

Abstract
Background
Microbial genomes at the National Center for Biotechnology Information (NCBI) represent a large collection of more than 35,000 assemblies. There are several complexities associated with the data: a great variation in sampling density since human pathogens are densely sampled while other bacteria are less represented; different protein families occur in annotations with different frequencies; and the quality of genome annotation varies greatly. In order to extract useful information from these sophisticated data, the analysis needs to be performed at multiple levels of phylogenomic resolution and protein similarity, with an adequate sampling strategy.
Results
Protein clustering is used to construct meaningful and stable groups of similar proteins to be used for analysis and functional annotation. Our approach is to create protein clusters at three levels. First, tight clusters in groups of closely-related genomes (species-level clades) are constructed using a combined approach that takes into account both sequence similarity and genome context. Second, clustroids of conservative in-clade clusters are organized into seed global clusters. Finally, global protein clusters are built around the the seed clusters. We propose filtering strategies that allow limiting the protein set included in global clustering.
The in-clade clustering procedure, subsequent selection of clustroids and organization into seed global clusters provides a robust representation and high rate of compression. Seed protein clusters are further extended by adding related proteins. Extended seed clusters include a significant part of the data and represent all major known cell machinery. The remaining part, coming from either non-conservative (unique) or rapidly evolving proteins, from rare genomes, or resulting from low-quality annotation, does not group together well. Processing these proteins requires significant computational resources and results in a large number of questionable clusters.
Conclusion
The developed filtering strategies allow to identify and exclude such peripheral proteins limiting the protein dataset in global clustering. Overall, the proposed methodology allows the relevant data at different levels of details to be obtained and data redundancy eliminated while keeping biologically interesting variations.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-016-1112-8) contains supplementary material, which is available to authorized users.
Keywords: Protein, Cluster, Clustering, Microbial, Procaryotic, Core-periphery, Multiscale, Multiresolution, Knowledge discovery, Data mining, Parallel processing, Parallel computing
Background
Microbial genomes at the National Center for Biotechnology Information (NCBI) represent a large collection of more than 35,000 assemblies from more than 5,000 species, with almost 40M unique proteins [1, 2]. Protein clustering is used to construct meaningful and stable groups of similar proteins to be analyzed and annotated, and serve as targets for efficient searching. There are several complexities associated with the data: the genomes in the dataset have different levels of sequence and assembly quality and large variation in sampling density; certain sets of related genomes, usually human pathogens, are densely sampled while other bacteria are less represented and sometimes sampled very coarsely (genomic and proteomic structure of a densely-sampled group of related strains is usually described by the concept of pan-genome [3–9]). Another factor contributing to the complexity of the analysis is a large variation in frequencies with which proteins from different families appear in genomes: “core proteins” occur at one end of the spectrum, unique proteins at another end, and “accessory proteins” in between (with some proteins partial in draft assemblies). In order to extract useful information from these complex data, the analysis needs to be performed at multiple levels of phylogenomic resolution and protein similarity, and an adequate sampling strategies.
Protein clusters are groups of similar (homologous) proteins that most likely share the same or similar function. Clustering procedure must possess a certain degree of stability and robustness and allow compression of information in comparison to the non-clustered representation. It is desirable that clusters consist of orthologs (protein coding regions that evolved from a common ancestral gene by speciation), while paralogs (genes related by duplication within a genome) stay in different clusters [10]. However, the ortholog-paralog distinction does not completely reflect the complexity of group relationships of homologous genes [11]. We make an effort to separate paralogs at the level of species-level genome groups (clades) using genomic context [12–18]. Since most microbial genomes at NCBI are draft genomes, local genomic context is utilized [19]. At the global level, we do not make a distinction between orthologous and paralogous proteins.
Here we present an efficient approach utilizing hierarchical clustering at several resolution levels. While large-scale hierarchical protein clustering is well-described in the literature [20–22], and methods for redundancy-elimination have been described by several authors [23–25], brute-force hierarchical clustering, even with a step of redundancy-elimination, becomes more expensive and less robust with the growth in the amount and complexity of data.
We construct protein clusters at three levels. First, in-clade protein clusters - tight protein clusters in groups of closely-related genomes (clades) are built. Then representaive proteins (clustroids) of conservative in-clade clusters are organized into seed global clusters. Clustroids of inclade clusters were selected as protein sequences providing minimal weighted average distance to other protein sequences in the clusters, where weight of each protein sequence was a number of coding regions in non-clonal genomes in the cluster encoding it. Finally, global protein clusters are built around the seed clusters. In-clade clustering with subsequent selection of clustroids and organizing them into seed global clusters provides a robust representation and high rate of compression in extended seed clusters. However, the proteins that are outside of the extended seed clustering set do not group together well. Processing of these proteins requires significant computational resources and results in a large number of questionable clusters. Such a pervasive behavior known as the core-periphery problem has been observed in many other areas of network analysis [26–28] where peripheral objects behaved very different from ones with high degree of centrality. We propose filtering strategies that allow limiting the protein set included in global clustering.
Methods
Microbial genomes with full and nearly-full genome representation and good quality are organized in groups of closely-related genomes (species-level clades) constructed using ribosomal protein markers [1, 29, 30], Non-redundant representative genomes are selected in the groups of near-clonal genomes in each clade using the complete-linkage hierarchical clustering algorithm based on pairwise genomic BLAST with 95 % identity cut-off (there is the following order of preferences in selection of a representative genome: (1) clade (species) reference or representative; (2) included in KEGG database; (3) an annotated genome).
We extended our basic clustering procedure described in [31]. The similarity of proteins is determined from the aggregated BLAST hits obtained by BLASTp [32, 33] with e-value 10−3. The sequences are considered related if the minimum coverage and minimum similarity conditions are satisfied. We required at least 80 % similarity with 85 % coverage in in-clade clustering and at least 50 % similarity with 70 % coverage in all global clustering steps.
In-clade clusters are constructed using a combined approach that takes into account both sequence similarity and local genome context [19]. First, sequence similarity clusters are calculated. Then, the genomic neighborhoods of proteins in each sequence-similarity cluster are analyzed using a moving window of 5-protein-length. Consequently, sub-clusters providing at least 3 out of 5 protein-similarity-cluster matches are selected (a protein map of local genomic neighborhood of the protein cluster containing the GTP-binding protein LepA (elongation factor) in Salmonella is shown in Fig. 1). Representaive proteins of inclade clusters (clustroids) were selected as protein sequences providing minimal weighted average distance to other protein sequences in the clusters, where weight of each protein sequence was a number of coding regions in non-clonal genomes in the cluster encoding it.
Fig. 1.
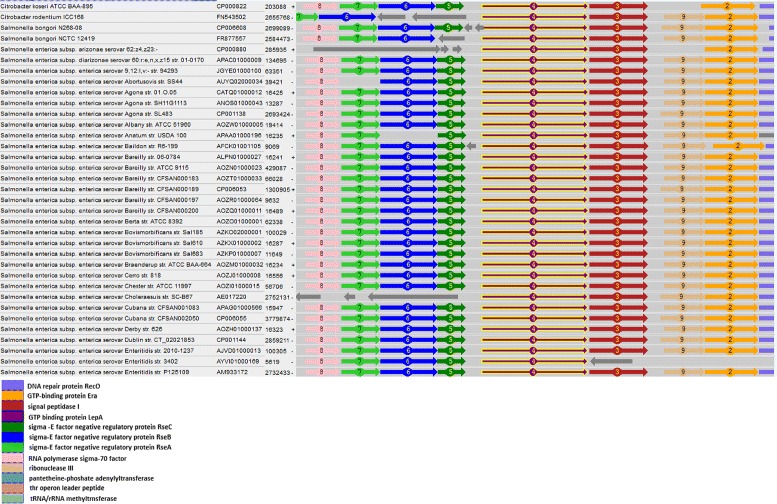
Local genomic neighborhood of the protein cluster containing the GTP-binding protein LepA (elongation factor) in Salmonella
Two algorithms were considered for building global clusters around the seed clusters. The modified hierarchical clustering algorithm utilized our basic procedure with the following modification: when two sub-clusters, one containing seed proteins and another one not, are merged, the latter is not used when new distances are determined. The second procedure allowed extension of the seed clusters by adding non-seed proteins to the nearest seed cluster if they are compatible with seed clustroids there.
UCLUST and USEARCH [25] were used at different proceeding stages for redundancy elimination. In all cases we use values wordlength 16, slots 400000009, maxrejects 64, maxaccepts 8. The coverage and identity thresholds are selected differently for different steps: (1) Representatives from groups of near-identical sequences are selected before in-clade clustering is performed using coverage 100 % with identity 98 %; (2) Tight groups of proteins are formed for global clustering using coverage 85 % and identity 80 % approximately corresponding to parameters used in in-clade clustering. (3) Filtering which allows to find distant neighbors of the seed proteins, is performed using coverage 70 % with identity as low as 10 % (The built-in limitations of USEARCH prevent it from obtaining overly weak hits even if the the identity threshold is not set or set too low).
Many processing steps, such as computing BLAST hits, are naturally parallel. However, parallelization of clustering algorithms is a challenging problem which has attracted attention of computer scientists for years [20–22, 34–39]. While the single-linkage clustering algorithm can be run in parallel on a variety of architectures, other clustering algorithms require intensive communication between parallel processes. An alternative to an intensive exchange of data between the parallel processes is an iterative approach with an exchange of data between iterations [37]. However, in some cases, it is possible to partition data using a single-linkage-type algorithm and then concurrently perform clustering in each partition using a serial algorithm. Although the latter approach naturally produces a workload which is imbalanced to a certain degree, it does not require communication between the processes and is well-suited for large weakly-coupled distributed computer systems [40] as long as the load imbalance is tolerable. The hardware available at NCBI (a UGE Grid-Engine-based computer farm [41] and PanFS scalable storage system [42] connected through a powerful router), requires coarse-grained parallelization.
In our case, dataset reductions through selection of representative genomes in near-clonal groups and representative proteins in clade-level protein clusters allow to use the latter simplified approach, with differences in the partition sizes and resulting load balance to be acceptable. Our parallel clustering procedure is performed in three stages, each allowing concurrent processing: (1) The dataset is partitioned in disjoint sets using a parallel implementation based on a disjoint-set forest with union-by-rank heuristics [43, 44]; (2) Data are redistributed according to the partitioning; (3) Clustering is performed in each partition.
Results
Since NCBI production databases are updated in real time, the clustering analysis was performed on a snapshot created in November 2014. Prior to protein clustering, the groups of closely-related genomes (species-level clades) were constructed using ribosomal protein markers [1, 30] (Fig. 2 shows parts of the NCBI clade tree around Salmonella,Bacillus and Streptococcus). Within each clade, genomes are organized in tight (near-clonal) groups calculated using whole-genome BLAST alignment, and a non-redundant representative is selected in each tight genome group (see Methods). Table 1 shows the statistics for the most abundant clades (the statistics for all 131 abundant clades is shown in Additional file 1: Table S1).
Fig. 2.
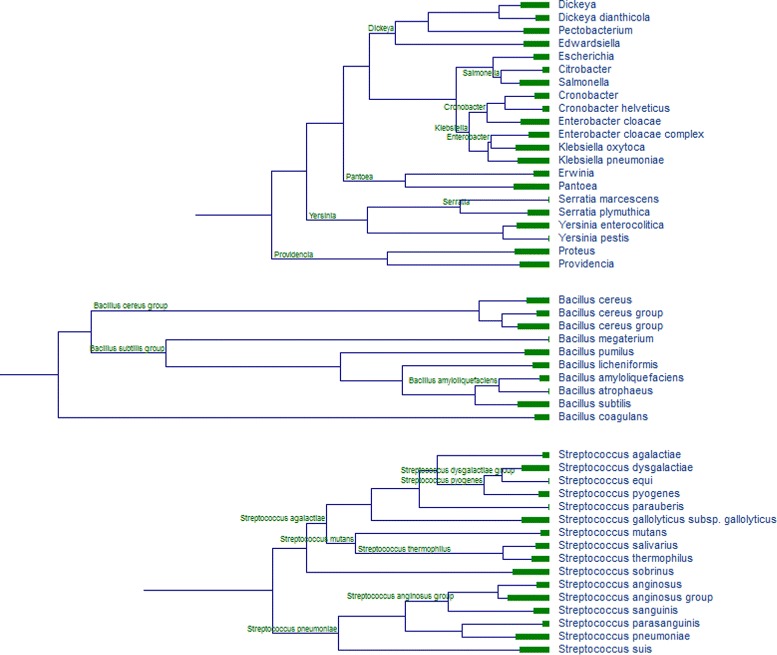
Parts of the clade tree around Salmonella, Bacillus and Streptococcus
Table 1.
Statistics for the most abundant clades. The information for all 131 abundant clades is provided in Additional file 1: Table S1
| Clade | Taxonomic content | No. annotated | No. nonclonal | No. protein | No. protein | No. conservative |
|---|---|---|---|---|---|---|
| Id | genomes | annotated genomes | coding regions | sequences | inclade clusters | |
| 19668 | Escherichia, Shigella | 2277 | 929 | 3303114 | 310023 | 3894 |
| 19507 | Acinetobacter | 749 | 280 | 774670 | 133653 | 3034 |
| 19252 | Helicobacter pylori | 309 | 216 | 254806 | 191419 | 1244 |
| 20139 | Enterococcus genus | 242 | 155 | 306721 | 33249 | 2106 |
| 20104 | Streptococcus genus | 347 | 139 | 163066 | 61589 | 1394 |
| 20137 | Enterococcus genus | 300 | 139 | 309061 | 45809 | 2314 |
| 19669 | Salmonella, Citrobacter | 638 | 134 | 478093 | 112833 | 3940 |
| 19672 | Enterobacter, Escherichia, Klebsiella | 350 | 132 | 593750 | 84168 | 4726 |
| 19537 | Pseudomonas | 229 | 118 | 622138 | 100992 | 5511 |
| 21194 | Vibrio | 271 | 118 | 433416 | 150390 | 4015 |
| 19400 | Neisseria genus | 204 | 109 | 162808 | 29688 | 1596 |
| 19988 | Staphylococcus aureus | 3827 | 108 | 235562 | 43260 | 2309 |
| 20122 | Streptococcus agalactiae | 285 | 103 | 165898 | 17943 | 1704 |
| 19671 | Enterobacter Lelliottia | 80 | 70 | 229896 | 102783 | 3476 |
| 20021 | Bacillus | 101 | 70 | 250224 | 101171 | 3919 |
| 20103 | Streptococcus suis | 92 | 69 | 97200 | 48055 | 1541 |
| 19543 | Pseudomonas | 108 | 68 | 219354 | 114229 | 3551 |
| 19270 | Campylobacter jejuni | 97 | 63 | 85618 | 29112 | 1444 |
| 20116 | Streptococcus mutans | 165 | 62 | 100740 | 28671 | 1672 |
| 19993 | Staphylococcus genus | 92 | 59 | 114655 | 23197 | 2014 |
The dataset contains 23,491 annotated assemblies, with 11,012 of them selected as representatives in near-clonal groups. The representative assemblies contain 40,362,750 protein-coding regions encoding 26,501,327 non-identical protein sequences, among them 25,021,987 marked as complete.
Protein clusters are built at three levels. First, tight protein clusters (80 % similarity with 85 % coverage) are built in large clades containing 10 or more non-clonal genomes using a combined approach that takes into account both sequence similarity and local genome context, and representative proteins (called clustroids) are selected in in-clade clusters. Then clustroids of conservative in-clade clusters are organized into medium-size (50 % similarity with 70 % coverage) seed global clusters, and global protein clusters are built around the seed clusters. The details of the algorithms are described in Methods.
In-clade clusters were built in 131 abundant clades containing 10 or more non-clonal assemblies. The results are summarized in Table 2.
Table 2.
Summary of in-clade clustering for abundant clades
| No. abundant clades | 131 |
| No. protein coding regions encoding complete | |
| proteins | 19,740,968 |
| No. non-identical protein sequences | 7,604,425 |
| No. clustroids | 1,566,371 |
| No. clustroids of conservative in-clade clusters | 351,881 |
| No. protein coding regions encoding complete | |
| proteins represented by clustroids | |
| of conservative in-clade clusters | 14,612,418 |
| No. seed global clusters | 144, 415 |
As a result of seed global clustering, 144,415 seed clusters have been produced. They represent complete proteins encoded by 14,612,418 protein coding regions - 67 % in-clade coding regions. With the seed clusters we observe a substantial 10-fold level of data compression (with even higher level of compression in the largest clades).
The remaining proteins come either from non-conservative (unique) or rapidly evolving proteins, or from rare genomes. The input dataset for extended global clustering contains 19,473,537 non-identical protein sequences: 351,881 sequences are clustroids of conservative protein clusters and the rest contains clustroids of non-conservative in-clade clusters and sequences coming from the outside of the large clades. Straightforward global clustering by the modified hierarchical clustering algorithm required calculating pairwise 19,473,537×19,473,537 BLAST hits and produced 5,595,941 global clusters (where only 2.5 % of them are extended seed clusters, while the most of the remaining 97.5 % are low-informative groups).
Since the critical factor in processing is the calculation of BLAST hits, we first looked for ways to further decrease the number of sequences to be processed by selecting representatives in tight groups of sequences using UCLUST [25] (tight UCLUST parameters approximately correspond to the parameters used in in-clade clustering, see Methods). As a result, 1,263,175 protein sequences were directly assigned the clustroids in the seed clusters, while remaining 17,858,401 sequences were grouped by UCLUST in tight groups allowing selection of 11,185,110 representatives. The described reduction allows to decrease the BLAST hit calculation from 19,473,537×19,473,537 to 11,536,991×11,536,991.
The effectiveness of processing can be tremendously increased, and the amount of work dramatically reduced, if we limit ourselves to extending the seed clusters. In this case, we could use an approximate procedure when non-seed proteins are added to the nearest seed cluster if they are compatible with seed clustroids there. Since non-seed proteins are compared only to seed proteins (and are not compared to each other) in the extension procedure, only BLAST hits of 11,185,110 representatives to 351,881 seed sequences need to be computed. Finally, the extension procedure could be accelerated by the following filtering. UCLUST search procedure with very liberal parameters (see Methods) is used to find a subset of 11,185,110 proteins containing distant neighbors of the seeds. This subset contains 4,174,038 proteins. When we compared this subset to the elements of extended clusters, we found that 99.5 % were assigned, with a loss rate of 0.5 %. As a result, we need to calculate BLAST hits of only 4,174,038 representatives to 351,881 seed sequences, providing 2-fold additional acceleration in comparison to the extension procedure without filtering.
By using 50 % similarity with 70 % coverage, we considered well-established medium-size global clusters that could be further aggregated or neighbor relationships between them could be established (indeed, decrease of the minimal similarity parameter from 50 to 30 % to consider the number of seed clusters decreases from 144,415 seed clusters to 77,532 (larger) seed clusters).
Discussion
We proposed a method to reduce redundancy in the 40 million prokaryotic proteins in the NCBI Microbial Genomes database. Protein clusters were created at the level of clades (organisms grouped by similarity at the species level) and the most conserved clusters were merged between the clades. Highly conserved proteins, for example those involved in cellular machinery, are conserved across taxa. Other proteins are highly conserved within well-studied large clades, for example human pathogens with extensive sequence data. This method has allowed a substantial reduction in redundancy within the microbial protein database.
The developed multilevel approach utilizing the in-clade clustering procedure, subsequent selection of clustroids, and organizing them into seed global clusters provides a robust representation and high rate of compression. Seed protein clusters are efficiently extended by adding related proteins. Extended seed clusters include a significant part of the data and represent all major known cell machinery. Medium-size extended seed clusters could be either organized in wider clusters (super-clusters) or linked together if they are related.
The remaining part of the protein dataset, known in the network theory as network periphery, comes from either non-conservative (unique) or rapidly evolving proteins, or from rare genomes, or resulting from low-quality annotations, requires significant computational resources to be processed in the clustering procedure, and results in a large number of questionable clusters. We propose filtering strategies limiting the protein dataset included in global clustering. The excluded proteins can be related as neighbors to the core clustering data through the links.
Conclusion
The proposed method allows the analysis the relevant data at different levels of details and eliminating data redundancy while keeping biologically interesting variations.
Acknowledgments
This research was supported by the Intramural Research Program of the National Library of Medicine, NIH. The work has been presented at the 11th International Symposium on Bioinformatics Research and Applications (ISBRA 2015), Norfolk, USA, June 7-10, 2015 and the abstract has been published in the conference proceedings [45].
The authors are thankful to David J. Lipman, James Ostell, Eugene Koonin, Yuri Wolf, Teresa Przytycka, Joshua Cherry, Vyacheslav Chetvernin, Kathleen O’Neill, Alexander Souvorov and Jan Hoinka for productive discussions, and to Igor Tolstoy and Vyacheslav Brover for database support.
Declaration
This article has been published as part of BMC Bioinformatics Volume 17 Supplement 8, 2016. Selected articles from the 11th International Symposium on Bioinformatics Research and Applications (ISBRA ’15): bioinformatics. The full contents of the supplement are available online https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-17-supplement-8.
Funding
The publication funding came from the Intramural Research Program of the National Library of Medicine, NIH.
Availability of data and materials
Genome and protein data used in this study are publicly available at NCBI. Per-clade statistics for abundant clades is provided in Additional file 1. Any other information is available from NCBI upon request.
Authors’ contributions
TAT and LZ proposed the approach. LZ developed protein clustering algorithms and software. BF developed the algorithm and software for selecting representative genomes in the groups of clonal genomes. SC performed protein cluster curation and quality-control. All authors worked on the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional file
Table S1. Shows per-clade statistics for 131 abundant clades; number of proteins represents non-redundant set of non-identical protein sequences. (PDF 38 kb)
Contributor Information
Leonid Zaslavsky, Email: zaslavsk@ncbi.nlm.nih.gov.
Stacy Ciufo, Email: ciufo@ncbi.nlm.nih.gov.
Boris Fedorov, Email: fedorov@ncbi.nlm.nih.gov.
Tatiana Tatusova, Email: tatiana@ncbi.nlm.nih.gov.
References
- 1.Tatusova T, Ciufo S, Federhen S, Fedorov B, McVeigh R, O’Neill K, Tolstoy I, Zaslavsky L. Update on refseq microbial genomes resources. Nucl Acids Res. 2015;43(Database Issue):599–605. doi: 10.1093/nar/gku1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tatusova T, Ciufo S, Fedorov B, O’Neill K, Tolstoy I. Refseq microbial genomes database: new representation and annotation strategy. Nucl Acids Res. 2014;42(Database Issue):553–9. doi: 10.1093/nar/gkt1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pangenome. Curr Opin Genet. 2005;15(6):589–94. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Tettelin H, Masignani V, Cieslewicz M, Donati C, Medini D, Ward N, Angiuoli S, Crabtree J, Jones A, Durkin A, Deboy R, Davidsen T, Mora M, Scarselli M, Margarit y Ros I, Peterson J, Hauser C, Sundaram J, Nelson W, Madupu R, Brinkac L, Dodson R, Rosovitz M, Sullivan S, Daugherty S, Haft D, Selengut J, Gwinn M, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O’Connor K, Smith S, Utterback T, White O, Rubens C, G G, Madoff L, Kasper D, Telford J, Wessels M, Rappuoli R, Fraser C. Genome analysis of multiple pathogenic isolates of streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci USA. 2005;102(39):13950–55. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muzzi A, Masignani V, Rappuoli R. The pan-genome: towards a knowledge-based discovery of novel targets for vaccines and antibacterials. Drug Discov Today. 2007;12(11–12):429–39. doi: 10.1016/j.drudis.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11(5):472–7. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Lapierre P, Gogarten J. Estimating the size of the bacterial pan-genome. Trends Genet. 2009;25(3):107–10. doi: 10.1016/j.tig.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Gillings M. Evolutionary consequences of antibiotic use for the resistome, mobilome and microbial pangenome. Front Microbiol. 2013;4(4):1–10. doi: 10.3389/fmicb.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vernikos G, Medini D, Riley D, Tettelin H. Ten years of pan-genome analyses. Curr Opin Microbiol. 2015;23:148–54. doi: 10.1016/j.mib.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 10.Fitch W. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–106. doi: 10.2307/2412448. [DOI] [PubMed] [Google Scholar]
- 11.Koonin E. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–48. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 12.Wolf Y, Rogozin I, Kondrashov A, Koonin E. Genome alignment, evolution of prokaryotic genome organization, and prediction of gene function using genomic context. Genome Res. 2001;11(3):356–72. doi: 10.1101/gr.GR-1619R. [DOI] [PubMed] [Google Scholar]
- 13.Rogozin I, Makarova K, Wolf Y, Koonin E. Computational approaches for the analysis of gene neighbourhoods in prokaryotic genomes. Brief Bioinform. 2004;5(2):131–49. doi: 10.1093/bib/5.2.131. [DOI] [PubMed] [Google Scholar]
- 14.Yelton A, Thomas B, Simmons S, Wilmes P, Zemla A, Thelen M, Justice N, Banfield J. A semi-quantitative, synteny-based method to improve functional predictions for hypothetical and poorly annotated bacterial and archaeal genes. PLoS Comput Biol. 2011;7(10):1002230. doi: 10.1371/journal.pcbi.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mavromatis K, Chu K, Ivanova N, Hooper S, Markowitz V, Kyrpides N. Gene context analysis in the integrated microbial genomes (img) data management system. PLoS ONE. 2009;4(11):7979. doi: 10.1371/journal.pone.0007979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Studer R, Robinson-Rechavi M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009;25(5):210–6. doi: 10.1016/j.tig.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Gabaldon T, Koonin E. Functional and evolutionary implications of gene orthology. Nat Rev Gen. 2013;15(5):360–6. doi: 10.1038/nrg3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonnhammer E, Gabaldón T, Sousa da Silva A, Martin M, Robinson-Rechavi M, Boeckmann B, Thomas P, Dessimoz C. Big data and other challenges in the quest for orthologs. Bioinformatics. 2014;30(21):2993–8. doi: 10.1093/bioinformatics/btu492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fouts D, Brinkac L, Beck E, Inman J, Sutton G. PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 2012;40(22):172. doi: 10.1093/nar/gks757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krause A, Stoye J, Vingron M. Large scale hierarchical clustering of protein sequences. BMC Bioinformatics. 2005;6:15. doi: 10.1186/1471-2105-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan N, Sasson O, Inbar U, Friedlich M, Fromer M, Fleischer H, Portugaly E, Linial N, Linial M. Protonet 4.0: a hierarchical classification of one million protein sequences. Nucleic Acids Res. 2005;33(Database Issue):216–8. doi: 10.1093/nar/gki007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loewenstein Y, Portugaly E, Fromer M, Linial M. Efficient algorithms for accurate hierarchical clustering of huge datasets: tackling the entire protein space. Bioinformatics. 2008;24(13):41–9. doi: 10.1093/bioinformatics/btn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm L, Sander C. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics. 1988;14(5):423–9. doi: 10.1093/bioinformatics/14.5.423. [DOI] [PubMed] [Google Scholar]
- 24.Cameron M, Bernstein Y, Williams H. Clustered sequence representation for fast homology search. J Comput Biol. 2007;14(5):594–614. doi: 10.1089/cmb.2007.R005. [DOI] [PubMed] [Google Scholar]
- 25.Edgar R. Search and clustering orders of magnitude faster than blast. Bioinformatics. 2010;26(19):2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 26.Borgatti SP, Everett MG. Models of core/periphery structures. Soc Netw. 2000;21(4):466–84. doi: 10.1016/S0378-8733(99)00019-2. [DOI] [Google Scholar]
- 27.Holme P. Core-periphery organization of complex networks. Phys Rev E. 2005;72(4):046111. doi: 10.1103/PhysRevE.72.046111. [DOI] [PubMed] [Google Scholar]
- 28.Rombach MP, Porter MA, Fowler JH, Mucha PJ. Core-periphery structure in networks. SIAM J Appl Math. 2014;74(1):167–90. doi: 10.1137/120881683. [DOI] [Google Scholar]
- 29.Ciccarelli F, Doerks T, von Mering C, Creevey C, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311(5765):1283–7. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 30.Zaslavsky L, Ciufo S, Fedorov B, Kiryutin B, Tolstoy I, Tatusova T. Dealing with the data deluge: new strategies in prokaryotic genome analysis. In: Kulski JK, editor. Next Generation Sequencing - Advances, Applications and Challenges. Croatia: Intech; 2016. [Google Scholar]
- 31.Tatusova T, Zaslavsky L, Fedorov F, Haddad D, Vatsan A, Ako-adjei D, Blinkova O, Ghazal H. The NCBI Handbook [Internet] Bethesda, Maryland, USA: National Center for Biotechnology Information; 2013. Protein clusters. [Google Scholar]
- 32.Altschul S, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Altschul S, Madden T, Schäffer A, Zhang J, Zhang Z, Miller W, Lipman D. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasmussen E, Willett E. Efficiency of hierarchic agglomerative clustering using the icl distributed array processor. J Doc. 1988;45:1–24. doi: 10.1108/eb026836. [DOI] [Google Scholar]
- 35.Olson C. Parallel algorithms for hierarchical clustering. Parallel Comput. 1995;21(8):1313–25. doi: 10.1016/0167-8191(95)00017-I. [DOI] [Google Scholar]
- 36.Dahlhaus E. Parallel algorithms for hierarchical clustering and applications to split decomposition and parity graph recognition. J Algorithm. 2000;36:205–40. doi: 10.1006/jagm.2000.1090. [DOI] [Google Scholar]
- 37.Zhao W, Ma H, He Q. Parallel k-means clustering based on mapreduce. In: Jaatun M, Zhao G, Rong C, editors. Cloud Computing. Lecture Notes in Computer Science, vol. 5931. Berlin: Springer; 2009. [Google Scholar]
- 38.Zhang J. A parallel clustering algorithm with mpi - mkmeans. J Comput. 2013;8(1):10–17. [Google Scholar]
- 39.Rytsareva I, Chapman T, Kalyanaraman A. Parallel algorithms for clustering biological graphs on distributed and shared memory architectures. Int J High Perform Comput Netw. 2014;7(4):241–57. doi: 10.1504/IJHPCN.2014.062724. [DOI] [Google Scholar]
- 40.Patterson DA, Hennessy JL. Computer Organization and Design, Fifth Edition: The Hardware/Software Interface (The Morgan Kaufmann Series in Computer Architecture and Design) New York: Morgan Kaufmann; 2013. [Google Scholar]
- 41.Grid Engine Software. Univa Corporation. http://www.univa.com/products/grid-engine.php.
- 42.Panasas ActiveStor. Panasas, Inc. http://www.panasas.com/solutions/lifesciences.
- 43.Tarjan R. Data Structures and Network Algorithms, CBMS 44. Philadelphia: Society for Industrial and Applied Mathematics; 1983. [Google Scholar]
- 44.Cormen T, Leiserson C, Rivest R, Stein C. Introduction to Algorithms. Cambridge, MA: The MIT Press; 2009. [Google Scholar]
- 45.Zaslavsky L, Tatusova T. Clustering analysis of proteins from microbial genomes at multiple levels of resolution In: Harrison R, et al., editors. ISBRA 2015, Bioinformatics Research and Applications, Lecture Notes of Computer Science, vol. 9096. Springer: 2015. p. 438–9.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Genome and protein data used in this study are publicly available at NCBI. Per-clade statistics for abundant clades is provided in Additional file 1. Any other information is available from NCBI upon request.