

Abstract
The world’s genomics data will never be stored in a single repository – rather, it will be distributed among many sites in many countries. No one site will have enough data to explain genotype to phenotype relationships in rare diseases; therefore, sites must share data. To accomplish this, the genetics community must forge common standards and protocols to make sharing and computing data among many sites a seamless activity. Through the Global Alliance for Genomics and Health, we are pioneering the development of shared application programming interfaces (APIs) to connect the world’s genome repositories. In parallel, we are developing an open source software stack (ADAM) that uses these APIs. This combination will create a cohesive genome informatics ecosystem. Using containers, we are facilitating the deployment of this software in a diverse array of environments. Through benchmarking efforts and big data driver projects, we are ensuring ADAM’s performance and utility.
Keywords: computational genomics, genomics, big data, APIs, genome informatics
MISSION
The National Institutes for Health Big Data to Knowledge (NIH BD2K) Center for Big Data in Translational Genomics (CBDTG) has a single, overarching aim: to help the biomedical community achieve the statistical power needed to understand the complex relationships between genotypes and phenotypes in human health and disease. This will only happen if geneticists can gather and compare many genomes – at minimum, hundreds of thousands and, likely, millions – because there are a vast number of genomic variants and each genome variant must be viewed against various genetic and epigenetic backgrounds. The need for such a large quantity of data presents a challenge, one that is compounded when combinations of variations are taken into account. Variants that are rare in most genetic or epigenetic contexts may have limited effects individually but enormous cumulative effects in combination with other variants, which may account for the majority of phenotypes. We will never have the statistical power to recognize rare variants with phenotypic impacts without analyzing large, shared datasets. Similarly, to study these correlations, robust and extensive phenotypic data will need to be readily available. The CBDTG is therefore building partnerships with other BD2K centers (eg, Mobile Data To Knowledge) to facilitate access to and the compatibility of such data.
SHARING BETWEEN GENOME SILOS USING GA4GH APIS
The culture of data sharing that began with the Bermuda Accord and the Human Genome Project1 has been replaced by a culture of genome data silos isolated by ethical, legal, social, and technical roadblocks. Recognizing this, the Global Alliance for Genomics and Health (GA4GH)2 has been formed to help clear these roadblocks and facilitate sharing and interoperability between individual data silos. The CBDTG is working with the GA4GH to solve the technical issues by creating application programming interfaces (APIs), which make interoperability between implementations possible through common communication protocols (Figure 1).
Figure 1:
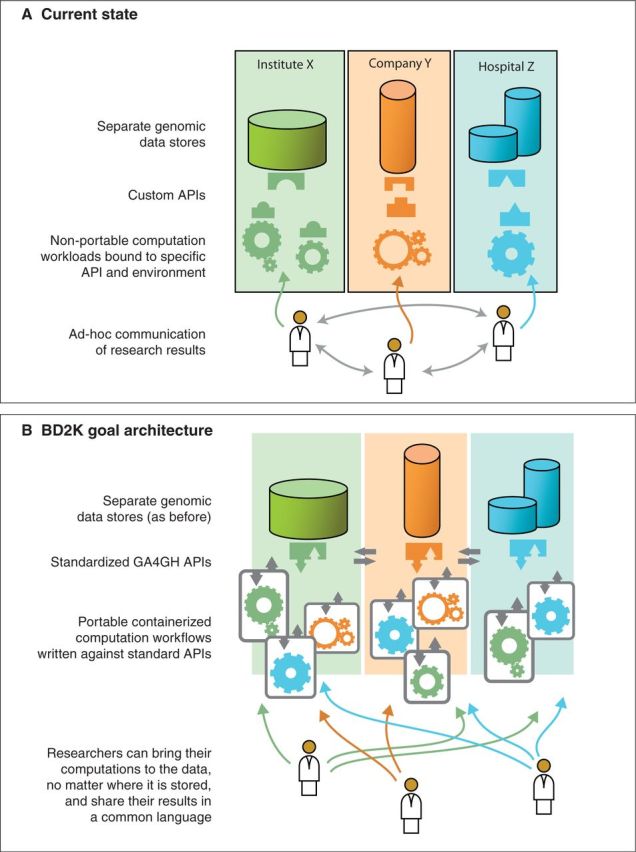
Through the development of standard APIs and the deployment of container technologies, the CBDTG aims to facilitate the move from the status quo (A) to a state in which interoperability and data sharing is the norm (B).
API development cannot be done effectively in isolation, because the success of an API relies on its acceptance by the entire community. The GA4GH has therefore created task teams, each composed of a globally distributed set of members of a specific genomics subcommunity and each charged with contributing to a shared set of API schemas on behalf of that subcommunity. Because community API development requires engineering support to flourish, members of the CBDTG are integrated into each task team The GA4GH task teams are focused on topics such as sequencing reads, genetic variations, RNA, genetic variant effect annotations, metadata, and genotype-to-phenotype associations. A stable version 0.5.1 API is available,3 with implementations by multiple groups, including Google, the European Bioinformatics Institute (EBI), and the National Center for Biotechnology Information (NCBI). Currently, version 0.5.1 provides support for managing sequencing reads and genomic variants, with a richer set of genomic data types on the way as the task teams progress.
To facilitate its distributed development model, the GA4GH is using the collaborative version control system GitHub for all its projects. Each change to the API is proposed by means of a “pull request” – essentially, a set of edits and additions to the existing API. Anyone can make pull requests, which are democratically assessed and discussed prior to acceptance. To be accepted, at least two members of the team must agree that the proposed change is positive, and all team members’ objections need to have been resolved by discussion or by amending the pull request. A pull request can be accepted and integrated into the API by any team member not organizationally associated with the request’s author. In addition to developing APIs, members of the CBDTG are, in accordance with good practice,4 helping lead the development of a reference server for prototyping API concepts and a compliance suite for testing API implementations.
The APIs use the popular Representational State Transfer (REST) software architecture,5 which emphasizes a stateless client-server relationship. Methods map to web requests and data is exchanged in JavaScript Object Notation (JSON). The data objects are described using Apache Avro, an interface description language (IDL) that allows for precise, strongly typed specification of JSON formats. Because the APIs are web-based, it is possible to layer on standard web security protocols for authentication and authorization. A separate security working group at the GA4GH is piloting the application of security standards.
To make the GA4GH API ecosystem useful, the CBDTG is supporting the integration of client applications with APIs. For data visualization, these include the University of California, Santa Cruz (UCSC) Genome and Cancer Genome browsers,6,7 which will allow API data to be viewed as custom tracks, integrated with UCSC’s wealth of existing data. In addition, by developing the reference server, the CBDTG will enable a number of public, web accessible servers for popular genomics datasets, in addition to those already being served by partners such as the EBI and Google.
BUILDING THE BIG DATA GENOMICS STACK
Although the volume of sequencing data has grown exponentially since the completion of the Human Genome Project, genomics processing pipelines are still built around monolithic tools that link cutting-edge analysis methods to flat file format parsers and single-node processing paradigms. While monolithic architectures may slightly improve the efficiency of some tasks, they lock the system into specific implementation choices. Failure to properly abstract out data representation from applications entails constant reinvention of basic components, such as file parsers, writers, and format converters, and makes it difficult to implement new and innovative computational techniques. While it is difficult to quantify the cost of these inefficiencies, these old-fashioned design decisions directly translate into wasted money and wasted time and inhibit innovation. Considering that a substantial fraction of the approximately $70 million that the National Human Genome Research Institute (NHGRI) alone currently invests in informatics each year (pers. comm.), goes toward the development of tools, it is likely significant.
Along with the advances in sequencing technologies that have enabled the development of many novel genomic assays, advances in computing technologies have made it easier to analyze petabytes of data. To link advances in both sequencing and computing, the CBDTG is developing the open source ADAM project.8 This project is in the process of building genomics-specific APIs on top of the Berkeley Data Analytics Stack (BDAS),9 a distributed computing framework.
ADAM is decomposed into the software layers shown in Figure 2. In this configuration, data storage layers are separated from the higher-level application layers that process the data. Such abstraction is increasingly important, because sequencing technology can now generate data in volumes that require its distribution across many machines. Separating the storage and retrieval layers from the application layer frees application developers from being concerned with both how and where the data is stored. The ADAM project is built on top of the Apache Spark and Apache Hadoop frameworks, which are industry-standard parallel distributed execution engines.
Figure 2:

ADAM’s stack decouples “under-the-hood” implementation details from the abstractions used to implement genomic analyses. Thus, ADAM provides bioinformaticians with a wide variety of fast programming abstractions and can be deployed flexibly across a variety of systems, including a single machine, a Hadoop cluster, an HPC grid, or a cloud. The explicit use of a simple, small, yet efficient, data schema is the “narrow waist” of the stack.
While the GA4GH REST APIs are appropriate for the transfer and sharing of genomics data over the internet, they are not themselves explicitly designed for distributed computation (ie, bringing the computation to the data). The ADAM project’s current data models predate the GA4GH APIs; however, a harmonization effort is underway, to make the ADAM data schemas compatible with the GA4GH API’s data model. When this effort is complete, ADAM will provide a platform for the GA4GH data model using the APIs of existing distributing computing engines to enable the efficient execution of algorithms.
GENOMICS EVERYWHERE VIA CONTAINERS
To make ADAM, the GA4GH reference server, and associated applications easy to deploy across a diverse array of platforms, we are using containers. A container platform makes it simple to configure an application or service and have it run anywhere the container platform is supported. This is achieved by encapsulating the software dependencies. Containers are isolated from one another, but multiple containers can share the same operating system, and, where appropriate, the same libraries and binaries; as a result, container-based systems generally outperform systems in which a virtual machine is run for each application. Docker10 is increasingly the standard container platform and allows us to deploy the software developed by the CBDTG on popular commercial clouds, such as those run by Amazon, Google, and Microsoft, as well as within the computing environments of private institutions. Current container platforms are designed for deploying applications and services on a single, physical machine. To deploy applications that run across multiple machines – for example, applications built on ADAM – we are exploring novel technologies such as Google’s Kubernetes.11 In the near future, it should be possible to deploy a cluster of containers within a diverse array of environments to execute a big data computation.
CONTINUOUS BENCHMARKING TO ESTABLISH BEST-OF-BREED INNOVATIONS
Fostering the development of a robust infrastructure for big genomics data requires more than a few tests, it requires establishing and popularizing benchmarks with which the effectiveness of new methods, tools, and implementation strategies can be assessed. Rather than evaluating new developments only once, it is preferable for the community to continuously use these benchmarks to constantly reevaluate the state of the field’s innovations. This necessitates making such benchmarks easy to run and making the benchmark results easy to disseminate. Our center is working to make running a benchmark a simple and reproducible exercise through the use of containers. Such containers encapsulate the benchmark dataset, the benchmark evaluation code, and the algorithm being tested. The combination can be executed on a standardized compute node, such as an Amazon EC2 instance of a certain size, allowing the runtime and memory usage to be recorded in a standard way. In this way, new algorithms can be submitted within a container and evaluated identically, and, optionally, the results can be shared via an online server. To engage with the community and thus popularize these benchmarks, our team includes scientists who lead the organization of crowd-sourced “collaborative challenges” through the popular DREAM Challenges initiative.12
DRIVING PROJECTS
To keep development focused, the CBDTG has identified a number of driving projects in which to apply the developed software. These projects both create useful research and feed back into the development process and motivate further specification of the software. We have identified a range of genomics projects of different sizes and types and involving several different subareas of biomedicine that are undertaking this work.
The first project, the Human Genome Variation Map (HGVM), is a comprehensive new representation of the human genome. The HGVM’s graph-based structure will augment the existing human reference genome with all common human germline variations, integrating many publicly available datasets and providing a means of naming, identifying, and analyzing variations precisely and reproducibly. The GA4GH APIs and reference server are being developed to accommodate the HGVM project’s data model, which has thus far resulted in the addition of a graph-based reference genome model to the GA4GH APIs (shortly to define v0.6 of the API). This graph model allows variations to be specified as part of the reference itself – from single nucleotide variations to complex genome rearrangements. Ultimately, the HGVM aims to supplement the existing human reference genome with a similar standard reference graph. Toward this goal, the HGVM is piloting the construction of algorithms to build such reference graphs, the result being that new genetics methodologies are being developed (in part, by the CBDTG).
The CBDTG has also adopted driving projects identified by the GA4GH, including projects on data sharing (the Beacon project), rare diseases (the Matchmaker project), and breast cancer (the BRCA1/2 project). These projects are being used in the development of the GA4GH APIs. For example, we are learning through each project what metadata, phenotype, and clinical data we need to include make the APIs useful. We are also gaining experience with the process of data normalization and cleaning and will ultimately be able to provide the community with feedback on and tools to aid the import of data into the GA4GH API-compliant data stores.
In addition to the GA4GH projects, the Cancer Genome Atlas (TCGA),13 the I-SPY 2 adaptive breast cancer trial,14 and the BeatAML genomics-guided leukemia therapy project are also all being supported by CBDTG developments. These cancer genomics projects are poster children for big data genomics. For example, the TCGA project contains more than 1.5 petabytes of compressed sequencing data from more than 10 000 tumor-normal samples. Such cancer samples cannot be analyzed independently; we must generally integrate data across all the samples to confidently capture low-frequency, recurrent variations.
FUTURE VISION
Genomics data is a major driver of new innovation and a contender for the largest source of big data as well as the largest challenge in the life science field. Genomics is also changing rapidly. The bulk of genomics data is no longer generated by a few large sequencing centers and is no longer being used only for research. The sheer volume of new data necessitates new approaches. Computational genomics must progress from file formats to APIs, from local hardware to the elasticity of the cloud, from a cottage industry of poorly maintained academic software to professional-grade, scalable code, and from one-time evaluation by publication to continuous evaluation by online benchmarks.
The infrastructure that the CBDTG is building will open the field of big data genomics to laboratories all over the world. It is not economically feasible, efficient, or effective for each separate laboratory or institution to build its own infrastructure from scratch. By creating APIs and open source software on which an active, market-driven, cloud-based genomics big data infrastructure can thrive, we will open genomics data up to the entire biomedical community for a reasonable investment and draw in talent from the engineering, mathematics, and computer science fields that would otherwise be shut out.
CONTRIBUTORS
BP and DH wrote the manuscript with assistance from other authors. FN and MS created the figures. All the authors edited the manuscript.
FUNDING
This work was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U54HG007990. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
COMPETING INTERESTS
None.
ACKNOWLEDGEMENTS
We would like to thank the reviewers for their helpful comments and suggestions.
REFERENCES
- 1.Lander E. S., et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 2.Global Alliance For Genomics And Health. at <http://genomicsandhealth.org/>
- 3.GA4GH. at <http://ga4gh.org/>
- 4.Wilson G., et al. Best practices for scientific computing. PLoS Biol. 2014;12:e1001745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fielding R. T., Taylor R. N. Principled design of the modern Web architecture. in ICSE’00. 2000; 407–416 (ACM Press). doi:10.1145/337180.337228. [Google Scholar]
- 6.Rosenbloom K. R., et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015;43:D670–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldman M., et al. The UCSC Cancer Genomics Browser: update 2015. Nucleic Acids Res. 2015;43:D812–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Massie M., Nothaft F., Hartl C., Kozanitis C. ADAM: Genomics Formats and Processing Patterns for Cloud Scale Computing. 2013. [Google Scholar]
- 9.Berkeley Data Analytics Stack. at <https://amplab.cs.berkeley.edu/software/>
- 10.Docker. at <https://www.docker.com/>
- 11.Kubernetes. at <http://kubernetes.io/>
- 12.DREAM Challenges. at http://dreamchallenges.org/
- 13.Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed]
- 14.I-SPY 2 Trial. at http://ispy2.org/