

Abstract
Key points
Endothelial expression and the release of endothelin‐1 (ET‐1) in levels sufficient to initiate vasoconstriction is considered to be a hallmark feature of pathological endothelial dysfunction.
During the immediate postnatal period, arterial endothelial cells undergo remarkable structural and functional changes as they transition to a mature protective cell layer, which includes a marked increase in NO dilator activity.
The present study demonstrates that endothelial cells lining newborn central arteries express high levels of ET‐1 peptides and, in response to endothelial stimulation, rapidly release ET‐1 and initiate powerful ET‐1‐mediated constriction. This activity is lost as the endothelium matures in the postnatal period.
Heightened activity of ET‐1 in the neonatal endothelium might contribute to inappropriate responses of immature arteries to stress or injury. Indeed, the immature endothelium resembles dysfunctional endothelial cells, and retention or re‐emergence of this phenotype may contribute to the development of vascular disease.
Abstract
Endothelial cells lining fetal and newborn arteries have an unusual phenotype, including reduced NO activity, prominent actin stress fibres and poorly developed cellular junctions. Experiments were performed to determine whether the immature endothelium of newborn arteries also expresses and releases endothelin‐1 (ET‐1) and initiates endothelium‐dependent constriction. Carotid arteries were isolated from newborn (postnatal day 1; P1), postnatal day 7 (P7) and postnatal day 21 (P21) mice and assessed in a pressure myograph system. Endothelial stimulation with A23187 or thrombin caused constriction in P1 arteries, no significant change in diameter of P7 arteries, and dilatation in P21 arteries. In P1 arteries, constriction to thrombin or A23187 was inhibited by endothelial‐denudation, by ET‐1 receptor antagonists (BQ123 plus BQ788) or by inhibition of endothelin‐converting enzyme (phosphoramidon or SM19712). ET‐1 receptor antagonism did not affect responses to thrombin or A23187 in more mature arteries. Exogenous ET‐1 caused similar concentration‐dependent constrictions of P1, P7 and P21 arteries. Endothelial stimulation with thrombin rapidly increased the endothelial release of ET‐1 from P1 but not P21 aortas. Endothelial expression of ET‐1 peptides, as assessed by immunofluorescence analysis, was increased in P1 compared to P21 arteries. Therefore, newborn endothelial cells express high levels of ET‐1 peptides, rapidly release ET‐1 in response to endothelial stimulation, and initiate ET‐1‐mediated endothelium‐dependent constriction. This activity is diminished as the endothelium matures in the immediate postnatal period. Heightened activity of ET‐1 in neonatal endothelium probably reflects an early developmental role of the peptide, although this might contribute to inappropriate responses of immature arteries to stress or injury.
Key points
Endothelial expression and the release of endothelin‐1 (ET‐1) in levels sufficient to initiate vasoconstriction is considered to be a hallmark feature of pathological endothelial dysfunction.
During the immediate postnatal period, arterial endothelial cells undergo remarkable structural and functional changes as they transition to a mature protective cell layer, which includes a marked increase in NO dilator activity.
The present study demonstrates that endothelial cells lining newborn central arteries express high levels of ET‐1 peptides and, in response to endothelial stimulation, rapidly release ET‐1 and initiate powerful ET‐1‐mediated constriction. This activity is lost as the endothelium matures in the postnatal period.
Heightened activity of ET‐1 in the neonatal endothelium might contribute to inappropriate responses of immature arteries to stress or injury. Indeed, the immature endothelium resembles dysfunctional endothelial cells, and retention or re‐emergence of this phenotype may contribute to the development of vascular disease.
Abbreviations
- ELISA
enzyme‐linked immunosorbent assay
- ET‐1
endothelin‐1
- NOS
NO synthase
- P1
postnatal day 1 or newborn
- P7
postnatal day 7
- P21
postnatal day 21
- VWF
von Willebrand factor
Introduction
The maintenance of a normal protective endothelial cell layer is an active rather than a passive process (Murakami et al. 2008; Dejana et al. 2009) and appears to be initiated during the early postnatal period (Flavahan et al. 2013; Flavahan & Flavahan, 2014). Compared to more mature arteries, endothelial cells lining fetal and newborn arteries have a highly unusual phenotype, including a markedly reduced ability to initiate endothelial NO‐mediated dilatation, as well as immature cellular junctions and prominent transcytoplasmic stress fibres (Abman et al. 1991; Zellers & Vanhoutte, 1991; Kobayashi & Sakai, 1994; Sugimoto et al. 1997; Flavahan et al. 2013; Flavahan & Flavahan, 2014). The unusual structural and functional features of newborn endothelium are mediated by heightened activity of Rho/Rho kinase signalling (Flavahan & Flavahan, 2014). During the early postnatal period, arterial endothelial cells acquire characteristics normally associated with mature protective cells, including exuberant NO‐mediated dilation, complex endothelial cellular junctions, reorganization of stress fibres to a cortical actin network and reduced Rho/Rho kinase signalling (Abman et al. 1991; Zellers & Vanhoutte, 1991; Kobayashi & Sakai, 1994; Sugimoto et al. 1997; Flavahan et al. 2013; Flavahan & Flavahan, 2014).
Endothelin‐1 (ET‐1) plays an important role in heart and vascular development (Kurihara et al. 1995; Yanagisawa et al. 1998; Kedzierski & Yanagisawa, 2001). Indeed, there is prominent arterial expression of ET‐1 throughout mouse embryonic and fetal development (Chan et al. 1995), which appears to decrease to normal adult levels in the early postnatal period (McLean et al. 2005). The present study aimed to further investigate the unusual phenotype of newborn endothelium and to determine whether these immature endothelial cells could initiate endothelin‐mediated constriction, which is normally considered to be a hallmark feature of pathological endothelial dysfunction (Kirkby et al. 2008; Goel et al. 2010; Feletou et al. 2012).
Methods
Ethical approval
Ethical approval was obtained for all animal procedures from the Institutional Animal Care and Use Committee of the Johns Hopkins University and all procedures complied with the NIH Guide for the Care and Use of Laboratory Animals.
Animals and tissue preparation
Newborn (postnatal day 1; P1), 1 week (postnatal day 7; P7) and 3 week (postnatal day 21; P21) old male and female mice (C57BL6, Jackson Labs, ME, USA) were killed by CO2 asphyxiation. Carotid arteries and aortas were rapidly removed and placed in cold Krebs‐Ringer bicarbonate solution containing (in mm) 118.3 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, 25.0 NaHCO3 and 11.1 glucose (control solution).
Analysis of vascular responses
Neonatal carotid arteries (∼1 mm in length) were cannulated at both ends with glass micropipettes, secured using 12‐0 nylon monofilament suture, and placed in a microvascular chamber (Living Systems, Burlington, VT, USA) (Flavahan et al. 2013). Carotid arteries were selected as a model of the central arterial system and because of our previous studies with these neonatal arteries (Flavahan et al. 2013; Flavahan & Flavahan, 2014). We are particularly interested in the structural development of central arteries and their role in physiological and pathological processes, including central arterial stiffening associated with vascular ageing (Goel et al. 2010; Santhanam et al. 2010). Unless stated otherwise, the arteries were maintained in the absence of flow at a constant transmural pressure (P TM) of 20 mmHg, which represents the mean arterial BP of P1 mice (Flavahan et al. 2013). The chamber was superfused with control solution (37°C, pH 7.4, 16% O2–5% CO2, balance N2) and placed on the stage of an inverted microscope (TMS‐F; Nikon, Tokyo, Japan) connected to a video camera (CCTV camera; Panasonic, Osaka, Japan). The internal diameter, determined using a video dimension analyser (Living Systems), was continuously monitored using a data acquisition system (BIOPAC, Goleta, CA, USA) (Flavahan et al. 2013). At a P TM of 20 mmHg, arterial diameters were 183.1 ± 3.1 μm for P1 arteries (significantly different from P21, P < 0.001 and P7, P < 0.05), 195.1 ± 2.2 μm for P7 arteries (significantly different from P21, P < 0.001) and 261.3 ± 4.4 μm for P21 arteries (n = 17 for each group).
Neonatal carotid arteries (P1 to P21) have minimal basal tone. Therefore, to allow endothelial agonists to evoke constriction or dilatation, arterial segments were pre‐constricted with the thromboxane receptor agonist U46619 by ∼10% to 25% of baseline diameter. The concentration of U46619 required to constrict the arteries was not significantly different between P1, P7 and P21 arteries (log m concentrations: P1, −8.00 ± 0.06; P7, −7.90 ± 0.07; P21 −8.04 ± 0.08, n = 17 in each group). After a stable constriction was achieved, concentration–response curves to A23187 or ET‐1 were generated by increasing agonist concentration in half‐log increments once the response to the previous concentration had stabilized. With thrombin, a single concentration of the agent was administered (1 U ml−1) to avoid desensitization of thrombin receptors (Goel et al. 2010). Only one set of responses was assessed on each artery. Agonist responses were determined in paired carotid arteries, with one artery analysed under control conditions and the other artery studied after a pharmacological or mechanical intervention. When pharmacological inhibitors were studied (100 μm l‐NAME, 30 μm phosphoramidon, 200 μm SM19712, 1 μm BQ123, 1 μm BQ788), the preparations were incubated for 30 min with the drugs before and during exposure of the arteries to the agonists. When evaluating the effects of ET‐1 receptor antagonists, the aim was to eliminate the biological activity of endogenous ET‐1 and so accurately determine the role of the peptide. Therefore, most experiments were conducted using combined blockade of ETA and ETB receptors (1 μm BQ123 and 1 μm BQ788). Endothelial denudation was achieved by inserting a 70 μm wire into the lumen of the carotid artery, and was confirmed by processing the arteries for nuclear staining with Draq5 followed by imaging using laser scanning microscopy.
Endothelial imaging
Neonatal carotid arteries were processed for imaging as described previously (Flavahan et al. 2013; Flavahan & Flavahan, 2014). Briefly, arteries were opened longitudinally during fixation with paraformaldehyde (3%, 4°C, 30 min). They were permeabilized (Triton‐X, 0.5%) and incubated in donkey serum (1.5%) to reduce non‐specific binding. Arteries were incubated overnight with a primary mouse monoclonal antibody to ET‐1 (clone TR.ET.48.5, dilution 1:200; Thermo Scientific, Waltham, MA, USA) and a primary polyclonal rabbit antibody to von Willebrand factor (VWF; dilution 1:50; Santa Cruz Biotechnology, Santa Cruz, CA, USA), then incubated (120 min) with an AlexaFluor 488 donkey anti‐mouse antibody (dilution 1:200; Jackson ImmunoResearch, West Grove, PA, USA), a Cy3‐labelled donkey anti‐rabbit antibody (dilution 1:200; Jackson ImmunoResearch) and with Draq5 (5 μm; Biostatus Ltd, Shepshed, UK), which labels nuclei. The antibody used for immunofluorescence imaging of ET‐1 has a similar affinity for ET‐1 and its immediate precursor, Big ET‐1 (Traish et al. 1992; Wolf et al. 2001). The endothelial surface was viewed using an AOBS‐equipped SP5 LSM (Leica Microsystems, Wetzlar, Germany) with a 63× objective (NA 1.4). Images (1024 × 1024 pixels) were obtained using a pinhole of 1 Airy unit, a scan speed of 400 Hz, six‐line averaging, an optical zoom of 3.0 and appropriate settings for Alexa 488 (excitation 488 nm, emission 492–541 nm), Cy3 (excitation 543 nm, emission 561–629 nm) and Draq5 (excitation 633 nm, emission 659–758 nm). Images are presented in their original unprocessed condition. Endothelial cells were carefully imaged using a fluorescence intensity filter and optical slices were captured at the highest level of staining for ET‐1 immunofluorescence. For quantitative comparison of fluorescence intensity, P1 and P21 arteries were processed at the same time using the same instrument settings. When comparing fluorescence signals between different groups, the mean of the average signal intensities in P1 control arteries was set as 100%, and the intensities of all images were expressed relative to that value (Goel et al. 2010). To confirm specificity of the ET‐1 immunofluorescence signal, staining was repeated following pre‐incubation of the ET‐1 primary antibody (1:200 or 12.5 μg ml−1) with ET‐1 peptide (20.7 μg ml−1), representing ∼100‐fold molar excess. Under these conditions, the immunofluorescence signal was abolished (0 ± 0% of control, n = 4).
ET‐1 release
Two to four neonatal descending aortas, comprising thoracic and abdominal segments, were split open and equilibrated together in 120 μl of control solution for 30 min at 37°C in a tissue culture incubator (5% CO2, balance air). During this time, some arteries were incubated with l‐NAME (100 μm) to inhibit NO synthase (NOS). Some arteries were mechanically denuded of endothelium by rubbing their luminal surface with curved forceps. After 30 min of incubation, the segments were stimulated with thrombin (1 U ml−1 for 15 min). On completion of incubation, the buffer was removed and assayed for ET‐1 in duplicate using a commercial enzyme‐linked immunosorbent assay (ELISA) kit in accordance with the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). ET‐1 release was expressed as pg (g dry weight)–1. Aortas, rather than carotid arteries, were used in this analysis to access a larger source of neonatal endothelial cells. The morphological and functional development of neonatal endothelium appears to be similar between these central arteries (Flavahan et al. 2013; Flavahan & Flavahan, 2014).
Drugs
A23187, l‐NAME, phosphoramidon, SM19712 and thrombin were obtained from Sigma‐Aldrich (St Louis, MO, USA). BQ123, BQ788 and ET‐1 were from EMD‐Millipore (Billerica, MA, USA). U46619 was from Cayman (Ann Arbor, MI, USA).
Statistical analysis
Vasomotor responses were expressed as a percentage change in baseline diameter before administrating the agents. Data are expressed as the mean ± SEM of n experiments, where ‘n’ equals the number of animals from which blood vessels were studied. For the ET‐1 ELISA assay, each ‘n = 1’ represents the average of duplicate readings from a single experiment comprising multiple aortas from different animals. Statistical evaluation of the data was performed by Student's t test for paired or unpaired observations. When more than two means were compared, ANOVA was used (repeated measures ANOVA for paired samples or one‐way ANOVA for unpaired samples). If a significant F value was found, a Tukey–Kramer multiple comparisons test was then employed to identify differences among groups. When comparing concentration–effect curves, the entire curve including the pre‐constriction to U46619 and each concentration of the agonist was included in the ANOVA analysis. P < 0.05 was considered statistically significant.
Results
Endothelial endothelin‐mediated constriction in neonatal arteries
The endothelial secretagogue and calcium ionophore A23187 (31.6 to 316 nm) caused constriction of P1 arteries, no significant effect on arterial diameter in P7 arteries and dilatation in P21 arteries (Fig. 1). Similar results were observed with a distinct endothelial secretagogue thrombin (1 U ml−1), which constricted P1 arteries, had no significant vasomotor effect in P7 arteries and dilated P21 arteries (Fig. 2). The constrictor responses in P1 arteries and dilator responses in P21 arteries to these agonists were rapid and stabilized within 10–15 min (Fig. 3). To compare neonatal arteries under similar experimental conditions, this analysis were performed at a P TM of 20 mmHg, which is the mean arterial BP for newborn mice (Ishii et al. 2001; Flavahan et al. 2013; Flavahan & Flavahan, 2014). When P7 and P21 arteries were assessed at 50 and 60 mmHg, respectively, which is the mean arterial BP for these age groups (Ishii et al. 2001), responses to A23187 were not significantly different from those observed at 20 mmHg (Fig. 4).
Figure 1. Effect of combined endothelin ETA and ETB receptor antagonism on vasomotor responses to calcium ionophore A23187 (31.6 to 316 nM) in isolated carotid arteries from neonatal mice .
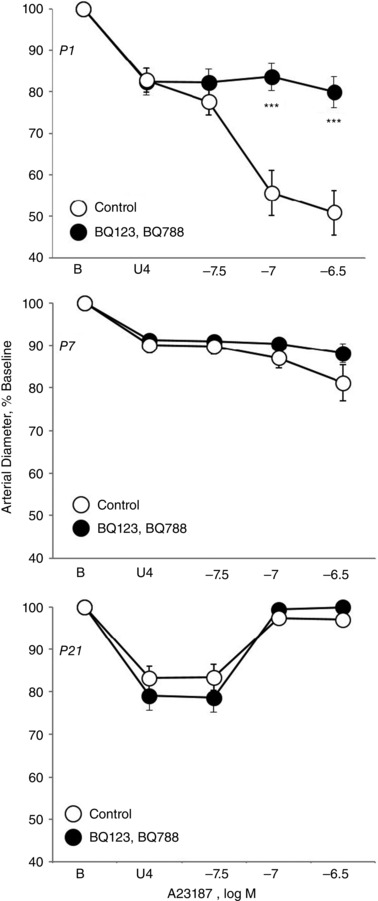
Arteries were isolated from P1 (newborn) (top), P7 (middle) and P21 (bottom) mice, analysed in a microperfusion system at a P TM of 20 mmHg, and studied under control conditions or following inhibition of ETA and ETB receptors (BQ123 1 μm plus BQ788 1 μm). Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 5 (P1) or 6 (P7, P21). Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of A23187. ***Significant difference between control and antagonist‐treated arteries (P < 0.001).
Figure 2. Effect of combined endothelin ETA and ETB receptor antagonism on vasomotor responses to thrombin (1 U ml−1) in isolated carotid arteries from neonatal mice .
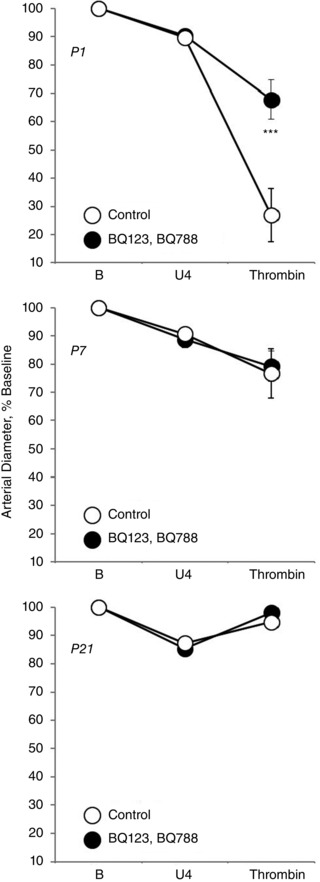
Arteries were isolated from P1 (newborn) (top), P7 (middle) and P21 (bottom) mice, analysed in a microperfusion system at a P TM of 20 mmHg, and studied under control conditions or following inhibition of ETA and ETB receptors (BQ123 1 μm plus BQ788 1 μm). Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 5. Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of thrombin. ***Significant difference between control and antagonist‐treated arteries (P < 0.001).
Figure 3. Representative tracings of arterial diameter demonstrating the contrasting vasomotor effects of calcium ionophore A23187 (31.6 to 316 nM) (left) and thrombin (1 U ml−1) (right) on isolated carotid arteries from newborn (P1) (top) and P21 (bottom) mice .

Arteries were analysed in a microperfusion system at a P TM of 20 mmHg and responses to thrombin and A23187 were assessed during constriction with U46619 (U4). A downward movement in the trace indicates constriction, with the dashed line representing 100% constriction (or 0% baseline diameter). Blue traces represent control arteries, whereas the red traces represent paired arteries treated with endothelin ETA and ETB receptor antagonists (BQ123 1 μm plus BQ788 1 μm).
Figure 4. Effect of altered PTM on vasomotor responses to calcium ionophore A23187 (31.6 to 316 nM) in isolated carotid arteries from neonatal mice .
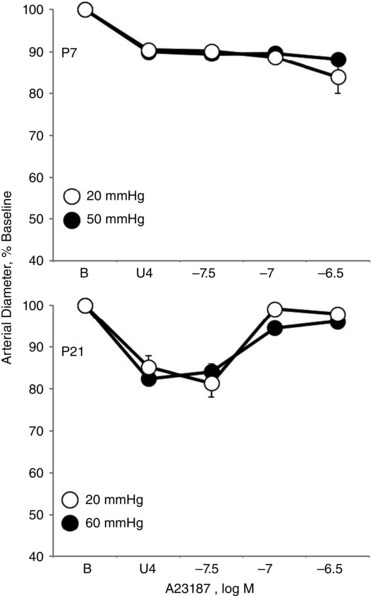
Arteries were isolated from P7 (top) and P21 (bottom) mice and analysed in a microperfusion system at a P TM of 20 mmHg and 50 mmHg (P7) or 60 mmHg (P21); 20, 50 and 60 mmHg represent the mean BP of newborn, P7 and P21 mice, respectively (Ishii et al. 2001). Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 5. Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of A23187.
Combined antagonism of endothelin ETA and ETB receptors (with BQ123 1 μm plus BQ788 1 μm, respectively) abolished constriction to A23187 or thrombin in P1 arteries but did not significantly influence the activity of A23187 or thrombin in P7 or P21 arteries (Figs 1, 2, 3). In P1 arteries, ETA antagonism alone (BQ123 1 μm) abolished constriction to A23187 (Fig. 5), indicating that the constriction was mediated by ETA receptors. Constriction to A23187 in P1 arteries was also abolished by inhibition of endothelin‐converting enzyme (phosphoramidon 30 μm or SM19712 200 μm) (Fig. 6) or by endothelial denudation (Fig. 7).
Figure 5. Effect of endothelin ETA receptor antagonism on vasomotor responses to calcium ionophore A23187 (31.6 to 316 nM) in isolated carotid arteries from newborn (P1) mice .
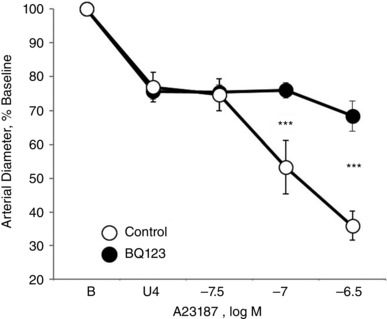
Arteries were analysed in a microperfusion system at a P TM of 20 mmHg and studied under control conditions or following BQ123 (1 μm) to inhibit ETA receptors. Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 5. Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of A23187. ***Significant difference between control and BQ123‐treated arteries (P < 0.001).
Figure 6. Effect of inhibiting endothelin‐converting enzyme (ECE) on vasomotor responses to calcium ionophore A23187 (31.6 to 316 nM) in isolated carotid arteries from newborn (P1) mice .
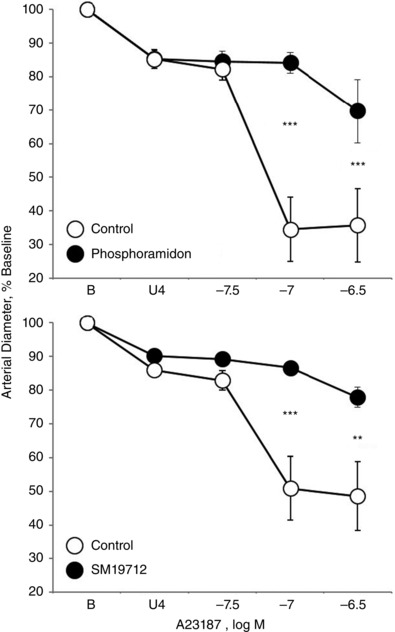
Arteries were analysed in a microperfusion system at a P TM of 20 mmHg and studied under control conditions or following inhibition of ECE with phosphoramidon (30 μm) (top) or SM19712 (200 μm) (bottom). Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 5 (top) or 6 (bottom). Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of A23187. *Significant difference between control and inhibitor‐treated arteries (** P < 0.01; *** P < 0.001).
Figure 7. Effect of endothelial‐denudation on vasomotor responses to calcium ionophore A23187 (31.6 to 316 nM) in isolated carotid arteries from newborn (P1) mice .
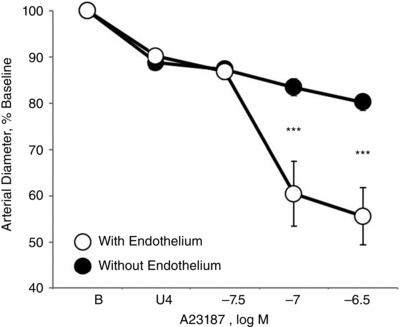
Arteries were analysed in a microperfusion system at a P TM of 20 mmHg and studied under control conditions or following mechanical denudation of the endothelium. Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 6. Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of A23187. ***Significant difference between endothelium‐containing and endothelium‐denuded arteries (P < 0.001).
Endothelial‐derived NO is the most important endogenous inhibitor of endothelin release and constriction (Goel et al. 2010). Its activity is minimal in P1 carotid arteries and increases progressively during the immediate postnatal period (P7 and P21 arteries) (Flavahan et al. 2013). Experiments were performed to determine whether this emerging activity of NO in P7 and P21 arteries might acutely contribute to reduced endothelium‐dependent constriction in these arteries. Indeed, in P7 arteries, l‐NAME uncovered constriction to A23187 (response to 316 nm; control: no significant change in diameter; l‐NAME: 54.3 ± 11.3% constriction of baseline diameter, P < 0.001, n = 6) (Figs 1 and 8). Although ET receptor antagonism had no effect in control P7 arteries (Fig. 1), the constriction that was uncovered to A23187 in l‐NAME‐treated P7 arteries was abolished by ET receptor blockade (BQ123 1 μm plus BQ788 1 μm) (Fig. 8). In P21 arteries, l‐NAME inhibited dilatation to A23187 (response to 316 nm; 91.2 ± 8.2% and 69.4 ± 12.9% dilatation of U46619‐induced constriction in control and l‐NAME‐treated arteries, respectively, n = 6, P < 0.001) (Figs 1 and 8). Therefore, as observed previously (Flavahan et al. 2013), there is prominent l‐NAME‐resistant, NO independent endothelial dilatation in P21 carotid arteries (Fig. 5). In P21 arteries, the dilator responses to A23187 occurring in control or l‐NAME‐treated arteries were not significantly affected by endothelin receptor antagonism (Figs 1 and 8). As expected, in P1 arteries, the NOS inhibitor, l‐NAME (100 μm), did not significantly affect responses to low concentrations of A23187 but increased the maximal‐observed constriction to the agonist (from 46.2 ± 8.8% to 81.7 ± 2.9% of baseline diameter, P < 0.01, n = 7).
Figure 8. Effect of endothelin receptor antagonism on the vasomotor responses to calcium ionophore A23187 (31.6 to 316 nM) in l‐NAME‐treated isolated neonatal carotid arteries .
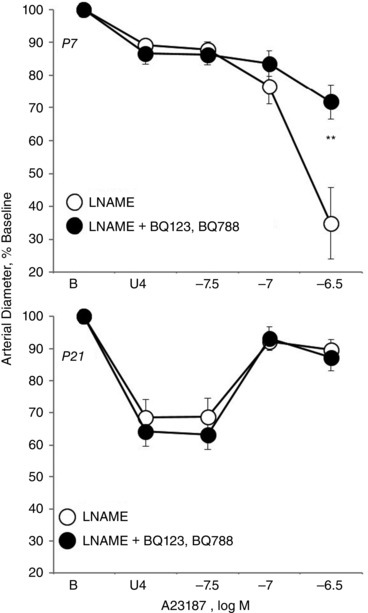
Arteries were isolated from P7 (top) and P21 mice (bottom), and analysed in a microperfusion system at a P TM of 20 mmHg and studied in the presence of l‐NAME (100 μm) to inhibit NOS and in the absence and presence of the ETA and ETB receptor antagonists, BQ123 1 μm plus BQ788 1 μm. Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 6. Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of A23187. **Significant difference between control and antagonist‐treated arteries (P < 0.01).
Constriction to exogenous ET‐1 was not significantly different between P1, P7 and P21 arteries (Fig. 9).
Figure 9. Constriction to exogenous ET‐1 (0.3 to 10 nM) in isolated carotid arteries from neonatal mice .
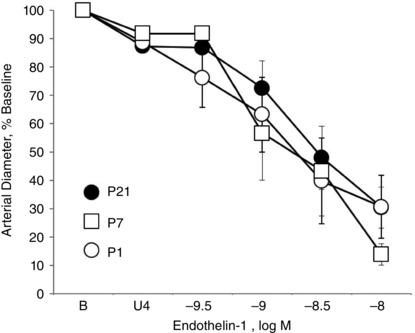
Arteries were isolated from P1 (newborn), P7 and P21 mice and analysed in a microperfusion system at a P TM of 20 mmHg. Responses are expressed as a percentage of the baseline diameter of the arteries (B) and presented as the mean ± SEM for n = 6 (P1), 7 (P7) or 8 (P21). Arteries were stably constricted with U46619 (U4) prior to evaluating the effects of endothelin‐1.
ET‐1 expression and release in neonatal arterial endothelium
Endothelial ET‐1 expression was assessed by immunofluorescence imaging of endothelial cells lining P1 and P21 arteries by laser scanning microscopy. Because the antibody recognizes both ET‐1 and its immediate precursor, Big ET‐1 (Traish et al. 1992), the immunofluorescence signal is described as (Big)ET‐1 to account for potential contributions from ET‐1 and Big ET‐1. (Big)ET‐1 immunofluorescence was characterized by prominent granular and perinuclear staining in P1 arterial endothelium (Fig. 10). Compared to P1 arteries, the immunofluorescence intensity for (Big)ET‐1 was significantly reduced by more than 2.5‐fold in P21 arteries (39.3 ± 3.0% of the intensity of P1 arteries, n = 4, P < 0.001) (Fig. 10).
Figure 10. Representative laser scanning microscopy fluorescence images demonstrating the expression and localization of ET‐1 (green) and VWF (red) immunofluorescence in endothelial cells lining carotid arteries from P1 (newborn) and P21 mice .
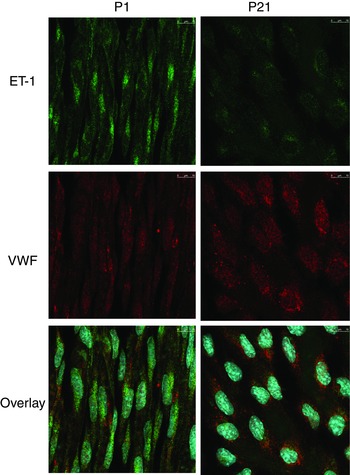
The bottom images present overlays of ET‐1, VWF and nuclear staining (blue, Draq5).
The basal release of ET‐1 was not significantly different between P1 and P21 arteries (305.0 ± 52.0 pg g−1 and 218.6 ± 36.0 pg g−1, respectively, n = 6). In P1 arteries, thrombin significantly increased ET‐1 release from arteries with endothelium but not in arteries that were denuded of endothelium (Fig. 11). Endothelial denudation did not significantly affect the basal release of ET‐1 from P1 arteries. In P21 arteries, thrombin did not significantly affect the release of ET‐1 under control conditions or following inhibition of NOS with l‐NAME (100 μm) (Fig. 11). l‐NAME did not significantly affect the basal release of ET‐1 in P21 arteries (Fig. 11).
Figure 11. Analysis of the release of ET‐1 from aortic segments of neonatal mice .
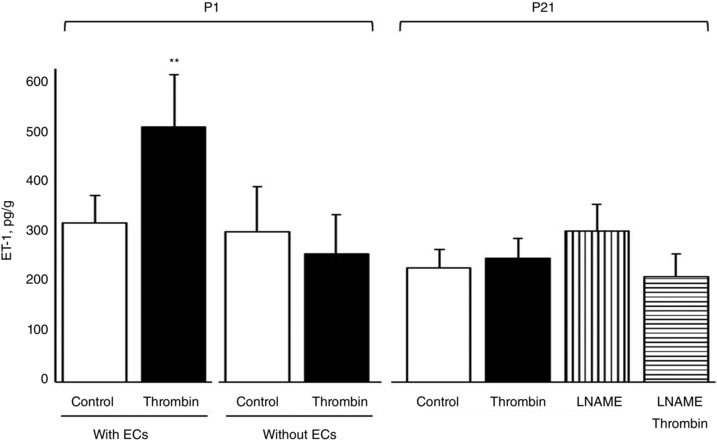
ET‐1 release from P1 (newborn) and P21 aortas was assessed under basal conditions and following endothelial stimulation with thrombin (1 U mL−1, 15 min), determined by ELISA, and expressed as pg (g dry weight)–1. In P1 arteries, the effect of thrombin was assessed in arteries with and without endothelium (ECs). In P21 arteries, the effect of thrombin was assessed in the absence and presence of l‐NAME (100 μm), an inhibitor of NOS. Data are presented as the mean ± SEM for n = 5 (P1 without endothelium) or 6 (all other conditions). **Significant difference from P1 Control with ECs (P < 0.01).
Discussion
Compared to healthy mature arteries, endothelial cells lining fetal and newborn arteries have a highly unusual phenotype, including diminished NO activity, heightened Rho/ROCK signalling, the presence of transcytoplasmic stress fibres and poorly developed cellular junctions (Kobayashi & Sakai, 1994; Sugimoto et al. 1997; Flavahan et al. 2013; Flavahan & Flavahan, 2014). This unusual phenotype is transformed during the immediate postnatal period, when endothelial cells acquire normal protective characteristics including exuberant NO activity, minimal Rho/ROCK signalling, a cortical actin cytoskeleton and well‐developed cellular junctions (Kobayashi & Sakai, 1994; Sugimoto et al. 1997; Flavahan et al. 2013; Flavahan & Flavahan, 2014). We now extend this paradigm by demonstrating that newborn endothelium can initiate powerful endothelium‐dependent constriction through the stimulated release of ET‐1, and that this activity is quickly lost in the immediate postnatal period. Although generally considered to be a prototypical pathological mediator (Goel et al. 2010; Feletou et al. 2012), the present study emphasizes that ET‐1 has important physiological activity during development.
Cultured endothelial cells release ET‐1 through two distinct processes: a constitutive pathway that is regulated principally by gene transcription and translation and a stimulated exocytotic pathway that rapidly releases peptide from endothelial storage granules (Russell et al. 1998; Kedzierski & Yanagisawa, 2001). Native endothelial cells lining healthy mature arteries do not express sufficient ET‐1 to initiate endothelium‐dependent constriction (Goel et al. 2010). However, expression and storage of ET‐1 precursors is increased in dysfunctional endothelium of senescent arteries, and, following appropriate stimulation, ET‐1 is released and generates endothelium‐dependent constriction (Goel et al. 2010). ET‐1 is actually formed during the exocytotic process and the associated endothelium‐dependent constriction is reduced by acute inhibition of ECE, which converts Big ET‐1 to ET‐1 (Goel et al. 2010).
A23187 and thrombin are efficacious endothelial secretagogues. Although normally associated with stimulation of endothelial‐dependent dilatation, they can also stimulate mediator release from endothelial storage granules, including ET‐1 peptides (Russell et al. 1998; Goel et al. 2010). As occurs in healthy mature arteries (Flavahan et al. 1989), A23187 and thrombin each caused endothelium‐dependent dilatation in P21 arteries. By contrast, in P1 arteries, they caused rapid constriction that was abolished by inhibition of ET receptors or ECE or by endothelial denudation. These endothelium‐dependent constrictions are therefore consistent with the rapid, stimulated generation of ET from newborn endothelial cells. Exogenous ET‐1 caused similar constriction in P1, P7 and P21 arteries, suggesting that the differing responses of neonatal arteries to endothelial stimuli probably reflected differences in endothelial ET generation. Indeed, thrombin stimulated ET‐1 release from P1 but not P21 arteries. This stimulated release of ET‐1 was abolished following endothelial denudation, consistent with the endothelial production of ET‐1. By contrast, the basal release of ET‐1 was not unaffected by endothelial denudation, indicating a non‐endothelial source for this basal activity. The suppression of endothelium‐dependent constriction in P1 arteries by acute inhibition of ECE suggests that active ET‐1 is generated during the exocytosis process, as observed in senescent arteries (Goel et al. 2010). This could reflect the distinct localization of ET‐1 precursors and ECE‐1 or steric hindrance of these molecules in endothelial storage granules (Goel et al. 2010). (Big)ET‐1 immunofluorescence in newborn endothelium was distinct from that of VWF, confirming that ET‐1 peptides are stored in separate granules from Weibel‐Palade bodies, the storage site for VWF (Goel et al. 2010). The expression of ET‐1 peptides assessed by immunofluorescence staining was reduced in P21 compared to P1 arteries, which is consistent with the loss of stimulated ET‐1 release and the loss of endothelium‐dependent constriction in maturing neonatal arteries.
Endothelium‐derived NO is a key endogenous inhibitor of endothelial exocytosis and ET‐1‐mediated constriction (Matsushita et al. 2003; Goel et al. 2010). NO dilator activity in response to endothelial activation with acetylcholine was almost absent in newborn carotid arteries but increased gradually during the immediate postnatal period (P7 and P21 arteries) (Flavahan et al. 2013; Flavahan & Flavahan, 2014). During this same developmental period, responses to thrombin and A23187 changed from constriction in P1, to no overall response in P7, and to dilatation in P21 arteries. The emergence of endothelial NO activity during this period could therefore contribute to a reduced ability of these agents to release ET‐1 and initiate endothelium‐dependent constriction. Indeed, in P7 arteries, NOS inhibition uncovered constriction to A23187, which was abolished by inhibition of ET receptors. This suggests that the emergence of endothelial NO activity in neonatal arteries contributes to functional antagonism of ET‐mediated constriction. However, in P21 arteries, acute NOS inhibition did not uncover ET‐1 release in response to thrombin or reveal ET‐1‐mediated constriction in response to A23187. This is consistent with the reduced expression of ET‐1 peptides in P21 arteries. In addition to inhibiting endothelial exocytosis, NO also suppresses ET‐1 expression in cultured endothelium (Kourembanas et al. 1993; Yokokawa et al. 1993). Therefore, the increasing activity of endothelium‐derived NO in maturing neonatal endothelium could contribute to inhibition of ET‐1 expression, ET‐1 release and ET‐1 constrictor activity. However, the present study did not assess whether a chronic increase in NO activity contributes to reduced ET‐1 expression in maturing arteries. Furthermore, although heightened activity of Rho/ROCK signalling together with reduced NO activity in newborn endothelium might contribute to increased ET‐1 expression (Hernandez‐Perera et al. 2000), this was also not assessed in the present study.
The present results are consistent with the important role of ET‐1 in embryonic and fetal development, including the development of the heart and great vessels (Kurihara et al. 1995; Yanagisawa et al. 1998; Kedzierski & Yanagisawa, 2001). There is strong expression of ET‐1 in the endothelium, particularly in central arteries, throughout mouse development (Chan et al. 1995). The high fetal expression of mouse aorta ET‐1 decreases in the early postnatal period (McLean et al. 2005). Similarly, circulating human fetal levels of ET‐1 in the second half of gestation are significantly higher than maternal levels (Radunovic et al. 1995). Although plasma levels of ET‐1 remain elevated in newborn babies, they decrease rapidly during postnatal life (Kojima et al. 1992; Radunovic et al. 1995; Endo et al. 1996; Endo et al. 2000). A similar rapid postnatal reduction in plasma ET‐1 levels is also observed in preclinical models (Levy et al. 1995; Nankervis & Nowicki, 2000). Mice lacking ET‐1, ECE or ETA receptors have central arterial defects, and endothelium‐derived ET‐1 was considered to activate ETA receptors on adjacent mesenchymal cells to ensure the correct development of arterial structures (Yanagisawa et al. 1998; Kedzierski & Yanagisawa, 2001). However, loss of endothelial ET‐1 was not associated with structural arterial defects (Kisanuki et al. 2010). The developmental role of endothelial‐derived ET‐1 has therefore not been clearly defined.
ET‐1 is generally considered to be a pathological mediator and biomarker for endothelial dysfunction (Kirkby et al. 2008; Goel et al. 2010; Feletou et al. 2012). Interestingly, constriction to endothelium‐derived ET‐1 in newborn carotid arteries far exceeds that observed with dysfunctional endothelium of senescent arteries, which was detected only after inhibition of residual NO activity (Goel et al. 2010). The exuberant activity of ET‐1 in newborn endothelium could therefore predispose these arteries to inappropriate constriction. Indeed, previous studies suggested that ET‐1 contributes to a greater sensitivity of the neonatal circulation to pathological stress. For example, brain injury caused increased cerebral constriction in newborn compared to juvenile pigs, which was associated with increased cerebrospinal fluid levels of ET‐1 and was attenuated by ETA antagonism (Armstead, 1999 b,a). Likewise, ischaemia‐reperfusion caused intestinal vasoconstriction in neonatal but not juvenile pigs, which was reduced by ETA antagonism (Nankervis et al. 2000). Although the cellular source of ET‐1 was not identified in these studies, it may reflect a pathological role for neonatal endothelium‐derived ET‐1.
In conclusion, endothelial cells lining central newborn arteries express high levels of ET‐1 peptides and rapidly release ET‐1 and initiate ET‐1‐mediated endothelium‐dependent constriction in response to stimulation by endothelial secretagogues. This probably reflects the physiological role of ET‐1 in fetal vascular development. In the immediate postnatal period, ET‐1 peptide expression decreases and the maturing arteries no longer release ET‐1 or initiate constriction in response to endothelial stimulation. The normal protective role of mature endothelium is an active rather than a passive process and appears to be initiated during this early postnatal period (Murakami et al. 2008; Dejana et al. 2009; Flavahan et al. 2013). The newborn endothelial phenotype, including heightened ET‐1 activity, is reminiscent of dysfunctional endothelial cells (Flavahan et al. 2013; Flavahan & Flavahan, 2014). This activity could therefore contribute to inappropriate responses of immature arteries to stress or injury. Furthermore, disruption of the normal maturation process by causing retention or reemergence of the immature endothelial phenotype could contribute to the development of vascular disease (Flavahan et al. 2013; Flavahan & Flavahan, 2014).
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
The experiments were all performed in the laboratory of Nicholas A. Flavahan. FC, SF and NAF conceived and designed the experiments. FC, SF, NAF collected, assembled, analysed and interpreted the data. FC, SF, NAF drafted the article or revised it critically for important intellectual content. All authors approved the final version of the manuscript, all authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
NIH award to NAF (HD078639).
Linked articles This article is highlighted by a Perspective by Bulley & Jaggar. To read this Perspective, visit http://dx.doi.org/10.1113/JP272564.
References
- Abman SH, Chatfield BA, Rodman DM, Hall SL & McMurtry IF (1991). Maturational changes in endothelium‐derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Physiol Lung Cell Mol Physiol 260, L280–L285. [DOI] [PubMed] [Google Scholar]
- Armstead WM (1999. a). Cerebral hemodynamics after traumatic brain injury of immature brain. Exp Toxicol Pathol 51, 137–142. [DOI] [PubMed] [Google Scholar]
- Armstead WM (1999. b). Role of endothelin‐1 in age‐dependent cerebrovascular hypotensive responses after brain injury. Am J Physiol Heart Circ Physiol 277, H1884–H1894. [DOI] [PubMed] [Google Scholar]
- Chan TS, Lin CX, Chan WY, Chung SS & Chung SK (1995). Mouse preproendothelin‐1 gene. cDNA cloning, sequence analysis and determination of sites of expression during embryonic development. Eur J Biochem 234, 819–826. [DOI] [PubMed] [Google Scholar]
- Dejana E, Tournier‐Lasserve E & Weinstein BM (2009). The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell 16, 209–221. [DOI] [PubMed] [Google Scholar]
- Endo A, Ayusawa M, Minato M, Takada M, Takahashi S & Harada K (2000). Physiologic significance of nitric oxide and endothelin‐1 in circulatory adaptation. Pediatr Int 42, 26–30. [DOI] [PubMed] [Google Scholar]
- Endo A, Shimada M, Ayusawa M, Minato M, Takada M, Takahashi S, Harada K, Masaoka N & Sato K (1996). Nitric oxide and endothelin 1 during postnatal life. Biol Neonate 70, 15–20. [DOI] [PubMed] [Google Scholar]
- Feletou M, Kohler R & Vanhoutte PM (2012). Nitric oxide: orchestrator of endothelium‐dependent responses. Ann Med 44, 694–716. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Shimokawa H & Vanhoutte PM (1989). Pertussis toxin inhibits endothelium‐dependent relaxations to certain agonists in porcine coronary arteries. J Physiol (Lond) 408, 549–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan S & Flavahan NA (2014). The atypical structure and function of newborn arterial endothelium is mediated by Rho/Rho kinase signaling. Am J Physiol Heart Circ Physiol 307, H628–H632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan S, Mozayan MM, Lindgren I & Flavahan NA (2013). Pressure‐induced maturation of endothelial cells on newborn mouse carotid arteries. Am J Physiol Heart Circ Physiol 305, H321–H329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Su B, Flavahan S, Lowenstein CJ, Berkowitz DE & Flavahan NA (2010). Increased endothelial exocytosis and generation of endothelin‐1 contributes to constriction of aged arteries. Circ Res 107, 242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Perera O, Perez‐Sala D, Soria E & Lamas S (2000). Involvement of Rho GTPases in the transcriptional inhibition of preproendothelin‐1 gene expression by simvastatin in vascular endothelial cells. Circ Res 87, 616–622. [DOI] [PubMed] [Google Scholar]
- Ishii T, Kuwaki T, Masuda Y & Fukuda Y (2001). Postnatal development of blood pressure and baroreflex in mice. Auton Neurosci 94, 34–41. [DOI] [PubMed] [Google Scholar]
- Kedzierski RM & Yanagisawa M (2001). Endothelin system: the double‐edged sword in health and disease. Annu Rev Pharmacol Toxicol 41, 851–876. [DOI] [PubMed] [Google Scholar]
- Kirkby NS, Hadoke PW, Bagnall AJ & Webb DJ (2008). The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? Br J Pharmacol 153, 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Emoto N, Ohuchi T, Widyantoro B, Yagi K, Nakayama K, Kedzierski RM, Hammer RE, Yanagisawa H, Williams SC, Richardson JA, Suzuki T & Yanagisawa M (2010). Low blood pressure in endothelial cell‐specific endothelin 1 knockout mice. Hypertension 56, 121–128. [DOI] [PubMed] [Google Scholar]
- Kobayashi N & Sakai T (1994). Postnatal reorganization of actin filaments and differentiation of intercellular boundaries in the rat aortic endothelial cells. Cell Tissue Res 278, 471–482. [DOI] [PubMed] [Google Scholar]
- Kojima T, Isozaki‐Fukuda Y, Takedatsu M, Hirata Y & Kobayashi Y (1992). Circulating levels of endothelin and atrial natriuretic factor during postnatal life. Acta Paediatr 81, 676–677. [DOI] [PubMed] [Google Scholar]
- Kourembanas S, McQuillan LP, Leung GK & Faller DV (1993). Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J Clin Invest 92, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Oda H, Maemura K, Nagai R, Ishikawa T & Yazaki Y (1995). Aortic arch malformations and ventricular septal defect in mice deficient in endothelin‐1. J Clin Invest 96, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Tulloh RM, Komai H, Stuart‐Smith K & Haworth SG (1995). Maturation of the contractile response and its endothelial modulation in newborn porcine intrapulmonary arteries. Pediatr Res 38, 25–29. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, Hara MR, Quick RA, Cao W, O'Rourke B, Lowenstein JM, Pevsner J, Wagner DD & Lowenstein CJ (2003). Nitric oxide regulates exocytosis by S‐nitrosylation of N‐ethylmaleimide‐sensitive factor. Cell 115, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean SE, Mecham BH, Kelleher CM, Mariani TJ & Mecham RP (2005). Extracellular matrix gene expression in the developing mouse aorta. Adv Dev Biol 15, 81–128. [Google Scholar]
- Murakami M, Nguyen LT, Zhuang ZW, Moodie KL, Carmeliet P, Stan RV & Simons M (2008). The FGF system has a key role in regulating vascular integrity. J Clin Invest 118, 3355–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankervis CA & Nowicki PT (2000). Role of endothelin‐1 in regulation of the postnatal intestinal circulation. Am J Physiol Gastrointest Liver Physiol 278, G367–G375. [DOI] [PubMed] [Google Scholar]
- Nankervis CA, Schauer GM & Miller CE (2000). Endothelin‐mediated vasoconstriction in postischemic newborn intestine. Am J Physiol Gastrointest Liver Physiol 279, G683–G691. [DOI] [PubMed] [Google Scholar]
- Radunovic N, Lockwood CJ, Alvarez M, Nastic D, Petkovic S & Berkowitz RL (1995). Fetal and maternal plasma endothelin levels during the second half of pregnancy. Am J Obstet Gynecol 172, 28–32. [DOI] [PubMed] [Google Scholar]
- Russell FD, Skepper JN & Davenport AP (1998). Human endothelial cell storage granules: a novel intracellular site for isoforms of the endothelin‐converting enzyme. Circ Res 83, 314–321. [DOI] [PubMed] [Google Scholar]
- Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM & Berkowitz DE (2010). Decreased S‐nitrosylation of tissue transglutaminase contributes to age‐related increases in vascular stiffness. Circ Res 107, 117–125. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Fujii S, Takemasa T & Yamashita K (1997). Factors inducing codistribution of marginal actin fibers and fibronectin in rat aortic endothelial cells. Am J Physiol 272, H2188–H2194. [DOI] [PubMed] [Google Scholar]
- Traish AM, Moran E, Daley JT, de las Morenas A & Saenz de Tejada I (1992). Monoclonal antibodies to human endothelin‐1: characterization and utilization in radioimmunoassay and immunocytochemistry. Hybridoma 11, 147–163. [DOI] [PubMed] [Google Scholar]
- Wolf WP, Weis M & von Scheidt W (2001). Endothelin immunocytochemistry: indications of false‐positive labeling patterns and non‐detectable antigen concentrations. Histochem Cell Biol 116, 411–426. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Hammer RE, Richardson JA, Williams SC, Clouthier DE & Yanagisawa M (1998). Role of endothelin‐1/endothelin‐A receptor‐mediated signaling pathway in the aortic arch patterning in mice. J Clin Invest 102, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokokawa K, Tahara H, Kohno M, Mandal AK, Yanagisawa M & Takeda T (1993). Heparin regulates endothelin production through endothelium‐derived nitric oxide in human endothelial cells. J Clin Invest 92, 2080–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellers TM & Vanhoutte PM (1991). Endothelium‐dependent relaxations of piglet pulmonary arteries augment with maturation. Pediatr Res 30, 176–180. [DOI] [PubMed] [Google Scholar]