

Abstract
Key points
The mechanism(s) that regulate hypoxia‐induced blood flow through intrapulmonary arteriovenous anastomoses (Q IPAVA) are currently unknown.
Our previous work has demonstrated that the mechanism of hypoxia‐induced Q IPAVA is not simply increased cardiac output, pulmonary artery systolic pressure or sympathetic nervous system activity and, instead, it may be a result of hypoxaemia directly.
To determine whether it is reduced arterial () or O2 content () that causes hypoxia‐induced Q IPAVA, individuals were instructed to breathe room air and three levels of hypoxic gas at rest before (control) and after was reduced by 10% by lowering the haemoglobin concentration (isovolaemic haemodilution; Low [Hb]).
Q IPAVA, assessed by transthoracic saline contrast echocardiography, significantly increased as decreased and, despite reduced (via isovolaemic haemodilution), was similar at iso‐.
These data suggest that, with alveolar hypoxia, low causes the hypoxia‐induced increase in Q IPAVA, although where and how this is detected remains unknown.
Abstract
Alveolar hypoxia causes increased blood flow through intrapulmonary arteriovenous anastomoses (Q IPAVA) in healthy humans at rest. However, it is unknown whether the stimulus regulating hypoxia‐induced Q IPAVA is decreased arterial () or O2 content (). is known to regulate blood flow in the systemic circulation and it is suggested that IPAVA may be regulated similar to the systemic vasculature. Thus, we hypothesized that reduced would be the stimulus for hypoxia‐induced Q IPAVA. Blood volume (BV) was measured using the optimized carbon monoxide rebreathing method in 10 individuals. Less than 5 days later, subjects breathed room air, as well as 18%, 14% and 12.5% O2, for 30 min each, in a randomized order, before (CON) and after isovolaemic haemodilution (10% of BV withdrawn and replaced with an equal volume of 5% human serum albumin–saline mixture) to reduce [Hb] (Low [Hb]). was measured at the end of each condition and Q IPAVA was assessed using transthoracic saline contrast echocardiography. [Hb] was reduced from 14.2 ± 0.8 to 12.8 ± 0.7 g dl−1 (10 ± 2% reduction) from CON to Low [Hb] conditions. was no different between CON and Low [Hb], although was 10.4%, 9.2% and 9.8% lower at 18%, 14% and 12.5% O2, respectively. Q IPAVA significantly increased as decreased and, despite reduced , was similar at iso‐. These data suggest that, with alveolar hypoxia, low causes the hypoxia‐induced increase in Q IPAVA. Whether the low is detected at the carotid body, airway and/or the vasculature remains unknown.
Key points
The mechanism(s) that regulate hypoxia‐induced blood flow through intrapulmonary arteriovenous anastomoses (Q IPAVA) are currently unknown.
Our previous work has demonstrated that the mechanism of hypoxia‐induced Q IPAVA is not simply increased cardiac output, pulmonary artery systolic pressure or sympathetic nervous system activity and, instead, it may be a result of hypoxaemia directly.
To determine whether it is reduced arterial () or O2 content () that causes hypoxia‐induced Q IPAVA, individuals were instructed to breathe room air and three levels of hypoxic gas at rest before (control) and after was reduced by 10% by lowering the haemoglobin concentration (isovolaemic haemodilution; Low [Hb]).
Q IPAVA, assessed by transthoracic saline contrast echocardiography, significantly increased as decreased and, despite reduced (via isovolaemic haemodilution), was similar at iso‐.
These data suggest that, with alveolar hypoxia, low causes the hypoxia‐induced increase in Q IPAVA, although where and how this is detected remains unknown.
Abbreviations
- A–aDO2
alveolar‐to‐arterial difference in
arterial O2 content
- HbCO%
carboxyhaemoglobin
- Hct
haematocrit
- HR
heart rate
- HPV
hypoxic pulmonary vasoconstriction
- HSA
human serum albumin
- IPAVA
intrapulmonary arteriovenous anastomoses
- LVOT
left ventricular outflow tract
- LVOTVTI
LVOT velocity time integral
- PASP
pulmonary artery systolic pressure
alveolar
arterial
arterial
- PFO
patent foramen ovale
- QIPAVA
blood flow through IPAVA
- QT
cardiac output
arterial O2 saturation
peripheral estimate of arterial O2 saturation
- TTSCE
transthoracic saline contrast echocardiography
Introduction
Blood flow through intrapulmonary arteriovenous anastomoses (Q IPAVA) is known to occur in humans and animals under a number of physiological and experimental conditions, including exercise (Eldridge et al. 2004; Stickland et al. 2004; Dujic et al. 2005; Stickland et al. 2007; Lovering et al. 2008 a,b, 2009; Elliott et al. 2011; Bates et al. 2014), i.v. inotropic drug infusion (Bryan et al. 2012; Laurie et al. 2012; Elliott et al. 2014 a) and hypoxia (Lovering et al. 2008 a; Laurie et al. 2010; Bates et al. 2012; Bates et al. 2014; Norris et al. 2014). Despite such findings, as well as many others, the precise mechanism(s) regulating Q IPAVA under each respective condition remain incompletely known (Lovering et al. 2015).
Transthoracic saline contrast echocardiography (TTSCE) is a non‐invasive method for studying Q IPAVA because it allows for serial injections in the same individual (Duke et al. 2015) and has demonstrated findings identical to those those obtained using quantitative methods in humans (Whyte et al. 1992; Lovering et al. 2009; Bates et al. 2014) and animals (Stickland et al. 2007; Bates et al. 2012). Using TTSCE in humans, we have recently demonstrated that Q IPAVA is increased in healthy humans in normoxia during i.v. inotropic drug infusion via an increase in cardiac output (Q T), with little or no dependence on pulmonary artery systolic pressure (PASP) (Elliott et al. 2014 a). With respect to breathing hypoxic gas at rest, the mechanism(s) regulating Q IPAVA are unknown, although we have suggested that IPAVA could be passively recruited via an active redistribution of pulmonary blood flow (i.e. hypoxic pulmonary vasoconstriction or another unknown mechanism) (Lovering et al. 2015).
Acutely breathing hypoxic gas at rest elicits a number of physiological changes depending on the severity and duration of the hypoxic stimulus [e.g. increased Q T, increased PASP, increased sympathetic nervous system activity, hypoxic pulmonary vasoconstriction (HPV), and both reduced arterial () and reduced arterial O2 content ()]. Hypoxia‐induced increases in Q IPAVA, as detected with TTSCE in humans, strongly correlate with decreased arterial O2 saturation measured via pulse oximetry (), such that the greatest degree of Q IPAVA occurs with the lowest and is present in the absence of a significant increase in PASP (Laurie et al. 2010). Additionally, Tremblay et al. (2015), using acetazolamide to block HPV, found that isocapnic hypoxia‐induced Q IPAVA occurred independently of significant increases in PASP. i.v. infusion of a non‐specific β‐receptor antagonist (propranolol) when subjects were breathing 10% O2 at rest did not abolish or reduce the hypoxia‐induced increase in Q IPAVA as detected with TTSCE (Laurie et al. 2012). In summary, this previous work suggests that hypoxia‐induced Q IPAVA is not a result of increased PASP, increased β‐receptor activation and/or exclusively HPV and, instead, may be a result of hypoxaemia directly because of its association with .
We have recently suggested that IPAVA are regulated similar to the systemic vasculature because hypoxia increases and hyperoxia decreases Q IPAVA, which is the same direction as that by which the systemic vasculature responds to hypoxia/hyperoxia (Lovering et al. 2015). With respect to the systemic circulation, vasodilatation and blood flow distribution in response to hypoxia in skeletal muscle has been demonstrated to be regulated by decreases in rather than decreases in (Roach et al. 1999; Calbet, 2000; Gonzalez‐Alonso et al. 2002; Gonzalez‐Alonso et al. 2006). The signal by which the effect of affects vasodilatation and thus increased skeletal muscle blood flow is probably via ATP being released from erythrocytes (Ellsworth & Sprague, 2012; Gonzalez‐Alonso, 2012). Again, because of the similarities in responses to hypoxia/hyperoxia between the systemic vasculature (i.e. increased skeletal muscle blood flow) and Q IPAVA, the possibility that hypoxia‐induced Q IPAVA is also regulated by reduced , rather than , is an intriguing and provocative hypothesis.
Accordingly, the present study aimed to determine whether it is a reduction in or that regulates hypoxia‐induced Q IPAVA. Testing the effect of reduced on Q IPAVA independent of the simultaneous reduction of requires an experimental paradigm to uncouple these related variables. To do so, in the present study, we detected Q IPAVA with TTSCE when individuals were breathing room air and under various levels of alveolar hypoxia (18%, 14% and 12.5% O2) where and were simultaneously reduced (CON; Part A) (Fig. 1). We then uncoupled these variables by reducing and keeping constant when individuals were breathing room air and at each level of hypoxia. This was carried out via an isovolaemic haemodilution of 10% (Low [Hb]; Part B) (Fig. 1). This experimental paradigm allowed us to compare hypoxia‐induced Q IPAVA via TTSCE in conditions of reduced but iso‐, as well as iso‐ but reduced (e.g. CON 14% O2 vs. Low [Hb] 18% O2 and CON 12.5% O2 vs. Low [Hb] 14% O2). We hypothesized that a reduction in , independent of , would regulate hypoxia‐induced Q IPAVA.
Figure 1. Schematic of protocol .
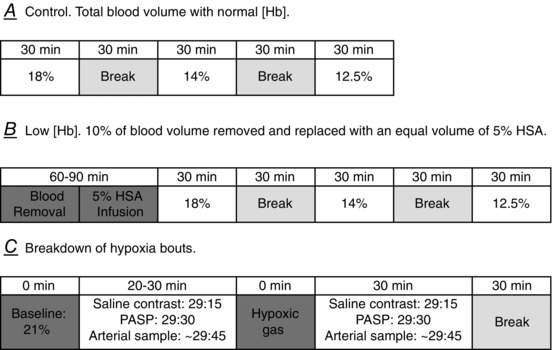
A, protocol used for the control condition with normal [Hb] (CON) (A). B, protocol used for the Low [Hb] condition following isovolaemic haemodilution (B). C, each individual bout of hypoxia. Saline contrast signifies an injection of agitated saline contrast (3 ml of saline + 1 ml of air) to obtain bubble score. PASP signifies an injection of agitated saline contrast (3 ml of saline + ∼0.1 ml of air) to obtain an enhanced tricuspid regurgitation signal to quantify PASP. Arterial sample signifies when an arterial blood sample was obtained.
Methods
Ethical approval
Eighteen college‐aged individuals (12 males and six females) volunteered to participate in the present study after being advised both verbally and in writing about the nature of the experiments. The informed consent form was approved by the University of Oregon Research Compliance Services. All studies were performed in accordance with the Declaration of Helsinki. Of the initial 18 individuals that volunteered to participate, five dropped out before completing all study visits for reasons not related to the study protocol (e.g. time commitment) and three did not qualify because the presence of a patent foramen ovale (PFO). The prevalence of PFO in the present study is much lower than that reported by ourselves and others (Woods & Patel, 2006; Woods et al. 2010; Elliott et al. 2013; Marriott et al. 2013) in that 12 individuals who volunteered for the study were invited to participate because they had previously been identified as not having a PFO.
Visits 1–3
Echocardiographic screening, fasting serum iron and ferritin, and pulmonary function
An initial echocardiographic screening and bubble study was performed on all subjects as reported previously (Lovering & Goodman, 2012; Elliott et al. 2013). Briefly, saline contrast was injected through an i.v. catheter in a peripheral vein with and without a Valsalva manoeuvre to confirm that a PFO was not present (i.e. bubbles appear in the left ventricle ≤3 cardiac cycles) as described previously (Lovering & Goodman, 2012; Elliott et al. 2013).
Because of the known effect of iron‐deficiency on the pulmonary vascular pressure response to hypoxia (Smith et al. 2008; Smith et al. 2009), we needed to ensure that participants were not iron‐deficient. Therefore, approximately 7 days before the main study visit (visit 4; see below), and on a separate day, serum iron and ferritin were assessed in each subject following a 12 h overnight fast. Blood was drawn from an antecubital vein into a sterile tiger‐top vaccutainer and immediately sent to PeaceHealth Laboratories in the Sacred Heart Medical Centre at RiverBend (Springfield, OR, USA) for iron and ferritin determination. All 10 subjects had values within the normal clinical range (Wu, 2006) (Table 1).
Table 1.
Anthropometric, serum iron and ferretin, and blood volume data
| Age (years) | 28 ± 8 | |
|---|---|---|
| Height (cm) | 180 ± 7 | |
| Mass (kg) | 81 ± 10 | |
| Serum iron (μg dl−1) | 107.9 ± 44.6 | (50–170) |
| Serum ferritin (ng ml−1) | 50.2 ± 14.0 | (10–300) |
| Blood volume (mL) | 6455 ± 1166 |
Anthropometric, iron, ferritin and blood volume data are shown as the mean ± SD. Values in parentheses are normal values according to a clinical reference textbook (Wu, 2006).
During a separate visit, each subject performed complete spirometry including forced and slow vital capacity manoeuvres in accordance with American Thoracic Society/European Respiratory Society standards (Miller et al. 2005; Wanger et al. 2005). Whole body plethysmography (MedGraphics Elite Plethysmograph, St Paul, MN, USA) was performed to determine lung volumes and capacities (Miller et al. 2005; Wanger et al. 2005). The single‐breath, breath‐hold technique was used for the determination of lung diffusion capacity for carbon monoxide (CO) (MacIntyre et al. 2005). All subjects had pulmonary function measurements ≥90% of predicted.
Visit 4
Blood volume determination
Haemoglobin mass and blood volume were measured using the optimized CO rebreathing method (Schmidt & Prommer, 2005; Prommer & Schmidt, 2007). Upon arrival at the laboratory, an i.v. catheter was placed in an antecubital vein and subjects rested when seated upright for 15–20 min. Then, a 5 ml venous blood sample was obtained to measure haemoglobin concentration ([Hb]), baseline carboxyhaemoglobin (HbCO%) and haematocrit (Hct) in sextuplicate. Hct was measured via microcentrifugation (M24 Centrifuge; LW Scientific, Lawrenceville, GA, USA) and [Hb] and HbCO% were measured via CO‐oximetry (OSM3; Radiometer, Copenhagen, Denmark). HbCO% values were corrected for oxygen saturation using the equation of Hutler et al. (2001). End‐tidal CO was measured by having subjects exhale slowly and completely into a portable CO detector (Draeger Pac 7000; Draeger, Lübeck, Germany).
Prior to the test, the custom‐built spirometer (Spico‐CO Respirations‐Applikator; Blood Tec, Bayreuth, Germany) was flushed and prefilled with 3–5 litres of 100% O2. A bolus of 99.9% pure CO (males: 1.0 ml kg−1 body mass; females: 0.8 ml kg−1 body mass) was administered into the spirometer from a calibrated syringe as the subjects breathed from residual volume to total lung capacity on the rebreathing circuit. After CO administration, subjects held their breath for 10 s and began breathing normally for an additional 1 min 50 s. At the end of the 2 min period, subjects exhaled to residual volume and continued resting while seated. Post‐rebreathing end‐tidal CO was measured 4 min after initial CO inhalation in the same manner as above. Seven minutes after the initial CO inhalation another venous blood sample was obtained from the i.v. catheter to determine the post‐rebreathing HbCO%, again in sextuplicate. The volume of CO remaining in the spirometer was measured using a calibration syringe and the portable CO detector. During the rebreathing procedure, the portable CO detector was used to monitor for potential CO leaks (Ryan et al. 2011). A leak was detected at the mouth in one subject; this test was discarded and we administered the rebreathing test a second time (1 week later) prior to proceeding with the study.
All data were compiled and used to calculate haemoglobin mass in accordance with previously published formulas (Schmidt & Prommer, 2005; Prommer & Schmidt, 2007). Blood volume was calculated from haemoglobin mass, [Hb] and Hct using equations in accordance with previous studies (Burge & Skinner, 1995; Schmidt et al. 2002; Ryan et al. 2014):
Haematocrit was multiplied by 0.96 to account for trapped plasma. The constant of 0.91 is included in the blood volume calculation to account for the ratio of body haematocrit to peripheral haematocrit (Chaplin et al. 1953).
Visit 5
Comparison of the effect of arterial O2 content and arterial on Q IPAVA
Protocol
Subjects reported to the laboratory in the morning (06.00–07.00 h) at least 24 h, but less than 5 days, following the blood volume determination visit (see above) to be re‐familiarized with the study procedures and to begin instrumentation (see below). After instrumentation was complete, subjects were positioned in a reclining chair in the left lateral decubitus position, where they would remain for the duration of the protocol, with the exception of a break between Parts A and B (Fig. 1).
After resting quietly in a chair for ∼15 min when breathing room air through a mouthpiece connected to a non‐rebreathing valve (Series 2700; Hans Rudolph, Shawnee, KS, USA), the first measurements were made (see below) to obtain resting, baseline data. Next, subjects were connected to large bore tubing connected to a 70 litre non‐diffusing gas bag (Hans Rudolph) that was pre‐filled with the prescribed hypoxic gas mixture (18%, 14% and 12.5% O2) assigned in a random order for Parts A and B. The non‐diffusing gas bag containing hypoxic gas was connected to an airtight bucket filled with warm water to humidify the gas. Subjects breathed each gas for 30 min and then had a break, during which they breathed room air for 20–30 min before breathing the next gas. A description of each data collection method is described in detail below. Metabolic data were collected continuously throughout each hypoxic bout, TTSCE was used to detect Q IPAVA at time 29 min 15 s, PASP was measured at 29 min 30 s, and a 3 ml arterial blood sample was drawn anaerobically from the radial artery catheter immediately after PASP was obtained (∼29 min 45 s). Q T was assessed simultaneously with the arterial blood sample (see below).
The first portion of the protocol (Part A) was performed with normal [Hb] (CON) and the second portion of the protocol (Part B) was performed with Low [Hb] following 10% isovolaemic haemodilution. For the Low [Hb] condition, we removed 10% of each subject's blood volume (calculated from the blood volume measured above) and immediately infused an equal volume of a 5% human serum albumin (HSA)‐saline mixture to maintain isovolaemia. The 5% HSA and saline solution was mixed immediately prior to each study from a 25% HSA solution (Buminate; Baxter, Deerfield, IL, USA) and sterile saline (0.9% NaCl). Removal occurred over a period of ∼60 min and infusion of the 5% HSA‐saline solution was conducted over a period of ∼15–20 min. For obvious technical reasons, Part B was always performed after Part A. In Part A, subjects breathed three levels of hypoxic gas (18%, 14% and 12.5% O2) for 30 min each in a random order. In Part B, subjects breathed the same three levels of hypoxic gas, also in a random order, with the exception of two subjects who did not breathe 12.5% O2 in the Low [Hb] condition.
To determine whether decreased is the mechanism regulating hypoxia‐induced Q IPAVA, our experimental protocol created four comparisons at iso‐ under CON and Low [Hb] conditions: room air (21%), 18%, 14% and 12.5% O2. To determine whether decreased is the mechanism regulating hypoxia‐induced Q IPAVA, we chose these specific hypoxic gas mixtures to create two comparisons at which between CON and Low [Hb] was approximately equivalent depending on the subject's individual hypoxic ventilatory response, but with a different . For example, using typical values for a male breathing 14% O2 in the CON condition, it could be approximated that = 50 Torr, [Hb] = 14 g dl−1 and = 85%, and thus = 16.1 ml O2 (dl blood)–1. Following isovolaemic haemodilution in this same individual breathing 18% O2, they would have = 77 Torr, [Hb] = 12.6 g dl−1 and = 94%, and thus = 16.1 ml O2 (dl blood)–1. Thus, we have a comparison at iso‐, but a different . Our second comparison with comparable but differing was 12.5% O2 in the CON condition compared to 14% O2 in the Low [Hb] condition (example math not shown).
Instrumentation
Following local anaesthesia [1% lidocaine, 2% by volume nitro‐glycerine (5 mg ml−1) to minimize vasospasm], a 20 Ga × 1.75 inch radial artery catheter (Arrow International, Reading, PA, USA) was placed under aseptic conditions by a board certified cardiologist (JAH). Patency of the arterial catheter was maintained using a pressurized flush system of normal saline. The volume of saline delivered was minimal as reflected by the minimal change in Hct and [Hb] across CON and across Low [Hb] conditions. An i.v. catheter (18–22 Ga) was placed into an antecubital vein for the injection of agitated saline contrast, removal of whole blood and infusion of the 5% HSA‐saline mixture (see above). Measurement of core body temperature was performed using an oesophageal temperature probe (Mon‐A‐Therm; Medtronic, Dublin, Ireland). The temperature probe was inserted into the oesophagus via nasal intubation following the application of an anaesthetic gel (1 ml of 2% lidocaine) to numb the naris and throat. The depth of the probe was determined as 0.479 × (sitting height in cm) as performed previously (Mekjavic & Rempel, 1990; Davis et al. 2015). In two subjects, the placement of an oesophageal probe was not possible because of a lidocaine allergy (n = 1) and intolerance of the placement of the probe (n = 1). In these subjects, we measured the core temperature using an ingestible temperature pill (CorTemp; HQInc., Palmetto, FL, USA). Unpublished observations from our laboratory demonstrate that the temperatures obtained using an oesophageal probe and the ingestible core temperature pill are in good agreement, particularly at rest (r 2 = 0.85).
Measurements
Metabolic, respiratory variables, and heart rate
Breath‐by‐breath metabolic data were collected (CardiO2; MedGraphics, St Paul, MN, USA) and presented as the mid 5 of 7. Oxygen consumption (), carbon dioxide production (), minute ventilation () and tidal volume () were collected continuously throughout each hypoxic bout. Alveolar ventilation () was calculated using the directly measured and the temperature‐ and tonometry‐corrected using the equation:
Gases with known O2 and CO2 concentrations within the physiological range were used to calibrate the gas analyser before each bout. Peripheral estimate of arterial oxygen saturation () and heart rate (HR) were continuously measured using a forehead sensor (Nellcor Oximax N‐600 pulse oximeter; Tyco, Mansfield, MA, USA).
Q T and PASP
Q T was estimated using echocardiography and determined at the level of the left ventricular outflow tract (LVOT) as reported previously (Elliott et al. 2014 b). Briefly, at rest in the left lateral decubitus position, LVOT was determined and recorded in each subject. This value was used to estimate the cross‐sectional area of the outflow tract. Pulse‐waved Doppler ultrasound (ie33; Philips, Eindhoven, The Netherlands) was used in all studies to determine LVOT velocity time integral (LVOTVTI) of blood flow through the outflow tract. LVOTVTI measures were recorded in triplicate, averaged and multiplied by the cross‐sectional area of the LVOT. The product is equal to stroke volume, which is then multiplied by HR to obtain Q T. Strong agreement has been reported between transesophageal LVOT and pulmonary artery thermodilution (r > 0.95 and SE of estimate = 0.87–0.97 litres min−1) (Stoddard et al. 1993). Unpublished data collected as part of recent work from our laboratory (Elliott et al. 2014 a) showed a strong agreement between transthoracic LVOT and the open‐circuit acetylene wash‐in method (r = 0.78), with LVOT consistently estimating Q T to be 1–2 litres min−1 greater than the acetylene wash‐in method.
PASP was determined via ultrasound (ie33; Philips) by measuring the peak velocity (v) of the tricuspid regurgitation jet and estimating right atrial pressure (P RA) based on the collapsibility of the inferior vena cava, and applying these to the modified Bernoulli equation 4v 2 + P RA (Yock & Popp, 1984; Currie et al. 1985; Himelman et al. 1989; Rudski et al. 2010). Doppler ultrasound estimates of PASP have been shown to be strongly correlated with direct measurements of PASP (r = 0.88–0.97) (Yock & Popp, 1984; Berger et al. 1985; Currie et al. 1985; Allemann et al. 2000) with Allemann et al. (2000) reporting a difference of less than 1 mmHg between estimates and direct measurements.
Q IPAVA detection
TTSCE was used to detect blood flow through IPAVA as reported previously (Lovering et al. 2008 a; Laurie et al. 2010; Elliott et al. 2011, 2014 a,b; Norris et al. 2014). Briefly, agitated saline contrast was produced by combining 3 ml of saline with 1 ml of room air and agitating for ∼15 s prior to the injection. Each agitated saline contrast injection was visualized in the apical, four‐chamber view and recorded at >30 frames s–1 for 20 cardiac cycles post‐appearance of saline contrast in the right ventricle. The single frame within the 20 cardiac cycle recording with the greatest density and spatial distribution of contrast was qualitatively assessed using a previously published scoring system (Lovering et al. 2008 b), similar to other studies (Barzilai et al. 1991) (0 = no bubbles; 1 = 1–3 bubbles; 2 = 4–12 bubbles; 3 = >12 bubbles appearing as a bolus; 4 = >12 bubbles heterogeneously filling the left ventricle; and 5 = >12 bubbles homogeneously filling the left ventricle). At present, TTSCE provides only an estimate of Q IPAVA and direct comparisons between Q IPAVA detected via TTSCE and quantified using radiolabelled macroaggregates in humans or microspheres in animals are yet to be performed. Nevertheless, hypoxia‐induced and exercise‐induced Q IPAVA studied using all of these methods in a number of studies all demonstrate almost identical findings as discussed in recent reviews (Duke et al. 2015; Lovering et al. 2015). Additionally, there is strong agreement between bubble score and PFO size measured invasively by intracardiac echocardiography (Fenster et al. 2014).
Arterial blood gas analysis
Arterial blood samples (3 ml) were drawn anaerobically via the radial artery over 10–15 s, immediately after each PASP measurement, into a heparinized syringe and immediately analysed in duplicate (and triplicate if necessary) for , arterial () and pH. The blood‐gas analyser (RAPIDLab 248; Siemens, Munich, Germany) was calibrated daily with tonometered whole human blood by equilibrating three 6 ml samples of human blood, positioned within a heated block (37°C), with gases of known concentrations of O2 and CO2 for ∼120 min. The gases were chosen to span the expected range of and anticipated in subjects breathing room air and hypoxic gas at rest ( = 100–42 Torr; = 49–15 Torr). Each sample was run in triplicate, and the values were used to create a predicted vs. measured slope. A correction factor based off of the inverse slope of this relationship was applied to measured values in the present study. Arterial blood gases were also corrected for body temperature (Kelman & Nunn, 1966; Severinghaus, 1966; Dempsey & Wagner, 1999). Direct measures of arterial O2 saturation () and [Hb] were measured with co‐oximetry. Haematocrit was analysed using the microcapillary tube centrifugation method.
Calculations
Alveolar () was calculated using the ideal gas equation as reported previously (Lovering et al. 2013; Duke et al. 2014; Elliott et al. 2014 a). Briefly, we used temperature‐ and tonometry‐corrected , barometric pressure, core body temperature (for correction of water vapour pressure) and respiratory exchange ratio from a 15 s average of breath‐by‐breath metabolic data corresponding to the time and duration of the arterial blood sampling. The alveolar‐to‐arterial difference in (A–aDO2) was calculated as – . Additionally, we calculated as reported previously (Lovering et al. 2013; Duke et al. 2014; Elliott et al. 2014 a) via the equation:
using an O2‐carrying capacity of 1.34 ml O2 (g haemoglobin)–1, measured via co‐oximetry that was corrected for the portion of haemoglobin that cannot effectively bind O2 (i.e. [Hb] = total [Hb] – HbCO – methaemoglobin) and temperature‐ and tonometry‐corrected .
Statistical analysis
All statistical analyses were performed using Prism, version 6.0f (GraphPad Software Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant for all tests. We compared [Hb] in the CON condition (before the start of the first hypoxia bout in Part A) and Low [Hb] condition (before the start of the first hypoxia bout in Part B) using a paired samples t test. To compare bubble scores across each level of hypoxia within CON and Low [Hb], we used a Friedman's test (non‐parametric repeated measures ANOVA). To determine whether bubble scores differed within a specific level of hypoxia between CON and Low [Hb], we computed a total of four paired samples non‐parametric t tests (Wilcoxon signed rank test) and adjusted α for multiple comparisons within a family of comparisons using the Bonferroni correction, such that α is divided by the number of comparisons (i.e. 0.05/4 = 0.0125). To determine whether there was a significant difference in bubble scores between CON and Low [Hb] conditions in the two iso‐ comparisons (14% O2 in CON vs. 18% O2 in Low [Hb] and 12.5% O2 in CON vs. 14% O2 in Low [Hb]), we computed two non‐parametric t tests (Wilcoxon signed rank test). Because these pairwise comparisons were made within the same family, we adjusted α via the Holm–Bonferroni correction to control the Type I error rate. To test for differences in our continuous variables (i.e. all non‐bubble score data) across levels of hypoxia and between CON and Low [Hb], we computed a two‐way repeated measures ANOVA with a Holm–Sidak multiple comparison test when appropriate.
Results
Anthropometrics, fasting iron and ferritin, blood volume, and pulmonary function
Anthropometrics, fasted serum iron and ferritin, and blood volume for all 10 subjects are presented in Table 1. Pulmonary function, lung volumes, and lung diffusion capacity for CO data for all 10 subjects are presented in Table 2. All pulmonary function parameters were within the normal range.
Table 2.
Pulmonary function, lung volumes, and diffusion capacity data
| FVC (litres) | 5.7 ± 0.9 | (101 ± 7) |
|---|---|---|
| SVC (litres) | 5.9 ± 0.8 | (105 ± 8) |
| FEV1 (litres) | 4.6 ± 0.7 | (99 ± 8) |
| FEV1/FVC (%) | 81 ± 6 | (97 ± 8) |
| FEF25—75 (litres s–1) | 4.4 ± 1.1 | (93 ± 20) |
| FRC pleth (litres) | 3.5 ± 0.6 | (101 ± 12) |
| IC (litres) | 3.8 ± 0.6 | (106 ± 10) |
| ERV (litres) | 2.1 ± 0.4 | (102 ± 12) |
| TLC (litres) | 7.3 ± 0.9 | (104 ± 5) |
| RV (litres) | 1.4 ± 0.4 | (87 ± 19) |
| DLCO (ml min–1 Torr–1) | 41.5 ± 7.0 | (124 ± 19) |
| DLCO/VA (ml min–1 Torr–1 litre–1) | 5.7 ± 0.7 | (119 ± 15) |
Pulmonary function data are shown as the mean ± SD and values in parentheses are the mean ± SD % of predicted. FVC, forced vital capacity; SVC, slow vital capacity; FEV1, forced expiratory volume in 1 s; FEV1/FVC, ratio of forced expiratory volume in 1 s to forced vital capacity; FEF25–75, forced expired flow rate from 25% to 75% of FVC; IC, inspiratory capacity; ERV, expiratory reserve volume; FRC pleth, functional residual capacity determined by whole body plethysmography; TLC, total lung capacity; RV, residual volume; DLCO, diffusion capacity for carbon monoxide; DLCO/VA, diffusion capacity for carbon monoxide per litre alveolar volume.
Arterial vs. arterial O2 content
Haemoglobin concentration
In CON, [Hb] was 14.2 ± 0.8 g dl−1 and decreased significantly to 12.8 ± 0.7 g dl−1 in the Low [Hb] condition (P < 0.0001). The magnitude of this decrease was 10 ± 2%, demonstrating that we successfully induced a 10% isovolaemic haemodilution in our subjects.
and
and data in CON and Low [Hb] conditions are shown in Fig. 2. For , there was no significant interaction effect between the level of hypoxia and condition (P = 0.9), nor was there a significant main effect for condition (CON vs. Low [Hb]; P = 0.24). However, there was a significant main effect for the level of hypoxia (P < 0.001). in hypoxia (all levels) was significantly lower (P < 0.05) than for room air in CON and Low [Hb] conditions. For , there was no significant interaction effect between the level of hypoxia and condition (P = 0.29). However, there was a significant main effect for condition (P < 0.0001) and the level of hypoxia (P < 0.0001). was significantly lower (P < 0.05) in the Low [Hb] condition compared to CON breathing room air and every level of hypoxia. In CON and Low [Hb] conditions, was significantly lower when breathing 14% and 12.5% O2 compared to room air (P < 0.05), although there was no difference between 18% O2 and room air (P > 0.05). For our two iso‐ comparisons (i.e. 14% CON vs. 18% Low [Hb] and 12.5% CON vs. 14% Low [Hb]), was significantly lower in the CON condition for both (P < 0.001), although there was no difference in between CON and Low [Hb] (P = 0.15 for both).
Figure 2. Arterial O2 tension and O2 content .
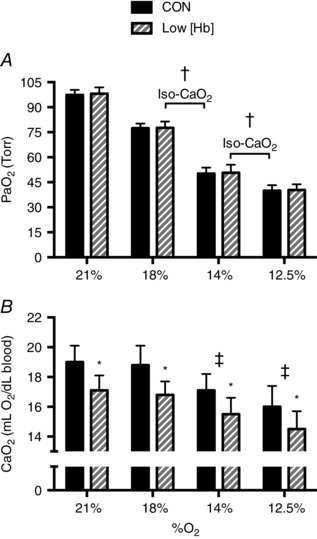
Arterial () in (A) and arterial O2 content () in (B) in the CON (solid bars) and Low [Hb] conditions (hashed bars). There was no difference in between CON and Low [Hb] at any % O2. †Significantly lower in CON compared to Low [Hb] in the iso‐ comparisons (14% O2 CON vs. 18% O2 Low [Hb] and 12.5% O2 CON vs. 14% O2 Low [Hb]). *Significantly lower at all % O2 between CON and Low [Hb]. There was no difference in in either iso‐ comparison. ‡Significantly lower compared to room air (21% O2) in CON and Low [Hb].
Bubble scores
Bubble scores in CON and Low [Hb] are shown in Fig. 3. In both CON and Low [Hb], there was a statistically significant effect of the level of hypoxia on bubble scores (P < 0.0001 for both conditions), which parallels our previous findings (Laurie et al. 2010). Under both CON and Low [Hb] conditions, bubble scores when breathing 14% and 12.5% O2 were significantly greater compared to room air, although bubbles scores when breathing 18% O2 did not differ from those when breathing room air. There was no difference in bubble scores at any level of hypoxia between CON and Low [Hb] conditions (P = 0.38–0.99). Additionally, at iso‐ (14% O2 in CON vs. 18% O2 in Low [Hb] and 12.5% O2 in CON vs. 14% O2 in Low [Hb]) (Fig. 3), bubble scores were significantly greater in the CON condition (P = 0.03 and P = 0.04, respectively). These data demonstrate that there is an effect of , but not , on Q IPAVA as detected with TTSCE.
Figure 3. Bubble scores in room air and in each level of hypoxia in CON (solid circles) and Low [Hb] (open squares) .

*Significantly greater bubble scores in 18%, 14% and 12.5% O2 compared to room air (21% O2) in CON and Low [Hb]. There was no difference in bubbles scores between CON and Low [Hb] at any level of hypoxia or room air. Iso‐ comparisons are denoted by the brackets (14% O2 CON vs. 18% O2 Low [Hb] and 12.5% O2 CON vs. 14% O2 Low [Hb]). †Significantly greater bubble score in CON compared to Low [Hb].
Arterial blood gas, cardiopulmonary and metabolic data
There was a significant main effect for the level of hypoxia on A–aDO2, , , pH and , although no differences between CON and Low [Hb] on any of these variables, with the exception of , which was significantly lower in the Low [Hb] condition when breathing room air (Table 3). For the iso‐ comparisons, was significantly lower in CON compared to Low [Hb]. Hct was significantly lower in Low [Hb] compared to CON at every level of hypoxia and there were no differences in lactate between any level of hypoxia or between conditions (Table 3). There was a significant main effect for level of hypoxia on Q T, PASP, HR, respiratory exchange ratio, , and respiratory rate. There was a significant difference between CON and Low [Hb] on Q T (all levels of hypoxia), PASP (all but room air), HR (all levels of hypoxia and room air) and (12.5% O2 only) (Table 4).
Table 3.
Arterial blood gas data
| CON | Low [Hb] | |||||||
|---|---|---|---|---|---|---|---|---|
| % O2 | ||||||||
| 21% | 18% | 14% | 12.5% | 21% | 18% | 14% | 12.5%2 | |
| A–aDO2 (Torr) | 1 ± 1 | 3 ± 1 | 5 ± 2 | 6 ± 2 | 1 ± 1 | 3 ± 2 | 4 ± 2 | 6 ± 2† |
| (Torr) | 98 ± 2 | 81 ± 3 | 55 ± 4** | 46 ± 3** | 99 ± 3 | 81 ± 3 | 55 ± 4 | 46 ± 2† |
| (Torr) | 40 ± 2 | 39 ± 2 | 37 ± 2 | 35 ± 3 | 38 ± 2* | 38 ± 2 | 36 ± 2 | 34 ± 2† |
| pH | 7.40 ± 0.01 | 7.41 ± 0.02 | 7.42 ± 0.02 | 7.43 ± 0.03 | 7.39 ± 0.02 | 7.40 ± 0.03 | 7.41 ± 0.02 | 7.43 ± 0.03† |
| (%) | 98 ± 1 | 97 ± 1 | 89 ± 2 | 81 ± 4 | 98 ± 1 | 97 ± 1 | 89 ± 3 | 82 ± 3† |
| Hct (%) | 41 ± 2 | 41 ± 2 | 41 ± 2 | 42 ± 2 | 36 ± 2* | 36 ± 2* | 36 ± 2* | 37 ± 2* |
| Lactate (mmol l−2) | 0.7 ± 0.2 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.8 ± 0.3 | 0.7 ± 0.2 | 0.7 ± 0.2 | 1.3 ± 1.8 |
Arterial blood gas data are shown as the mean ± SD. pH = the arterial pH. Lactate = arterial lactate concentration. 1 n – 1. 2 n – 2. *Significant difference between CON and Low [Hb]. †Significant main effect for level of hypoxia. **Significant difference between CON and Low [Hb] at iso‐ (i.e. 14% O2 in CON vs. 18% O2 in Low [Hb] and 12.5% O2 in CON vs. 14% O2 in Low [Hb]).
Table 4.
Cardiopulmonary and metabolic data
| CON | Low [Hb] | |||||||
|---|---|---|---|---|---|---|---|---|
| % O2 | ||||||||
| 21% | 18% | 14% | 12.5% | 21% | 18% | 14% | 12.5%2 | |
| Q T (litres min−1) | 4.8 ± 0.7 | 4.6 ± 0.4 | 5.2 ± 0.9 | 5.8 ± 0.9 | 5.6 ± 0.9* | 5.5 ± 0.9* | 6.0 ± 1.1* | 6.5 ± 1.1*, † |
| PASP (mmHg) | 27.0 ± 3.3 | 28.6 ± 4.1 | 34.0 ± 7.6 | 37.2 ± 8.8 | 26.3 ± 2.3 | 33.2 ± 6.1* | 39.3 ± 7.8* | 41.3 ± 9.5*, † |
| HR (beats min–1) | 58 ± 9 | 56 ± 5 | 60 ± 7 | 66 ± 12 | 62 ± 8* | 61 ± 8* | 67 ± 9* | 71 ± 71*, † |
| (litres min−1) | 0.37 ± 0.08 | 0.36 ± 0.10 | 0.36 ± 0.12 | 0.37 ± 0.07 | 0.37 ± 0.10 | 0.36 ± 0.07 | 0.39 ± 0.10 | 0.39 ± 0.06 |
| RER | 0.78 ± 0.06 | 0.83 ± 0.06 | 0.87 ± 0.18 | 0.85 ± 0.06 | 0.77 ± 0.05 | 0.82 ± 0.08 | 0.84 ± 0.08 | 0.83 ± 0.06† |
| (litres min−1) | 6.4 ± 1.4 | 6.7 ± 1.8 | 7.3 ± 2.2 | 8.0 ± 1.1 | 6.5 ± 2.1 | 7.0 ± 1.3 | 7.5 ± 2.0 | 8.0 ± 0.8† |
| (litres breath–1) | 0.60 ± 0.11 | 0.62 ± 0.16 | 0.65 ± 0.21 | 0.72 ± 0.18 | 0.65 ± 0.12 | 0.64 ± 0.14 | 0.70 ± 0.16 | 0.86 ± 0.25*, † |
| RR (breaths min–1) | 20 ± 5 | 20 ± 4 | 21 ± 6 | 18 ± 5 | 20 ± 4 | 21 ± 5 | 20 ± 4 | 17 ± 6† |
Cardiopulmonary and metabolic data are show as the mean ± SD. RER, respiratory exchange ratio; RR, respiratory rate. 1 n – 1. 2 n – 2. *Significant difference between CON and Low [Hb]. †Significant main effect for level of hypoxia.
Discussion
The present study aimed to determine whether hypoxia‐induced Q IPAVA is associated with a reduction in or a reduction in . Previously, we have demonstrated that Q IPAVA increases at rest with alveolar hypoxia and during exercise with alveolar normoxia or hypoxia, but decreases during exercise with alveolar hyperoxia (Lovering et al. 2008 b; Laurie et al. 2010; Elliott et al. 2011). Therefore, we have suggested that IPAVA may be regulated similarly to the peripheral systemic vasculature and opposite to the conventional pulmonary circulation (Lovering et al. 2015). Skeletal muscle vasodilatation and increased blood flow was demonstrated to be a response mediated by rather than (Roach et al. 1999; Calbet, 2000; Gonzalez‐Alonso et al. 2002; Gonzalez‐Alonso et al. 2006). Thus, we hypothesized that hypoxia‐induced Q IPAVA would be the result of a reduction in rather than . However, our data do not support this hypothesis and, instead, they demonstrate that reduced or , by some currently unknown mechanism(s), stimulates the hypoxia‐induced increase in Q IPAVA.
As discussed above, there are a number of changes that occur when an individual acutely breathes hypoxic gas at rest and all of them, independently or in total, could play a role in hypoxia‐induced Q IPAVA. For reasons discussed previously (Lovering et al. 2015) and as outlined above, we have ruled out elevated pulmonary blood flow, elevated PASP and increased β‐receptor activation in acute hypoxia as the primary mechanism regulating hypoxia‐induced Q IPAVA. However, there are a number of other possible mechanisms that relate directly to the data obtained in the present study.
Role of hypoxaemia independently
We hypothesized that a reduction in would be the mechanism regulating hypoxia‐induced Q IPAVA because of the similarities between IPAVA and the peripheral systemic vasculature. Specifically, skeletal muscle blood flow is regulated via (Roach et al. 1999; Calbet, 2000; Gonzalez‐Alonso et al. 2002; Gonzalez‐Alonso et al. 2006) with the effect probably mediated by ATP being released from erythrocytes (Ellsworth et al. 2009; Ellsworth & Sprague, 2012). Although we did not observe an effect of on hypoxia‐induced Q IPAVA, an effect of ATP on pulmonary blood flow distribution cannot be entirely ruled out. ATP release from erythrocytes is known to occur with a reduction in O2 tension in various animals (Ellsworth et al. 2009), as well as mechanical deformation of the erythrocyte, which is known to occur in the pulmonary circulation (Sprague et al. 1996). ATP released from the erythrocyte would not circulate as a mediator, but would act on the pulmonary vascular endothelium and regulate local vessel calibre (Sprague et al. 1996). This is supported by the identification of several ATP receptors located in the pulmonary vasculature (Communi et al. 1995).
Therefore, alveolar hypoxia sensed either in the airways/alveoli (see below) and/or the of the mixed venous blood entering the pulmonary circulation, in combination with mechanical deformation of the erythrocyte, would cause ATP release and subsequent vasodilatation of IPAVA and/or the region(s) of the lung where IPAVA are located. ATP causes smooth muscle dilatation/relaxation, pulmonary vascular resistance is decreased, and blood flow is directed to areas of the lung that are theoretically well ventilated (Walley, 1996). Our hypothesis was that IPAVA are present in these areas of the pulmonary circulation where blood is allowed to flow as a result of ATP‐mediated reduced pulmonary vascular resistance. This is highly speculative and will require studies to be performed where NO synthesis is blunted or blocked completely (e.g. by infusion of l‐NAME) in the animal as has been reported previously (Sprague et al. 1996).
Effect of pulmonary blood flow
Under normoxic conditions, we have demonstrated that Q IPAVA is a result of increased Q T (Laurie et al. 2012; Elliott et al. 2014 a) and so it is conceivable that this is also the case under hypoxic conditions. Our previous work has suggested that, under normoxic conditions, a Q T ‘threshold’ of ∼10 litres min−1 may need to be exceeded before significant Q IPAVA can occur (Laurie et al. 2012). Similarly, Lovering et al. (2008 a) did not observe an onset of Q IPAVA during exercise in normoxia until the workload exceeded ∼40% of VO2peak, which corresponds to an estimated Q T of 12–13 litres min−1. In the present study, Q T never exceeded our proposed necessary threshold (5.8 ± 0.9 and 6.2 ± 1.0 litres min−1 in CON and Low [Hb] when breathing 12.5% O2) to induce significant Q IPAVA as detected via TTSCE. This is consistent with our previous work, where Q T increased from 4.2 litres min−1 in normoxia to 5.0 litres min−1 in hypoxia (12% O2) in young individuals (Norris et al. 2014). Although hypoxaemia may alter the proposed necessary Q T threshold, the existing data do not support Q T as a primary mechanism causing hypoxia‐induced Q IPAVA primarily because we observed a significantly greater Q T in Low [Hb] at all levels of hypoxia and room air, but no difference in Q IPAVA between conditions.
Effect of hypoxic pulmonary vasoconstriction and blood flow redistribution
HPV is a phenomenon occurring during hypobaric or normobaric alveolar hypoxia where pulmonary arteries constrict and redistribute pulmonary blood flow, as first demonstrated by von Euler and Liljestrand (1946). We have concluded previously that hypoxia‐induced Q IPAVA was not caused by HPV per se because hypoxia‐induced Q IPAVA occurs in the absence of a significant increase in PASP (Laurie et al. 2010). Increased PASP is the hallmark manifestation of HPV occurring 2–4 h after the onset of hypoxaemia (Talbot et al. 2008). Support for this is provided in the present study with respect to the significant effect of condition (i.e. decreasing [Hb]) on PASP. Although PASP was significantly greater in Low [Hb], there was no effect on Q IPAVA. Additionally, we have observed significant hypoxia‐induced Q IPAVA even when HPV was blocked with acetazolamide (Tremblay et al. 2015). However, it is important to note that HPV is known to begin within 5 min of the onset of hypoxaemia (Talbot et al. 2005) and could therefore still contribute to a redistribution of pulmonary blood flow without any vascular changes occurring in IPAVA. Accordingly, hypoxia‐induced Q IPAVA may occur as a result of the passive recruitment of IPAVA secondary to any active redistribution of pulmonary blood flow that would be expected when breathing hypoxic gas at rest (Lovering et al. 2015).
HPV occurs in the absence of neural connections because it occurs in the isolated lung preparation and following lung transplantation (Robin et al. 1987), and is the result of a ‘stimulus’ that is a combination of and mixed venous , with having the greatest effect (Marshall et al. 1992). Therefore, it is possible that , instead of or in combination with , is the mechanism regulating hypoxia‐induced Q IPAVA. Our data support this potential conclusion because the data, unsurprisingly, mirror that of (Fig. 2A and Table 3). An important aspect of the existing literature to consider in this context is the comparison of isolated lung and intact animal preparations as it pertains to hypoxia‐induced Q IPAVA. Specifically, hypoxia‐induced Q IPAVA in isolated rat lungs was <0.01% of Q T, but was 1.1% of Q T in the intact rat under the same hypoxic conditions (Bates et al. 2012). If the mechanism were only , then one would expect Q IPAVA to be equivalent between the isolated lung and intact animal preparations. Additionally, Q IPAVA under normoxic conditions measured in isolated lungs from healthy humans and baboons, ventilated and perfused under physiological conditions equivalent to rest, was <0.1% of Q T (Lovering et al. 2007). This is less than the 0.4% of Q T measured using macroaggregates of albumin in healthy humans at rest in normoxia (Lovering et al. 2009). These data demonstrate that there is at least some reliance on arterial and/or mixed venous . Nevertheless, the present study was not designed to separate and to identify which is the regulation mechanism. To address such a question specifically, a study would be needed in an intact animal to independently quantify the effects of alveolar hypoxia and arterial hypoxaemia on Q IPAVA (i.e. by ventilating an animal with hypoxic gas, but perfusing it with normoxic blood and vice versa).
Based on the discussion above, the role of HPV in hypoxia‐induced Q IPAVA is appealing but is probably an incomplete explanation for hypoxia‐induced Q IPAVA. Although HPV is a complex system, it is clear that the change in pulmonary vascular tone is a result of the contraction of pulmonary artery smooth muscle. The contractile response in pulmonary artery smooth muscle cells can occur via non‐L‐type Ca2+ channels (Shimoda et al. 2007), L‐type Ca2+ channels (Franco‐Obregon & Lopez‐Barneo, 1996) and reduced nitric oxide and/or endothelin receptors (Sylvester et al. 2012). Recently Tremblay et al. (2015) detected Q IPAVA in isocapnic hypoxia using TTSCE and used oral acetazolamide to blunt the hypoxia‐induced increase in PASP. It was found that the degree of hypoxia‐induced Q IPAVA did not differ between isocapnic hypoxia with and without acetazolamide, suggesting little or no role for non‐L‐type Ca2+ channels in hypoxia‐induced Q IPAVA. We have recently investigated the independent effect of sildenafil (nitric oxide), nifedipine (L‐type Ca2+ channels) and acetazolamide (non‐L‐type Ca2+ channels) with respect to the hyperoxia‐induced reduction in Q IPAVA during exercise (Elliott et al. 2014 b). In combination, data from Elliott et al. (2014 b) and Tremblay et al. (2015) suggest that there is no independent effect of these smooth muscle cell vasoconstrictor pathways with respect to the increase or decrease in Q IPAVA observed during alveolar hypoxia and hyperoxia.
An important aspect of the existing literature to consider is the comparison of isolated lung and intact animal studies with respect to Q IPAVA. Q IPAVA has been measured using hypoxic, isolated rat lungs, as being less than 0.01% of Q T but as 1.1% of Q T in the intact rat under the same hypoxic condition (Bates et al. 2012). Furthermore, Q IPAVA was reported to increase by 4.6% of Q T from normoxic rest to hypoxic rest (10% O2) in healthy humans (Bates et al. 2014). Similarly, under normoxic conditions, Q IPAVA measured in isolated lungs from healthy humans and baboons, ventilated and perfused under physiological conditions equivalent to rest, was < 0.1% of Q T (Lovering et al. 2007), which is less than the 0.4% of Q T measured using macroaggregates of albumin in healthy humans at rest in normoxia (Lovering et al. 2009). This is important because HPV is not dependent upon lung neural innervation (Robin et al. 1987), which suggests that HPV does not completely explain hypoxia‐induced Q IPAVA.
Role of the peripheral chemoreceptors (carotid bodies)
Reduced corresponded with an increase in Q IPAVA in CON and Low [Hb], with no difference in between the two conditions (Fig. 2); therefore, it is possible that hypoxia‐induced Q IPAVA is mediated via the peripheral chemoreceptor. Glomus cells (type I) of the carotid body sense a reduction in and cause a subsequent increase in ventilation. In addition to its association with , there is also a known interaction between peripheral chemoreceptors and the pulmonary vasculature. Specifically, pulmonary vascular pressure and resistance are decreased with peripheral chemoreceptor stimulation via hypoxia (Daly & Daly, 1959; Levitzky et al. 1977; Naeije et al. 1989; Wilson & Levitzky, 1989; Fitzgerald et al. 1992) and peripheral chemoreceptor denervation exacerbates the increase in pulmonary vascular pressure and resistance observed in acute hypoxia (Levitzky et al. 1977; Chapleau et al. 1988; Naeije et al. 1989; Wilson & Levitzky, 1989). However it is important to note that peripheral chemoreceptor denervation, without maintaining a constant alveolar ventilation to the control condition, results in a lower (Bisgard et al. 1976), which would probably result in a greater stimulus .
Nevertheless, there is some indirect support for hypoxia‐induced Q IPAVA being a carotid body mediated response in intact healthy humans. It is well known that there is a hyperbolic relationship between peripheral chemoreceptor output (i.e. ventilation) and /, such that there is an ‘effective hypoxia’ necessary to result in a meaningful chemoreceptor response (Rahn & Otis, 1949). There is also considerable intra‐individual variability in the ventilatory responsiveness to hypoxia where some have a ‘brisk’ response and others have a ‘slow’ response (Weil et al. 1970). We have previously reported similar findings with respect to the degree of hypoxia‐induced Q IPAVA where some subjects are ‘high shunters’ and others are ‘low shunters’ and some individuals have Q IPAVA with a mildly hypoxic gas and others do not have Q IPAVA until more severe levels of hypoxic gas are breathed (Laurie et al. 2010; Norris et al. 2014). Coincidentally, almost all individuals (>99%) have some degree of hypoxia‐induced Q IPAVA when their measured or estimated is less than 50 Torr, which corresponds to the break point in the relationship between peripheral chemoreceptor output (i.e. ventilation) and /. The present study was not designed specifically to address the potential role of the peripheral chemoreceptors, although we can calculate a Spearman correlation between the change in ventilation from baseline to 30 min when breathing 12.5% and the bubble score at the same time point. Doing so resulted in a significant correlation (r = 0.76; P = 0.03), which suggests that an increase in chemoreceptor output (i.e. ventilation) is related to hypoxia‐induced Q IPAVA. This is far from definitive proof, although does provide a rationale to investigate the potential role of the peripheral chemoreceptor in mediating hypoxia‐induced Q IPAVA.
Limitations
There are several limitations associated with the present study. One limitation is that we were only able to obtain two iso‐ comparisons between bubble scores. Ideally, we would have progressively removed whole blood throughout the study and obtained several iso‐ comparisons to enhance the strength of our findings. However, logistically, this was not possible because it was imperative to maintain isovolaemia between both arms of the study. Because we have demonstrated that Q IPAVA increases via catecholamine‐induced increases in cardiac output (Laurie et al. 2012; Elliott et al. 2014 a), it is conceivable that hypovolemia would have the opposite effect. We could have used CO loading to alter as has been reported previously (Ekblom et al. 1975) but, because CO is a known signalling molecule, its use could have impacted our data interpretation (i.e. Q IPAVA might be mediated via increased HbCO or decreased ). Specific to the present study, low dose CO inhalation has been demonstrated to alter the pulmonary vascular response to hypoxia in sheep (Nachar et al. 2001). An additional limitation was that we found significant variability in our bubble scores. Ideally, all subjects would have had significant Q IPAVA (i.e. bubble score of 4–5) when breathing 14% and 12.5% O2, which we may have observed had we used a lower level of hypoxia (i.e. 10% O2) as employed previously (Laurie et al. 2010). Doing so would have provided us with an additional comparison between iso‐ but would have not provided us with an additional iso‐ comparison because the content when breathing 10% O2 in the Low [Hb] would not have matched up with a in CON (see example math above).
Summary
The present study aimed to determine whether hypoxia‐induced Q IPAVA was a result of reduced or . Our data demonstrate that hypoxia‐induced Q IPAVA is mediated by a reduction in and/or that is not dependent on acting through a number of potential mechanisms: (1) HPV in the presence or absence of increased PASP; (2) carotid body efferent influence over pulmonary vascular tone because, without lung innervation (i.e. isolated lung preparation), there is little or no Q IPAVA; and (3) hypoxaemia stimulating ATP release from erythrocytes causing a reduction in pulmonary vascular resistance. One important point is that the changes in mirrored those of and so it could be either or a combination of both. Studies in isolated lungs compared to those in intact animals suggest that it may not be solely a mechanism (Bates et al. 2012), although the present study is unable to completely rule out a potential role of . However, these considerations are speculative at this point and will require additional follow‐up studies for clarification. In conclusion, the data obtained in the present study support our working hypothesis that Q IPAVA is the consequence of active or passive changes in pulmonary blood flow distribution (Lovering et al. 2015).
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
JWD, BJR, JEE and ATL conceived and designed the experiments. ATL, JTD and JEE were responsible for financial support. JWD, JTD, BJR, JEE, KMB, JAH and ATL collected and assembled data. JWD, JTD, JEE, KMB and ATL were responsible for data analysis and interpretation. JWD, JTD, BJR, JEE, KMB, JAH, WCB and ATL were responsible for writing the manuscript. JWD, JTD, BJR, JEE, KMB, JAH, WCB and ATL approved the final manuscript submitted for publication. All authors agree to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by the Defense Medical Research & Development Program, Department of Defense Grant W81XWH‐10–2‐0114; Eugene & Clarissa Evonuk Memorial Graduate Fellowship in Environmental, Exercise, or Stress Physiology (JTD and JEE).
Acknowledgements
The authors would like to extend their gratitude to the subjects for their enthusiastic participation in this study. We would also like to thank the invaluable and expert sonography performed by Randy Goodman and Eben Futral from Sacred Heart Medical Centre and the Oregon Heart & Vascular Institute. We would like express our appreciation to Adrianne Huxtable for loaning us equipment and for the assistance of Frank Pretrassi, MS, Charlie Hodgeman, Alberto Cristobal, Alyssa Hardin, Alex Chang and Madeline Hay for help with study preparation and data collection.
References
- Allemann Y, Sartori C, Lepori M, Pierre S, Melot C, Naeije R, Scherrer U & Maggiorini M (2000). Echocardiographic and invasive measurements of pulmonary artery pressure correlate closely at high altitude. Am J Physiol Heart Circ Physiol 279, H2013–H2016. [DOI] [PubMed] [Google Scholar]
- Barzilai B, Waggoner AD, Spessert C, Picus D & Goodenberger D (1991). Two-dimensional contrast echocardiography in the detection and follow-up of congenital pulmonary arteriovenous malformations. Am J Cardiol 68, 1507–1510. [DOI] [PubMed] [Google Scholar]
- Bates ML, Farrell ET, Drezdon A, Jacobson JE, Perlman SB & Eldridge MW (2014). Hypoxia and exercise increase the transpulmonary passage of 99mTc‐labeled albumin particles in humans. PloS ONE 9, e101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates ML, Fulmer BR, Farrell ET, Drezdon A, Pegelow DF, Conhaim RL & Eldridge MW (2012). Hypoxia recruits intrapulmonary arteriovenous pathways in intact rats but not isolated rat lungs. J Appl Physiol 112, 1915–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, Haimowitz A, Van Tosh A, Berdoff RL & Goldberg E (1985). Quantitative assessment of pulmonary hypertension in patients with tricuspid regurgitation using continuous wave Doppler ultrasound. J Am Coll Cardiol 6, 359–365. [DOI] [PubMed] [Google Scholar]
- Bisgard GE, Forster HV, Orr JA, Buss DD, Rawlings CA & Rasmussen B (1976). Hypoventilation in ponies after carotid body denervation. J Appl Physiol 40, 184–190. [DOI] [PubMed] [Google Scholar]
- Bryan TL, van Diepen S, Bhutani M, Shanks M, Welsh RC & Stickland MK (2012). The effects of dobutamine and dopamine on intrapulmonary shunt and gas exchange in healthy humans. J Appl Physiol 113, 541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge CM & Skinner SL (1995). Determination of hemoglobin mass and blood volume with CO: evaluation and application of a method. J Appl Physiol 79, 623–631. [DOI] [PubMed] [Google Scholar]
- Calbet JA (2000). Oxygen tension and content in the regulation of limb blood flow. Acta Physiol Scand 168, 465–472. [DOI] [PubMed] [Google Scholar]
- Chapleau MW, Wilson LB, Gregory TJ & Levitzky MG (1988). Chemoreceptor stimulation interferes with regional hypoxic pulmonary vasoconstriction. Respir Physiol 71, 185–200. [DOI] [PubMed] [Google Scholar]
- Chaplin H Jr, Mollison PL & Vetter H (1953). The body/venous hematocrit ratio: its constancy over a wide hematocrit range. J Clin Invest 32, 1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Communi D, Raspe E, Pirotton S & Boeynaems JM (1995). Coexpression of P2Y and P2U receptors on aortic endothelial cells. Comparison of cell localization and signaling pathways. Circ Res 76, 191–198. [DOI] [PubMed] [Google Scholar]
- Currie PJ, Seward JB, Chan KL, Fyfe DA, Hagler DJ, Mair DD, Reeder GS, Nishimura RA & Tajik AJ (1985). Continuous wave Doppler determination of right ventricular pressure: a simultaneous Doppler‐catheterization study in 127 patients. J Am Coll Cardiol 6, 750–756. [DOI] [PubMed] [Google Scholar]
- Daly ID & Daly MD (1959). The effects of stimulation of the carotid body chemoreceptors on the pulmonary vascular bed in the dog: the ‘vasosensory controlled perfused living animal’ preparation. J Physiol 148, 201–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JT, Ng CY, Hill SD, Padgett RC & Lovering AT (2015). Higher oesophageal temperature at rest and during exercise in humans with patent foramen ovale. J Physiol 593, 4615–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JA & Wagner PD (1999). Exercise‐induced arterial hypoxemia. J Appl Physiol 87, 1997–2006. [DOI] [PubMed] [Google Scholar]
- Dujic Z, Palada I, Obad A, Duplancic D, Brubakk AO & Valic Z (2005). Exercise‐induced intrapulmonary shunting of venous gas emboli does not occur after open‐sea diving. J Appl Physiol 99, 944–949. [DOI] [PubMed] [Google Scholar]
- Duke JW, Elliott JE, Laurie SS, Beasley KM, Mangum TS, Hawn JA, Gladstone IM & Lovering AT (2014). Pulmonary gas exchange efficiency during exercise breathing normoxic and hypoxic gas in adults born very preterm with low diffusion capacity. J Appl Physiol 117, 473–481. [DOI] [PubMed] [Google Scholar]
- Duke JW, Elliott JE & Lovering AT (2015). Clinical consideration for techniques to detect and quantify blood flow through intrapulmonary arteriovenous anastomoses: Lessons from physiological studies. Echocardiography 32, S195–S204. [DOI] [PubMed] [Google Scholar]
- Ekblom B, Huot R, Stein EM & Thorstensson AT (1975). Effect of changes in arterial oxygen content on circulation and physical performance. J Appl Physiol 39, 71–75. [DOI] [PubMed] [Google Scholar]
- Eldridge MW, Dempsey JA, Haverkamp HC, Lovering AT & Hokanson JS (2004). Exercise‐induced intrapulmonary arteriovenous shunting in healthy humans. J Appl Physiol 97, 797–805. [DOI] [PubMed] [Google Scholar]
- Elliott JE, Choi Y, Laurie SS, Yang X, Gladston IM & Lovering AT (2011). Effect of initial gas bubble composition on detection of inducible intrapulmonary arteriovenous shunt during exercise in normoxia, hypoxi, or hyperoxia. J Appl Physiol 110, 35–45. [DOI] [PubMed] [Google Scholar]
- Elliott JE, Duke JW, Hawn JA, Halliwill JR & Lovering AT (2014. a). Increased cardiac output, not pulmonary artery systolic pressure, increases intrapulmonary shunt in healthy humans breathing room air and 40% O2 . J Physiol 592, 4537–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JE, Friedman JM, Futral JE, Goodman RD & Lovering AT (2014. b). Sildenafil, nifedipine, and acetazolamide do not allow for blood flow through intrapulmonary arteriovenous anastomoses during exercise breathing 100% oxygen. Exp Physiol 99, 1636–1647. [DOI] [PubMed] [Google Scholar]
- Elliott JE, Nigam SM, Laurie SS, Beasley KM, Goodman RD, Hawn JA, Gladstone IM, Chesnutt MS & Lovering AT (2013). Prevalence of left heart contrast in healthy, young, asymptomatic humans at rest breathing room air. Respir Physiol Neurobiol 188, 71–78. [DOI] [PubMed] [Google Scholar]
- Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH & Sprague RS (2009). Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology 24, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth ML & Sprague RS (2012). Regulation of blood flow distribution in skeletal muscle: role of erythrocyte‐released ATP. J Physiol 590, 4985–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenster BE, Curran‐Everett D, Freeman AM, Weinberger HD, Kern Buckner J & Carroll JD (2014). Saline contrast echocardiography for the detection of patent foramen ovale in hypoxia: a validation study using intracardiac echocardiography. Echocardiography 31, 420–427. [DOI] [PubMed] [Google Scholar]
- Fitzgerald RS, Dehghani GA, Sham JS, Shirahata M & Mitzner WA (1992). Peripheral chemoreceptor modulation of the pulmonary vasculature in the cat. J Appl Physiol 73, 20–29. [DOI] [PubMed] [Google Scholar]
- Franco‐Obregon A & Lopez‐Barneo J (1996). Low PO2 inhibits calcium channel activity in arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 271, H2290–H2299. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Alonso J (2012). ATP as a mediator of erythrocyte‐dependent regulation of skeletal muscle blood flow and oxygen delivery in humans. J Physiol 590, 5001–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Alonso J, Mortensen SP, Dawson EA, Secher NH & Damsgaard R (2006). Erythrocytes and the regulation of human skeletal muscle blood flow and oxygen delivery: role of erythrocyte count and oxygenation state of haemoglobin. J Physiol 572, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Alonso J, Olsen DB & Saltin B (2002). Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res 91, 1046–1055. [DOI] [PubMed] [Google Scholar]
- Himelman RB, Stulbarg M, Kircher B, Lee E, Kee L, Dean NC, Golden J, Wolfe CL & Schiller NB (1989). Noninvasive evaluation of pulmonary artery pressure during exercise by saline‐enhanced Doppler echocardiography in chronic pulmonary disease. Circulation 79, 863–871. [DOI] [PubMed] [Google Scholar]
- Hutler M, Beneke R, Littschwager A & Boning D (2001). Measured fraction of carboxyhaemoglobin depends on oxygen saturation of haemoglobin. Scand J Clin Lab Invest 61, 83–87. [DOI] [PubMed] [Google Scholar]
- Kelman GR & Nunn JF (1966). Nomograms for correction of blood PO2, PCO2, pH, and base excess for time and temperature. J Appl Physiol 21, 1484–1490. [DOI] [PubMed] [Google Scholar]
- Laurie SS, Elliott JE, Goodman RD, Lovering AT (2012). Catecholamine‐induced opening of intrapulmonary arteriovenous anastomoses in healthy humans at rest. J Appl Physiol 113, 1213–1222. [DOI] [PubMed] [Google Scholar]
- Laurie SS, Yang X, Elliott JE, Beasley KM & Lovering AT (2010). Hypoxia‐induced intrapulmonary arteriovenous shunting at rest in healthy humans. J Appl Physiol 109, 1072–1079. [DOI] [PubMed] [Google Scholar]
- Levitzky MG, Newell JC, Krasney JA & Dutton RE (1977). Chemoreceptor influence on pulmonary blood flow during unilateral hypoxia in dogs. Respir Physiol 31, 345–356. [DOI] [PubMed] [Google Scholar]
- Lovering AT & Goodman RD (2012). Detection of intracardiac and intrapulmonary shunts at rest and during exercise using saline contrast echocardiography. In Applied Aspects of Ultrasonography in Humans, ed. Ainslie P, pp. 159–174. InTech publications. [Google Scholar]
- Lovering AT, Duke JW & Elliott JE (2015). Intrapulmonary arteriovenous anastomoses in humans – response to exercise and the environment. J Physiol 593, 507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering AT, Haverkamp HC, Romer LM, Hokanson JS & Eldridge MW (2009). Transpulmonary passage of 99mTc macroaggregated albumin in healthy humans at rest and during maximal exercise. J Appl Physiol 106, 1986–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering AT, Laurie SS, Elliott JE, Beasley KM, Yang X, Gust CE, Mangum TS, Goodman RD, Hawn JA & Gladstone IM (2013). Normal pulmonary gas exchange efficiency and absence of exercise‐induced arterial hypoxemia in adults with bronchopulmonary dysplasia. J Appl Physiol 115, 1050–1056. [DOI] [PubMed] [Google Scholar]
- Lovering AT, Romer LM, Haverkamp HC, Pegelow DF, Hokanson JS & Eldridge MW (2008. a). Intrapulmonary shunting and pulmonary gas exchange during normoxic and hypoxic exercise in healthy humans. J Appl Physiol 104, 1418–1425. [DOI] [PubMed] [Google Scholar]
- Lovering AT, Stickland MK, Amann M, Murphy JC, O'Brien MJ, Hokanson JS & Eldridge MW (2008. b). Hyperoxia prevents exercise‐induced intrapulmonary arteriovenous shunt in healthy humans. J Physiol 586, 4559–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering AT, Stickland MK, Kelso AJ, Eldridge MW (2007). Direct demonstration of 25‐ and 50‐μm arteriovenous pathways in healthy human and baboon lungs. Am J Physiol Heart Circ Physiol 292, H1777–H1781. [DOI] [PubMed] [Google Scholar]
- MacIntyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CPM, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, Gustafsson P, Hankinson J, Jensen R, McKay R, Miller MR, Navajas D, Pedersen OF, Pellegrino R & Wanger J (2005). Standardisation of the single‐breath determination of carbon monoxide uptake in the lung. Eur Respir J 26, 720–735. [DOI] [PubMed] [Google Scholar]
- Marriott K, Manins V, Forshaw A, Wright J & Pascoe R (2013). Detection of right‐to‐left atrial communication using saline contrast imaging: experience with 1162 patients and recommendations for echocardiography. J Am Soc Echocardiogr 26, 96–102. [DOI] [PubMed] [Google Scholar]
- Marshall BE, Marshall C & Frasch HF (1992). Control of the pulmonary circulation In Anesthesia and the Lung, ed. Stanley TH, and Sperry RJ, pp. 9–18. Kluwer, Dodrecht. [Google Scholar]
- Mekjavic IB & Rempel ME (1990). Determination of esophageal probe insertion length based on standing and sitting height. J Appl Physiol 69, 376–379. [DOI] [PubMed] [Google Scholar]
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CPM, Gustafsson P, Jensen R, Johnson DC, MacIntyre N, McKay R, Navajas D, Pedersen OF, Pellegrino R, Viegi G, Wanger J & Force AE (2005). Standardisation of spirometry. Eur Respir J 26, 319–338. [DOI] [PubMed] [Google Scholar]
- Nachar RA, Pastene CM, Herrera EA, Riquelme RA, Sanhueza EM, Troncoso S & Llanos AJ (2001). Low‐dose inhaled carbon monoxide reduces pulmonary vascular resistance during acute hypoxemia in adult sheep. High Alt Med Biol 2, 377–385. [DOI] [PubMed] [Google Scholar]
- Naeije R, Lejeune P, Leeman M, Melot C & Closset J (1989). Pulmonary vascular responses to surgical chemodenervation and chemical sympathectomy in dogs. J Appl Physiol 66, 42–50. [DOI] [PubMed] [Google Scholar]
- Norris HC, Mangum TS, Duke JW, Strayley TS, Hawn JA, Goodman RD & Lovering AT (2014). Exercise‐ and hypoxia‐induced blood flow through intrapulmonary arteriovenous anastomoses is reduced in older adults. J Appl Physiol 116, 1324–1333. [DOI] [PubMed] [Google Scholar]
- Prommer N & Schmidt W (2007). Loss of CO from the intravascular bed and its impact on the optimised CO‐rebreathing method. Eur J Appl Phyiol 100, 383–391. [DOI] [PubMed] [Google Scholar]
- Rahn H & Otis AB (1949). Man's respiratory response during and after acclimatization to high altitude. Am J Physiol 157, 445–462. [DOI] [PubMed] [Google Scholar]
- Roach RC, Koskolou MD, Calbet JA & Saltin B (1999). Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol Heart Circ Physiol 276, H438–H445. [DOI] [PubMed] [Google Scholar]
- Robin ED, Theodore J, Burke CM, Oesterle SN, Fowler MB, Jamieson SW, Baldwin JC, Morris AJ, Hunt SA, Vankessel A & et al (1987). Hypoxic pulmonary vasoconstriction persists in the human transplanted lung. Clin Sci 72, 283–287. [DOI] [PubMed] [Google Scholar]
- Rudski LG, Lai WW, Afilalo J, Hua L, Handschumacher MD, Chandrasekaran K, Solomon SD, Louie EK & Schiller NB (2010). Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr 23, 685–713. [DOI] [PubMed] [Google Scholar]
- Ryan BJ, Brothers MD, Nelson JL, Doan BK, Zupan MF, Prommer N & Byrnes WC (2011). Influence of carbon monoxide leaks on the measurement error of total haemoglobin mass. Scand J Clin Lab Invest 71, 523–528. [DOI] [PubMed] [Google Scholar]
- Ryan BJ, Wachsmuth NB, Schmidt WF, Byrnes WC, Julian CG, Lovering AT, Subudhi AW & Roach RC (2014). AltitudeOmics: rapid hemoglobin mass alterations with early acclimatization to and de‐acclimatization from 5260 m in healthy humans. PloS ONE 9, e108788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt W, Heinicke K, Rojas J, Manuel Gomez J, Serrato M, Mora M, Wolfarth B, Schmid A & Keul J (2002). Blood volume and hemoglobin mass in endurance athletes from moderate altitude. Med Sci Sports Exerc 34, 1934–1940. [DOI] [PubMed] [Google Scholar]
- Schmidt W & Prommer N (2005). The optimised CO‐rebreathing method: a new tool to determine total haemoglobin mass routinely. Eur J Appl Phyiol 95, 486–495. [DOI] [PubMed] [Google Scholar]
- Severinghaus JW (1966). Blood gas calculator. J Appl Physiol 21, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Shimoda LA, Luke R, Sylvester JT, Shih HW, Jain A & Swenson ER (2007). Inhibition of hypoxia‐induced calcium responses in pulmonary arterial smooth muscle by acetazolamide is independent of carbonic anhydrase inhibition. Am J Physiol Lung Cell Mol Physiol 292, L1002–L1012. [DOI] [PubMed] [Google Scholar]
- Smith TG, Balanos GM, Croft QP, Talbot NP, Dorrington KL, Ratcliffe PJ & Robbins PA (2008). The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J Physiol 586, 5999–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Talbot NP, Privat C, Rivera‐Ch M, Nickol AH, Ratcliffe PJ, Dorrington KL, Leon‐Velarde F & Robbins PA (2009). Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: two randomized controlled trials. JAMA 302, 1444–1450. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH & Lonigro AJ (1996). ATP: the red blood cell link to NO and local control of the pulmonary circulation. Am J Physiol Heart Circ Physiol 271, H2717–H2722. [DOI] [PubMed] [Google Scholar]
- Stickland MK, Lovering AT & Eldridge MW (2007). Exercise‐induced arteriovenous intrapulmonary shunting in dogs. Am J Respir Crit Care Med 176, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickland MK, Welsh RC, Haykowsky MJ, Petersen SR, Anderson WD, Taylor DA, Bouffard M & Jones RL (2004). Intra‐pulmonary shunt and pulmonary gas exchange during exercise in humans. J Physiol 561, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard MF, Prince CR, Ammash N, Goad JL & Vogel RL (1993). Pulsed Doppler transesophageal echocardiographic determination of cardiac output in human beings: comparison with thermodilution technique. Am Heart J 126, 956–962. [DOI] [PubMed] [Google Scholar]
- Sylvester JT, Shimoda LA, Aaronson PI & Ward JP (2012). Hypoxic pulmonary vasoconstriction. Physiol Rev 92, 367–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot NP, Balanos GM, Dorrington KL & Robbins PA (2005). Two temporal components within the human pulmonary vascular response to approximately 2 h of isocapnic hypoxia. J Appl Physiol 98, 1125–1139. [DOI] [PubMed] [Google Scholar]
- Talbot NP, Balanos GM, Robbins PA & Dorrington KL (2008). Can intravenous endothelin‐1 be used to enhance hypoxic pulmonary vasoconstriction in healthy humans? Br J Anaesth 101, 466–472. [DOI] [PubMed] [Google Scholar]
- Tremblay JC, Lovering AT, Ainslie PN, Stembridge M, Burgess KR, Bakker A, Donnelly J, Lucas SJ, Lewis NC, Dominelli PB, Henderson WR, Dominelli GS, Sheel AW & Foster GE (2015). Hypoxia, not pulmonary vascular pressure induces blood flow through intrapulmonary arteriovenous anastomoses. J Physiol 593, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Euler US & Liljjestrand G (1946). Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 12, 301–320. [Google Scholar]
- Walley KR (1996). Heterogeneity of oxygen delivery impairs oxygen extraction by peripheral tissues: theory. J Appl Physiol 81, 885–894. [DOI] [PubMed] [Google Scholar]
- Wanger J, Clausen JL, Coates A, Pederson OF, Brusasco V, Burgos F, Casaburi R, Crapo R, Enright P, van der Grinten CPM, Gustafsson P, Hankinson J, Jensen R, Johnson D, MacIntyre N, McKay R, Miller MR, Navajas D, Pellegrino R & Viegi G (2005). Standardisation of the measurement of lung volumes. Eur Respir J 26, 511–522. [DOI] [PubMed] [Google Scholar]
- Weil JV, Byrne‐Quinn E, Sodal IE, Friesen WO, Underhill B, Filley GF & Grover RF (1970). Hypoxic ventilatory drive in normal man. J Clin Invest 49, 1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte MK, Peters AM, Hughes JM, Henderson BL, Billingan GJ, Jackson JE & Chilvers ER (1992). Quantification of right to left shunt at rest and during exercise in patients with pulmonary arteriovenous malformations. Thorax 47, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LB & Levitzky MG (1989). Chemoreflex blunting of hypoxic pulmonary vasoconstriction is vagally mediated. J Appl Physiol 66, 782–791. [DOI] [PubMed] [Google Scholar]
- Woods TD & Patel A (2006). A critical review of patent foramen ovale detection using saline contrast echocardiography: when bubbles lie. J Am Soc Echocardiogr 19, 215–222. [DOI] [PubMed] [Google Scholar]
- Woods TD, Harmann L, Purath T, Ramaurthy S, Subramanian S, Jackson S & Tarima S (2010). Small‐ and moderate‐ size right‐to‐left shunts identified by saline contrast echocardiography are normal and unrelated to migraine headaches. Chest 138, 264. [DOI] [PubMed] [Google Scholar]
- Wu AHB (2006). Tietz Clinical Guide to Laboratory Tests. Saunders, Philadelphia, PA. [Google Scholar]
- Yock PG & Popp RL (1984). Noninvasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation 70, 657–662. [DOI] [PubMed] [Google Scholar]