

Abstract
Members of the myocardin family bind to the transcription factor serum response factor (SRF) and act as coactivators controlling genes of relevance for myogenic differentiation and motile function. Binding of SRF to DNA is mediated by genetic elements called CArG boxes, found often but not exclusively in muscle and growth controlling genes. Studies aimed at defining the full spectrum of these CArG elements in the genome (i.e. the CArGome) have in recent years, unveiled unexpected roles of the myocardin family proteins in lipid and glucose homeostasis. This coactivator family includes the protein myocardin (MYOCD), the myocardin‐related transcription factors A and B (MRTF‐A/MKL1 and MRTF‐B/MKL2) and MASTR (MAMSTR). Here we discuss growing evidence that SRF‐driven transcription is controlled by extracellular glucose through activation of the Rho‐kinase pathway and actin polymerization. We also describe data showing that adipogenesis is influenced by MLK activity through actions upstream of peroxisome proliferator‐activated receptor γ with consequences for whole body fat mass and insulin sensitivity. The recently demonstrated involvement of myocardin coactivators in the biogenesis of caveolae, Ω‐shaped membrane invaginations of importance for lipid and glucose metabolism, is finally discussed. These novel roles of myocardin proteins may open the way for new unexplored strategies to combat metabolic diseases such as diabetes, which, at the current incidence, is expected to reach 333 million people worldwide by 2025.
This review highlights newly discovered roles of myocardin‐related transcription factors in lipid and glucose metabolism as well as novel insights into their well‐established role as mediators of stretch‐dependent effects in smooth muscle. As co‐factors for serum response factor (SRF), MKLs regulates transcription of genes involved in the contractile function of smooth muscle cells. In addition to mechanical stimuli, this regulation has now been found to be promoted by extracellular glucose levels in smooth muscle. Recent reports also suggest that MKLs can regulate a subset of genes involved in the formation of lipid‐rich invaginations in the cell membrane called caveolae. Finally, a potential role of MKLs in non‐muscle cells has been discovered as they negatively influence adipocyte differentiation.
Discovery and domain organization of myocardin family coactivators
Cell‐linage specification, and subsequently formation of a vast diversity of cell types, requires a coordinated interplay between intracellular and extracellular signalling events that converge on genomic activation. This coordination is also essential for proper cell function and requires the association of sequence‐specific DNA‐binding proteins like transcription factors and their co‐regulators to cognate sites on DNA. Co‐regulators are not generally considered to bind DNA directly; instead they help provide specificity of cellular transcription by interacting with DNA‐binding proteins (Spiegelman & Heinrich, 2004).
The Myocardin family of transcriptional coactivators (Fig. 1) provides a classic example of a group of proteins that control gene expression without directly binding to DNA. This family includes the protein myocardin (MYOCD), the myocardin‐related transcription factors A and B (MRTF‐A/MKL1 and MRTF‐B/MKL2) and MASTR (MAMSTR) (Wang et al. 2001, 2002). All members except MASTR bind to serum response factor (SRF; blue globules in Fig. 1), a highly interactive transcription factor controlling gene expression in a wide range of cell types. SRF is a member of the MADS family of transcription factors that share a conserved MADS‐box domain involved in regulating binding to DNA. Binding of SRF to DNA is mediated by A/T‐rich regulatory elements, known as CArG boxes, which are present in promoters and intronic sequences of target genes (Miano, 2003; Sun et al. 2006).
Figure 1. Domain organization of myocardin family coactivators and association with DNA‐binding transcription factors .
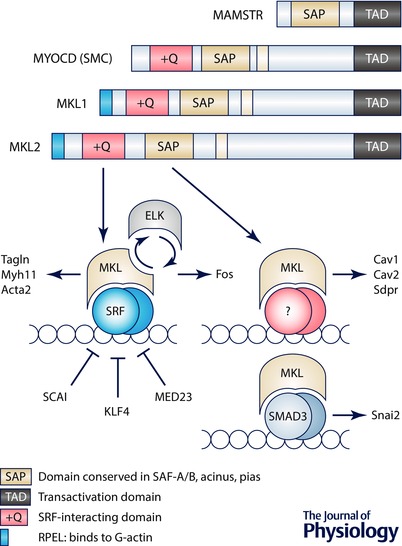
The key at the top shows the different functional domains. Below this are simplified schematic representations of the myocardin family proteins. Two splice variants of myocardin (MYOCD) exist and the smooth muscle (SMC) variant is shown. Lower left, binding of MKL1 to the serum response factor (SRF) occurs in competition with ternary complex factors such as ELK. The MKL–SRF complex controls cytoskeletal genes such as TAGLN, MYL11 and ACTA2, whereas ELK–SRF controls genes of relevance for growth, such as FOS. Several proteins may influence MKL–SRF complex formation, including SCAI, KLF4 and MED23. Some effects of myocardin family coactivators appear to be mediated by the SAP domain. Those transcriptional influences are funnelled through direct binding to DNA or, alternatively, and as illustrated, via an as yet unidentified transcription factor. Here we discuss data showing that CAV1, CAV2 and SDPR depend on the SAP domain of myocardin. One study suggested that MKL may exert effects through SMAD3 (lower right).
Myocardin was discovered in a bioinformatics‐based screen (Wang et al. 2001). Whereas myocardin expression is largely confined to the cardiovascular and gastrointestinal systems, MKL1 and MKL2 are expressed in a wide range of embryonic and adult tissues (Wang et al. 2002). MEF2‐activating SAP transcriptional regulator (MASTR or MAMSTR) is a more divergent member of the myocardin family that is expressed in skeletal muscle and in the adrenal gland. It encompasses a unique peptide sequence that allows association with myocyte enhancer factor 2 (MEF2), a MADS‐box transcription factor related to SRF that is involved in the regulation of muscle and growth‐associated gene expression (Black & Olson, 1998; Creemers et al. 2006). MASTR lacks an SRF‐binding domain (orange domain named +Q in Fig. 1) and hence, does not interact with SRF (Wang et al. 2001).
As schematically depicted in Fig. 1, myocardin and MKL share homology in several domains that facilitate their function as coactivators of SRF. These include a basic and a glutamine‐rich domain (+Q, orange), which is required for binding to SRF, and a transcriptional activation domain (TAD), which is important for stimulating SRF activity. Furthermore, all family members encompass a highly conserved SAF‐A, Acinus, PIAS (SAP, green in Fig. 1) domain, implicated in communicating specificity to target gene activation and chromatin remodelling (Aravind & Koonin, 2000). A particularly strongly conserved domain, located at the N terminus, consists of a series of motifs known as RPELs (blue in Fig. 1). The RPEL repeats are central for the ability of MKLs to bind monomeric globular actin (G‐actin), and thus for their retention in the cytosol (Miralles et al. 2003). Nuclear MKL translocation is mediated by activation of the Rho‐actin signalling pathway that releases MKL from monomeric G‐actin as a consequence of actin polymerization, leading to transportation into the nucleus where it stimulates SRF activity (Sotiropoulos et al. 1999; Vartiainen et al. 2007). The association of MKL with SRF potently stimulates the transcription of a group of genes involved in myogenic differentiation and cytoskeletal organization (Blank et al. 1992; Solway et al. 1995; Li et al. 1997; Esnault et al. 2014). By contrast, myocardin is nuclear and constitutively active due to lack of RPEL domains, rendering it insensitive to changes in actin dynamics (Miralles et al. 2003).
SRF versus SAP domain targets
As ‘coactivators’, the myocardin family interact with at least three factors. SRF (Fig. 1) is the archetypal and most widely studied factor (Wang et al. 2001; Posern & Treisman, 2006; Olson & Nordheim, 2010), but interactions with SMAD3 (Qiu et al. 2005) and MEF2 (Creemers et al. 2006) have also been described. An intriguing domain that is conserved in all myocardin family proteins is the SAP domain, and some transcriptional effects depend on the SAP domain and occur independently of SRF. An example is the matrix protein tenascin C, which is induced by mechanical tension via MKL1 independently of SRF (Asparuhova et al. 2011). We recently discovered that caveolins (CAV1, CAV2, CAV3) and cavin‐2 (SDPR) are induced by myocardin and that knock‐down of SRF had little effect on this transcriptional response (Krawczyk et al. 2015). There are many conceivable ways in which the SAP domain may act. One is via binding to an unidentified transcription factor (Fig. 1). Another is through direct interaction with DNA. Available evidence is insufficient to distinguish between these possibilities, but it is clear that the SAP domain is important, if not critical, for the function of myocardin family coactivators.
A ‘subtractive transcript profiling screen’ was recently devised to distinguish between SRF‐dependent and SAP‐dependent targets using mammary epithelial cells (Gurbuz et al. 2014). These authors overexpressed full length MKL1 and domain‐deletion mutants (mutB: unable to bind SRF; ΔSAP: lacking SAP domain) and performed microarrays. By subtracting the ΔSAP and mutB gene lists, respectively, from the MKL1 full‐length gene list, groups of SRF‐dependent (blue circle in Fig. 2 A) and SAP‐dependent (red circle in Fig. 2 A) targets were defined. As expected, a number of targets depended on both domains. In Fig. 2 A targets of potential relevance for lipid metabolism are listed within each group, some of which are discussed in greater detail below. It is important to point out that independent ChiP‐Seq analyses have indicated that only a low fraction (5%) of MKL1 binding to DNA occurs independently of SRF (Esnault et al. 2014). This is much less than the fraction of targets that are SAP domain‐dependent (and thus independent of an important SRF‐binding domain, Fig. 2 A). Whether this apparent discrepancy is due to use of different cell types, to the fact that overexpression versus serum stimulation was used for MKL activation, or to use of different experimental strategies is not currently known.
Figure 2. SRF‐ versus SAP‐dependent targets .
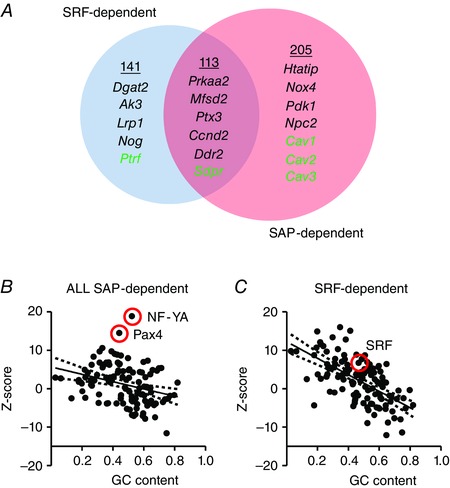
Gene targets of myocardin coactivators may broadly be defined as SRF‐dependent (blue circle), SAP‐dependent (red circle) and those that depend on both domains. A recent subtractive profiling approach (Gurbus et al. 2014) was used to define these groups. Targets of relevance for lipid homeostasis in each group are indicated in A. Panels B and C show transcription factor binding site analyses using oPOSSUM. In B, all SAP‐dependent targets identified by Gurbus et al. were entered into the analysis and in C all SRF‐dependent targets were used. Enrichment (Z‐scores) of binding sites for different transcription factors are shown relative to promoter GC content.
Despite the growing amount of transcriptomic data available, it has been difficult to identify a specific transcription factor that mediates the effects of the SAP domain. Results of analyses of transcription factor binding sites using oPOSSUM (http://opossum.cisreg.ca/) are shown in Fig. 2 B and C. When all SAP‐dependent targets (Gurbuz et al. 2014) are entered into the analysis (Fig. 2 B) two transcription factors deviate considerably from the overall relationship between Z‐score and promoter GC content: NF‐YA and Pax4. NF‐YA often stands out in such analyses, as the sequence motif to which it binds is exceptionally frequent. Pax4 shows little promise as this factor is primarily expressed in testis and intestine (http://www.gtexportal.org/), making it a less likely candidate. This leaves approximately 30 potential transcription factors that could be involved. However, it may be difficult to further pinpoint the transcription factor mediating the effects of the SAP domain by only using in silico analyses. This is further illustrated by the fact that when SRF‐dependent gene targets (Gurbuz et al. 2014) are entered into the analysis, SRF, albeit deviating from the overall relationship between GC content and Z‐score, clusters with at least 30 other factors (Fig. 2 C). This emphasizes a need for direct experimental approaches to determine how the SAP domain mediates its transcriptional effects.
Selected myocardin family targets of potential relevance for lipid metabolism
As discussed below, MKL1 expression is reduced by adipogenic cues. Paradoxically, a number of the MKL targets identified (Gurbuz et al. 2014) would nonetheless seem to promote adipogenesis. One example of an MKL‐regulated and SRF‐dependent target is diacylglycerol O‐acyltransferase 2 (Dgat2), which plays a key role in triglyceride synthesis (Stone et al. 2004). Another example in this group is Lrp1, which is a lipoprotein receptor that belongs to the LDL receptor family. Lrp1 has been found to be important for adipocyte differentiation and is upregulated in adipocytes of obese mice (Masson et al. 2009). Moreover, the MKL1‐target Noggin is involved in differentiation of mesenchymal stem cells (MSC) to adipocytes. By direct binding to bone morphogenetic protein (BMP), Noggin inhibits osteoblast and promotes adipocyte differentiation (Sawant et al. 2012).
Several SAP‐dependent MKL1 targets of potential relevance for adipogenesis have also been identified, including phosphoinositide‐dependent protein kinase‐1 (PDK1) and Major facilitator superfamily domain‐containing protein 2 (Mfds2) (Gurbuz et al. 2014) (Fig. 2 A). PDK1 is an enzyme involved in the insulin signalling pathway by activating PKB/Akt. PDK1 is also involved in adipocyte differentiation, by activating peroxisome proliferator‐activated receptor γ (PPARγ) (Yin et al. 2006). Mfsd2 belongs to the ‘major facilitator superfamily’ of transporters and is highly expressed in brain endothelial cells where it contributes to the blood–brain barrier function (Ben‐Zvi et al. 2014). Mfsd2a has been directly demonstrated to transport lysophosphatidylcholine in brain endothelial cells, and mice lacking Mfsd2a have smaller brains and reduced omega‐3 fatty acid content (Nguyen et al. 2014). A recent study has shown a central role of MKL/SRF signalling in blood–brain barrier regulation (Weinl et al. 2015) and one may therefore speculate that Mfsd2a is a relevant target of MKL1 in this regard.
Induction of caveolae by myocardin family coactivators
Caveolae are 50–100 nm cave‐like structures in the cell membrane that are enriched in cholesterol and sphingolipids. The core protein components of caveolae are caveolins (CAV1, CAV2, CAV3) and cavins (PTRF, SDPR, PRKCDBP, MURC). Caveolins are 20–24 kDa proteins embedded in the inner leaflet of the plasma membrane and whose N‐ and C‐termini are intracellular. Caveolins bind cholesterol and have an extreme propensity to oligomerize. It is thought that clusters of 140 or so caveolin monomers are required for formation of one caveola. Cavins on the other hand are cytosolic proteins that form homo‐ and heterotrimers (Kovtun et al. 2015) that associate with caveolins at the membrane.
Caveolae are involved in a number of cellular processes, including lipid metabolism and insulin signalling (Stralfors, 2012; Echarri & Del Pozo, 2015; Kovtun et al. 2015). These critical functions are underscored by rare congenital diseases caused by mutations in caveolins and cavins, the structural proteins of caveolae, that cause lipodystrophy in combination with a host of other changes (Kim et al. 2008; Hayashi et al. 2009; Rajab et al. 2010). Immunoelectron microscopy has demonstrated that insulin receptors are concentrated in caveolae (Gustavsson et al. 1999), and, in keeping with a functional role of this localization, insulin resistance has been reported in both caveolin‐3 (CAV3) (Oshikawa et al. 2004) and caveolin‐1 (CAV1) (Cohen et al. 2003) knockout mice. The latter study demonstrated that insulin resistance was due to destabilization of insulin receptor‐β subunit in adipose tissue. Recent work also demonstrated that caveolin‐1 is targeted by two microRNAs (miR‐103 and miR‐107) that are induced by obesity in mice and humans (Trajkovski et al. 2011). Overexpression of miR‐103/107 caused insulin resistance and this effect was absent in CAV1 knockout mice. This supports the view that CAV1 may be targeted to ameliorate insulin resistance. Obesity‐induced CAV1 repression is not a universal finding, however, and it has been reported that CAV1 increases with some diet protocols (Gomez‐Ruiz et al. 2010).
Because CAV1 and CAV3, and SDPR (cavin‐2) and PRKCDBP (cavin‐3), can substitute for each other, caveolae with distinct protein compositions exist in different cell types (Bastiani et al. 2009). Indeed, the ratio between SDPR and PRKCDBP mRNAs differs by at least one order of magnitude between visceral adipose tissue, where the ratio is high, and the aorta, where it is low (http://www.gtexportal.org/). CAV3 to CAV1 expression ratios vary even more extremely, with the highest ratio seen in skeletal muscle. Tissue‐specific transcriptional mechanisms are considered to underlie such expression differences but their nature is poorly defined. Motivated by the presence of SRF‐binding CArG‐boxes at the PTRF (cavin‐1) and SDPR (cavin‐2) loci, we recently addressed the hypothesis that myocardin family coactivators regulate cavins and caveolins (Krawczyk et al. 2015). Regulation by both myocardin and MKL1 was observed for both caveolins and cavins (induction of caveolae by myocardin is graphically illustrated in Fig. 3), suggesting that myocardin family coactivators may constitute something akin to a main switch for caveolae biogenesis. A promoter assay indicated that proximal sequences are involved in the case of CAV1 regulation. However, SRF silencing had little effect, suggesting involvement of the SAP domain. Given the function of caveolae in lipid homeostasis and insulin sensitivity, one would predict that regulation of caveolae genes is one way by which myocardin family coactivators could influence lipid homeostasis. Further work is, however, required to test if this regulatory mechanism is operative in adipose tissue and, if so, how it works together with PPARγ, which also activates the CAV1 gene (Matsusue et al. 2003). In fact, as discussed in further detail below, MKL1 and PPARγ are likely to be mutually antagonistic in adipocytes.
Figure 3. Myocardin promotes formation of caveolae .
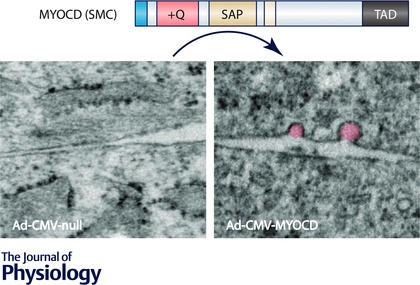
Electron micrographs from cells infected with control adenovirus (null, left) and adenovirus encoding myocardin (MYOCD, right) are shown below the domain diagram for myocardin. Caveolae (highlighted in pink in the micrograph to the right), which are omega‐shaped organelles of relevance for lipid metabolism and mechanoprotection, are more numerous in cells transduced with myocardin (Krawczyk et al. 2015). SRF knockdown experiments and promoter analysis suggested that this effect is mediated by the SAP domain.
A newly discovered role of MKL1 in adipogenesis
Adipocyte differentiation (or adipogenesis) is a tightly orchestrated process including various transcription factors, of which PPARγ is considered the master regulator (Rosen et al. 2000). Expression of PPARγ is controlled by several pro‐adipogenic factors including CCAAT‐enhancer binding proteins (C/EBPs) and Kruppel‐like factors (KLFs) as well as anti‐adipogenic factors like GATA‐binding (GATA2/3) and forkhead box (FOXO) proteins. Transcriptional coactivators play a central role in regulation of PPARγ. For example, histone acetyltransferases (HATs) modify the chromatin structures (Grunstein, 1997), whereas others (like PGC‐1α and p160/SRC family) participate by forming PPARγ‐containing protein complexes that influence adipocyte differentiation.
Adipogenesis requires a drastic remodelling of cell morphology, where cells change their shape from elongated to spherical. One may envision that this cell shape change has an impact on the actin cytoskeleton and consequently the activity of myocardin family coactivators. A first observation in a recent study on adipogenesis was that the cell rounding associated with a disruption of actin stress fibres, and that interventions stabilizing the actin cytoskeleton suppress adipogenesis. The authors (Nobusue et al. 2014) then went on to show that the increased G‐actin pool caused by stress fibre disassembly caused cytosolic sequestration of MKL1. This was associated with increased expression of PPARγ and its target genes (Fabp4, Cebpa1, Plin1 and Slc2a4). Interestingly, suppression of MKL1 expression triggered spontaneous adipocyte differentiation without adipogenic stimuli. Overexpression of PPARγ moreover decreased the level of MKL1, establishing a mutually antagonistic relationship between these transcription factors. Earlier work identified enrichment of SRF‐binding sites in regions undergoing chromatin remodelling during adipogenesis, and demonstrated that overexpression of SRF directly antagonizes adipogenesis (Mikkelsen et al. 2010). In all, these findings suggested that adipocyte differentiation is initiated by a disruption of actin stress fibres, leading to cytosolic retention of MKL1, subsequent PPARγ expression, and lipid storage, as illustrated in the upper part of Fig. 4.
Figure 4. MKL1 repression controls adipogenesis and browning of fat .
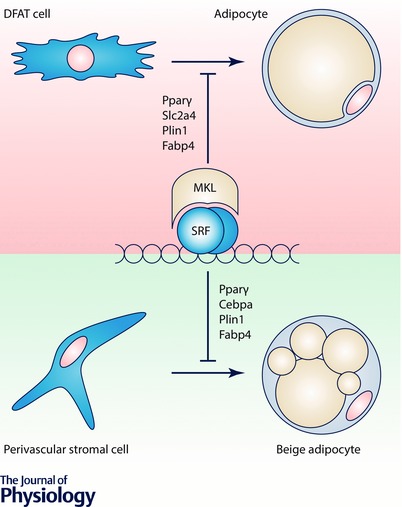
Recent studies have shown that MKL1 represses Pparγ, a key adipogenic factor. Adipogenic cues cause rounding of DFAT cells (Nobusue et al. 2014) associated with depolymerization of actin and resultant MKL1 inhibition, leading to de‐repression of Pparγ. Pparγ target genes (Slc2a4, Plin1, Fabp4) are subsequently transcribed and adipogenesis ensues. Another study showed that MKL1 knockout mice develop increased ‘brite’ or ‘beige’ fat depots with increased UCP1 expression and have a favourable metabolic profile (McDonald et al. 2015). These effects were mediated by BMP7 and p38MAPK‐dependent commitment of mesenchymal stem cells to the UCP1+ lineage via suppression of ROCK and MKL1 activity.
Also, of considerable interest, the MED23 subunit of the mediator complex, a protein complex which regulates cellular processes and gene transcription, plays a role for directing cell linage commitments of mesenchymal cells into adipocytes or SMCs (Yin et al. 2012). Expression of MED23 promoted binding of ELK1 to SRF, leading to expression of genes associated with adipogenesis, while MED23 deficiency destabilized the ELK–SRF complex, favouring MKL1–SRF binding and expression of SMC‐related genes. Thus MED23 performs a balancing act in terms of directing differentiation of mesenchymal cells into either SMCs or adipocytes.
In another recent study, MKL1 was shown to regulate commitment of mesenchymal stem cells (MSCs) into beige adipocytes in white adipose tissue through a bone morphogenic protein 7 (BMP7)–Rho‐associated protein kinase (ROCK)‐dependent pathway (McDonald et al. 2015). BMP7 suppressed ROCK activity causing cell rounding and this induced browning. Transforming growth factor β (TGFβ), on the other hand, promoted formation of actin filaments and promoted smooth muscle cell (SMC) differentiation (McDonald et al. 2015). Pharmacological ROCK inhibition enhanced the BMP7‐induced increase of Fabp4 and PPARγ. Treatment with the MKL inhibitor CCG‐1423 moreover enhanced adipocyte differentiation and uncoupling protein 1 (UCP1) expression without affecting the actin filament arrangement, arguing that BMP7 stimulates brown adipocyte differentiation in a ROCK and MKL/SRF‐dependent fashion. Interestingly, reduced body weight, increased multilocular adipocytes, smaller adipocyte size and decreased expression of angiotensinogen (Atg) argued for increased browning of adipose progenitors in MKL1 knockout mice (McDonald et al. 2015). MKL1 deficiency moreover protected against high fat diet‐induced obesity and insulin resistance, and this was associated with increased energy expenditure. In all, these findings argue for favourable metabolic effects of reduced adipocyte MKL1 activity via effects on adipocyte browning (illustrated in the lower part of Fig. 4).
The studies by Nobusue et al. (2014) and McDonald et al. (2015) point to important roles of MKL1 in adipocytes, but they also raise novel questions. What, for instance, is the role of MKL2 in adipocytes and how can positive regulation of mediators in adipogeneis (Dgat2, Noggin, etc.) be reconciled with MKL1 suppression as adipogenesis commences. One may speculate that a high MKL2 expression in mature adipocytes might allow for actin‐dependent regulation of gene expression later in life.
MKL as glucose sensor
Diabetes is strongly associated with hypertension, which is one of the major risk factors for cardiovascular disease (Chobanian et al. 2003). The cause of this effect is, at least in part, due to increased vascular smooth muscle contractility as demonstrated in a number of studies using hyperglycaemic animal models, as well as in diabetic patients (White & Carrier, 1988; Abebe et al. 1990; Fleischhacker et al. 1999; Ungvari et al. 1999; Guo et al. 2005). One of the underlying mechanisms is increased activation of Rho‐kinase in both endothelial cells (Rikitake & Liao, 2005) and smooth muscle (Xie et al. 2006, 2010). In smooth muscle, activation of the Rho‐kinase pathway results in a calcium sensitization, a phenomenon where phosphorylation of the myosin light chains is increased via inhibition of the light chain phosphatase (Uehata et al. 1997). This mechanism can mediate a rapid increase in smooth muscle contractility under hyperglycaemic conditions. However, elevated Rho‐kinase activation may also result in transcriptional effects in vascular smooth muscle by promoting actin polymerization and nuclear translocation of MKL1/2.
In a recent study we found that long‐term (> 1 week) glucose‐induced activation of the Rho‐kinase signalling pathway subsequently results in an increased F/G‐actin ratio and elevated expression of contractile markers in cultured mouse aortic smooth muscle cells (Fig. 5) (Thi Hien et al. 2016). The effects of glucose on smooth muscle differentiation were completely blocked by MKL1/2 siRNA or CCG‐1423 (Thi Hien et al. 2016), an inhibitor of MKL1/2 nuclear translocation (Hayashi et al. 2014). High glucose also reduced the expression of KLF4, a known repressor of myocardin expression (Liu et al. 2005). Furthermore, we found that human mammary arteries from diabetic patients and mesenteric arteries from hyperglycaemic mice expressed significantly higher levels of several contractile smooth muscle markers (Thi Hien et al. 2016). Another recent study demonstrated elevated levels of the MKL‐regulated, smooth muscle‐enriched microRNAs miR‐143/145 in saphenous veins from diabetic patients (Riches et al. 2014). Together, these results suggest that hyperglycaemia can activate MKL1/2‐dependent transcription in smooth muscle. However, in skeletal muscle, activation of the Rho–actin–MKL1/SRF pathway in normoglycaemic type 2 diabetic patients has been demonstrated to be linked to insulin resistance, suggesting that additional factors may be involved in MKL1/2‐activation in diabetes (Jin et al. 2011).
Figure 5. Glucose‐induced changes in the G‐ to F‐actin ratio leads to MKL activation .
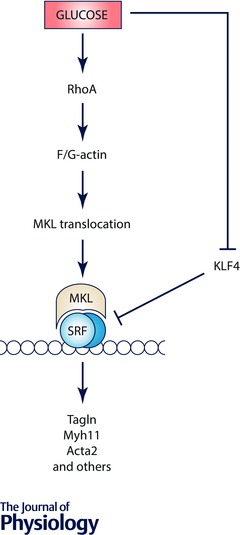
Schematic illustration of MKL1 activation by high extracellular glucose via RhoA/ROCK and changes in the F‐ to G‐actin ratio. MKL1 and MKL2 have RPEL motifs that allow for binding to G‐actin and that confer a cytosolic localization. When actin is polymerized, MKLs dissociate from actin and move into the nucleus. In addition, high glucose somehow reduces Klf4. Klf4 is a repressor of myocardin expression and impairs binding of SRF to CArG boxes via GC‐rich repressor elements. In this manner, high glucose potently stimulates SRF target gene expression in smooth muscle and in diabetic nephropathy.
Several reports have suggested that smooth muscle cells exposed to hyperglycaemic or diabetic conditions undergo a phenotypic shift towards an osteogenic phenotype (Towler et al. 1998). This is mainly defined by an increased osteopontin expression, which is stimulated by hyperglycaemia via the glucose‐sensitive transcription factor nuclear factor of activated T cells (NFAT) (Nilsson‐Berglund et al. 2010). Interestingly, glucose‐induced osteopontin expression is promoted by PKC/Rho‐kinase activation (Kawamura et al. 2004), which is activated by multiple mechanisms including increased glucose metabolism via the hexosamine biosynthetic pathway and formation of advanced glycation end products (Schleicher & Weigert, 2000; Thi Hien et al. 2016). In breast cancer cells, MKL1 can directly activate the transcription of osteopontin suggesting that similar mechanisms can regulate osteogenic and contractile smooth muscle differentiation. However, despite similarities in the regulatory mechanisms, these transcriptional programmes are often activated in different situations, suggesting a more complex control, possibly including epigenetic mechanisms.
Chronic exposure to high glucose is strongly associated with microvascular complications of diabetes, including diabetic nephropathy, retinopathy and neuropathy. Several recent studies have established a link between the epigenetic machinery and hyperglycaemia‐induced cellular dysfunction (Brasacchio et al. 2009; Villeneuve & Natarajan, 2010; Villeneuve et al. 2010; Zhong & Kowluru, 2013; De Marinis et al. 2016). One of the hallmarks of diabetic nephropathy and of a variety of chronic kidney diseases is the accelerated synthesis of extracellular matrix leading to tubulointerstitial fibrosis. This is a well‐orchestrated epithelial‐to‐mesenchymal transition (EMT) process that involves the down‐regulation of the epithelial programme, the activation of the mesenchymal–fibrogenic programme and finally the activation of the myogenic programme (Zeisberg & Neilson, 2010). Interestingly, suppressor of cancer cell invasion (SCAI), which had been identified as a negative regulator of MKL1 in the context of cancer (Brandt et al. 2009), was shown to act also as an endogenous inhibitor of SMA expression and renal fibrosis (Fintha et al. 2013). More recently, Xu et al. showed that MKL1 expression was induced in the kidney by high glucose and synergized with glucose to promote the transcription of collagen type I, the major component of extracellular matrix. The authors elegantly demonstrated that MKL1 recruited the histone acetyltransferase p300 and WDR5, a key component of the histone H3K4 methyltransferase complex to collagen promoters and engaged them in transcriptional activation (Xu et al. 2015). That study portrays MKL1 as a glucose‐responsive regulator of renal fibrosis and suggests that targeting the epigenetic machinery associated with MKL1 might be an attractive therapeutic approach for the treatment of diabetic nephropathy.
Myocardin family coactivators as mechanosensors
Building on the prior demonstration (Sotiropoulos et al. 1999) that actin dynamics controls SRF activity, Treisman and colleagues demonstrated in 2003 (Miralles et al. 2003) that this involves MKL1. Since the actin cytoskeleton is ideally suited to sense and transmit mechanical forces, MKL1 constitutes an appealing nodal point in the conversion of mechanical force to a transcriptional response. Mechanical stretch of the vascular wall promotes the expression of smooth muscle markers that are regulated by the myocardin family (Zeidan et al. 2003, 2004; Albinsson et al. 2004). This transcriptional regulation requires Rho‐activation and an increased polymerization of actin filaments suggesting involvement of MKL1/2 rather than myocardin (Albinsson et al. 2004; Albinsson & Hellstrand, 2007). The earliest demonstration, to our knowledge, that MKL1 translocates to the nucleus as a consequence of pulling forces in vivo was made in Drosophila (Somogyi & Rorth, 2004). This finding has subsequently been confirmed in numerous mammalian cell types (e.g. Cui et al. 2015, and reviewed in Mendez & Janmey, 2012). Recent in vivo evidence for a role of MKL1 in ‘mechano‐transcription’ coupling is the exclusion of MKL1 from muscle cell nuclei following denervation (Collard et al. 2014). In that model, reciprocal manipulation of MKL1 protects and worsens, respectively, the ensuing muscle atrophy. Two recent studies have fuelled the concept that caveolae are mechanoprotective organelles that can flatten in response to membrane forces and provide a membrane reservoir that prevents membrane rupture (Cheng et al. 2015; Lo et al. 2015). In our view, it makes perfect sense that MKL1, which responds to mechanical forces, control caveolae genes (Krawczyk et al. 2015), and hence upregulates mechanoprotective systems. Such adaptation would prime cellular defences against future mechanical insult.
Concluding remarks
Studies on myocardin family proteins were originally focused on the cardiovascular system due to the prominent cardiovascular phenotypes of corresponding transgenic mice. However, recent work has uncovered novel and unexpected roles of myocardin family coactivators in adipogenesis and adipocyte browning. This effect is funnelled, in part, through repression of PPARγ. Paradoxically, a number of MKL targets would seem to promote adipogenesis, while MKL1 itself is downregulated during adipocyte differentiation. A unique role of MKL2 in adipocytes may reconcile these disparate findings. Another research frontier relates to formation of the blood–brain barrier. Indeed, caveolae and Mfsd2a, a lysophosphatidyl transporter, may be at a crossroads between blood–brain barrier regulation and lipid turnover. MKL1 and MKL2 moreover respond to both metabolic and mechanical cues, transforming these into genomic responses. This is likely to be of relevance for several disease states, including hypertension and diabetic nephropathy.
Additional information
Competing interests
The authors have declared that no competing interests exist
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the Swedish Research Council (No. 2014‐3352 to M.F.G., No. 2014‐3326 to K.Sw., No. 2013‐3542 to K.St., No. 2012‐2197 to S.A.), the Novo Nordisk Foundation (No. 16144 to S.A.), the Crafoord Foundation (C.R. and S.A.), the Swedish Heart and Lung Foundation (No. 20130451 and No. 20120298 to S.A.), the Royal Physiographic Society (S.A.), the Per‐Eric and Ulla Schyberg Foundation (S.A.) and the Swedish Diabetes Foundation (K.St.)
Biography
Sebastian Albinsson’s group, where Azra Alajbegovic is a PhD student, focuses on the role of microRNAs and MRTF transcription factors for the regulation of smooth muscle function. A major goal of this group is to clarify the mechanism underlying mechanotransduction in the vascular wall. Maria Gomez’s research focuses on excitation–transcription coupling mechanisms in the context of diabetes and cardiovascular disease. Karin Stenkula’s research group is centred on glucose transport and protein trafficking in adipocytes. Karl Swärd’s research group, which includes Catarina Rippe, specializes in vascular physiology with emphasis on the regulation and function of caveolae. The groups are situated at the Biomedical Center in Lund or at the Clinical Research Center in Malmö (Gomez group).

References
- Abebe W, Harris KH & MacLeod KM (1990). Enhanced contractile responses of arteries from diabetic rats to α1‐adrenoceptor stimulation in the absence and presence of extracellular calcium. J Cardiovasc Pharmacol 16, 239–248. [DOI] [PubMed] [Google Scholar]
- Albinsson S & Hellstrand P (2007). Integration of signal pathways for stretch‐dependent growth and differentiation in vascular smooth muscle. Am J Physiol Cell Physiol 293, C772–C782. [DOI] [PubMed] [Google Scholar]
- Albinsson S, Nordstrom I & Hellstrand P (2004). Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J Biol Chem 279, 34849–34855. [DOI] [PubMed] [Google Scholar]
- Aravind L & Koonin EV (2000). SAP – a putative DNA‐binding motif involved in chromosomal organization. Trends Biochem Sci 25, 112–114. [DOI] [PubMed] [Google Scholar]
- Asparuhova MB, Ferralli J, Chiquet M & Chiquet‐Ehrismann R (2011). The transcriptional regulator megakaryoblastic leukemia‐1 mediates serum response factor‐independent activation of tenascin‐C transcription by mechanical stress. FASEB J 25, 3477–3488. [DOI] [PubMed] [Google Scholar]
- Bastiani M, Liu L, Hill MM, Jedrychowski MP, Nixon SJ, Lo HP, Abankwa D, Luetterforst R, Fernandez‐Rojo M, Breen MR, Gygi SP, Vinten J, Walser PJ, North KN, Hancock JF, Pilch PF & Parton RG (2009). MURC/Cavin‐4 and cavin family members form tissue‐specific caveolar complexes. J Cell Biol 185, 1259–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H & Gu C (2014). Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature 509, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL & Olson EN (1998). Transcriptional control of muscle development by myocyte enhancer factor‐2 (MEF2) proteins. Annu Rev Cell Dev Biol 14, 167–196. [DOI] [PubMed] [Google Scholar]
- Blank RS, McQuinn TC, Yin KC, Thompson MM, Takeyasu K, Schwartz RJ & Owens GK (1992). Elements of the smooth muscle α‐actin promoter required in cis for transcriptional activation in smooth muscle. Evidence for cell type‐specific regulation. J Biol Chem 267, 984–989. [PubMed] [Google Scholar]
- Brandt DT, Baarlink C, Kitzing TM, Kremmer E, Ivaska J, Nollau P & Grosse R (2009). SCAI acts as a suppressor of cancer cell invasion through the transcriptional control of β1‐integrin. Nat Cell Biol 11, 557–568. [DOI] [PubMed] [Google Scholar]
- Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, Calkin AC, Brownlee M, Cooper ME & El‐Osta A (2009). Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene‐activating epigenetic marks that coexist on the lysine tail. Diabetes 58, 1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JP, Mendoza‐Topaz C, Howard G, Chadwick J, Shvets E, Cowburn AS, Dunmore BJ, Crosby A, Morrell NW & Nichols BJ (2015). Caveolae protect endothelial cells from membrane rupture during increased cardiac output. J Cell Biol 211, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr & Roccella EJ (2003). The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 289, 2560–2572. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE & Lisanti MP (2003). Caveolin‐1‐deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol 285, C222–C235. [DOI] [PubMed] [Google Scholar]
- Collard L, Herledan G, Pincini A, Guerci A, Randrianarison‐Huetz V & Sotiropoulos A (2014). Nuclear actin and myocardin‐related transcription factors control disuse muscle atrophy through regulation of Srf activity. J Cell Sci 127, 5157–5163. [DOI] [PubMed] [Google Scholar]
- Creemers EE, Sutherland LB, Oh J, Barbosa AC & Olson EN (2006). Coactivation of MEF2 by the SAP domain proteins myocardin and MASTR. Mol Cell 23, 83–96. [DOI] [PubMed] [Google Scholar]
- Cui Y, Hameed FM, Yang B, Lee K, Pan CQ, Park S & Sheetz M (2015). Cyclic stretching of soft substrates induces spreading and growth. Nat Commun 6, 6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marinis Y, Cai M, Bompada P, Atac D, Kotova O, Johansson ME, Garcia‐Vaz E, Gomez MF, Laakso M & Groop L (2016). Epigenetic regulation of the thioredoxin‐interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int 89, 342–353. [DOI] [PubMed] [Google Scholar]
- Echarri A & Del Pozo MA (2015). Caveolae – mechanosensitive membrane invaginations linked to actin filaments. J Cell Sci 128, 2747–2758. [DOI] [PubMed] [Google Scholar]
- Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N & Treisman R (2014). Rho‐actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev 28, 943–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fintha A, Gasparics A, Fang L, Erdei Z, Hamar P, Mozes MM, Kokeny G, Rosivall L & Sebe A (2013). Characterization and role of SCAI during renal fibrosis and epithelial‐to‐mesenchymal transition. Am J Pathol 182, 388–400. [DOI] [PubMed] [Google Scholar]
- Fleischhacker E, Esenabhalu VE, Spitaler M, Holzmann S, Skrabal F, Koidl B, Kostner GM & Graier WF (1999). Human diabetes is associated with hyperreactivity of vascular smooth muscle cells due to altered subcellular Ca2+ distribution. Diabetes 48, 1323–1330. [DOI] [PubMed] [Google Scholar]
- Gomez‐Ruiz A, Milagro FI, Campion J, Martinez JA & de Miguel C (2010). Caveolin expression and activation in retroperitoneal and subcutaneous adipocytes: influence of a high‐fat diet. J Cell Physiol 225, 206–213. [DOI] [PubMed] [Google Scholar]
- Grunstein M (1997). Histone acetylation in chromatin structure and transcription. Nature 389, 349–352. [DOI] [PubMed] [Google Scholar]
- Guo Z, Su W, Allen S, Pang H, Daugherty A, Smart E & Gong MC (2005). COX‐2 up‐regulation and vascular smooth muscle contractile hyperreactivity in spontaneous diabetic db/db mice. Cardiovasc Res 67, 723–735. [DOI] [PubMed] [Google Scholar]
- Gurbuz I, Ferralli J, Roloff T, Chiquet‐Ehrismann R & Asparuhova MB (2014). SAP domain‐dependent Mkl1 signaling stimulates proliferation and cell migration by induction of a distinct gene set indicative of poor prognosis in breast cancer patients. Mol Cancer 13, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson J, Parpal S, Karlsson M, Ramsing C, Thorn H, Borg M, Lindroth M, Peterson KH, Magnusson KE & Stralfors P (1999). Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J 13, 1961–1971. [PubMed] [Google Scholar]
- Hayashi K, Watanabe B, Nakagawa Y, Minami S & Morita T (2014). RPEL proteins are the molecular targets for CCG‐1423, an inhibitor of Rho signaling. PLoS One 9, e89016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino‐Fukuyo N, Haginoya K, Sugano H & Nishino I (2009). Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest 119, 2623–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Goldfine AB, Boes T, Henry RR, Ciaraldi TP, Kim EY, Emecan M, Fitzpatrick C, Sen A, Shah A, Mun E, Vokes V, Schroeder J, Tatro E, Jimenez‐Chillaron J & Patti ME (2011). Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J Clin Invest 121, 918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura H, Yokote K, Asaumi S, Kobayashi K, Fujimoto M, Maezawa Y, Saito Y & Mori S (2004). High glucose‐induced upregulation of osteopontin is mediated via Rho/Rho kinase pathway in cultured rat aortic smooth muscle cells. Arterioscler Thromb Vasc Biol 24, 276–281. [DOI] [PubMed] [Google Scholar]
- Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M & Magre J (2008). Association of a homozygous nonsense caveolin‐1 mutation with Berardinelli‐Seip congenital lipodystrophy. J Clin Endocrinol Metab 93, 1129–1134. [DOI] [PubMed] [Google Scholar]
- Kovtun O, Tillu VA, Ariotti N, Parton RG & Collins BM (2015). Cavin family proteins and the assembly of caveolae. J Cell Sci 128, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk KK, Yao Mattisson I, Ekman M, Oskolkov N, Grantinge R, Kotowska D, Olde B, Hansson O, Albinsson S, Miano JM, Rippe C & Sward K (2015). Myocardin family members drive formation of caveolae. PLoS One 10, e0133931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu Z, Mercer B, Overbeek P & Olson EN (1997). Evidence for serum response factor‐mediated regulatory networks governing SM22α transcription in smooth, skeletal, and cardiac muscle cells. Dev Biol 187, 311–321. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH & Owens GK (2005). Kruppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. J Biol Chem 280, 9719–9727. [DOI] [PubMed] [Google Scholar]
- Lo HP, Nixon SJ, Hall TE, Cowling BS, Ferguson C, Morgan GP, Schieber NL, Fernandez‐Rojo MA, Bastiani M, Floetenmeyer M, Martel N, Laporte J, Pilch PF & Parton RG (2015). The caveolin‐cavin system plays a conserved and critical role in mechanoprotection of skeletal muscle. J Cell Biol 210, 833–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson O, Chavey C, Dray C, Meulle A, Daviaud D, Quilliot D, Muller C, Valet P & Liaudet‐Coopman E (2009). LRP1 receptor controls adipogenesis and is up‐regulated in human and mouse obese adipose tissue. PLoS One 4, e7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B Jr, Reitman ML & Gonzalez FJ (2003). Liver‐specific disruption of PPARγ in leptin‐deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 111, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ME, Li C, Bian H, Smith BD, Layne MD & Farmer SR (2015). Myocardin‐related transcription factor A regulates conversion of progenitors to beige adipocytes. Cell 160, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MG & Janmey PA (2012). Transcription factor regulation by mechanical stress. Int J Biochem Cell Biol 44, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM (2003). Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol 35, 577–593. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES & Rosen ED (2010). Comparative epigenomic analysis of murine and human adipogenesis. Cell 143, 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI & Treisman R (2003). Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113, 329–342. [DOI] [PubMed] [Google Scholar]
- Nguyen LN, Ma D, Shui G, Wong P, Cazenave‐Gassiot A, Zhang X, Wenk MR, Goh EL & Silver DL (2014). Mfsd2a is a transporter for the essential omega‐3 fatty acid docosahexaenoic acid. Nature 509, 503–506. [DOI] [PubMed] [Google Scholar]
- Nilsson‐Berglund LM, Zetterqvist AV, Nilsson‐Ohman J, Sigvardsson M, Gonzalez Bosc LV, Smith ML, Salehi A, Agardh E, Fredrikson GN, Agardh CD, Nilsson J, Wamhoff BR, Hultgardh‐Nilsson A & Gomez MF (2010). Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes‐induced hyperglycemia. Arterioscler Thromb Vasc Biol 30, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobusue H, Onishi N, Shimizu T, Sugihara E, Oki Y, Sumikawa Y, Chiyoda T, Akashi K, Saya H & Kano K (2014). Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat Commun 5, 3368. [DOI] [PubMed] [Google Scholar]
- Olson EN & Nordheim A (2010). Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa J, Otsu K, Toya Y, Tsunematsu T, Hankins R, Kawabe J, Minamisawa S, Umemura S, Hagiwara Y & Ishikawa Y (2004). Insulin resistance in skeletal muscles of caveolin‐3‐null mice. Proc Natl Acad Sci USA 101, 12670–12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G & Treisman R (2006). Actin’ together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol 16, 588–596. [DOI] [PubMed] [Google Scholar]
- Qiu P, Ritchie RP, Fu Z, Cao D, Cumming J, Miano JM, Wang DZ, Li HJ & Li L (2005). Myocardin enhances Smad3‐mediated transforming growth factor‐β1 signaling in a CArG box‐independent manner: Smad‐binding element is an important cis element for SM22α transcription in vivo. Circ Res 97, 983–991. [DOI] [PubMed] [Google Scholar]
- Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, Schulze A, Lucke B, Lutzkendorf S, Karbasiyan M, Bachmann S, Spuler S & Schuelke M (2010). Fatal cardiac arrhythmia and long‐QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF‐CAVIN mutations. PLoS Genet 6, e1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riches K, Alshanwani AR, Warburton P, O'Regan DJ, Ball SG, Wood IC, Turner NA & Porter KE (2014). Elevated expression levels of miR‐143/5 in saphenous vein smooth muscle cells from patients with Type 2 diabetes drive persistent changes in phenotype and function. J Mol Cell Cardiol 74, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikitake Y & Liao JK (2005). Rho‐kinase mediates hyperglycemia‐induced plasminogen activator inhibitor‐1 expression in vascular endothelial cells. Circulation 111, 3261–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, Walkey CJ, Puigserver P & Spiegelman BM (2000). Transcriptional regulation of adipogenesis. Genes Dev 14, 1293–1307. [PubMed] [Google Scholar]
- Sawant A, Chanda D, Isayeva T, Tsuladze G, Garvey WT & Ponnazhagan S (2012). Noggin is novel inducer of mesenchymal stem cell adipogenesis: implications for bone health and obesity. J Biol Chem 287, 12241–12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleicher ED & Weigert C (2000). Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl 77, S13–18. [DOI] [PubMed] [Google Scholar]
- Solway J, Seltzer J, Samaha FF, Kim S, Alger LE, Niu Q, Morrisey EE, Ip HS & Parmacek MS (1995). Structure and expression of a smooth muscle cell‐specific gene, SM22 alpha. J Biol Chem 270, 13460–13469. [DOI] [PubMed] [Google Scholar]
- Somogyi K & Rorth P (2004). Cortactin modulates cell migration and ring canal morphogenesis during Drosophila oogenesis. Mech Dev 121, 57–64. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos A, Gineitis D, Copeland J & Treisman R (1999). Signal‐regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 98, 159–169. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM & Heinrich R (2004). Biological control through regulated transcriptional coactivators. Cell 119, 157–167. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM & Farese RV Jr (2004). Lipopenia and skin barrier abnormalities in DGAT2‐deficient mice. J Biol Chem 279, 11767–11776. [DOI] [PubMed] [Google Scholar]
- Stralfors P (2012). Caveolins and caveolae, roles in insulin signalling and diabetes. Adv Exp Med Biol 729, 111–126. [DOI] [PubMed] [Google Scholar]
- Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ Jr & Miano JM (2006). Defining the mammalian CArGome. Genome Res 16, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi Hien T, Turczynska KM, Dahan D, Ekman M, Grossi M, Sjogren J, Nilsson J, Braun T, Boettger T, Garcia Vaz E, Stenkula K, Sward K, Gomez MF & Albinsson S (2016). Elevated glucose levels promote contractile and cytoskeletal gene expression in vascular smooth muscle via Rho/protein kinase C and actin polymerization. J Biol Chem 291, 3552–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler DA, Bidder M, Latifi T, Coleman T & Semenkovich CF (1998). Diet‐induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor‐deficient mice. J Biol Chem 273, 30427–30434. [DOI] [PubMed] [Google Scholar]
- Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH & Stoffel M (2011). MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474, 649–653. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M & Narumiya S (1997). Calcium sensitization of smooth muscle mediated by a Rho‐associated protein kinase in hypertension. Nature 389, 990–994. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Pacher P, Kecskemeti V, Papp G, Szollar L & Koller A (1999). Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase C. Cardiovasc Res 43, 1018–1028. [DOI] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA & Olson EN (2001). Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862. [DOI] [PubMed] [Google Scholar]
- Wang DZ, Li S, Hockemeyer D, Sutherland L, Wang Z, Schratt G, Richardson JA, Nordheim A & Olson EN (2002). Potentiation of serum response factor activity by a family of myocardin‐related transcription factors. Proc Natl Acad Sci USA 99, 14855–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen MK, Guettler S, Larijani B & Treisman R (2007). Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752. [DOI] [PubMed] [Google Scholar]
- Weinl C, Castaneda Vega S, Riehle H, Stritt C, Calaminus C, Wolburg H, Mauel S, Breithaupt A, Gruber AD, Wasylyk B, Olson EN, Adams RH, Pichler BJ & Nordheim A (2015). Endothelial depletion of murine SRF/MRTF provokes intracerebral hemorrhagic stroke. Proc Natl Acad Sci USA 112, 9914–9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RE & Carrier GO (1988). Enhanced vascular alpha‐adrenergic neuroeffector system in diabetes: importance of calcium. Am J Physiol Heart Circ Physiol 255, H1036–H1042. [DOI] [PubMed] [Google Scholar]
- Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L & Natarajan R (2010). Enhanced levels of microRNA‐125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 59, 2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve LM & Natarajan R (2010). The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol 299, F14–F25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Gong MC, Su W, Xie D, Turk J & Guo Z (2010). Role of calcium‐independent phospholipase A2β in high glucose‐induced activation of RhoA, Rho kinase, and CPI‐17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. J Biol Chem 285, 8628–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Su W, Guo Z, Pang H, Post SR & Gong MC (2006). Up‐regulation of CPI‐17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovasc Res 69, 491–501. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu X, Qin H, Tian W, Chen J, Sun L, Fang M & Xu Y (2015). Myocardin‐related transcription factor A epigenetically regulates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol 26, 1648–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JW, Liang Y, Park JY, Chen D, Yao X, Xiao Q, Liu Z, Jiang B, Fu Y, Bao M, Huang Y, Liu Y, Yan J, Zhu MS, Yang Z, Gao P, Tian B, Li D & Wang G (2012). Mediator MED23 plays opposing roles in directing smooth muscle cell and adipocyte differentiation. Genes Dev 26, 2192–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Yuan H, Wang C, Pattabiraman N, Rao M, Pestell RG & Glazer RI (2006). 3‐Phosphoinositide‐dependent protein kinase‐1 activates the peroxisome proliferator‐activated receptor‐γ and promotes adipocyte differentiation. Mol Endocrinol 20, 268–278. [DOI] [PubMed] [Google Scholar]
- Zeidan A, Nordstrom I, Albinsson S, Malmqvist U, Sward K & Hellstrand P (2003). Stretch‐induced contractile differentiation of vascular smooth muscle: sensitivity to actin polymerization inhibitors. Am J Physiol Cell Physiol 284, C1387–C1396. [DOI] [PubMed] [Google Scholar]
- Zeidan A, Sward K, Nordstrom I, Ekblad E, Zhang JC, Parmacek MS & Hellstrand P (2004). Ablation of SM22α decreases contractility and actin contents of mouse vascular smooth muscle. FEBS Lett 562, 141–146. [DOI] [PubMed] [Google Scholar]
- Zeisberg M & Neilson EG (2010). Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 21, 1819–1834. [DOI] [PubMed] [Google Scholar]
- Zhong Q & Kowluru RA (2013). Regulation of matrix metalloproteinase‐9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes 62, 2559–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]