

Abstract
Immunotherapeutic approaches are currently the most advanced treatments for Alzheimer's disease (AD). Antibodies against amyloid β‐peptide (Aβ) bind to amyloid plaques and induce their clearance by microglia via Fc receptor‐mediated phagocytosis. Dysfunctions of microglia may play a pivotal role in AD pathogenesis and could result in reduced efficacy of antibody‐mediated Aβ clearance. Recently, heterozygous mutations in the triggering receptor expressed on myeloid cells 2 (TREM2), a microglial gene involved in phagocytosis, were genetically linked to late onset AD. Loss of TREM2 reduces the ability of microglia to engulf Aβ. We have now investigated whether loss of TREM2 affects the efficacy of immunotherapeutic approaches. We show that anti‐Aβ antibodies stimulate Aβ uptake and amyloid plaque clearance in a dose‐dependent manner in the presence or absence of TREM2. However, TREM2‐deficient N9 microglial cell lines, macrophages as well as primary microglia showed significantly reduced uptake of antibody‐bound Aβ and as a consequence reduced clearance of amyloid plaques. Titration experiments revealed that reduced efficacy of amyloid plaque clearance by Trem2 knockout cells can be compensated by elevating the concentration of therapeutic antibodies.
Keywords: Alzheimer's disease, immunotherapy, neurodegeneration, phagocytosis, TREM2
Subject Categories: Immunology, Neuroscience, Pharmacology & Drug Discovery
Introduction
Alzheimer's disease (AD) is the most abundant neurodegenerative disorder and threatens our aging society. Therapeutic treatment is desperately required to slow progression of dementia. The amyloid cascade (Hardy & Selkoe, 2002) provides a number of opportunities to therapeutically interfere with disease onset and progression. Obvious targets are β‐ and γ‐secretases, the two proteases, which generate the amyloid β‐peptide (Aβ) from its precursor, the β‐amyloid precursor protein (APP) (Haass, 2004). γ‐Secretase inhibition caused major side effects in a clinical trial, which were at least partially due to inhibition of its biological activity in Notch signaling (Doody et al, 2013). Similarly, recent findings may also indicate that inhibition of β‐secretase may be problematic due to the accumulation of alternative neurotoxic APP‐derived fragments (Willem et al, 2015). Moreover, β‐secretase has numerous brain‐specific substrates and their biological function may be disturbed upon inhibition of shedding (Kuhn et al, 2012; Zhou et al, 2012; Barao et al, 2016). In fact, neuregulin‐1 signaling depends on biologically active β‐secretase (Willem et al, 2006; Cheret et al, 2013). Nevertheless, several β‐secretase inhibitors are currently in late‐stage clinical development and if demonstrated to be safe may be of therapeutic value. The most advanced and very promising therapeutic approach is currently the anti‐Aβ immunotherapy (Wisniewski & Goni, 2015), a treatment strategy initiated after the pivotal report by Schenk et al (Schenk et al, 1999) demonstrating its efficacy in amyloid plaque removal and reduction in memory decline in an animal model (Morgan et al, 2000). After crossing the blood–brain barrier, anti‐Aβ antibodies like 3D6 and others bind to amyloid plaques and trigger Fc receptor‐mediated phagocytosis of plaques by microglia cells (Bard et al, 2000). Indeed, 11C‐PiB positron emission tomography (PET) revealed a significant reduction in amyloid deposition in patients treated with anti‐Aβ antibodies (Rinne et al, 2010; Ostrowitzki et al, 2012). Although initially there was no obvious beneficial effect on memory, data from two recent clinical trials now suggest that immunotherapy may significantly slow memory decline (Reardon, 2015). Moreover, a recent clinical trial using Aducanumab indicated that significant amyloid removal is required for clinical benefits suggesting a critical role of amyloid removal by microglia. Indeed, antibody‐mediated clearance of amyloid plaques requires biologically active microglia cells (Bard et al, 2000). Recently, it was reported that heterozygous mutations in the triggering receptor expressed on myeloid cells 2 (TREM2) increase the risk for AD, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease (Guerreiro et al, 2013; Jonsson et al, 2013; Rayaprolu et al, 2013; Borroni et al, 2014; Cady et al, 2014; Cuyvers et al, 2014; Colonna & Wang, 2016; Ulrich & Holtzman, 2016; Villegas‐Llerena et al, 2016). We have shown that certain mutations in TREM2 such as p.T66M reduce the ability of microglia to phagocytose Aβ fibers, bacteria, and beads due to impairment of TREM2 maturation and cell surface transport (Kleinberger et al, 2014). Furthermore, a complete knockout of TREM2 also reduces the ability of primary microglia to phagocytose Aβ (Kleinberger et al, 2014). Similarly, in a cuprizone model for acute demyelination, TREM2‐deficient mice showed reduced phagocytic removal of myelin debris (Cantoni et al, 2015). However, in mouse models for AD pathology, TREM2 deficiency resulted in inconsistent findings. Jay et al (2015) reported a detrimental role of TREM2 in AD by demonstrating that its knockout leads to a reduction in the amyloid plaque load, inflammation, astrogliosis, and tau phosphorylation. On the other hand, Wang et al (2015) demonstrated that TREM2 deficiency enhanced amyloid plaque load. This discrepancy may be due to the use of different mouse models, but also due to the fact that microglial function may be differentially compromised depending on the time point one investigates amyloid pathology (Tanzi, 2015).
It is well known that antibodies bound to amyloid plaques trigger Fc receptor‐mediated Aβ clearance by microglia cells (Bard et al, 2000). We were interested whether antibody‐mediated Aβ clearance may be influenced by TREM2 function. To avoid confounding effects of different mouse models, time points of analysis and differences in progression of disease pathology, which in aggressive APP/presenilin overexpressing mouse models may even override microglial amyloid plaque clearance, we choose several independent models to quantitatively investigate TREM2‐dependent antibody‐mediated Aβ clearance under controlled conditions. We used CRISPR/Cas9‐modified N9 microglial cell lines as well as bone marrow‐derived macrophages (BMDM) and primary microglia cells from wild‐type (wt) or Trem2 knockout (ko) mice and investigated the potential of these cells for antibody‐dependent phagocytosis of pre‐formed Aβ fibrils or engulfment of antibody covered amyloid plaques from brain cryosections obtained from a mouse model for AD pathology.
Results
TREM2 deficiency reduces uptake efficacy of antibody‐bound Aβ by phagocytic cells
To investigate a potential influence of TREM2 deficiency on antibody‐mediated Aβ clearance, we first studied Aβ uptake in the microglial cell line N9 (Sessa et al, 2004). Trem2 mutant cell lines were generated using the CRISPR/Cas9 technology (Ran et al, 2013). We obtained a mutant cell line with a single nucleotide insertion (93_94insG) within the target site, which leads to a frame shift and an early stop codon that eliminates TREM2 expression (Fig 1A). Immunoblotting confirmed the loss of full‐length, membrane‐bound TREM2 as well as soluble TREM2 (sTREM2) in Trem2 mutant N9 cells (N9 mu) (Fig 1B). In line with our previous findings (Kleinberger et al, 2014), loss of TREM2 significantly reduced uptake of pre‐aggregated HiLyte™ Fluor 488‐labeled Aβ42 (fAβ42) (Fig 1C). Cytochalasin D, an inhibitor of actin polymerization, was used as negative control. Addition of 5 μg/ml of antibody 2D8, an antibody raised to the N‐terminus of the Aβ domain (Shirotani et al, 2007), significantly stimulated Aβ uptake in both N9 wt and N9 mu; however, phagocytosis remained less efficient in N9 mu (Fig 1C). As a negative control, we used antibody 6687 raised to the C‐terminus of APP (Capell et al, 2000). Although this antibody interacts with APP, it fails to increase Aβ uptake demonstrating that the specific interaction of 2D8 with Aβ is required to trigger the uptake (Fig 1C).
Figure 1. TREM2 deficiency reduces efficacy of antibody‐stimulated Aβ uptake by phagocytic cells.
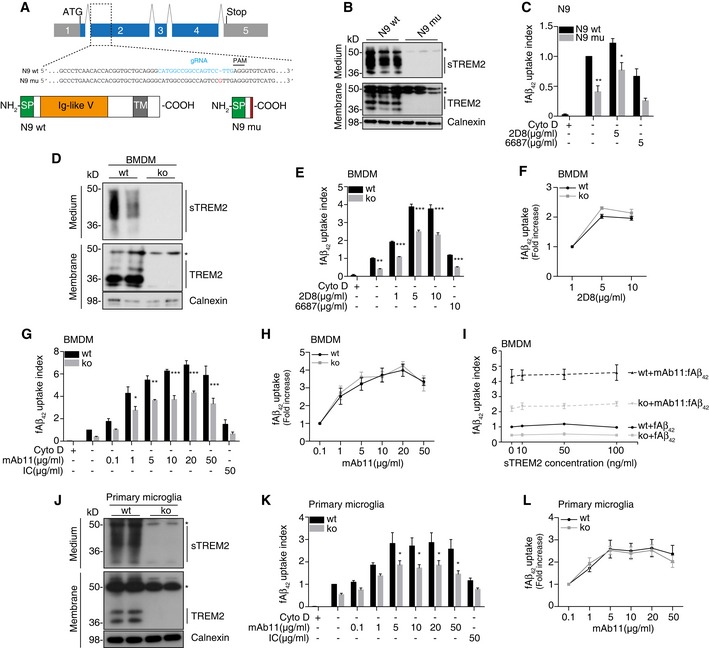
- Schematic of the mouse Trem2 locus and the TREM2 protein. Sequence alignment of wild‐type N9 (N9 wt) and TREM2 mutant N9 (N9 mu) surrounding the gRNA target site. The gRNA sequence is in cyan, and protospacer‐adjacent motif (PAM) is marked with a line. The single nucleotide insertion is labeled in red. Schematic representation of wild‐type TREM2 (NP_112544.1) and CRISPR/Cas9‐modified TREM2 (N9 mu). TM, transmembrane domain; SP, signal peptide.
- Western blot analysis of lysates and media from wt and mutant N9 cells (N9 wt /mu) using the antibody anti‐murine TREM2 (clone 5F4), which is raised against the murine TREM2 extracellular domain. sTREM2, soluble TREM2. *indicate unspecific bands. Calnexin was used as a loading control.
- Phagocytosis of 1 μM HiLyte™ Fluor 488 Aβ1‐42 (fAβ42) by N9 wt and N9 mu in the presence or absence of antibody 2D8 or the non‐binding antibody 6687. Cytochalasin D (CytoD, 10 mM) was used as control to verify phagocytic uptake. (n = 4, ± SEM; two‐way ANOVA, interaction P = 0.61, genotype P < 0.0001, treatment P = 0.0001; post hoc tests wt vs. mu for the following conditions: fAβ42 P = 0.0043, fAβ42‐2D8 P = 0.0436).
- Western blot of BMDM derived from wt and Trem2 knockout (ko) animals using antibody 5F4. *indicate unspecific bands.
- Phagocytosis of fAβ42 by BMDM from wt and Trem2 ko animals in the presence or absence of 2D8, or the non‐binding control antibody 6687. (n = 3, ± SEM; two‐way ANOVA, interaction P = 0.0005, genotype P < 0.0001, treatment P < 0.0001; post hoc tests wt vs. ko for the following conditions: fAβ42 P = 0.0021, fAβ42‐2D8 1 μg/ml P < 0.0001, fAβ42‐2D8 5 μg/ml P < 0.0001, fAβ42‐2D8 10 μg/ml P < 0.0001, fAβ42/6687 10 μg/ml P = 0.0007).
- Quantification of relative fAβ42 uptake to lowest antibody concentration used (n = 3, ± SEM).
- Phagocytosis of fAβ42 by BMDM from wt and Trem2 ko animals in the presence or absence of mAb11, or an isotype control antibody (IC). (n = 4, ± SEM; two‐way ANOVA, interaction P = 0.0223, genotype P < 0.0001, treatment P < 0.0001; post hoc tests wt vs. ko for the following conditions: fAβ42‐mAb11 1 μg/ml P = 0.0391, fAβ42‐mAb11 5 μg/ml P = 0.0069, fAβ42‐mAb11 10 μg/ml P < 0.0001, fAβ42‐mAb11 20 μg/ml P = 0.0001, fAβ42‐mAb11 50 μg/ml P < 0.0001).
- Quantification of relative fAβ42 uptake to lowest antibody concentration used (n = 4, ± SEM).
- Recombinant mouse sTREM2 does not rescue fAβ42 uptake in Trem2‐deficient BMDM. Increasing amounts of sTREM2 were added to the media of wt or Trem2 ko BMDM in the presence or absence of mAb11 (10 μg/ml) (n = 4, ± SEM).
- Western blot of primary microglia from wt or Trem2 ko animals using antibody 5F4. *indicate unspecific bands.
- Phagocytosis of fAβ42 by primary microglia from wt and Trem2 ko animals in the presence or absence of mAb11, or an isotype control antibody (IC). (n = 5, ± SEM; two‐way ANOVA, interaction P = 0.4797, genotype P < 0.0001, treatment P < 0.0001; post hoc tests wt vs. ko for the following conditions: fAβ42‐mAb11 5 μg/ml P = 0.0449, fAβ42‐mAb11 10 μg/ml P = 0.0370, fAβ42‐mAb11 20 μg/ml P = 0.0299, fAβ42‐mAb11 50 μg/ml P = 0.0120).
- Quantification of relative fAβ42 uptake to lowest antibody concentration used (n = 5, ± SEM).
To investigate antibody‐stimulated Aβ clearance in primary phagocytic cells, we first used BMDM derived from Trem2 ko animals (Turnbull et al, 2006) or wt controls. Western blotting confirmed the absence of sTREM2 and full‐length, membrane‐bound TREM2 in cells derived from Trem2 ko mice (Fig 1D). We then studied fAβ42 uptake in the presence of 0, 1, 5, or 10 μg/ml of antibody 2D8 or a non‐binding control antibody 6687. BMDM readily internalized fAβ42, which could be blocked entirely by addition of cytochalasin D (Fig 1E). Consistent with our previous findings (Kleinberger et al, 2014), BMDM derived from Trem2 ko mice showed a significantly reduced phagocytic activity compared to BMDM derived from wt mice (Fig 1E). Phagocytosis of fAβ42 in wt BMDM could be intensively stimulated by antibody 2D8 with a maximum stimulation at 5 μg/ml. Antibody 6687 even at a high concentration of 10 μg/ml had only a very minor effect (Fig 1E). Although antibody 2D8 significantly stimulated uptake of fAβ42 even in Trem2 ko BMDM, phagocytosis was less efficient compared to wt at all antibody concentrations used (Fig 1E). These findings suggest that fAβ42 uptake is greatly stimulated upon antibody binding in both wt and Trem2 ko BMDM; however, the overall uptake capacity is reduced in Trem2 ko BMDM. In line with that, there was no significant change in the relative increase of antibody‐stimulated uptake in both genotypes (Fig 1F) suggesting that antibody‐stimulated uptake per se is not reduced due to TREM2 deficiency.
Next, we used the monoclonal antibody mAb11, a murine IgG2a antibody, which has similar amyloid binding properties like the therapeutically used human IgG1 anti‐Aβ antibody Gantenerumab (Bohrmann et al, 2012; Lathuiliere et al, 2016) and performed titration experiments in the fAβ42 uptake assay using BMDM derived from wt or Trem2 ko mice. mAb11 but not an IgG2a isotype control (50 μg/ml) strongly stimulated phagocytosis of fAβ42 (Fig 1G). Interestingly, even very low concentrations of 0.1 μg/ml, which may be reached in the brain by peripheral antibody administration, were sufficient to trigger fAβ42 uptake. Uptake plateaued at 20 μg/ml and could not be further enhanced by using antibody concentrations up to 50 μg/ml (Fig 1G). In line with the data shown in Fig 1C and E, fAβ42 uptake by BMDM derived from Trem2 ko mice could be efficiently stimulated by increasing the amounts of mAb11 (Fig 1G and H). However, again the phagocytic capacity never reached the level of wt BMDM even at the highest antibody concentration used (Fig 1G). Thus, a monoclonal antibody with efficient target engagement similar to the therapeutically used Gantenerumab stimulates both TREM2‐dependent and TREM2‐independent engulfment of fAβ42.
To investigate whether sTREM2 could rescue reduced fAβ42 uptake of BMDM derived from Trem2 ko mice in a non‐cell autonomous manner, we supplemented the culture media of wt and ko cells with increasing amounts of recombinant mouse sTREM2. sTREM2 even added at a 10‐fold higher concentration as compared to its physiologically concentration in plasma of mice (approximately 10 ng/ml) did not rescue fAβ42 uptake efficacy of Trem2 ko cells in the presence or absence of mAb11 (Fig 1I). Thus, receptor‐mediated signaling in a cell autonomous manner appears to trigger fAβ42 uptake.
Finally, we further confirmed our findings in primary microglia derived from wt or Trem2 ko mice. Western blotting confirmed the absence of sTREM2 and full‐length, membrane‐bound TREM2 in primary microglia from Trem2 ko mice (Fig 1J). In line with our previous findings (Kleinberger et al, 2014), microglia from Trem2 ko mice showed significantly less uptake of fAβ42 (Fig 1K). Again, phagocytosis was similarly stimulated by mAb11 in a concentration‐dependent manner in both microglia derived from wt and microglia derived from Trem2 ko mice (Fig 1K and L). In line with the above data derived from N9 cells and BMDM (Fig 1C, E, and G), total uptake capacity was reduced at each antibody concentration in Trem2 ko microglia (Fig 1K).
Increased Fcγ‐receptors expression and enhanced Syk phosphorylation in TREM2‐deficient BMDM
To investigate whether reduced phagocytic capacity but similar increases in antibody‐stimulated uptake in the absence of TREM2 may be due to compensatory mechanisms, we first investigated expression of Fcγ‐receptors (FcγR), which are of central importance for antibody‐mediated phagocytosis. Compared to wt BMDM, cell surface FcγRI, FcγRIIB, and (or) FcγRIII were significantly increased in Trem2 ko cells (Fig 2A and B). Correspondingly, mRNA levels of the respective FcγR (I, IIB, III) were all significantly increased in Trem2 ko cells (Fig 2C). mRNA level of FcγRIV was also increased, but it did not result in enhanced cell surface protein level (Fig 2A–C). To further validate the compensatory mechanisms, downstream signaling was studied. It is well known that upon stimulation of TREM2 as well as Fcγ‐receptors, Syk gets phosphorylated and activates a variety of downstream signaling cascades (Crowley et al, 1997; Paradowska‐Gorycka & Jurkowska, 2013; Ulrich & Holtzman, 2016). As expected, Syk phosphorylation strongly increases upon incubation of wt BMDM with 2D8‐bound Aβ, which was not the case when using an isotype control antibody together with Aβ (Fig 2D and E). Interestingly, phosphorylation of Syk is further increased upon stimulation with antibody‐bound Aβ in the absence of TREM2 (Fig 2D and E).
Figure 2. Increased Fcγ‐receptors expression and enhanced Syk phosphorylation in TREM2‐deficient BMDM .
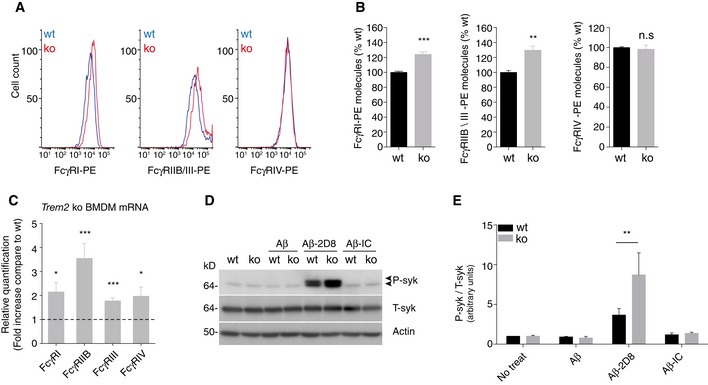
- Representative histograms for Fcγ‐receptors‐PE (FcγR‐PE) expression levels as used for quantification. Stacked histograms for log PE fluorescence intensity of wt and Trem2 ko BMDM are shown for the respective FcγR (I, IIB/III, and IV).
- Relative quantification of cell surface levels of FcγR molecules. Absolute number of cell surface FcγR‐PE molecules was determined by the BD QuantiBRITE® method (see methods section for details) and normalized to expression levels of the respective wt control. (n = 4, ± SEM, t‐test, two‐tailed; wt vs. ko: FcγRI‐PE P = 0.0008, FcγRIIB/III‐PE P = 0.0033, FcγRIV‐PE P = 0.7001).
- mRNA levels of FcγR are increased in Trem2 ko BMDM. Fold changes of the respective FcγR (I, IIB, III, IV) mRNA levels in Trem2 ko BMDM were determined by quantitative real‐time PCR. (n = 6, ± SEM; one‐sample t‐test, two‐tailed; wt vs. ko: FcγRI P = 0.0118, FcγRIIB P = 0.0006, FcγRIII P = 0.0004, FcγRIV P = 0.0284)
- Phosphorylated Syk (P‐Syk) and total Syk (T‐Syk) levels were determined by Western blotting in lysates from wt and Trem2 ko BMDM after 1 h treatment with Aβ alone, together with antibody 2D8 (Aβ‐2D8), or an isotype control (Aβ‐IC). Actin was used as a loading control.
- Quantification of P‐Syk normalized to T‐Syk. (n = 4, ± SEM; 2 way ANOVA, interaction P = 0.0490, genotype P = 0.0898, treatment P < 0.0001. Bonferroni‐corrected pair‐wise post hoc tests, **P = 0.0075 vs. wt).
Source data are available online for this figure.
Together with increased levels of FcγR, this may suggest a compensatory increase in TREM2‐independent antibody/antigen uptake pathways probably explaining why Trem2 ko cells still respond to increasing concentrations of anti‐Aβ antibodies.
TREM2 deficiency reduces antibody‐mediated amyloid plaque clearance
Next, we used an ex vivo model (Fig 3A), which would allow monitoring antibody‐dependent amyloid plaque clearance under controlled and comparable conditions (Bard et al, 2000). We used cryosections of whole brains from APP/PS1 mice (Radde et al, 2006) at 6 months of age, a time point where these mice are known to exhibit a high amyloid plaque burden within the brain (Radde et al, 2006). mAb11, but not the isotype control, binds selectively to the methoxy‐X04‐positive amyloid plaques (Fig 3B). When wt BMDM (Fig 3C) or primary microglia (Fig 3D) were added to brain sections pre‐incubated with mAb11, we observed a clustering of CD68‐positive cells around methoxy‐X04‐labeled amyloid plaques. Moreover, cells clustering around amyloid plaques engulfed Aβ fibers, as shown by the intracellular methoxy‐X04 staining, which partially co‐localized with the lysosomal protein CD68 (Fig 3C and D).
Figure 3. Engulfment of fibrillar Aβ by BMDM and primary microglia.
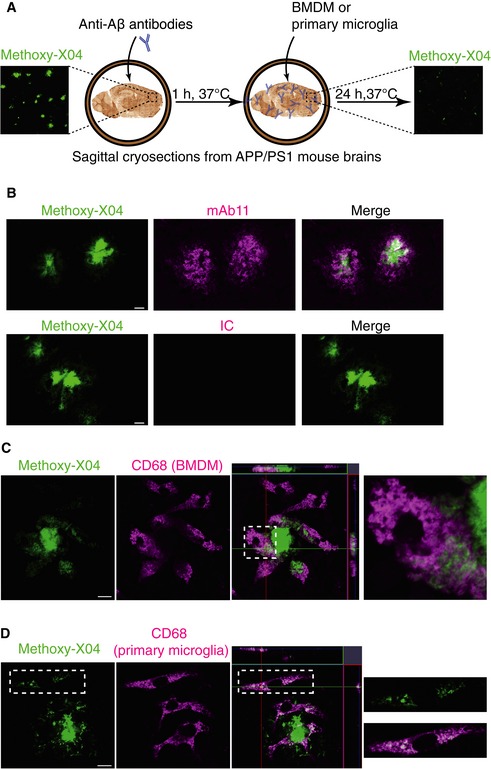
-
AA schematic shows 10‐μm cryosections of unfixed brain from 6‐month‐old APP/PS1 mice, which show a high amyloid plaque burden (right panel with methoxy‐X04 staining), were incubated with antibody for 1 h, followed by adding BMDM or primary microglia on top of the sections. After 24 h incubation, sections were analyzed by immunostaining or immunoblotting.
-
BmAb11 but not the isotype control (IC) co‐localized with methoxy‐X04. Scale bar: 10 μm.
-
C, DBMDM (C) or primary microglia (D) were cultured on cryosections pre‐incubated with mAb11 (1 μg/ml). After 24 h, sections were processed for immunostaining using antibody against CD68 to identify myeloid cells and methoxy‐X04 staining to visualize Aβ. Note that both cell types internalize Aβ into intracellular vesicles (right panels show enlargement of insets). Scale bar: 10 μm.
We next used the ex vivo assay to investigate TREM2‐dependent antibody‐stimulated amyloid plaque clearance. When cryosections of APP/PS1 mice were incubated with BMDM derived from wt or Trem2 ko mice, a significant TREM2‐dependent reduction in amyloid plaque clearance was observed (Fig 4A and B). Upon pre‐incubation of cryosections with mAb11 (1 μg/ml), amyloid plaque clearance by wt BMDM was significantly stimulated (Fig 4A and B). In line with the results in Fig 1, TREM2‐deficient BMDM were significantly less efficient in clearing amyloid plaques than wt BMDM under the same experimental conditions (Fig 4A and B). Cell densities in the different experiments were similar, as assessed by CD68 staining on consecutive slices (Fig 4C and D). Reduced amyloid plaque clearance by BMDM derived from the Trem2 ko animals upon antibody stimulation was confirmed by Western blotting of total protein lysates generated from cryosections after termination of the experiment. In line with the experiments in Fig 4A and B, mAb11 caused a stronger reduction in Aβ signals on Western blots in the presence than in the absence of TREM2. However, even in the absence of TREM2, a reduction in Aβ was observed with mAb11 pre‐incubation as compared to the no‐antibody control (Fig 4E).
Figure 4. TREM2 deficiency reduces antibody‐mediated amyloid plaque clearance.
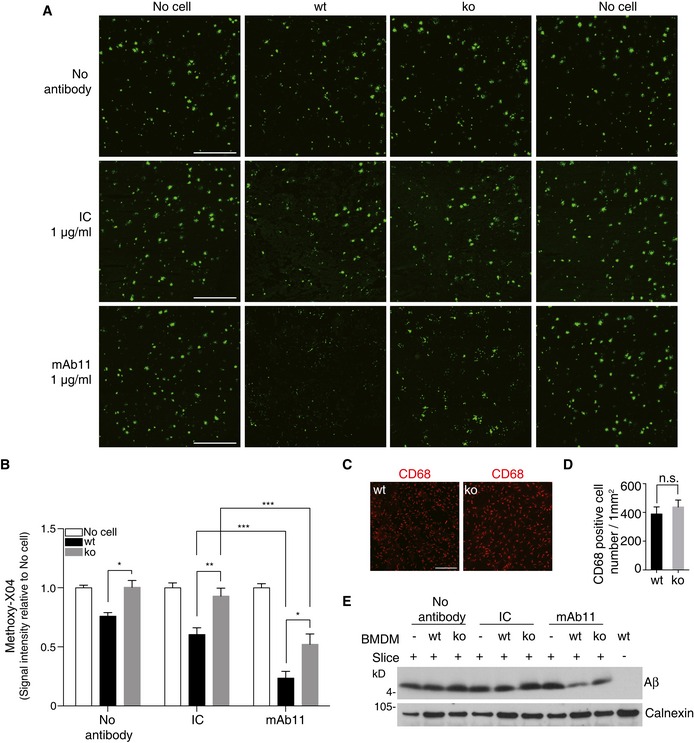
-
ABMDM from wt or Trem2 ko mice were cultured on APP/PS1 mice brain cryosections incubated with or without mAb11 (1 μg/ml) or an isotype control (IC; 1 μg/ml) for 24 h. Sections were then probed with methoxy‐X04. Scale bar, 500 μm.
-
BThe amyloid plaque load was quantified from the entire sagittal section. Sections incubated with medium (no cell) were set as baseline. (n = 6, ± SEM; two‐way ANOVA, interaction P < 0.0001, genotype P < 0.0001, treatment P < 0.0001; Tukey's multiple comparisons tests; wt vs. ko for the following conditions: no antibody P = 0.0304, IC P = 0.0049, mAb11 P = 0.0212; wt: IC vs. wt: mAb11 P = 0.0008; ko: IC vs. ko: mAb11 P = 0.0001).
-
C, DEqual numbers of wt and Trem2 ko BMDM were added, and cell numbers were analyzed after termination of experiments by quantifying the CD68‐positive cells on top of the sections. (n = 4, ± SEM; t‐test; n.s., non‐significant, P = 0.5004). Scale bar, 200 μm.
-
EAβ was extracted by urea buffer from replicate slices of the experiment shown in (A), and total Aβ was identified by Western blotting.
Source data are available online for this figure.
Improvement of amyloid plaque clearance by elevated antibody concentrations
We next titrated mAb11 with the aim to compare the efficacy of antibody‐mediated TREM2‐dependent amyloid plaque clearance. Cryosections from brains of APP/PS1 mice were pre‐incubated with increasing concentrations of mAb11 from 0.001 to 5 μg/ml before incubation with BMDM derived from wt or Trem2 ko mice. In line with the fAβ42 uptake assays described in Fig 1, this revealed a concentration‐dependent clearance of amyloid plaques (Fig 5A and B). Comparison of the extend of methoxy‐X04‐positive amyloid labeling after clearance by BMDM derived from wt or Trem2 ko mice demonstrates that antibody‐mediated clearance can occur in the absence of TREM2 (Fig 5A and B), similar to the uptake of fAβ42 shown in Fig 1. However, the total capacity to engulf amyloid plaques is reduced in Trem2 ko BMDM (Fig 5A and B). Of note, a statistically significant effect on amyloid plaque clearance is observed in Trem2 ko BMDM at 0.1 μg/ml, a concentration which is therapeutically reachable in brain by appropriate dose adjustment (Bohrmann et al, 2012; Lathuiliere et al, 2016). Taken together, our finding suggests that patients with compromised TREM2 function may require a higher dose of the therapeutic antibody to achieve efficient Aβ clearance.
Figure 5. Compensation of reduced amyloid plaque clearance by elevated antibody dose.
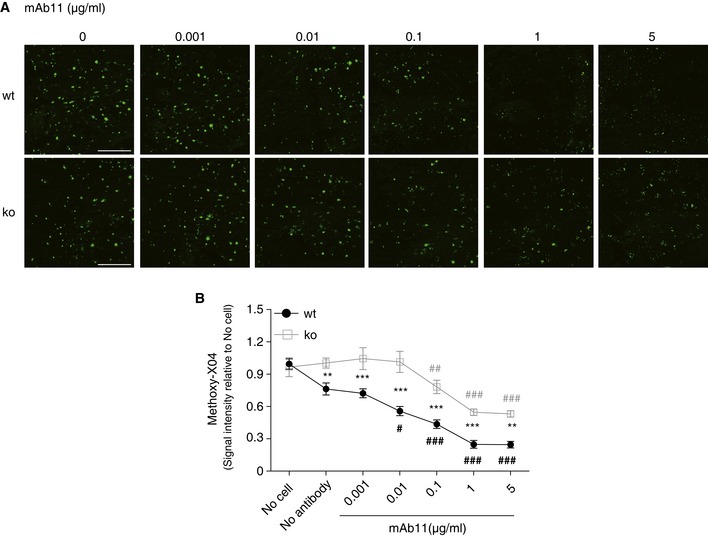
- Cryosections from unfixed brain of 6‐month‐old APP/PS1 mice were pre‐incubated with increasing concentrations of mAb11 (0.001, 0.01, 0.1, 1, 5 μg/ml). BMDM from wt or Trem2 ko mice were added for 24 h. Sections were stained with methoxy‐X04. Scale bar, 500 μm.
- Methoxy‐X04 signals were quantified from the entire sagittal section. (n = 5, ± SEM; two‐way ANOVA, interaction P = 0.0082, genotype P < 0.0001, treatment P < 0.0001. Fisher's LSD post hoc comparisons; * show statistics between wt and ko under the same experimental condition. # in black shows wt compares to no‐antibody stimulation; # in gray shows ko compares to no‐antibody stimulation; wt vs. ko for the following conditions: no antibody P = 0.0053, mAb11 0.001 μg/ml P = 0.0003, mAb11 0.01 μg/ml P < 0.0001, mAb11 0.1 μg/ml P = 0.0001, mAb11 1 μg/ml P = 0.0007, mAb11 5 μg/ml P = 0.0011; following conditions compare to wt/no antibody: wt/mAb11 0.01 μg/ml P = 0.0166, wt/mAb11 0.1 μg/ml P = 0.0002, wt/mAb11 1 μg/ml P < 0.0001, wt/mAb11 5 μg/ml P < 0.0001; following conditions compare to ko/no antibody: ko/mAb11 0.1 μg/ml P = 0.0099, ko/mAb11 1 μg/ml P < 0.0001, ko/mAb11 5 μg/ml P < 0.0001.
Discussion
A pivotal role of microglia and inflammatory mechanisms in AD and other neurodegenerative disorders, which was already emphasized early during AD research (McGeer & Rogers, 1992), was recently strongly supported by genetic evidence (Villegas‐Llerena et al, 2016). Specifically, rare heterozygous mutations in TREM2, which within the brain is exclusively expressed in microglia cells, dramatically increase the risk for late onset AD in a magnitude similar to ApoE ε4 (Guerreiro et al, 2013; Jonsson et al, 2013). Although TREM2 is well known to be involved in phagocytosis and removal of apoptotic neurons (Takahashi et al, 2005; Colonna et al, 2007; Hsieh et al, 2009), the pathological consequences of a TREM2 loss of function in the context of AD pathogenesis are highly controversial. While Aβ was shown to be engulfed at least to some extend in a TREM2‐dependent manner in cultured cells (Kleinberger et al, 2014), experiments on mouse models for AD pathology reached surprising and opposite results. On the one hand, reduction in TREM2 ameliorated several aspects of AD pathology, including inflammation, astrocytosis, and amyloid plaque burden (Jay et al, 2015), while on the other hand, Trem2 deficiency leads to exacerbated disease pathology including increased amyloid plaque burden (Wang et al, 2015). However, attenuated Aβ engulfment by microglia was thought to be due to reduced migration and survival and thus not directly associated with phagocytic activity of microglial cells (Wang et al, 2015). Our findings may now provide direct evidence that TREM2 is required at least partially for Aβ engulfment and clearance. Even in the absence of antibody stimulation, we consistently find reduced Aβ uptake and amyloid plaque clearance in all model systems used. Potential survival deficits in Trem2 ko cells do not impact our results, since uptake was measured after 2 h of incubation in the phagocytosis assay. Moreover, equal amounts of cells were used in the ex vivo plaque clearance assay and cell density was assessed after termination of experiments ruling out differences due to survival or the total number of phagocytic cells.
After antibody binding to Aβ, uptake and amyloid plaque clearance increase in a concentration‐dependent manner in the presence or absence of functional TREM2, although the total uptake capacity of cells lacking TREM2 is reduced. This indicates that TREM2‐independent Fcγ‐receptor‐mediated pathways are intact and used in addition to TREM2‐dependent uptake mechanisms. Indeed, it has been shown previously that Syk phosphorylation is an important signaling event upon Fcγ‐receptors activation in macrophage phagocytosis (Greenberg et al, 1994; Crowley et al, 1997). Higher levels of FcγR and upregulated phosphorylated Syk in Trem2 ko BMDM upon stimulation with Aβ‐2D8 immune complexes may therefore suggest a compensatory upregulation of TREM2‐independent phagocytosis.
Our findings might also indicate that direct effects of TREM2 on amyloid plaque clearance are difficult to assess in vivo since in the absence of an antibody stimulus Aβ uptake is 4–5 fold lower and probably difficult to quantify. In addition, the mouse models used to study the effects of TREM2 on amyloid plaque clearance not only highly overexpress APP but also express rather aggressive familial AD‐associated mutations, which may override modulatory effects of TREM2. Moreover, since TREM2 affects survival of microglial cells (Wang et al, 2015; Wu et al, 2015), the age as well as the specific mouse model used for the investigation must be carefully considered. In that regard, it is interesting to note that at very early time points of amyloid plaque deposition, Aβ accumulation was similar in wt and Trem2 ko mice, but Trem2 ko mice showed reduced microglia accumulations around amyloid plaques (Wang et al, 2016).
Finally, our findings may be valid for future immunotherapeutic approaches. Anti‐Aβ immunotherapy is currently a promising and clinically advanced approach (Wisniewski & Goni, 2015). It not only lowers the amyloid plaque load, but also prevents de novo deposition of plaques and stabilized memory deficits at least to some extend in two recently reported clinical trials (Reardon, 2015). We now demonstrate that mAb11, a murine IgG2a antibody, which has similar amyloid binding properties like Gantenerumab (Lathuiliere et al, 2016) which is currently explored in clinical trials (Bohrmann et al, 2012), significantly stimulates Aβ engulfment even in the absence of TREM2. For successful immunotherapy, knowledge on microglial activity and survival in individual patients may be crucial for optimal outcome of the treatment. In that regard, we recently demonstrated that in AD patients, sTREM2 levels significantly increase very early before onset of AD and tend to decrease in later phases of the disease (Suarez‐Calvet et al, 2016). In addition to CSF biomarkers, microglial PET imaging may be required similar to amyloid PET imaging used in the clinic to select patients for enrollment into clinical trials and to in vivo prove the effects of treatment on amyloid plaque load. If microglial function and survival are reduced during aging in a TREM2‐dependent manner, one may also speculate to modulate TREM2 activity. Moreover, in case of attenuated TREM2 function, an increased dose of therapeutic antibodies may be required to compensate impaired plaque clearance.
Materials and Methods
Mice
All animal experiments were performed in accordance with local animal handling laws. APP/PS1 (Radde et al, 2006) and Trem2 knockout mice (Trem2 ko) (Turnbull et al, 2006) were maintained on a C57BL/6J background. For bone marrow extraction, adult mice (aged from 2 to 12 months, mixed gender) were euthanized by CO2 and then sacrificed by cervical dislocation. For primary microglia culture, postnatal day P0‐2 mice (mixed gender) were sacrificed by decapitation.
CRISPR/Cas9‐mediated genome engineering in N9
The murine microglia cell line N9 (Sessa et al, 2004) was maintained in Dulbecco's modified Eagle's medium (DMEM) + GlutaMAX™ (Life Technologies) with 10% (v/v) fetal calf serum (FCS; Sigma‐Aldrich) and 100 U/ml penicillin, 100 μg/ml streptomycin. All cells were mycoplasmas free. N9 cells were transfected with pSpCas9(BB)‐2A‐Puro V2.0 (PX459; gift from Feng Zhang; Addgene plasmid # 62988) using the Nucleofector™ SF‐Kit for 4D‐Nucleofector™ (Lonza) according to manufacturer's recommendations. Twenty‐four hours post‐transfection, cells were selected with 4 μg/ml puromycin for 2 days. Single cell clones were then cultured with normal culture medium, followed by screening for genetic modifications in Trem2 by PCR amplification and missense detection using T7 endonuclease I (NEB). Mutations were confirmed by direct sequencing (GATC‐Biotech).
Bone marrow‐derived macrophages culture
Bone marrow‐derived macrophages (BMDM) were prepared essentially as described before (Marim et al, 2010). Briefly, mice femurs and tibias were dissected and sterilized with 70% ethanol. The bones were flushed with a syringe filled with advanced RPMI 1640 (Life Technologies) to extrude bone marrow. Cell suspensions were filtered through a 100‐μm cell strainer and incubated for 2 min in ACK lysis solution (Thermo Fisher Scientific) to lyse red blood cells. The bone marrow cells were cultured and differentiated in advanced RPMI 1640 supplemented with 2 mM l‐glutamine, 10% (v/v) FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml murine M‐CSF (R&D System) using non‐tissue culture treated Petri dishes (BD Biosciences). Three days after seeding, fresh murine M‐CSF (final concentration of M‐CSF 50 ng/ml) was added and media was changed at day 5 of culture. BMDM were used for experiments at day 7 in vitro. Immunostaining with CD68 (Figs 3C and 4C) as well as flow cytometry (see below) using anti‐CD11b antibody (Fig EV1A) was used to confirm the identity of these cells.
Figure EV1. BMDM and primary microglia are positive for CD11b.
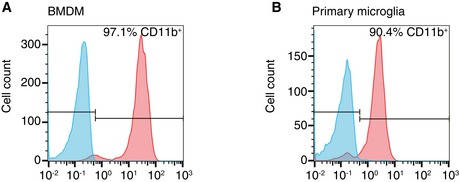
- About 97.1% bone marrow‐derived macrophages (BMDM) were positive for CD11b.
- Primary microglia from mixed glial culture were 90.4% positive for CD11b.
Primary microglial culture
Primary murine microglial cultures were prepared as previously described (Fleisher‐Berkovich et al, 2010). Briefly, mixed glial cultures were prepared from the whole brain devoid meninges and cerebellum of P0‐2 mice and cultured in DMEM with Glutamax I (Life Technologies), supplemented with 10% (v/v) FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. About 50% of the medium was replaced every other day, and 10 ng/ml murine M‐CSF (R&D System) was supplemented at day 7 in culture. After 10 days in culture, cells were shaken using horizontal orbital shaker at 200 rpm for 30 min to isolate microglia. The medium containing microglia was pelleted at 200 g for 8 min and reseeded to corresponding plates for experiments. Cell identity was confirmed using anti‐CD68 (Fig 3D) and anti‐CD11b (Fig EV1B) antibodies.
Flow cytometry
Bone marrow‐derived macrophages or primary microglial cells were suspended in FACS buffer (Hank's balanced salt solution, no calcium, no magnesium; 0.2% bovine serum albumin; Thermo Fisher Scientific). Samples were labeled with anti‐CD11b antibodies (M1/70), APC‐Cy7® conjugated (1:100; Life Technologies) for 30 min at 4°C. Data were acquired using MACSQuant® VYB (Miltenyi Biotec) and analyzed by FlowJo software.
To quantify absolute expression levels for the different cell surface Fcγ‐receptors (FcγR), BMDM were labeled with PE anti‐FcγRII/III (1:100; BD Bioscience), PE anti‐FcγRI (1:100; R&D System) or PE anti‐FcγRIV (1:100; BD Bioscience) at 4°C for 30 min. Data were acquired on a FACSVerse flow cytometer (BD, USA) and analyzed using FACSuite software (BD, USA). Absolute numbers of PE anti‐FcγR molecules per cell were determined using QuantiBRITE (BD Bioscience) PE calibration beads following the analysis strategy according to the manufacturer's protocol. Gates were set according to unlabeled and isotope controls. Absolute PE anti‐FcγR molecules counts were performed per individual sample and normalized to the mean of the wt control group.
Cell lysis and immunoblotting
Western blot analysis of membrane‐bound TREM2 was performed as previously described (Kleinberger et al, 2014). For Western blot analysis of Aβ, lysates were prepared with 8 M urea lysis buffer (8 M Urea, 50 mM Tris–HCL pH 7.4) freshly supplemented with a protease inhibitor cocktail (Sigma‐Aldrich) and incubated on ice for 30 min. The lysate was mixed with Laemmli sample buffer supplemented with beta mercaptoethanol, and equal amounts of protein were separated by SDS–PAGE. After transfer onto polyvinylidene difluoride membranes (Amersham Hybond P 0.45 PVDF, GE Healthcare Life Science) or nitrocellulose membranes (GE Healthcare Life Science), blots were blocked for 1 h with I‐Block™ (Thermo Fisher Scientific) and exposed to 5F4 for TREM2 and sTREM2 or 2D8 (Shirotani et al, 2007) for Aβ. Signals were visualized with HRP‐conjugated secondary antibodies using ECL kit (Thermo Fisher Scientific).
Antibodies
For in vitro and ex vivo phagocytosis assay, the following antibodies were used: rat monoclonal antibody 2D8 against Aβ1–16 (Shirotani et al, 2007), rabbit polyclonal antibody 6687 against cytosolic domain of APP (Capell et al, 2000), mouse IgG2a isotype control (Sigma‐Aldrich), and mouse monoclonal mAb11 (IgG2a (Bohrmann et al, 2012; Lathuiliere et al, 2016)). mAb11 is a closely related clone to the clinically used anti‐Aβ antibody Gantenerumab that binds preferentially to Aβ 40/42 aggregates, namely Aβ fibrils and oligomers via a conformational epitope comprising N‐terminal and central epitopes (more details on epitopes in (Bohrmann et al, 2012; Lathuiliere et al, 2016)). Affinity parameters were determined before for both antibodies by surface plasmon resonance (Biacore) and revealed equilibrium dissociation constant (KD) values for mAb11 between 0.14 and 0.67 nM, which were determined for Aβ40 and Aβ42, which is similar to Gantenerumab with a KD of 0.6 nM (Bohrmann et al, 2012; Lathuiliere et al, 2016). Binding and stability of bound antibody to soluble Aβ monomers is substantially lower. We measured a KD of 18 nM for Gantenerumab binding to Aβ monomers with a notably kinetically instable complex with monomers showing a rapid off‐rate. In more details, the kinetically most stable complex formation with Gantenerumab was observed for Aβ fibrils (kd 2.8 × 10−4 1/s) and Aβ oligomers (kd 4.9 × 10−4 1/s), whereas for monomeric Aβ, a higher dissociation rate was observed (kd 1.2 × 10−2 1/s) suggesting rapid exchange of antibody‐bound monomeric Aβ. Thus, mAb11 binding parameters can be considered almost identical to Gantenerumab. Selective binding to aggregated Aβ40/42 is in agreement with demonstrated amyloid plaque binding measured by immunofluorescence in vitro and in vivo. Selective and sensitive binding to amyloid plaques in human AD and transgenic mouse brain tissues is again very comparable for Gantenerumab and mAb11 with positive immundecoration detectable at low concentration of 10 ng/mL and plaque binding in vivo in transgenic mice (Bohrmann et al, 2012; Lathuiliere et al, 2016).
For immunofluorescence staining of BMDM and primary microglia, staining was performed using rat anti‐CD68 (1:500; AbD Serotec) and goat anti‐rat Alexa Fluor 555 (1:200; Thermo Fisher Scientific).
For Western blotting of TREM2, we raised a rat monoclonal antibody (clone 5F4; 1:50) against the extracellular domain of murine TREM2 (Creative Biomart; Trem2‐3276M). Further used antibodies were rat antibody 2D8 (1:100) (Shirotani et al, 2007) against Aβ, rabbit anti‐calnexin (1:3,000; Enzo Life Sciences), secondary antibodies HRP‐conjugated goat anti‐rat (1:5,000; Santa Cruz Biotechnology), and goat anti‐rabbit IgG (1:10,000; Promega).
In vitro Aβ phagocytosis assays
Phagocytosis assays with N9, BMDM, and primary microglia were performed as described (Kleinberger et al, 2014) with minor adjustments. Briefly, HiLyte™ Fluor 488 Aβ1‐42 (Anaspec) was aggregated overnight at 37°C with agitation. 3 × 104 N9 cells, BMDM, or 2 × 104 primary microglia were plated in poly‐D‐lysine‐coated black‐walled 96‐well plates (Greiner bio‐one). Pre‐aggregated HiLyte™ Fluor 488 Aβ1‐42 (fAβ42) was incubated with antibodies for 1 h at 37°C. Fibril Aβ42 or antibody‐fAβ42 complexes were added to a final concentration 1 μM and incubated for 2 h. As negative control, 10 mM cytochalasin D was added 30 min before addition of fAβ42 or antibody‐fAβ42. Before measurement, medium was removed and extracellular fAβ42 was quenched with 100 μl 0.2% trypan blue in phosphate‐buffered saline (PBS), pH 4.4 for 1 min. After aspiration, fluorescence signals were measured at 485‐nm excitation/538‐nm emission using a Fluoroskan Ascent™ Microplate Fluorometer (Lab Systems).
For the experiment shown in Fig 1I, BMDM were incubated with recombinant mouse sTREM2 (Creative Biomart) overnight before adding fAβ42 or antibody‐fAβ42 complexes.
Ex vivo plaque clearance assay
Ex vivo plaque clearance assays were performed as previously described (Bard et al, 2000) with the following slight modifications. 6‐month‐old APP/PS1 transgenic mice (mixed gender) were perfused with PBS. Brains were separated into two hemispheres and snap‐frozen using dry ice powder. Frozen brains were sectioned into 10‐μm sagittal sections using a cryostat (Leica) and collect onto poly‐D‐lysine‐coated round glass coverslips (15 mm). Sections were dried at room temperature for 2 h followed by incubation with antibodies (isotype control (IC) or anti‐Aβ) in culture medium for 1 h at 37°C. BMDM or primary microglia cells were seeded at densities of 3×105 cells per well in 12‐well plates and incubated at 37°C with 5% CO2 for 24 h. After incubation, cultures were fixed with 4% paraformaldehyde for 15 min and stained with methoxy‐X04 (2 μg/ml; Tocris Bioscience). In the indicated experiments, the fixed cultures were permeabilized with 0.1% Triton X‐100 for 3 min and stained with rat anti‐CD68 antibody overnight at 4°C followed by methoxy‐X04 staining.
Images were acquired on a LSM700 confocal microscope with Z stacks and tile scan to cover the entire sagittal sections using the Zen 2009 imaging software (Zeiss). Maximum intensity projection from each sagittal sections was then analyzed by ImageJ (Schneider et al, 2012) to obtain methoxy‐X04 signal intensities. In Fig 4A and B, four consecutive slices were set as a group, and signals from slices incubated with no cell were set as base line. Methoxy‐X04 signal intensities from slices either incubated with wt or incubated with Trem2 ko BMDM were compared to base line. In Fig 5A and B, eight consecutive slices (#1–8) were used as a group and signal intensities from slices #1 and 8 were set as base line. Signals from slices #2 to 6, which were incubated with increasing concentrations of mAb11 (0–5 μg/ml) in the presence of BMDM, were compared to base line.
Quantitative real‐time PCR analysis
Total RNA was isolated from BMDM using the RNeasy Mini Kit (Qiagen) according the manufacturer's instructions. The concentration and purity of RNA was determined using the NanoPhotomer (Implen). Equal amounts of RNA were reverse‐transcribed into cDNA using High‐capacity cDNA Reverse Transcription kit (Thermo Fisher). The expression level of Fcγ‐receptors (I, IIB, III, IV) was analyzed by Taqman® real‐time PCR assay. Probes target to FcγΙ (Mm00438874_m1, Thermo Fisher Scientific), or FcγΙΙB (Mm00438875_m1, Thermo Fisher Scientific), or FcγΙΙΙ (Mm00438882_m1, Thermo Fisher Scientific), or FcγΙV (Mm00519988_m1, Thermo Fisher Scientific) were mixed with 1:5 diluted cDNA, added to the Taqman® master mix (Thermo Fisher Scientific), and amplified with the 7500 fast real‐time PCR system (Applied Biosystems). Geometric mean of two housekeeping genes, Gusb (Mm01197698_m1, Thermo Fisher Scientific) and Hsp90ab1 (Mm00833431_g1, Thermo Fisher Scientific), which highly correlate with each other, was used as endogenous control. Fold change of mRNA in Trem2 ko BMDM was calculated using wt BMDM as reference sample.
Statistics
All statistical analysis was performed using Prism 6 (GraphPad). Data are presented as mean ± SEM with biological repeats. Data from the phagocytosis assays were analyzed by two‐way ANOVA with Bonferroni correction for multiple comparison. Cell surface FcγR relative levels were analyzed by Student's t‐test. Fold change of mRNA was analyzed by one‐sample t‐test of its log2 value. Western blot quantification in Fig 2C was analyzed by two‐way ANOVA with Bonferroni correction for multiple comparison. Methoxy‐X04 signal intensity presented in Figs 4B and 5B was analyzed by two‐way ANOVA, with Tukey's and Fisher's least significant difference (LSD) post hoc tests for pair‐wise comparisons, respectively. All tests were 2‐tailed, with a significant level of α = 0.05.
Author contributions
XX, GK, and CH designed the study and interpreted the results. XX performed all experiments except generating mutant N9 cells, which was performed by GW and AC. The quantification of cell surface Fcγ‐receptors expression level was done by AL. BB, and IK provided antibodies. RF generated monoclonal antibody against TREM2 (clone 5F4). FM and GK provided bone marrow cells. CH wrote the manuscript with input of all co‐authors.
Conflict of interest
C.H. is an advisor of F. Hoffmann‐La Roche. I.K. and B.B. are full‐time employees at Roche. All other authors declare that they have no conflict of interest.
The paper explained.
Problem
Immunotherapeutic approaches are currently the most advanced treatments for Alzheimer's disease (AD). Antibodies against amyloid β‐peptide (Aβ) bind to amyloid plaques and trigger their clearance by microglia via Fc receptor‐mediated phagocytosis. Rare variants in the triggering receptor expressed on myeloid cells 2 (TREM2) increase the risk for late onset AD. Since a loss of Trem2 function reduces the ability of microglia to engulf Aβ, we investigated whether antibody‐mediated Aβ clearance may be affected by TREM2 deficiency.
Results
Anti‐Aβ antibodies stimulate Aβ uptake and amyloid plaque clearance in a dose‐dependent manner in the presence or absence of TREM2. Elevated levels of Fc receptors in TREM2‐deficient cells indicate compensatory mechanisms, which stimulate the phagocytic activity in the absence of TREM2. Albeit compensatory increases of Fc receptor‐mediated phagocytosis, TREM2‐deficient phagocytic cells, showed significantly reduced uptake of antibody‐bound Aβ and as a consequence reduced clearance of amyloid plaques. Titration experiments revealed that reduced efficacy of amyloid plaque clearance by Trem2 knockout cells can be improved by elevating the concentration of therapeutic antibodies.
Impact
Our findings are of direct therapeutic relevance, since they suggest that patients with phagocytic deficits caused by loss of TREM2 function may need a higher immunotherapeutic antibody dose to efficiently clear amyloid plaques. Thus, monitoring microglia function in patients at risk for AD may facilitate efficient immunotherapeutic strategies. In addition, our study unambiguously proofs that TREM2 function is required for Aβ uptake and amyloid plaque clearance.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2D
Source Data for Figure 4E
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy), the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013)/ERC Grant Agreement No. 321366‐Amyloid, the general legacy of Mrs. Ammer, the MetLife award, and the Cure Alzheimer's Fund. We would like to thank Dr. Feng Zhang (Broad Institute) for providing PX459V2.0. We thank Dr. Marco Colonna for providing Trem2 knockout mice. We thank Samira Parhizkar for screening antibodies against TREM2 and Dr. Marc Suárez‐Calvet for critically reading the manuscript.
EMBO Mol Med (2016) 8: 992–1004
References
- Barao S, Moechars D, Lichtenthaler SF, De Strooper B (2016) BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer's Disease. Trends Neurosci 39: 158–169 [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K et al (2000) Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919 [DOI] [PubMed] [Google Scholar]
- Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF et al (2012) Gantenerumab: a novel human anti‐Abeta antibody demonstrates sustained cerebral amyloid‐beta binding and elicits cell‐mediated removal of human amyloid‐beta. J Alzheimer's Dis 28: 49–69 [DOI] [PubMed] [Google Scholar]
- Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, Fenoglio C, Piaceri I, Archetti S, Bonvicini C et al (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35: 934.e7–934.e10 [DOI] [PubMed] [Google Scholar]
- Cady J, Koval ED, Benitez BA, Zaidman C, Jockel‐Balsarotti J, Allred P, Baloh RH, Ravits J, Simpson E, Appel SH et al (2014) TREM2 variant p. R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol 71: 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, Yuede CM, Galimberti D, Olivecrona G, Klein RS et al (2015) TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 129: 429–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A, Steiner H, Romig H, Keck S, Baader M, Grim MG, Baumeister R, Haass C (2000) Presenilin‐1 differentially facilitates endoproteolysis of the beta‐amyloid precursor protein and Notch. Nat Cell Biol 2: 205–211 [DOI] [PubMed] [Google Scholar]
- Cheret C, Willem M, Fricker FR, Wende H, Wulf‐Goldenberg A, Tahirovic S, Nave KA, Saftig P, Haass C, Garratt AN et al (2013) Bace1 and Neuregulin‐1 cooperate to control formation and maintenance of muscle spindles. EMBO J 32: 2015–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Turnbull I, Klesney‐Tait J (2007) The enigmatic function of TREM‐2 in osteoclastogenesis. Adv Exp Med Biol 602: 97–105 [DOI] [PubMed] [Google Scholar]
- Colonna M, Wang Y (2016) TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 17: 201–207 [DOI] [PubMed] [Google Scholar]
- Crowley MT, Costello PS, Fitzer‐Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VL, DeFranco AL (1997) A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J Exp Med 186: 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, Engelborghs S, Vandenbulcke M, Van Dongen J, Geerts N et al (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging 35(726): e711–e729 [DOI] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG et al (2013) A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med 369: 341–350 [DOI] [PubMed] [Google Scholar]
- Fleisher‐Berkovich S, Filipovich‐Rimon T, Ben‐Shmuel S, Hulsmann C, Kummer MP, Heneka MT (2010) Distinct modulation of microglial amyloid beta phagocytosis and migration by neuropeptides (i). J Neuroinflammation 7: 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S, Chang P, Silverstein SC (1994) Tyrosine phosphorylation of the gamma subunit of Fc gamma receptors, p72syk, and paxillin during Fc receptor‐mediated phagocytosis in macrophages. J Biol Chem 269: 3897–3902 [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C (2004) Take five–BACE and the gamma‐secretase quartet conduct Alzheimer's amyloid beta‐peptide generation. EMBO J 23: 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356 [DOI] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109: 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL et al (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med 212: 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suarez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzierl A, Mazaheri F et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6: 243ra286 [DOI] [PubMed] [Google Scholar]
- Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A et al (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 31: 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathuiliere A, Laversenne V, Astolfo A, Kopetzki E, Jacobsen H, Stampanoni M, Bohrmann B, Schneider BL, Aebischer P (2016) A subcutaneous cellular implant for passive immunization against amyloid‐beta reduces brain amyloid and tau pathologies. Brain 139: 1587–1604 [DOI] [PubMed] [Google Scholar]
- Marim FM, Silveira TN, Lima DS Jr, Zamboni DS (2010) A method for generation of bone marrow‐derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE 5: e15263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Rogers J (1992) Anti‐inflammatory agents as a therapeutic approach to Alzheimer's disease. Neurology 42: 447–449 [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D et al (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982–985 [DOI] [PubMed] [Google Scholar]
- Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX et al (2012) Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol 69: 198–207 [DOI] [PubMed] [Google Scholar]
- Paradowska‐Gorycka A, Jurkowska M (2013) Structure, expression pattern and biological activity of molecular complex TREM‐2/DAP12. Hum Immunol 74: 730–737 [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S et al (2006) Abeta42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: 940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen‐Hoerth C, Kertesz A, Bigio EH et al (2013) TREM2 in neurodegeneration: evidence for association of the p. R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon S (2015) Antibody drugs for Alzheimer's show glimmers of promise. Nature 523: 509–510 [DOI] [PubMed] [Google Scholar]
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA et al (2010) 11C‐PiB PET assessment of change in fibrillar amyloid‐beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double‐blind, placebo‐controlled, ascending‐dose study. Lancet Neurol 9: 363–372 [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson‐Wood K, Khan K et al (1999) Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400: 173–177 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F, Colonna M, Panina P, Meldolesi J (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci 20: 2617–2628 [DOI] [PubMed] [Google Scholar]
- Shirotani K, Tomioka M, Kremmer E, Haass C, Steiner H (2007) Pathological activity of familial Alzheimer's disease‐associated mutant presenilin can be executed by six different gamma‐secretase complexes. Neurobiol Dis 27: 102–107 [DOI] [PubMed] [Google Scholar]
- Suarez‐Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD et al (2016) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early‐stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med 8: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells‐2. J Exp Med 201: 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi RE (2015) TREM2 and risk of Alzheimer's disease‐Friend or Foe? N Engl J Med 372: 2564–2565 [DOI] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM‐2 attenuates macrophage activation. J Immunol 177: 3520–3524 [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Holtzman DM (2016) TREM2 Function in Alzheimer's disease and neurodegeneration. ACS Chem Neurosci 7: 420–427 [DOI] [PubMed] [Google Scholar]
- Villegas‐Llerena C, Phillips A, Garcia‐Reitboeck P, Hardy J, Pocock JM (2016) Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol 36: 74–81 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH et al (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S et al (2016) TREM2‐mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C (2006) Control of peripheral nerve myelination by the beta‐secretase BACE1. Science 314: 664–666 [DOI] [PubMed] [Google Scholar]
- Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S, Hornburg D, Evans LD, Moore S, Daria A et al (2015) eta‐Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 526: 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Goni F (2015) Immunotherapeutic approaches for Alzheimer's disease. Neuron 85: 1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Byers DE, Jin X, Agapov E, Alexander‐Brett J, Patel AC, Cella M, Gilfilan S, Colonna M, Kober DL et al (2015) TREM‐2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med 212: 681–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Barao S, Laga M, Bockstael K, Borgers M, Gijsen H, Annaert W, Moechars D, Mercken M, Gevaert K et al (2012) The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem 287: 25927–25940 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2D
Source Data for Figure 4E