of weight for gestational age or below 2.5 kg at term, and accounts for ∼10% of births in the Western world (Martin et al. 2011, 2013). The clinical pattern of growth restriction varies depending on the nature of the causative factor, the stage of gestation in which it occurs, and the duration of the intrauterine insult (Lin & Santolaya‐Forgas, 1998; Gluckman & Hanson, 2004; Morrison, 2008). A suboptimal in utero environment can be caused by factors such as genetic predisposition, maternal malnutrition, smoking, alcohol consumption, drug abuse and stress, all of which can individually contribute to impaired fetal growth (Lin & Santolaya‐Forgas, 1998). Maternal undernutrition is the predominant cause of growth restriction in developing countries while in Western society, intrauterine growth restriction is largely attributed to uteroplacental insufficiency during late gestation (Henriksen & Clausen, 2002). Uteroplacental insufficiency impairs blood flow through the uterine vessels and placenta, which in turn compromises the supply of oxygen and nutrients to the fetus (Henriksen & Clausen, 2002), and is responsible for approximately 30% of the total cases of fetal growth restriction (Nardozza et al. 2012).
Numerous researchers have used sheep models to investigate the role of placental insufficiency in long‐term disease outcomes (reviewed by Morrison, 2008). In the sheep, placental insufficiency is induced by removal of placentation sites (carunclectomy) prior to mating or injection of microspheres in late pregnancy (Robinson et al. 1979; Cheung et al. 2004). The use of microspheres allows for specific stages of gestation to be investigated. These models have been very useful to study the effects of placental insufficiency on fetal development and function (Zhang et al. 2015). While the sheep is a robust model for studying long‐term physiological outcomes in the programming field, studies are costly and time consuming due to their size and the long gestation period. As a result rodents are frequently used to model this condition and uteroplacental insufficiency can be induced by bilateral ligation of uterine blood vessels, which impedes nutrient and oxygen delivery to the developing fetus. First developed to study the role of uterine blood supply in fetal growth (Wigglesworth, 1974), this is now a widely used technique to induce uteroplacental insufficiency and fetal growth restriction and is particularly useful for studies examining long‐term outcomes in offspring throughout the complete life course. We have established a model in Wistar–Kyoto rats in which surgery is performed on day 18 of a 22‐day pregnancy to replicate the late gestational onset of uteroplacental insufficiency commonly observed in humans. Our rat model, using bilateral uterine vessel ligation surgery, is known to cause disruption to the intrauterine environment resulting in offspring being consistently 10–15% lighter than control counterparts (Wlodek et al. 2005). Although inducing uteroplacental insufficiency through surgery results in sudden onset of the disease, the impact is similar to the diseases identified in humans and mimics their more gradual late gestation insufficiency, including offspring organ deficits, disease predisposition and reductions in birth weight (Table 1). However, as discussed in the following section, disease outcomes are highly dependent upon the sex of the offspring.
Table 1.
Sex‐specific offspring programming outcomes of uteroplacental insufficiency in rats
| Offspring disease | Growth restricted | Growth restricted | Growth restricted female |
|---|---|---|---|
| phenotype | male | female | during pregnancy |
| Low birth weight | ✓ | ✓ | — |
| (Wlodek et al. 2008) | (Gallo et al. 2012) | ||
| Nephron deficits | ✓6 months | ✓6 months | ✓4 months |
| (Wlodek et al. 2008) | (Moritz et al. 2009) | (Gallo et al. 2012) | |
| Glomerular hypertrophy | ✓6 months | ✗6 months | ✓4 months |
| (Wlodek et al. 2008) | (Moritz et al. 2009) | (Gallo et al. 2012) | |
| ✓18 months | |||
| (Moritz et al. 2009) | |||
| Cardiomyocyte deficits | ✓Day 7 | Not characterised | Not characterised |
| (Black et al. 2012) | |||
| High blood pressure | ✓6 months | ✗5 months | ✗4 months |
| (Wlodek et al. 2008) | (Moritz et al. 2009) | (Gallo et al. 2012) | |
| ✗18 months | |||
| (Moritz et al. 2009) | |||
| β‐cell deficits | ✓6 months | ✓4 months | ✗4 months |
| (Siebel et al. 2010; Laker et al. 2011) | (Gallo et al. 2012) | (Gallo et al. 2012) | |
| Glucose intolerance | ✓6 months | ✗4 months | ✓4 months |
| (Siebel et al. 2008; | (Gallo et al. 2012) | (Gallo et al. 2012) | |
| Laker et al. 2011) | ✗12 months | ||
| (Tran et al. 2015) | |||
| Impaired insulin response | ✓6 months | ✗4 months | ✗4 months |
| (Siebel et al. 2008; | (Gallo et al. 2012) | (Gallo et al. 2012) | |
| Laker et al. 2011) | ✗12 months | ||
| (Tran et al. 2015) |
Uteroplacental insufficiency in rats results in low birth weight and programs sex‐specific offspring dysfunction and deficits that often affect males more than females. Even with an additional hit of ageing, females are generally well protected from the development of adult diseases. However, this protection can be lost due to the physiological demands of pregnancy, and diseases are revealed for the first time even at a relatively young age.
Uteroplacental insufficiency results from impaired placental development that often arises as a consequence of maternal diseases such as preeclampsia, hypertension and metabolic diseases (Resnik, 2002). Since the formation of the placenta is an essential feature of mammalian pregnancy, if the spiral arteries fail to migrate into the trophoblast, blood supply to the growing placenta is compromised, potentially resulting in growth restriction and preeclampsia (Nayak & Giudice, 2003). Indeed, human studies have demonstrated associations between placental efficiency and the programming of diseases (Risnes et al. 2009; Wen et al. 2010). The shape of the placenta in particular can be a marker of maladaptive responses to adverse conditions. In general, a circular placenta is indicative of greater placental efficiency, whereas an oval placenta is indicative of a maladaption to adverse maternal conditions (Eriksson et al. 2011; Barker et al. 2011). The fact that placental shape reflects impaired fetal development and long‐term disease may be explained by the hypothesis that the placenta is polarised from the time of implantation. It has been suggested that the growth of the major axis of the placenta is aligned with the rostro‐caudal growth of the fetus whereas growth of the minor axis is associated with nutrient availability (Kajantie et al. 2010). As such, when a fetus grows to its maximal size despite poor nutrient availability, the placental diameter is likely to be much greater in its major axis than its minor axis. This may in turn impair placental efficiency and lead to programmed disease outcomes. Eriksson et al. studied the relationship between placental dimensions and later hypertension, revealing that males often compensate for impaired placentation by expanding the placental surface across its minor axis (Eriksson et al. 2010). The fact that this relationship was only evident in males suggests that the placentas of males and females respond differentially to adverse events during pregnancy.
Sexual dimorphism in the developmental origins of health and disease
More males are conceived than females but they are less likely to survive to term such that this sex bias is almost absent by birth (Kraemer, 2000). Male fetuses grow at a faster rate than do females and this accelerated growth trajectory makes male fetuses more vulnerable during disturbed pregnancies, with less favourable outcomes occurring throughout the life course of the individual (Eriksson et al. 2010; Ben‐Haroush et al. 2012). When faced with an adverse challenge, males often do not alter their growth trajectories, but instead continue to grow, ultimately attempting to maintain overall size. In contrast, the slower growth pattern of female fetuses provides flexibility for them to adapt to the surrounding environment by investing in long‐term survival (Clifton, 2010; Rosenfeld, 2015). These sexually dimorphic adaptations are regulated by the placenta, which is itself a sexually dimorphic organ (Buckberry et al. 2014; Myatt et al. 2014; Rosenfeld, 2015; Matheson et al. 2016). Placentas of male and female human fetuses express vastly different mRNA profiles (Buckberry et al. 2014) and rodent models also demonstrate sexually dimorphic placental and fetal responses to pregnancy perturbations. In mice, maternal dexamethasone exposure or magnesium deficiency reduces fetal weight in both males and females but causes sex‐specific changes in placental weight, morphology and the expression of growth factors and placental transporters (Cuffe et al. 2011; Schlegel et al. 2015). Maternal hypoxia has been shown to dysregulate the expression of nutrient transporters (solute carrier family 2, member 1 (Slc2a1) and solute carrier family 38, member 1 (Slc38a1)) and growth factors (insulin‐like growth factor 1 receptor (Igf1r) and insulin like growth factor 2 (Igf2)) in placentas of female fetuses only (Cuffe et al. 2014). Female‐specific placental adaptations similarly occur in rats following exposure to alcohol around conception, with the placentas of female fetuses having increased cell accumulation of glycogen and increased glucose transporter 3 (Glut3), kinase insert domain receptor (Kdr) and Igf2 mRNA expression while males had reduced Igf1r and Glut3 expression (Gardebjer et al. 2014). Human pregnancies complicated by maternal obesity result in sex‐specific impairment of mitochondrial function, although its functional consequences in the placenta are not yet understood (Mele et al. 2014; Muralimanoharan et al. 2015).
These sex‐specific placental adaptations are often associated with male offspring developing adult disease while females are minimally affected. In the mouse model of dexamethasone exposure, only male offspring developed hypertension (O'Sullivan et al. 2013). Similarly, periconceptional alcohol exposure programs more severe metabolic phenotypes in male offspring, an effect that is exacerbated following exposure to a high‐fat diet in postnatal life (Gardebjer et al. 2015). We have extensively studied the sex‐specific outcomes in our rat model of uteroplacental insufficiency (Table 1). Male but not female offspring develop increased systolic blood pressure in adult life. This is despite both sexes having decreased nephron number and males (females were not characterised) having a cardiomyocyte deficit early in life (Black et al. 2012). Interestingly, glomerular hypertrophy developed in males by 6 months of age but was only evident in females at 18 months (Wlodek et al. 2008; Moritz et al. 2009). Male offspring also had impaired glucose tolerance and reduced insulin secretion (Siebel et al. 2008), which was accompanied by a reduction in pancreatic β‐cell mass (Siebel et al. 2010; Laker et al. 2011). In contrast, females born small had a normal metabolic profile despite having pancreatic deficits. Together, these studies highlight that despite similar structural changes in both sexes (nephron and β‐cell deficits), the programmed disease phenotype is only evident in male offspring. While these differences could be attributed to the different growth strategies that male and female fetuses employ in utero as we discussed earlier, insults experienced in utero have been shown to induce sex‐specific changes in hormones in postnatal life that may contribute to sex‐specific disease phenotypes (Romano et al. 2015). In addition, sex hormones are known to regulate normal physiology and are likely to contribute to differences in programmed disease outcomes between males and females (Ojeda et al. 2013).
Pregnancy unmasks programmed disease in susceptible females
Although females are generally less susceptible to programmed disease development, under the physiological demands of pregnancy, various disease states are often unmasked (Table 1). Specifically, female rats born small following uteroplacental insufficiency develop glucose intolerance for the first time during gestation (Gallo et al. 2012). Indeed, studies have demonstrated that pregnancy complications such as gestational diabetes and preeclampsia are more prevalent in women that were themselves born small but were otherwise healthy before pregnancy (Zetterström et al. 2007; á Rogvi et al. 2012). This suggests maternal events or ‘second hits’ experienced during pregnancy may unmask predispositions to pregnancy complications. Thus it may be that latent programmed phenotypes can subsequently affect the mother either during or after pregnancy, and consequently her offspring (Fig. 1).
Figure 1. Transgenerational transmission of programmed outcomes through the maternal lineage .
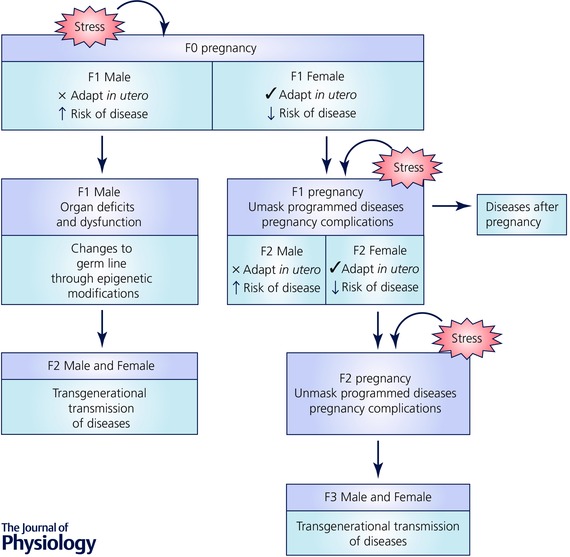
Female fetuses generally adapt well to perturbations in utero and alter their developmental strategy in accordance with their environment. However, when these F1 females become pregnant, programmed diseases can be unmasked leading to the development of pregnancy complications. This in turn creates a suboptimal intrauterine environment for the developing F2 fetus. Similarly, F2 female fetuses are programmed for adult disease, and when they become pregnant, pregnancy complications may arise leading to programming consequences in the subsequent F3 generation. If the cycle continues, this would result in transgenerational programming of disease that may persist across multiple generations. Furthermore, pregnancy complications impair the long‐term health of the mother, and she may experience long‐term diseases even after the conclusion of pregnancy. Although the maternal lineage has been thought to be mainly responsible for the transgenerational transmission of disease in the past, recent studies have demonstrated important roles of the paternal lineage. When the developing F1 male fetus is exposed to a suboptimal environment in utero, this increases the risk of postnatal organ deficits and dysfunction. This increased susceptibility to adverse health may impact on the germ cells of these males during reproductive age and when they mate with a normal female, the alterations in the germ line may be passed on to the offspring. As the germ cells that give rise to the F2 generation were not present during the initial insult, phenotypes observed in this generation are sufficient to classify this as transgenerational transmission of programmed disease.
In order to further characterise pregnancy adaptations of female rats born small following uteroplacental insufficiency, we conducted a series of physiological measurements (metabolic cage, tail cuff blood pressure, intraperitoneal glucose tolerance test) during pregnancy. Interestingly, the physiological measurements conducted during gestation induced fetal growth restriction in the developing rat, but only when their mothers themselves were small at birth (Gallo et al. 2012). This suggests a complex interaction between being born small, physiological adaptations associated with pregnancy and being stressed during pregnancy, which may translate at a population level to the transmission of disease vulnerability across generations. Stress in pregnancy is known to contribute towards myriad pregnancy complications and offspring disease outcomes, but rarely has the birth weight of the mother been considered. These aspects are often considered in isolation and rarely is stress considered a trigger for unmasking pregnancy complications that may result in impaired fetal development and programmed disease outcomes.
Interaction of stress and pregnancy in unmasking disease phenotype in programmed individuals
Numerous reviews have previously discussed how the type of ‘stress’ (or exposure to ‘stress’ hormones), the model species used and the timing of exposure all contribute to the type and severity of programmed disease outcomes (Drake et al. 2007; Singh et al. 2012; Giussani et al. 2013; Moisiadis & Matthews, 2014). Here, we use the term ‘maternal stress’ to refer to any external factor that acts as a second hit and places additional pressure on an already strained physiological system during pregnancy that can reveal pregnancy diseases (Table 2). The stress encountered following the death of a family member has been shown to increase the risk of preeclampsia (László et al. 2013) and the development of gestational diabetes (László et al. 2015). Similarly, women exposed to stress encountered through Hurricane Katrina had an increased incidence of pregnancy‐induced hypertension and gestational diabetes (Oni et al. 2015). Vitamin D deficiency has been shown to increase the risk of severe preeclampsia (Bodnar et al. 2014). Furthermore, women who become pregnant at an advance maternal age, have increased risk of stillbirths and preterm delivery (Kenny et al. 2013). Maternal low birth weight has also been implicated in the development of gestational diabetes (Seghieri et al. 2002). Another study demonstrated that being born small doubles a women's risk of developing preeclampsia. This risk was increased to fourfold if the women were overweight during pregnancy, demonstrating that the additional stress of being overweight exacerbates the preeclampsia risk (Dempsey et al. 2003; Oni et al. 2015). Furthermore, maternal obesity is correlated to a substantially higher risk of developing gestational diabetes, with the risk increasing with the severity of obesity (Chu et al. 2007).
Table 2.
Stress during pregnancy and associated pregnancy complications in humans
| Type of stress | Pregnancy complications | References |
|---|---|---|
| Exposure to Hurricane Katrina | Induction of labour | Oni et al. 2015 |
| Increased perceived stress | ||
| Death of close relative | Preeclampsia | László et al. 2013 |
| Gestational diabetes | László et al. 2015 | |
| Obesity during pregnancy | Gestational diabetes | Chu et al. 2007 |
| Overweight during pregnancy | Preeclampsia | Dempsey et al. 2003 |
| Maternal low birth weight | Preeclampsia | Dempsey et al. 2003 |
| Gestational diabetes | Seghieri et al. 2002 | |
| Vitamin D deficiency | Preeclampsia | Bodnar et al. 2014 |
Stressful maternal events that can contribute to the development of various pregnancy complications as demonstrated in epidemiological studies.
Of interest, while the mechanisms that regulate programmed pregnancy complications are unknown, they are likely to overlap with factors that program disease outcomes in women exposed to external stressors. Thus, women who are at an increased risk of pregnancy complications may have this complication unmasked by exposure to additional stress. This stress response is likely to involve both a systemic physiological stress response and a cellular stress response. The systemic stress response is largely mediated through hormonal and chemical messengers (such as glucocorticoids or oxygen and glucose) that can transfer the stress signal from the site of action to various tissues throughout the body. In the target tissues, a cellular stress response may ensue. Glucocorticoids regulate stress‐responsive pathways by binding to the glucocorticoid receptors to regulate gene transcription (Fowden & Forhead, 2015). Similarly, alterations in blood oxygen saturation can regulate transcription of genes including those involved in proliferation through the activation of hypoxia‐inducible factors (Semenza, 2014; Guimarães‐Camboa et al. 2015). In addition, changes in glucose levels can influence fetal outcome by altering cellular glucose uptake and impacting on embryonic adaptive responses via pathway such as hexosamine signalling and O‐linked glycosylation (Howerton & Bale, 2014; Pantaleon et al. 2015). Studies have also demonstrated that systemic stress can activate cellular stress pathways including but not limited to cellular oxidative stress (Spiers et al. 2015). Oxidative stress has been shown to play a major role in the aetiology of multiple pregnancy complications including preeclampsia (Sahay et al. 2015) and intrauterine growth restriction (Schneider et al. 2015), and is also known to contribute to programmned disease outcomes in offspring (Giussani et al. 2012). Oxidative stress is often associated with mitochondrial dysfunction, which has been demonstrated in the placentas of pregnancies complicated by factors such as maternal obesity (Mele et al. 2014; Muralimanoharan et al. 2015).
Other cellular stress responses include the heat shock response, the unfolded protein response and the DNA damage response (Fulda et al. 2010). Studies have demonstrated that cellular stress pathways converge to induce similar cellular outcomes, being predominantly related to cell survival or cell death. One factor central in the regulation of cellular stress is O‐linked‐N‐acetylglucosamine transferase (OGT) (Zachara et al. 2011). This enzyme is encoded by an X‐linked gene that escapes inactivation in the placenta, resulting in higher OGT levels in females compared with males. OGT regulates O‐linked glycosylation to control cellular functions, predominantly related to cellular stress. Of particular importance, OGT has been recognised as a key mediator of sex differences and placental stress responsivity (Nugent et al. 2015). Importantly, OGT has been shown to mediate the toxic effects of maternal hyperglycaemia on early embryonic development (Pantaleon et al. 2010). Furthermore, OGT has been suggested to play a major role in the sex‐specific programming of disease (Howerton & Bale, 2014) and is well characterised as being a significant factor in the regulation of adult diseases such as diabetes (Akimoto et al. 2005).
Effects of maternal pregnancy complications on the long‐term health of the mother
Pregnancy is likely to be the greatest physiological challenge in a woman's life, and it is unsurprising that adverse health conditions that develop for the first time during pregnancy often manifest as long‐term health conditions well after the completion of pregnancy. A study of more than 129,000 births demonstrated that women who had preeclampsia, preterm delivery or babies in the lowest quintile for birth weight had a significantly higher risk of developing ischaemic heart disease in later life. When a pregnancy was complicated by more than one of these factors, maternal risk increased with each additional factor (Smith et al. 2001). Other studies have supported these findings in women diagnosed with preeclampsia known to have increased risk of hypertension, ischaemic heart disease, stroke and venous thromboembolism post‐pregnancy (Bellamy et al. 2007). Hypertensive disorders diagnosed during pregnancy are also correlated with higher prevalence of metabolic syndrome after pregnancy (Hermes et al. 2013). Other studies have shown that women who develop gestational diabetes have increased lifetime risk of developing overt diabetes (Kim et al. 2002; Damm, 2009) and their health can be further compromised by second hits such as obesity (Lauenborg et al. 2005). In a study of over 5000 women, detection of hypothyroidism in early pregnancy strongly predicts subsequent morbidity related to thyroid conditions post‐pregnancy, and overt hypothyroidism during pregnancy has also been known to increase risk of later diabetes morbidity sixfold (Männistö et al. 2010). Since women born small have heightened risk of developing pregnancy complications and often have more severe disease phenotypes, it is likely that they also have an increased risk of developing long‐term diseases that persist post‐pregnancy. Rats that were born small and developed glucose intolerance during pregnancy did not have any long‐term cardiovascular or metabolic adverse consequences (Tran et al. 2012), but it is hypothesised that an additional ‘second hit’ of maternal stress may have deleterious effects on long‐term maternal health (Fig. 1). Thus far, there have not been any human studies linking low maternal birth weight to later‐life health outcomes unmasked by pregnancy. Understanding the underlying mechanisms may provide an opportunity for intervention before the onset of pregnancy or immediately following pregnancy, with the aim of preventing the development of overt disease phenotypes in the mother.
Importantly, if maternal health is compromised following a pregnancy, this may impact upon any future pregnancies she experiences. A women who experience spontaneous preterm delivery in her first pregnancy is at an increased risk of preterm delivery in her second pregnancy (Lykke et al. 2009). Furthermore, there is an increased risk of preeclampsia, small for gestational age and placental abruption in the second pregnancy (Lykke et al. 2009). This study highlights that the earlier the preterm delivery in the first pregnancy, the higher the risk of a pregnancy complication in the second pregnancy. Similarly, intrauterine growth restriction and hypertensive disorders in the first pregnancy are independent risk factors for increased hypertensive disorders in the second pregnancy (Zhang et al. 2001). These findings may reflect increased risk of disease in the second pregnancy due to unmasked disease from the first pregnancy. This warrants investigation in future studies.
Pregnancy complications and programmed offspring outcomes: intergenerational programming of disease
Given that pregnancy complications are more common and more severe in women who themselves were exposed to adverse pregnancy conditions in utero, the possibility arises of a transgenerational cycle of disease transmission (Fig. 1). Human and animal studies have supported this hypothesis, and maternal complications such as those mentioned above have been shown to program offspring disease. In a cohort study of over 280,000 pregnancies in Sweden, maternal diabetes was associated with increased BMI in male offspring (Lawlor et al. 2011). Similar findings have been reported following other pregnancy complications. Women with hypertension during pregnancy gave birth to offspring who had a greater risk for hypertension (Miettola et al. 2013). Grandmaternal depression is similarly predictive of early onset depression in youth and interpersonal difficulties, as well as higher rates of depression among their daughters who had children (Hammen et al. 2011).
In experimental studies, the nomenclature used for the various generations involved in intergenerational studies often defines the F0 generation as the pregnant mothers exposed to the initial insult, with the offspring of these mothers being the F1 generation, whilst the second generation offspring are termed the F2 generation and so forth for subsequent generations. In our rat model of uteroplacental insufficiency, the reduced nephron endowment previously observed in the males of the F1 generation (Wlodek et al. 2008) persisted to the F2 generation during fetal life in the absence of any additional insults, but was restored after birth (Gallo et al. 2014). Furthermore, these F2 males presented with high blood pressure and metabolic disturbances (Tran et al. 2013; Gallo et al. 2014). Another study has demonstrated that maternal bisphenol A (BPA) exposure is implicated in programming glucose intolerance in F1 and F2 male but not female offspring, once again demonstrating sex specificity of programming outcomes (Susiarjo et al. 2015). As the F2 glucose intolerance observed in male offspring cannot be attributed to metabolic disturbances of the mother as F1 females were not affected, this is suggestive of transmission through factors other than maternal environment, with the authors attributing it to epigenetic modifications (Susiarjo et al. 2015). In addition, maternal obesity in the mice has been shown to cause cardiac dysfunction and hypertrophy in the male offspring (Fernandez‐Twinn et al. 2012; Blackmore et al. 2014). Indeed, maternal obesity has been implicated in many programming outcomes (Mahizir et al. 2015).
Numerous studies have demonstrated that stress during pregnancy can program alterations in offspring glucocorticoid production that may reoccur in subsequent generations. In a model where F1 rat offspring and their lactating F0 mother were subjected to social stress (male intruder) from postnatal day 2 to 16, this led to altered social behaviours in the F2 generation, which were accompanied by reduced glucocorticoid levels (Babb et al. 2014). F2 rats whose grandmothers were exposed to restraint stress and forced swimming during gestation had a significantly shorter gestational length even if their mothers had a non‐stressful pregnancy, and this effect persisted to the F3 generation (Yao et al. 2014). Interestingly, the F3 generation was the first generation to be born of lower birth weight after the initial F0 exposure to stress during pregnancy. Of particular importance is that the developing F1 fetus and F2 germ‐line (developing within the F1 fetus) were present during the initial insult to the F0 pregnancy, and may have been directly affected by the perturbation. As the F3 generation is the first generation that is not exposed to the initial insult, it can be deduced that the phenotype is truly of a transgenerational nature and suggests that the transmission is at least partially through epigenetic mechanisms (Skinner, 2008).
Intergenerational programming of disease through paternal lineage
Although a large proportion of previous studies have focused on the maternal contributions to programming outcomes in the offspring, recent evidence in animals has indicated the importance of the paternal contribution. We have published that preimplantation blastocysts derived from fathers born small are altered, highlighting a potential pathway to F2 offspring deficits and dysfunction (Master et al. 2015). Ng et al. were amongst the first to show that paternal obesity in rodents programmed pancreatic deficits in the resultant female offspring through alterations in pancreatic islet genes. The significance of these findings was that they demonstrated non‐genetic transmission of disease of a high‐fat diet from father to offspring (Ng et al. 2010). The same authors later found evidence of premature ageing and chronic degenerative disorders in white adipose tissue and pancreatic islets of females born to fathers that consumed high‐fat diets (Ng et al. 2014). Male rats born to mothers who were fed a low‐protein diet were found to have reduced plasma corticosterone levels and altered behavioural patterns. Interestingly, when these male rats fathered daughters, these F2 females also had reduced corticosterone levels and altered behavioural patterns. In contrast male offspring of these programmed males were unaffected (Reyes‐Castro et al. 2015). As discussed above, changes in maternal physiology often contribute to long‐term outcomes that may impact on the pregnancy and also lactation in the early postnatal period (Siebel et al. 2008, 2010). The main advantage of studying programming outcomes through the paternal line is the elimination of confounders associated with the maternal line such as the maternal environment. In the paternal lineage, the maternal environment is eliminated, leaving genetic and epigenetic mechanisms as the predominant likely explanation (Fig. 1). As the sole mediator of disease transmission within the paternal linage is through the germ line, any impairment in the sperm could lead to persistent effects spanning multiple generations (Jiménez‐Chillarón et al. 2015). One way in which this could occur is through alterations to chromatin or RNA, especially small non‐coding RNAs in the sperm (Lane et al. 2014; Gapp et al. 2014). Importantly, studies have shown that these changes can be induced through environmental influences, with diet‐induced paternal obesity causing alternations in the sperm microRNA, which resulted in metabolic dysregulation in the F1 and F2 generation offspring (Fullston et al. 2013). In a model of undernutrition, F1 male mice that were undernourished in utero gave rise to F2 mice with altered lipogenic genes in their liver, which was attributed to altered patterns of DNA methylation in the Lxra locus (Martínez et al. 2014). Intriguingly, the same epigenetic change was observed in the sperm of F1 males that gave rise to the F2 progeny, providing compelling evidence that epigenetic modifications in the germ cells of the one generation can persist and be transmitted to the somatic cells of the next generation (Martínez et al. 2014). Male mice that were exposed to 6 weeks of chronic stress at various stages of life induced changes in the sperm microRNA environment. When these mice were bred with a normal female, their offspring had blunted stress responses to a 15 min restraint stress test and also had lower corticosterone levels, and this was observed regardless of whether the father was stressed early or later in life (Rodgers et al. 2013). A separate study that investigated the paternal effects of early prenatal stress on neurodevelopmental processes reported a shift in gene expression in the brain from a male‐typical to a more female‐typical pattern in the F2 male offspring of prenatally stressed fathers. Furthermore, this disrupted masculinisation process was also associated with altered stress responsivity in adulthood (Morgan & Bale, 2011).
An important point to consider in the transgenerational programming of disease through either the maternal or paternal line is the sex‐specific adaptations that occur in utero that may regulate whether programmed disease can be passed on to subsequent generations. A major confounding factor to consider is the fact that male fetuses exposed to a suboptimal environment are less likely to survive than their female counterparts (Kraemer, 2000). In contrast, female fetuses are likely to have survived the fetal insult, which may result in their developing disease in adulthood and during pregnancy, potentially inducing disease transmission to future generations.
Conclusions
Perturbations during fetal development have the ability to program disease in the offspring after birth and into adulthood, and these diseases are more severe and more frequently observed in males. Although females are generally well protected from developing adult diseases, this protection is often lost as a result of the physiological demands of pregnancy. Furthermore, exposure to ‘second hits’ such as maternal stress during pregnancy can exacerbate the severity of the pregnancy complications. The effects of these complications can be long lasting and maternal health may be compromised well beyond the completion of pregnancy. Hence, studies investigating pregnancy complications should recognise and take into consideration maternal birth weight as a risk factor. Diseases encountered during pregnancy can program the developing fetus for sex‐specific offspring diseases that may then be passed on to future generations through either the maternal or the paternal lines. The underlying mechanisms of disease transmission between these two lines are likely to differ. Future studies should aim to uncover the molecular mechanisms of different types of stress and their significance in programming transgenerational disease transmission and long‐term maternal health.
Additional information
Competing interests
None.
Author contributions
J.N.C., M.E.W., J.S.M.C. and K.M. were responsible for conception and design of the review. All authors were involved in drafting the review and revising it critically for important intellectual content. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Biographies
Jean Cheong is a final year PhD student at the University of Melbourne; her research encompasses the areas of fetal physiology and the programming of metabolic diseases.
Mary Wlodek is a Professor in the Department of Physiology at the University of Melbourne. She graduated with a BSc(Hons) and MSc from the University of Western Ontario, London, Canada and PhD in Physiology from Monash University, Australia. She is a global leader in developmental origins physiological research and Head of the Fetal, Postnatal & Adult Physiology & Disease Laboratory.
Karen Moritz completed her PhD at the University of Melbourne and is currently a Senior Research Fellow in the School of Biomedical Sciences at the University of Queensland. The aim of her work is to understand how prenatal perturbations contribute to an increased risk of developing cardiovascular, renal and metabolic disease in adulthood.
James Cuffe is Postdoctoral Research Fellow at the School of Biomedical Sciences at the University of Queensland. He has strong collaborations with Karen Moritz and Mary Wlodek and has extensive experience investigating the role of the placenta in regulating the sex‐specific programming of long‐term diseases. His current research investigates cellular stress responses to systemic physiological stressors.
References
- Acharya A, Santos J, Linde B & Anis K (2013). Acute kidney injury in pregnancy—current status. Adv Chronic Kidney Dis 20, 215–222. [DOI] [PubMed] [Google Scholar]
- Akimoto Y, Hart GW, Hirano H & Kawakami H (2005). O‐GlcNAc modification of nucleocytoplasmic proteins and diabetes. Med Mol Morphol 38, 84–91. [DOI] [PubMed] [Google Scholar]
- á Rogvi R, Forman JL, Damm P & Greisen G (2012). Women born preterm or with inappropriate weight for gestational age are at risk of subsequent gestational diabetes and pre‐eclampsia. PLoS One 7, e34001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb JA, Carini LM, Spears SL & Nephew BC (2014). Transgenerational effects of social stress on social behavior, corticosterone, oxytocin, and prolactin in rats. Horms Behav 65, 386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP (1994). The fetal origins of adult disease. Fetal Matern Med Rev 6, 71–80. [Google Scholar]
- Barker DJP, Osmond C, Golding J, Kuh D & Wadsworth MEJ (1989. a). Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP, Osmond C, Thornburg KL, Kajantie E & Eriksson JG (2011). The lifespan of men and the shape of their placental surface at birth. Placenta 32, 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP, Osmond C, Winter PD, Margetts B & Simmonds SJ (1989. b). Weight in infancy and death from ischaemic heart disease. Lancet 334, 577–580. [DOI] [PubMed] [Google Scholar]
- Bellamy L, Casas JP, Hingorani AD & Williams DJ (2007). Pre‐eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta‐analysis. BMJ 335, 974–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Haroush A, Melamed N, Oron G, Meizner I, Fisch B & Glezerman M (2012). Early first‐trimester crown‐rump length measurements in male and female singleton fetuses in IVF pregnancies. J Matern Fetal Neonatal Med 25, 2610–2612. [DOI] [PubMed] [Google Scholar]
- Black MJ, Siebel AL, Gezmish O, Moritz KM & Wlodek ME (2012). Normal lactational environment restores cardiomyocyte number after uteroplacental insufficiency: implications for the preterm neonate. Am J Physiol Regul Integr Comp Physiol 302, R1101–R1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore HL, Niu Y, Fernandez‐Twinn DS, Tarry‐Adkins JL, Giussani DA & Ozanne SE (2014). Maternal diet‐induced obesity programs cardiovascular dysfunction in adult male mouse offspring independent of current body weight. Endocrinology 155, 3970–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar LM, Simhan HN, Catov JM, Roberts JM, Platt RW, Diesel JC & Klebanoff MA (2014). Maternal vitamin D status and the risk of mild and severe preeclampsia. Epidemiology 25, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckberry S, Bianco‐Miotto T, Bent SJ, Dekker GA & Roberts CT (2014). Integrative transcriptome meta‐analysis reveals widespread sex‐biased gene expression at the human fetal‐maternal interface. Mo Hum Reprod 20, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CY, Bogic L & Gagnon R (2004). Morphologic alterations in ovine placenta and fetal liver following induced severe placental insufficiency. J Soc Gynecol Investig 11, 521–528. [DOI] [PubMed] [Google Scholar]
- Chu SY, Callaghan WM, Kim SY, Schmid CH, Lau J, England LJ & Dietz PM (2007). Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care 30, 2070–2076. [DOI] [PubMed] [Google Scholar]
- Clifton VL (2010). Review: sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta 31, S33–S39. [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, Dickinson H, Simmons DG & Moritz KM (2011). Sex specific changes in placental growth and MAPK following short term maternal dexamethasone exposure in the mouse. Placenta 32, 981–989. [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, Walton SL, Singh RR, Spiers JG, Bielefeldt‐Ohmann H, Wilkinson L, Little MH & Moritz KM (2014). Mid‐ to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex‐specific manner. J Physiol 592, 3127–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm P (2009). Future risk of diabetes in mother and child after gestational diabetes mellitus. Int J Gynaecol Obstet 104, S25–S26. [DOI] [PubMed] [Google Scholar]
- Dempsey JC, Williams MA, Luthy DA, Emanuel I & Shy K (2003). Weight at birth and subsequent risk of preeclampsia as an adult. Am J Obstet Gynecol 189, 494–500. [DOI] [PubMed] [Google Scholar]
- Drake AJ, Tang JI & Nyirenda MJ (2007). Mechanisms underlying the role of glucocorticoids in the early life programming of adult disease. Clin Sci 113, 219. [DOI] [PubMed] [Google Scholar]
- Eriksson JG, Kajantie E, Osmond C, Thornburg K & Barker DJ (2010). Boys live dangerously in the womb. Am J Hum Biol 22, 330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Kajantie E, Thornburg KL, Osmond C & Barker DJP (2011). Mother's body size and placental size predict coronary heart disease in men. Eur Heart J 32, 2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Twinn DS, Blackmore HL, Siggens L, Giussani DA, Cross CM, Foo R & Ozanne SE (2012). The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology 153, 5961–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowden AL & Forhead AJ (2015). Glucocorticoids as regulatory signals during intrauterine development. Exp Physiol 100, 1477–1487. [DOI] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O & Samali A (2010). Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullston T, Ohlsson Teague EMC, Palmer NO, DeBlasio MJ, Mitchell M, Corbett M, Print CG, Owens JA & Lane M (2013). Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J 27, 4226–4243. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Tran M & Cullen‐McEwen LA (2014). Transgenerational programming of fetal nephron deficits and sex‐specific adult hypertension in rats. Reprod Fertil Dev 26, 1032–1043. [DOI] [PubMed] [Google Scholar]
- Gallo LA, Tran M, Moritz KM, Mazzuca MQ, Parry LJ, Westcott KT, Jefferies AJ, Cullen McEwen LA & Wlodek ME (2012). Cardio‐renal and metabolic adaptations during pregnancy in female rats born small: implications for maternal health and second generation fetal growth. J Physiol 590, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, Farinelli L, Miska E & Mansuy IM (2014). Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nature 17, 667–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardebjer EM, Anderson ST, Pantaleon M & Wlodek ME (2015). Maternal alcohol intake around the time of conception causes glucose intolerance and insulin insensitivity in rat offspring, which is exacerbated by a postnatal high‐fat diet. FASEB J 29, 2690–2701. [DOI] [PubMed] [Google Scholar]
- Gardebjer EM, Cuffe JSM, Pantaleon M, Wlodek ME & Moritz KM (2014). Periconceptional alcohol consumption causes fetal growth restriction and increases glycogen accumulation in the late gestation rat placenta. Placenta 35, 50–57. [DOI] [PubMed] [Google Scholar]
- Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, Blake EZ, Horder KA, Thakor AS, Hansell JA, Kane AD, Wooding FBP, Cross CM & Herrera EA (2012). Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One 7, e31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA & Davidge ST (2013). Developmental programming of cardiovascular disease by prenatal hypoxia. J Dev Orig Health Dis 4, 328–337. [DOI] [PubMed] [Google Scholar]
- Gluckman PD & Hanson MA (2004). Maternal constraint of fetal growth and its consequences. Semin Fetal Neonatal Med 9, 419–425. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA & Beedle AS (2007). Early life events and their consequences for later disease: A life history and evolutionary perspective. Am J Hum Biol 19, 1–19. [DOI] [PubMed] [Google Scholar]
- Guimarães‐Camboa N, Stowe J, Aneas I, Sakabe N, Cattaneo P, Henderson L, Kilberg MS, Johnson RS, Chen J, McCulloch AD, Nobrega MA, Evans SM & Zambon AC (2015). HIF1a represses cell stress pathways to allow proliferation of hypoxic fetal cardiomyocytes. Dev Cell 33, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammen C, Brennan PA & Le Brocque R (2011). Youth depression and early childrearing: Stress generation and intergenerational transmission of depression. J Consult Clin Psychol 79, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen T & Clausen T (2002). The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well‐nourished populations. Acta Obstet Ggynecol Scand 81, 112–114. [DOI] [PubMed] [Google Scholar]
- Hermes W, Franx A, van Pampus MG, Bloemenkamp KWM, Bots ML, van der Post JA, Porath M, Ponjee GAE, Tamsma JT, Mol BWJ & de Groot CJM (2013). Cardiovascular risk factors in women who had hypertensive disorders late in pregnancy: a cohort study. Am J Obstet Gynecol 208, 474.e1–474.e8. [DOI] [PubMed] [Google Scholar]
- Howerton CL & Bale TL (2014). Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc Natl Acad Sci USA 111, 9639–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez‐Chillarón JC, Ramón‐Krauel M, Ribó S & Díaz R (2015). Transgenerational epigenetic inheritance of diabetes risk as a consequence of early nutritional imbalances. Proc Nutr Soc 75, 78–89. [DOI] [PubMed] [Google Scholar]
- Joshi D, James A, Quaglia A, Westbrook R & Heneghan MA (2010). Liver disease in pregnancy. Lancet 375, 594–605. [DOI] [PubMed] [Google Scholar]
- Kajantie E, Thornburg K, Eriksson JG, Osmond L & Barker DJP (2010). In preeclampsia, the placenta grows slowly along its minor axis. Int J Dev Biol 54, 469–473. [DOI] [PubMed] [Google Scholar]
- Kenny LC, Lavender T, McNamee R, O'Neill SM, Mills T & Khashan AS (2013). Advanced maternal age and adverse pregnancy outcome: evidence from a large contemporary cohort. PLoS One 8, e56583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Newton KM & Knopp RH (2002). Gestational diabetes and the incidence of type 2 diabetes a systematic review. Diabetes Care 25, 1862–1868. [DOI] [PubMed] [Google Scholar]
- Kraemer S (2000). The fragile male. BMJ 321, 1609–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker RC, Gallo LA, Wlodek ME, Siebel AL, Wadley GD & McConell GK (2011). Short‐term exercise training early in life restores deficits in pancreatic β‐cell mass associated with growth restriction in adult male rats. Am J Physiol Endocrinol Metab 301, E931–E940. [DOI] [PubMed] [Google Scholar]
- Lane M, Robker RL & Robertson SA (2014). Parenting from before conception. Science 345, 756–760. [DOI] [PubMed] [Google Scholar]
- László KD, Liu XQ, Svensson T, Wikström AK & Li J (2013). Psychosocial stress related to the loss of a close relative the year before or during pregnancy and risk of preeclampsia. Hypertension 62, 183–189. [DOI] [PubMed] [Google Scholar]
- László KD, Olsen J, Li J, Persson M, Vestergaard M, Svensson T, Obel C & Cnattingius S (2015). The risk of gestational diabetes mellitus following bereavement: a cohort study from Denmark and Sweden. Paediatr Perinat Epidemiol 29, 271–280. [DOI] [PubMed] [Google Scholar]
- Lauenborg J, Mathiesen E, Hansen T, Glümer C, Jørgensen T, Borch‐Johnsen K, Hornnes P, Pedersen O & Damm P (2005). The prevalence of the metabolic syndrome in a Danish population of women with previous gestational diabetes mellitus is three‐fold higher than in the general population. J Clin Endocrinol Metab 90, 4004–4010. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Lichtenstein P & Langstrom N (2011). Association of maternal diabetes mellitus in pregnancy with offspring adiposity into early adulthood: sibling study in a prospective cohort of 280 866 men from 248 293 families. Circulation 123, 258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C‐C & Santolaya‐Forgas J (1998). Current concepts of fetal growth restriction: part I. Causes, classification, and pathophysiology. Obstet Gynecol 92, 1044–1055. [DOI] [PubMed] [Google Scholar]
- Lykke JA, Paidas MJ & Langhoff‐Roos J (2009). Recurring complications in second pregnancy. Obstet Gynecol 113, 1217–1224. [DOI] [PubMed] [Google Scholar]
- Mahizir D, Briffa JF, Hryciw DH, Wadley GD, Moritz KM & Wlodek ME (2015). Maternal obesity in females born small: pregnancy complications and offspring disease risk. Mol Nutr Food Res 60, 8–17. [DOI] [PubMed] [Google Scholar]
- Matheson H, Veerbeek JHW, Charnock‐Jones DS, Burton GJ & Yung HW (2016). Morphological and molecular changes in the murine placenta exposed to normobaric hypoxia throughout pregnancy. J Physiol 594, 1371–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Ventura SJ, Osterman MJ & Matthews TJ (2013). Births: final data for 2011. Natl Vital Stat Rep 62, 1–90. [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Ventura SJ, Osterman MJ, Kirmeyer S, Mathews TJ & Wilson EC (2011). Births: final data for 2009. Natl Vital Stat Rep 60, 1–70. [PubMed] [Google Scholar]
- Martínez D, Pentinat T, Ribó S, Daviaud C, Bloks VW, Cebrià J, Villalmanzo N, Kalko SG, Ramón‐Krauel M, Díaz R, Plösch T, Tost J & Jiménez‐Chillarón JC (2014). In utero undernutrition in male mice programs liver lipid metabolism in the second‐generation offspring involving altered Lxra DNA methylation. Cell Metab 19, 941–951. [DOI] [PubMed] [Google Scholar]
- Master JS, Thouas GA, Harvey AJ, Sheedy JR, Hannan NJ, Gardner DK & Wlodek ME (2015). Fathers that are born small program alterations in the next‐generation preimplantation rat embryos. J Nutr 145, 876–883. [DOI] [PubMed] [Google Scholar]
- Männistö T, Vääräsmäki M, Pouta A, Hartikainen A‐L, Ruokonen A, Surcel H‐M, Bloigu A, Järvelin M‐R & Suvanto E (2010). Thyroid dysfunction and autoantibodies during pregnancy as predictive factors of pregnancy complications and maternal morbidity in later life. J Clin Endocrinol Metab 95, 1084–1094. [DOI] [PubMed] [Google Scholar]
- Mcmillen IC & Robinson JS (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85, 571–633. [DOI] [PubMed] [Google Scholar]
- Mele J, Muralimanoharan S, Maloyan A & Myatt L (2014). Impaired mitochondrial function in human placenta with increased maternal adiposity. Am J Physiol Endocrinol Metab 307, E419–E425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettola S, Hartikainen A‐L, Vääräsmäki M, Bloigu A, Ruokonen A, Järvelin M‐R & Pouta A (2013). Offspring's blood pressure and metabolic phenotype after exposure to gestational hypertension in utero. Eur J Epidemiol 28, 87–98. [DOI] [PubMed] [Google Scholar]
- Moisiadis VG & Matthews SG (2014). Glucocorticoids and fetal programming part 1: outcomes. Nature 10, 391–402. [DOI] [PubMed] [Google Scholar]
- Morgan CP & Bale TL (2011). Early prenatal stress epigenetically programs dysmasculinization in second‐generation offspring via the paternal lineage. J Neurosci 31, 11748–11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, Owens JA & Wlodek ME (2009). Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol 587, 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JL (2008). Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35, 730–743. [DOI] [PubMed] [Google Scholar]
- Muralimanoharan S, Guo C, Myatt L & Maloyan A (2015). Sexual dimorphism in miR‐210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int J Obes Relat Metab Disord 39, 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt L, Muralimanoharan S & Maloyan A (2014). Effect of preeclampsia on placental function: influence of sexual dimorphism, microRNA's and mitochondria In Advances in Experimental Medicine and Biology, ed. Zhang L. & Ducsay CA, pp. 133–146. Springer, New York. [DOI] [PubMed] [Google Scholar]
- Nardozza L, Júnior EA, Barbosa MM, Caetano A, Lee D & Moron AF (2012). Fetal growth restriction: current knowledge to the general Obs/Gyn. Arch Gynecol Obstet 286, 1–13. [DOI] [PubMed] [Google Scholar]
- Nayak NR & Giudice LC (2003). Comparative biology of the IGF system in endometrium, decidua, and placenta, and clinical implications for foetal growth and implantation disorders. Placenta 24, 281–296. [DOI] [PubMed] [Google Scholar]
- Ng S‐F, Lin RCY, Laybutt DR, Barres R, Owens JA & Morris MJ (2010). Chronic high‐fat diet in fathers programs β‐cell dysfunction in female rat offspring. Nature 467, 963–966. [DOI] [PubMed] [Google Scholar]
- Ng S‐F, Lin RCY, Maloney CA, Youngson NA, Owens JA & Morris MJ (2014). Paternal high‐fat diet consumption induces common changes in the transcriptomes of retroperitoneal adipose and pancreatic islet tissues in female rat offspring. FASEB J 28, 1830–1841. [DOI] [PubMed] [Google Scholar]
- Nugent BM & Bale TL (2015). The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol 39, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda NB, Intapad S & Alexander BT (2013). Sex differences in the developmental programming of hypertension. Acta Physiol 210, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oni O, Harville E, Xiong X & Buekens P (2015). Relationships among stress coping styles and pregnancy complications among women exposed to Hurricane Katrina. J Obstet Gynecol Neonatal Nurs 44, 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan L, Cuffe JSM, Paravicini TM, Campbell S, Dickinson H, Singh RR, Gezmish O, Black MJ & Moritz KM (2013). Prenatal exposure to dexamethasone in the mouse alters cardiac growth patterns and increases pulse pressure in aged male offspring. PLoS One 8, e69149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleon M (2015). The role of hexosamine biosynthesis and signaling in early development. Adv Exp Med Biol 843, 53–76. [DOI] [PubMed] [Google Scholar]
- Pantaleon M, Tan HY, Kafer GR & Kaye PL (2010). Toxic effects of hyperglycemia are mediated by the hexosamine signaling pathway and o‐linked glycosylation in early mouse embryos. Biol Reprod 82, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik R (2002). Intrauterine growth restriction. Obstet Gynecol 99, 490–496. [DOI] [PubMed] [Google Scholar]
- Reyes‐Castro LA, Rodríguez González GL, Chavira R, Ibáñez C, Lomas‐Soria C, Rodriguez JS, Nathanielsz PW & Zambrano E (2015). Paternal line multigenerational passage of altered risk assessment behavior in female but not male rat offspring of mothers fed a low protein diet. Physiol Behav 140, 89–95. [DOI] [PubMed] [Google Scholar]
- Risnes KR, Romundstad PR, Nilsen TIL, Eskild A & Vatten LJ (2009). Placental weight relative to birth weight and long‐term cardiovascular mortality: findings from a cohort of 31,307 men and women. Am J Epidemiol 170, 622–631. [DOI] [PubMed] [Google Scholar]
- Robinson JS, Kingston EJ, Jones CT & Thorburn GD (1979). Studies on experimental growth retardation in sheep: the effect of removal of endometrial caruncles on fetal size and metabolism. J Dev Physiol 1, 379–398. [PubMed] [Google Scholar]
- Rodgers AB, Morgan CP, Bronson SL, Revello S & Bale TL (2013). Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci 33, 9003–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano T, Hryciw DH, Westcott KT & Wlodek ME (2015). Puberty onset is delayed following uteroplacental insufficiency and occurs earlier with improved lactation and growth for pups born small. Reprod Fertil Dev, doi: 10.1071/RD15151. [DOI] [PubMed] [Google Scholar]
- Rosenfeld CS (2015). Sex‐specific placental responses in fetal development. Endocrinology 156, 3422–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay AS, Sundrani DP, Wagh GN, Mehendale SS & Joshi SR (2015). Regional differences in the placental levels of oxidative stress markers in pre‐eclampsia. Int J Gynaecol Obstet 129, 213–218. [DOI] [PubMed] [Google Scholar]
- Schlegel RN, Cuffe JSM, Moritz KM & Paravicini TM (2015). Maternal hypomagnesemia causes placental abnormalities and fetal and postnatal mortality. Placenta 36, 750–758. [DOI] [PubMed] [Google Scholar]
- Schneider D, Hernández C, Farías M, Uauy R, Krause BJ & Casanello P (2015). Oxidative stress as common trait of endothelial dysfunction in chorionic arteries from fetuses with IUGR and LGA. Placenta 36, 552–558. [DOI] [PubMed] [Google Scholar]
- Seghieri G, Anichini R, De Bellis A & Alviggi L (2002). Relationship between gestational diabetes mellitus and low maternal birth weight. Diabetes Care 25, 1761–1765. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2014). Oxygen sensing, hypoxia‐inducible factors, and disease pathophysiology. Annu Rev Pathol Mech Dis 9, 47–71. [DOI] [PubMed] [Google Scholar]
- Siebel AL, Gallo LA, Guan TC, Owens JA & Wlodek ME (2010). Cross‐fostering and improved lactation ameliorates deficits in endocrine pancreatic morphology in growth‐restricted adult male rat offspring. J Dev Orig Health Dis 1, 234–244. [DOI] [PubMed] [Google Scholar]
- Siebel AL, Mibus A, De Blasio MJ, Westcott KT, Morris MJ, Prior L, Owens JA & Wlodek ME (2008). Improved lactational nutrition and postnatal growth ameliorates impairment of glucose tolerance by uteroplacental insufficiency in male rat offspring. Endocrinology 149, 3067–3076. [DOI] [PubMed] [Google Scholar]
- Singh RR, Cuffe JS & Moritz KM (2012). Short‐ and long‐term effects of exposure to natural and synthetic glucocorticoids during development. Clin Exp Pharmacol Physiol 39, 979–989. [DOI] [PubMed] [Google Scholar]
- Skinner MK (2008). What is an epigenetic transgenerational phenotype?: F3 or F2. Reprod Toxicol 25, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G, Pell JP & Walsh D (2001). Pregnancy complications and maternal risk of ischaemic heart disease: a retrospective cohort study of 129 290 births. Lancet 357, 2002–2006. [DOI] [PubMed] [Google Scholar]
- Spiers JG, Chen H‐JC, Sernia C & Lavidis NA (2015). Activation of the hypothalamic‐pituitary‐adrenal stress axis induces cellular oxidative stress. Front Neurosci 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegers EA, von Dadelszen P, Duvekot JJ & Pijnenborg R (2010). Pre‐eclampsia. Lancet 376, 631–644. [DOI] [PubMed] [Google Scholar]
- Susiarjo M, Xin F, Bansal A, Stefaniak M, Li C, Simmons RA & Bartolomei MS (2015). Bisphenol A exposure disrupts metabolic health across multiple generations in the mouse. Endocrinology 156, 2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgersen CKL & Curran CA (2006). A systematic approach to the physiologic adaptations of pregnancy. Crit Care Nurs Q 29, 2–19. [DOI] [PubMed] [Google Scholar]
- Tran M, Gallo LA, Jefferies AJ, Moritz KM & Wlodek ME (2013). Transgenerational metabolic outcomes associated with uteroplacental insufficiency. J Endocrinol 217, 105–118. [DOI] [PubMed] [Google Scholar]
- Tran M, Gallo LA, Wadley GD, Jefferies AJ, Moritz KM & Wlodek ME (2012). Effect of pregnancy for females born small on later life metabolic disease risk. PLoS One 7, e45188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran M, Young ME, Jefferies AJ, Hryciw DH, Ward MM, Fletcher EL, Wlodek ME & Wadley GD (2015). Uteroplacental insufficiency leads to hypertension, but not glucose intolerance or impaired skeletal muscle mitochondrial biogenesis, in 12‐month‐old rats. Physiol Rep 3, e12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner M & Ozanne S (2010). Mechanisms involved in the developmental programming of adulthood disease. Biochem J 427, 333–347. [DOI] [PubMed] [Google Scholar]
- Weissgerber TL & Wolfe LA (2006). Physiological adaptation in early human pregnancy: adaptation to balance maternal‐fetal demands. Appl Physiol Nutr Metab 31, 1–11. [DOI] [PubMed] [Google Scholar]
- Wen X, Triche EW, Hogan JW, Shenassa ED & Buka SL (2010). Association between placental morphology and childhood systolic blood pressure. Hypertension 57, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigglesworth JS (1974). Fetal growth retardation. Animal model: uterine vessel ligation in the pregnant rat. Am J Pathol 77, 347. [PMC free article] [PubMed] [Google Scholar]
- Williams D (2003). Pregnancy: a stress test for life. Curr Opin Obstet Gynecol 15, 465–471. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Westcott KT, O'Dowd R, Serruto A, Wassef L, Moritz KM & Moseley JM (2005). Uteroplacental restriction in the rat impairs fetal growth in association with alterations in placental growth factors including PTHrP. Am J Physiol Regul Integr Comp Physiol 288, R1620–R1627. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Westcott K, Siebel AL, Owens JA & Moritz KM (2008). Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int 74, 187–195. [DOI] [PubMed] [Google Scholar]
- Yao Y, Robinson AM, Zucchi FC, Robbins JC, Babenko O, Kovalchuk O, Kovalchuk I, Olson DM & Metz GA (2014). Ancestral exposure to stress epigenetically programs preterm birth risk and adverse maternal and newborn outcomes. BMC Med 12, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE, Molina H, Wong KY, Pandey A & Hart GW (2011). The dynamic stress‐induced “O‐GlcNAc‐ome” highlights functions for O‐GlcNAc in regulating DNA damage/repair and other cellular pathways. Amino Acids 40, 793–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterström K, Lindeberg S, Haglund B, Magnuson A & Hanson U (2007). Being born small for gestational age increases the risk of severe pre‐eclampsia. BJOG 114, 319–324. [DOI] [PubMed] [Google Scholar]
- Zhang J, Troendle JF & Levine RJ (2001). Risks of hypertensive disorders in the second pregnancy. Paediatr Perinat Epidemiol 15, 226–231. [DOI] [PubMed] [Google Scholar]
- Zhang S, Regnault T, Barker P, Botting K, McMillen I, McMillan C, Roberts C & Morrison J (2015). Placental adaptations in growth restriction. Nutrients 7, 360–389. [DOI] [PMC free article] [PubMed] [Google Scholar]