

Abstract
A coregulated module of genes (“regulon”) can have evolutionarily conserved expression patterns and yet have diverged upstream regulators across species. For instance, the ribosomal genes regulon is regulated by the transcription factor (TF) TBF1 in Candida albicans, while in Saccharomyces cerevisiae it is regulated by RAP1. Only a handful of such rewiring events have been established, and the prevalence or conditions conducive to such events are not well known. Here, we develop a novel probabilistic scoring method to comprehensively screen for regulatory rewiring within regulons across 23 yeast species. Investigation of 1,713 regulons and 176 TFs yielded 5,353 significant rewiring events at 5% false discovery rate (FDR). Besides successfully recapitulating known rewiring events, our analyses also suggest TF candidates for certain processes reported to be under distinct regulatory controls in S. cerevisiae and C. albicans, for which the implied regulators are not known: 1) Oxidative stress response (Sc-MSN2 to Ca-FKH2) and 2) nutrient modulation (Sc-RTG1 to Ca-GCN4/Ca-UME6). Furthermore, a stringent screen to detect TF rewiring at individual genes identified 1,446 events at 10% FDR. Overall, these events are supported by strong coexpression between the predicted regulator and its target gene(s) in a species-specific fashion (>50-fold). Independent functional analyses of rewiring TF pairs revealed greater functional interactions and shared biological processes between them (P = 1 × 10−3).
Our study represents the first comprehensive assessment of regulatory rewiring; with a novel approach that has generated a unique high-confidence resource of several specific events, suggesting that evolutionary rewiring is relatively frequent and may be a significant mechanism of regulatory innovation.
Keywords: transcription factor binding, promoter, cis-regulation, rewiring, genome scale, yeasts
Introduction
Gene expression variability (a biomarker of phenotypic diversity) within and across species is largely brought about by the differences in transcriptional control mechanisms (King and Wilson 1975; Stranger et al. 2012) that are partly reflected in the sequences of regulatory elements, such as transcription factor (TF) binding sites (TFBS), and sequences that effect nucleosome positioning (Wray 2007; Wittkopp and Kalay 2011; Connelly et al. 2014). The converse is not necessarily true; it has been observed that genes with highly conserved spatiotemporal transcriptional patterns have highly divergent cis-regulatory configurations in different species (e.g., Endo16 in sea urchins [Romano and Wray 2003], eve and runt in Drosophila species, and many more [Weirauch and Hughes 2010]). Furthermore, a recent comparative study of TF footprints between human and mouse showed only a small (20%) fraction of the footprints to be shared between the two species indicating a large turnover of TFBS (Stergachis et al. 2014). Collectively, these observations support the idea that there is extensive plasticity in the cis-regulatory circuitry that is representative of both conserved and diverged expression programs across species—the extent of which is only beginning to be appreciated (Wray 2007; Weirauch and Hughes 2010).
TF rewiring is a prominent mechanism of evolutionary changes in cis-regulation, and can occur over relatively short evolutionary timescales (Tuch et al. 2008). Essentially, specific genes (or a set of coregulated genes) have undergone a switch in cis-regulation; whereby in the ancestral species the genes were regulated by a particular TF, but at a specific evolutionary lineage (represented by a subset of extant species) the genes are instead regulated by a different TF (fig. 1). Such evolutionary rewiring of TFs may, or may not result in changes in downstream expression patterns. A well-known example of the latter type of regulatory rewiring in yeast species occurred in a set of functionally related coexpressed genes, namely, the ribosomal regulon. This regulon in Candida albicans and related yeast species is under the control of the DNA binding factor TBF1, whereas in the more recently evolved Saccharomyces cerevisiae, the repressor-activator protein RAP1 regulates the transcription of the same regulon (Hogues et al. 2008). This switch in regulatory factors (from TBF1 to RAP1) is likely due to the loss of binding sites for TBF1, and the simultaneous gain of the RAP1 binding sites in the promoters of 60+ genes that comprise this regulon (Weirauch and Hughes 2010). As mentioned previously, in this case, the function and expression pattern of the regulon is maintained in the two species; however, because transcriptional output is the end point of signal transduction pathways, this rewiring has probably allowed the two species to respond differently to internal or external signals. Furthermore, such rewiring might even constitute changes essential for maintaining robustness in regulatory connections (Isalan et al. 2008).
Fig. 1.

Overview of the approach. The figure illustrates the rationale, and the search space. (A) Toy example: This sample tree shows four species (s1, s2, s3, s4) partitioned at a select branch b to produce the partition of two species in the left clade (s1, s2 Є S) and two species (s3, s4 Є T) in the right clade. Gene locus g represents the orthologous group of genes across all the four species (g1, g2, g3, g4 Є G) that hypothetically exhibit differential usage of regulating TFs X and Y, where X is used by species in the left clade and Y is used by species in the right clade, and not vice versa. (B) Phylogenetic tree of Ascomycetes: Tree shows relationships between the 23 yeast species surveyed in this analysis. Branches are numbered from 1 to 44. Six branches highlighted in bold and larger font numbering represent the chosen branches across which we partitioned the species to assess lineage-specific cis-regulatory rewiring.
Only a few examples of TF rewiring of coregulated genes (regulons) with conserved expression patterns across species have been reported. In addition to the above-mentioned RAP1-TBF1 switch in ribosomal genes, a GAL4/TYE7-GCR1 switch in glucose metabolism genes across yeast species has previously been characterized, and a GAL4-CPH1 switch in regulation was recently observed in the galactose metabolism regulon (Martchenko et al. 2007)—although these are expected to represent just the “tip of the iceberg.” Identification of additional cases of rewiring will facilitate comparative analysis of regulation, help discover clade/species-specific instances of regulatory innovation, inform the contribution of TF rewiring in genes/processes toward adaptability, and also enable investigations of evolutionary conditions conducive to such regulatory switching. Despite its importance, no genome-wide efforts to detect rewiring events have been reported.
Here we develop a genome-scale approach to identify potential TF rewiring events in 23 related species of yeast. We utilize comprehensive DNA binding motifs for 176 yeast TFs, annotation of gene promoters, and established orthology groups across 23 divergent yeast (ascomycetes) species (Matys et al. 2006; Wapinski et al. 2007a, 2007b), to inform a probabilistic function that tests for clade-specific and gene-specific rewiring of TFs. Briefly, for a TF pair (rewiring candidate) and a select evolutionary branch (that partitions 23 species into 2 groups), we compute a probabilistic score which assesses the proposition that a gene is regulated by one of the TFs (say, X) in one group of species and by another TF (say, Y) in the other group of species, as illustrated in figure 1A. We thus compute a “rewiring score” (RS) for every gene (more precisely, orthologous gene family) and every TF pair across six select partitions of the yeast evolutionary tree (only the branches numbered in bold/larger font in fig. 1B).
Next, we apply our novel method to detect rewiring events for groups of genes involved in the same biological process and whose expression are correlated in both Scer and Calb. Our broad application to 1,713 regulons detected 5,353 significant rewiring events (false discovery rate [FDR] < 0.05). While successfully recapitulating the known rewiring events discussed earlier, our results also suggest plausible TF candidates for certain processes reported to be under distinct regulatory controls in Scer and Calb but for which specific regulators are not known. Specifically, MSN2/4 are known to be major players in controlling the response to oxidative stress in Scer (Elfving et al. 2014), although these TFs possess no known roles in regulating the same in Calb (Nicholls et al. 2004); we present evidence for the co-option of FKH2 in regulating this process in Calb. Similarly, RTG1 plays a role in regulating the metabolism of intermediates in Scer such that its misregulation leads to amino acid auxotrophies (Homann et al. 2009), while the same does not occur in Calb. Our results indicate that the promoters of some of the genes involved in this process seem to have diverged to accommodate binding sites for Ca-GCN4/Ca-UME6, thereby potentially rewiring their transcriptional regulator.
Furthermore, independent functional analyses of TF pairs that tend to rewire among themselves revealed that they 1) possess greater functional connections (P < 1 × 10−4) and shared biological processes (P < 1 × 10−3), 2) occupy lower levels of the TF hierarchy, and 3) display strong coexpression between the predicted regulator and the target gene(s) in a species-specific fashion (>50-fold enrichment) across rewiring events. Next, to assess the significance of rewiring events at the level of individual genes, we applied a highly stringent control using a phylogeny-preserving permutation technique (called rotation test) to generate a suitable null expectation. At FDR < 0.1, we detected over 1,000 significant rewiring events at the individual gene level. Similar to regulon rewiring, gene-level rewiring events are also supported by species-specific coexpression of TFs and targets, as well as greater functional connections between rewiring TFs.
Altogether, the assessment of TF rewiring within regulons and individual genes across 23 yeast species suggests that evolutionary rewiring is relatively frequent and may be a significant mechanism of regulatory innovation.
Results
A Probabilistic Framework to Detect Rewiring Events
We define a probabilistic function called the “rewiring score”, henceforth referred to as RS to provide a metric indicative of how likely it is that a given gene locus (including all orthologs across 23 yeast species, or an orthogroup) has selectively switched its regulator in a particular lineage. The RS function is illustrated in figure 1A and described in the Methods section. Very briefly, consider orthogroup g and a phylogenetic tree branch b that partitions the 23 species into species set S comprising of the species descending from the internal branch b, and the complement species set T. For TFs X and Y, RS(X,Y,g,b) calculates the probability that X regulates g in the species set S (and Y does not), and Y regulates g in the species set T (and X does not). Following previous works (Levy and Hannenhalli 2002; Habib et al. 2012), the probability that a TF regulates a gene in a species is derived from the score of the TF’s DNA binding motif against the gene promoter (see Methods).
High-Throughput Computation of Rewiring Scores across All Orthogroups, TFs, and Lineages
Our goal was to comprehensively assess rewiring among all orthogroups across 23 extant yeast species, for all possible pairs of 176 TFs (annotated for DNA binding motif in S. cerevisiae). We chose 6 distinct lineages in the evolutionary tree of 23 ascomycetes to test for rewiring (fig. 1B). The internal branches defining these lineages were selected based on two criteria: 1) Each of the two species groups separated by the lineage comprised at least 3 species and 2) the partitioning is biologically meaningful, for example, non–sensu-stricto and sensu-stricto species, pre-WGD (whole genome duplication) and post-WGD, and so on.
We obtained the 3,844 orthogroups corresponding to protein-coding genes spanning 23 yeast species from the Fungal Orthogroups Repository (Wapinski et al. 2007b). The 600 bp promoter sequences for all genes in all 23 species were obtained from Wapinski et al. (2007a). Using the DNA binding motifs for 176 S. cerevisiae TFs from TRANSFAC (Matys et al. 2006), we obtained the binding probabilities (a value between 0 and 1) of all TFs in all promoters of 23 species. We thus computed an RS for all 176 × 175 = 30,800 TF pairs for 3,844 orthogroups at 6 lineages, resulting in over 118 million RSs per branch. The branch-wise distributions of RSs over all orthogroups and all TF pairs are shown in figure 2. It is evident that more rewiring has occurred on branch #19 than on other branches. In fact, due to the nature of the RS function, the distribution of RSs is dependent on the species partitioning into distinct clades, and is therefore branch specific. This is reflected in the variation in RS distributions across branches (see Methods).
Fig. 2.
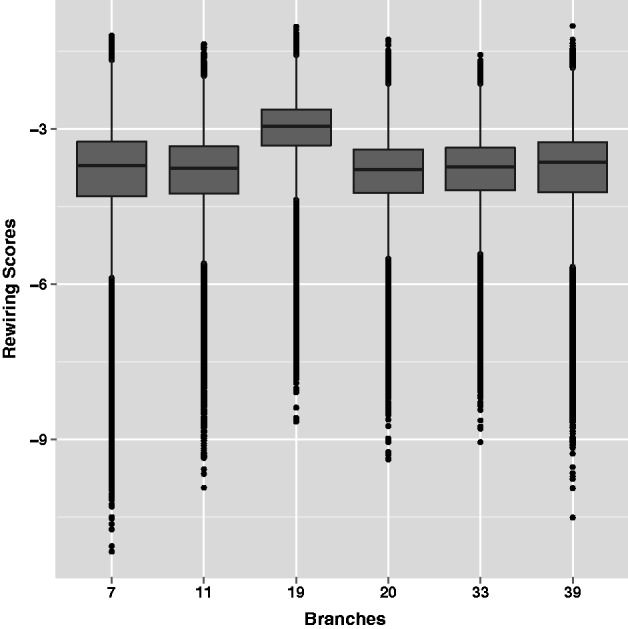
Distribution of all computed rewiring scores. Each boxplot here represents the distribution of RSs (log scale terms on the y-axis) across all triplets RS(X, Y, g), at a chosen branch b ϵ (7, 11, 19, 20, 33, 39) (shown on the x-axis).
In general, TF binding motifs with high information content (IC) yield a more skewed binding probability distribution relative to TF motifs with low IC. To ensure that this inherent difference in binding properties does not introduce a bias in the RSs, we categorized RSs based on IC values of the two TFs (supplementary fig. S1, Supplementary Material online). We found that the pooled distributions in different IC bins are not significantly different from each other, suggesting that the RSs are not sensitive to differences in IC of the TF motifs.
Another potential concern is that the TF DNA binding motifs derived from Scer are used to estimate binding probability in all yeast species. Divergence in DNA binding specificity of orthologous transcriptional regulators across related species is believed to occur infrequently because of the pleiotropic consequences of alterations to TF DNA binding specificity (Prud’homme et al. 2007). With some exceptions (e.g., Matα1 TFBS in yeast species; Baker et al. 2011), previous studies have observed a strong conservation of the regulatory lexicon (∼95% between mouse and human; Stergachis et al. 2014), as well as the function of several TFs across large evolutionary distances (McGinnis et al. 1990). Our approach cannot identify these exceptions, as we scan promoters for TFBS using known TF motifs, as opposed to de novo motif detection, which, however, is more error prone and difficult to interpret. Although in principle species-specific refinements of the motif can be derived, a recent work based on the same data sets used here showed that such a refinement step did not result in substantial differences in the detection of binding sites (Habib et al. 2012).
Regulon-Level Rewiring of Transcription Factor Usage
A regulon, as described earlier, is a collection of transcriptionally coregulated and presumably functionally related genes (Segal et al. 2003). Such coordinated regulation is evidenced by correlated expression patterns of the genes across multiple spatiotemporal conditions. Following our primary motivation of detecting coordinated changes in TF usage that are representative of conserved expression phenotypes across sets of related genes, (similar to ribosomal genes; Weirauch and Hughes 2010), we specifically assessed those sets of genes that shared a biological function and had strongly correlated expression both in Scer and Calb (separated by over 300 My) for rewiring of their TF regulators. Very briefly, starting with gene sets corresponding to 577 distinct biological functions (Gene Ontology term) or pathways, we identified disjoint subsets of genes that exhibit highly correlated expression across hundreds of spatiotemporal conditions, both in Scer and Calb (see Methods). A total of 1,713 gene groups with an average size of 32 genes were assessed for regulatory rewiring.
To assess regulatory rewiring of a regulon, we computed the RS for each gene in the regulon as described above, yielding a distribution of RSs. To estimate the significance of this distribution, we compared it with the distribution of RSs for all orthogroups (at the same lineage and for the same TF pair) using Wilcoxon test. A significantly higher RS distribution for the regulon genes was interpreted as evidence for rewiring. We thus estimated significance of rewiring for each of the 1,713 regulons, 176 × 175 = 30,800 TF pairs at 4 select lineages (descending from internal branches b ϵ 7, 11, 19, 20) shown in figure 1B. These branches were selected because they partition the two well-characterized species with expression data—Scer and Calb. After correcting for multiple testing (Storey 1995), we identified 5,353 significant rewiring events at FDR < 0.05. Given that our method for detecting TF binding is purely sequence based, it is possible that the apparent “multiplicity” in cases where multiple TFs rewire at the same gene(s) and the same branch is simply an artifact of motif similarity between detected TFs. We found that while this is true, it explains only a very small fraction of cases (supplementary fig. S2, Supplementary Material online) to be of any concern.
The detected rewiring events span regulons involved in 577 processes ranging from core processes (ex. sugar and amino acid metabolism, growth, sporulation, etc.) to more specialized ones (ex. response to drug, chemical stimulus etc.), suggesting that regulatory rewiring has occurred extensively across the evolution of divergent yeast species. We discuss the detected rewiring events in the following sections.
Our Method Recapitulates Previously Established Rewiring Events in Yeast
Rewiring of Ribosomal Genes
Ribosomal protein (RP) genes are crucial for cellular growth and viability. As described earlier, this fairly large regulon has a different regulator in Scer and its closely related species (RAP1) as compared with the ancestral species (TBF1). The switch is believed to have specifically occurred along branch 19, as is shown in figure 1B (Hogues et al. 2008). Although this branch represents an evolutionary period of time that precedes the WGD event, and there might be a possible link between this switch and WGD; there is currently no evidence to support this. This particular regulatory substitution is supported by the presence of binding sites of the rewired factor as well as the explicit loss of binding sites of the replaced factor, in the corresponding species (Weirauch and Hughes 2010). Furthermore, it has been shown that both RAP1 and TBF1 in their respective species are involved in recruiting the IFH1/FHL1 complex to the RP promoters (Mallick and Whiteway 2013), which are the primary regulators of RP genes. Despite the requirement of this dimer in both species, the cis-regulatory organization of RP genes in Calb is different from those in Scer; in Calb, these are mainly dominated by CBF1 binding sites while lacking discernible IFH1 sites, while the opposite pattern is observed in Scer (Hogues et al. 2008).
These differences in cis-element configurations (viz., RAP1/IFH1 binding sites in Scer vs. TBF1/CBF1 binding sites in Calb) of ribosomal genes are immediately apparent in the RSs of the TFs implicated in the above process. Because our method does not consider combinatorial relationships between TF binding within species, it detects all 4 pairwise combinations of TFs (viz., RAP1-TBF1, RAP1-CBF1, IFH1-TBF1, and IFH1-CBF1) as having significantly rewired at that lineage. We present in the main text results for the RAP1-TBF1 and IFH1-CBF1 rewiring events only, but the results are similar for all 4 cases (supplementary fig. S3, Supplementary Material online).
Figure 3 shows the distributions of RSs of the RP regulon versus the background (over all genes) for the implicated TFs. Figure 3A compares the RSs assessing the potential that RAP1 regulates the genes in species diverging from a given branch b, and TBF1 regulates the ancestral species. We observe that the differential in the RSs for RP genes and the background is indeed the greatest at branch #19 (FDR < 1 × 10−4). Figure 3B depicts plots analogous to figure 3A, but for the potential that TBF1 regulates the genes in species diverging from branch b, and RAP1 regulates the ancestral species. Because this is essentially the complementary configuration, we expected to see a negative shift in RSs of RP genes relative to background. Interestingly, the negative shift at branch #19 in figure 3B is far more extreme than their positive shift counterpart in figure 3A (compared with null; FDR < 1 × 10−16). This is consistent with the fact that this rewiring event was mainly driven by the loss of TBF1 sites in Scer and related species, rather than gain of RAP1 binding sites (Weirauch and Hughes 2010). Figure 3C and D show qualitatively similar trends for the IFH1-CBF1 rewiring event and is consistent with the rewiring between the two TFs at branch #19 (Hogues et al. 2008).
Fig. 3.

Rewiring scores of the ribosomal regulon for RAP1-TBF1 and IFH1-CBF1 switches across branches. The rewiring scores are shown on the y-axis, and the selected branches are shown on x-axis. (A) RAP1 in lineage and TBF1 in ancestral species: This plot compares the RS distribution of the background (all genes, in white) and that of ribosomal genes (in gray) for the potential that RAP1 regulates its member genes in species diverging from a given branch b, and TBF1 regulates the ancestral species. (B) TBF1 in lineage and RAP1 in ancestral species: This plot compares the rewiring score distribution of the background (in white) and that of ribosomal genes (in gray) for the potential that TBF1 regulates its member genes in species diverging from a given branch b, and RAP1 regulates the ancestral species. (C) and (D) are analogous to (A) and (B), respectively, for the IFH1-CBF1 switch in RP genes.
Rewiring of Galactose Metabolism Genes
Galactose metabolism is another process that has undergone rewiring of the transcriptional circuitry, such that the upstream regulatory regions of a subset of genes encoding enzymes of this pathway have significantly diverged (viz., GAL1, GAL2, GAL3, GAL7, and GAL10) in related fungi (Rokas and Hittinger 2007). In S. cerevisiae, the regulator GAL4 positively activates transcription of these genes in response to galactose through the recognition sequence CGG(N11)CCG (Martchenko et al. 2007). However, in C. albicans, GAL4-mediated regulation and the same recognition sequence is found in contexts unrelated to galactose metabolism. Martchenko et al. further suggested that the regulation of these genes in Calb is instead mediated by CPH1, the homolog of STE12 in Scer; these two factors share 86% sequence similarity in their DNA binding domain.
Indeed, analogous to RP genes, we detected significant support of rewiring in the galactose regulon genes for the two factors, GAL4 and STE12. Specifically, figure 4A compares the RS distribution of the background (over all genes) against that of galactose metabolism genes for the potential that GAL4 regulates the genes in species diverging from a given branch b, and STE12 (or CPH1) regulates the ancestral species. Here, we see positive shifts in RSs of the galactose regulon across all branches that separate Scer and Calb, with the highest shift in branch #19 (FDR < 0.02). See supplementary figure S4A, Supplementary Material online, for the potential that STE12 (or CPH1) regulates the genes in species diverging from a given branch b, and GAL4 regulates the ancestral species. Similar to the case of RP genes, we observed significantly lower regulon RSs when compared with the null background expectation in branch #19 (FDR < 0.002).
Fig. 4.

Rewiring scores of other known rewiring events across branches. See figure 3 legend for details. (A) Galactose regulon rewiring scores for GAL4 in lineage and CPH1 in ancestral species. (B) Glucose metabolism regulon (subset) rewiring scores for GCR1 in lineage and GAL4 in ancestral species.
Taken together, these results suggest that this change in cis-configuration, and thereby regulation, occurred at branch #19. Martchenko et al. (2007) hypothesized that this switch probably occurred as a consequence of WGD, but our analysis suggests that the gain in GAL4 binding sites, as well as loss in CPH1 binding sites initiated before the WGD event.
Rewiring of Glucose Metabolism Genes
In C. albicans, genes involved in glucose utilization are regulated by GAL4 and TYE7, whereas in S. cerevisiae this task has been taken over by GCR1 and GCR2 (Askew et al. 2009; Lavoie et al. 2009). Consistent with this event, we detect significant signal of rewiring in a subset of genes involved in glucose metabolism for the two factors, GCR1 and GAL4. Specifically, figure 4B compares the RS distribution of the background against that of glucose metabolism genes for the potential that GCR1 regulates the genes in species diverging from a given branch b, and GAL4 regulates the ancestral species. We see positive shifts in RSs of glucose metabolism genes across all branches that separate Scer and Calb, with the highest shift in branch #19 (FDR < 0.005). Similar to previous cases, we observe significantly lower regulon RSs when compared with the background in the complementary scenario as shown in supplementary figure S4B, Supplementary Material online (FDR < 0.05).
Identifying Candidate TFs for Known Rewiring Events
Next, we searched the literature for processes that are reported to be under distinct regulatory controls in S. cerevisiae and C. albicans, but for which specific regulators have not been implicated, and assessed whether our probabilistic method can help identify potential regulators in these cases.
Stress response
Zinc finger TFs MSN2/4 bind to highly similar motifs and are the primary regulators of response to a variety of stresses (nutritional, oxidative, etc.) in Scer. Here, MSN2 elicits a complex response to stress, whereby different cohorts of target genes respond differently, resulting in either gene expression activation or repression (Elfving et al. 2014). However, these TFs are not known to play a role in stress response in Calb; the disruption of Ca-MSN2/4 had no tangible effect on the resistance of the species to heat, oxidative, and osmotic stresses (Nicholls et al. 2004). Consistent with the rewiring of stress response regulators in the two species, we found that the regulon involved in oxidative stress response shows strong signals for regulatory rewiring of MSN2/MSN4 regulating these genes in Scer to being regulated by FKH2 in Calb. Specifically, figure 5A compares the RS distribution of the background (over all genes) against that of stress response genes for the potential that MSN2/4 regulates the genes in species diverging from a given branch b, and FKH2 regulates the ancestral species. Here, we see significant positive shifts in the RSs of the regulon in branch #19 (fig. 5A; FDR < 0.01), and significant negative shifts for the complementary scenario akin to previous cases (supplementary fig. S5A, Supplementary Material online; FDR < 0.01). Although short of a direct experimental validation, our finding is supported by a prior study showing that Ca-FKH2 mutants in C. albicans resulted in increased transcript levels of genes involved in stress response (Bensen et al. 2002).
Fig. 5.

Rewiring scores of predicted rewiring events across branches. See figure 3 legend for details. (A) Oxidative stress response regulon rewiring scores for MSN2 in lineage and FKH2 in ancestral species. (B) Glycolysis regulon rewiring scores for RTG1 in lineage and UME6 in ancestral species. (C) Glucose metabolism regulon (subset) rewiring scores for RTG1 in lineage and GCN4 in ancestral species.
Metabolism
Retrograde (RTG) signaling, triggered by lack of glutamine, modulates carbohydrate and nitrogen metabolism through nuclear accumulation of the heterodimeric TFs, RTG1/3 (Giannattasio et al. 2005). This accumulation and subsequent binding to metabolic gene targets allows cells to maintain synthesis of α-ketoglutarate, which is a precursor to glutamate and glutamine (Crespo and Powers 2002) (the latter is a preferred nitrogen source in yeast [Crespo and Hall 2002], lack of which leads to amino acid starvation [AAS]). It has been shown that deletion of TF RTG1 in Scer causes glutamate and aspartate auxotrophies, yet deletion of its ortholog in Calb does not result in the same phenotype (Homann et al. 2009).
Our results indicate that the promoters of some of the genes involved in carbohydrate metabolism (glycolysis regulon, as well as a subset of genes involved in glucose metabolism) display an aggregate loss of RTG1 binding sites in Calb, and a concomitant gain of binding sites for Ca-GCN4 and Ca-UME6, respectively, thereby potentially rewiring their regulation (fig. 5B and C). Figure 5B compares the RS distribution of the background (over all genes) against that of glycolysis genes for the potential that RTG1 regulates the genes in species diverging from a given branch b, and GCN4 regulates the ancestral species. The plot depicts positive shifts at branches separating Scer and Calb into distinct clades (FDR < 0.05). Supplementary figure S5B, Supplementary Material online, shows the complementary scenario with corresponding negative shifts (FDR < 0.05). Figure 5C (FDR < 0.05) and supplementary figure S5C, Supplementary Material online (FDR < 0.05) show qualitatively similar trends to figure 5B and supplementary figure S5B, Supplementary Material online, respectively, for the RTG1-UME6 rewiring event in glucose metabolism genes, and is consistent with the rewiring between the two TFs. These regulatory changes potentially result in Calb evolving alternate responses to the lack of glutamine, or to the lack of intermediates essential for amino acid synthesis to prevent starvation. GCN4 is known to be involved in AAS responses that include 1) amino acid biosynthesis, 2) increasing expression of autophagy genes, and 3) repressing genes encoding ribosome proteins (Hinnebusch 2005). Similarly, UME6 in Calb is part of a signaling cascade that regulates autophagy (Bartholomew et al. 2012) and is also involved in regulating hyphal (filamentous) growth (Banerjee and Thompson 2008)—a phenotype better suited for nutrient scavenging.
Note
Graphical illustrations of the binding site profile of rewiring TFs for all significant events described in the above two sections are shown in supplementary figure S6, Supplementary Material online. For conciseness, we only show TFBS profiles for regulons in Scer and Calb for each rewiring event.
Rewiring Events Are Strongly Supported by Coexpression between the Regulator and Targets
Even though the causal link between TF gene level and the target gene level is confounded by 1) low constitutive expression of many TFs and 2) regulatory mechanisms including posttranslational modifications, cofactors, and so on, in general, expression of TF genes and their target genes are expected to be correlated across different environments to some extent (Basso et al. 2005). We assessed if such correlated expression patterns are apparent among the 5,353 detected rewiring events. Specifically, we tested if the expression of the predicted regulator of a regulon correlates with the expression of the regulon’s component genes in a species-specific fashion (using expression data in Scer and Calb from Ihmels et al. 2005). For instance, in the case of RP regulon rewiring, we assessed whether RAP1 is coexpressed with the RP regulon genes in S. cerevisiae, but not in C. albicans, and whether the converse was true for TBF1?
For each significant rewiring event at the regulon level (say between TF X and TF Y in regulon R), we carried out the following analysis. We collected four different sets of Spearman correlations between 1) TF X and R genes in Scer, 2) TF Y and R genes in Calb, 3) TF Y and R genes in Scer, and 4) TF X and R in Calb. Of 5,353 events, complete expression correlation data for all 4 sets was available for 3,030 cases. Because the detected rewiring events predict which regulator is being used by which species, we simply calculated the number of cases in which the set of correlations between the TF are predicted in a given species and the target genes are significantly greater (Wilcoxon P ≤ 0.05) than those for the TF not being used in the species. We required this condition to be satisfied in both Scer and Calb. We found that in 493 of the 3,030 cases, the coexpression patterns support the predicted regulatory switch in both species, which is strong evidence in light of the null expectation (16% vs. 0.25%; 65-fold enrichment). In fact, most of this enrichment is localized to rewiring events specific to the WGD branch (32% vs. 0.25% at WGD branch #11).
In case of RP genes, the degree of coexpression in Scer of RAP1 and TBF1 with the RP genes (fig. 6A) was comparable (Wilcoxon P = 0.53). However, in Calb the coexpression between TBF1 and RP genes was significantly higher than that of RAP1 and RP genes (Wilcoxon P < 1 × 10−7). In case of glucose metabolism, coexpression of GCR1 as well as GAL4 with the glucose metabolism genes (fig. 6B) is consistent with the direction of TF rewiring in Scer (Wilcoxon P < 1 × 10−6); this could not be tested in Calb due to the absence of an annotated GCR1 homolog. Finally, in case of the galactose regulon, absence of sufficient coexpression data points for GAL4 and STE12 (or CPH1) (due to small regulon size, comprising of three genes) limits the analysis.
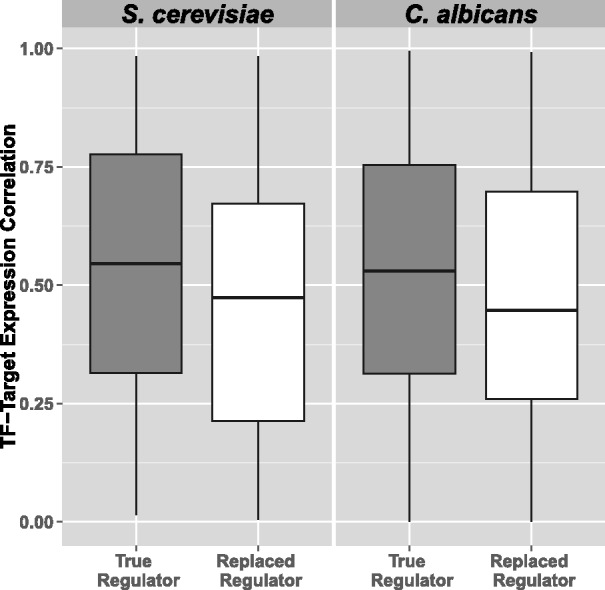
Fig. 6.

Coexpression analyses for regulon rewiring events. In each panel from A–F, we compare the TF-to-Target expression correlations on the y-axis for the candidate regulator (e.g., RAP1 in Scer) and the replaced regulator (e.g., TBF1 in Scer) on the x-axis. The distribution of correlations with regulon gene targets is shown in gray, while that with all genes (comprising the background) is shown in white. The facets in each panel represent individual species (Scer and Calb). (A) Ribosomal gene regulon (RAP1-TBF1). (B) Glucose metabolism regulon (GCR1-GAL4). (C) Oxidative stress regulon (MSN2-FKH2). (D) Glycolysis regulon (RTG1/3-UME6). (E) Glucose metabolism regulon (RTG1/3-GCN4).
In case of oxidative stress response genes, we observe coexpression support (fig. 6C) for the implicated TFs (MSN2/4 and FKH2) only in Scer (Wilcoxon P = 0.03), but not in Calb (Wilcoxon P = 0.22; although coexpression median of “True Regulator-Regulon” > “Replaced Regulator-Regulon” in Calb). On the other hand, coexpression of regulator and targets in RTG response is as follows: 1) For genes involved in glycolysis (fig. 6D), we observe strong coexpression support for the implicated factors (RTG1 and UME6) in Scer (Wilcoxon P < 1 × 10−55) as well as Calb (Wilcoxon P < 2 × 10−4). 2) For a subset of genes involved in glucose metabolism (fig. 6E), we observe strong coexpression support for the implicated factors (RTG1 and GCN4) only in Calb (Wilcoxon P < 0.01), but not in Scer. Furthermore, it can be seen from figure 6 that even when the coexpression between replaced regulator and regulon genes is relatively low, the coexpression of the replaced regulator with all genes is still high. This suggests that the differences in coexpression levels of true and replaced regulators with their putative gene targets across species is not simply due to an overall reduced expression of the replaced regulator in a given species, and in most cases the opted-out regulator still functions to regulate genes involved in other processes. For example, although CPH1 (or its homolog STE12 in Scer) does not regulate galactose metabolism genes in Scer anymore, it is still involved in regulating genes involved in mating-type determination.
Thus, although coexpression of TFs and targets consistent with the direction of rewiring in the member species is not a necessary condition for rewiring (as illustrated for RP genes), we observe strong overall support for target regulation in a species-specific fashion. Interestingly, as mentioned above, this support is the highest for rewiring events occurring at a branch associated with WGD (branch #11); WGD is linked with higher degrees of expression and protein sequence divergence (Ha et al. 2009; Li et al. 2015).
Functional Connections between Rewiring TFs and Their Properties
Next, we investigated the functional characteristics of rewired TF pairs that might have enabled or facilitated rewiring. It is plausible that aspects such as protein domain similarities, an increased propensity for physical interaction, coordinated expression across conditions for the two TFs, as well as their mutual involvement in common biological processes/pathways could individually or synergistically facilitate rewiring between the two TFs. For example, RAP1, like TBF1, is a Myb family protein (Bhattacharya and Warner 2008) and has similar GC-rich binding specificities (Weirauch and Hughes 2010); this could have predisposed RAP1 to acquire the competency for RP regulation. We assessed the extent to which these different features are enriched among rewiring TFs.
Physical Interaction Potential
First, we compared the propensity for physical interaction between rewired TFs relative to randomly selected TFs. We used protein-protein interaction (PPI) annotations from BioGRID database (Chatr-Aryamontri et al. 2015) for proteins known to physically interact in S. cerevisiae and binned TF pairs in a 2 × 2 contingency table based on whether or not they interact, and whether or not they rewire. Based on a Fisher’s test, we did not observe a greater propensity for direct interaction between rewiring TF pairs (fig. 7A; odds ratio = 1.02; Fisher’s P = 0.93). We obtained qualitatively similar results using PPI annotations from STRING database (Franceschini et al. 2013) (supplementary fig. S7, Supplementary Material online). Although such direct interaction potential between rewiring TFs is absent, it has previously been suggested that if members of rewiring TFs can bind and colocalize with a common cofactor to cooperatively regulate a target(s), then a series of small successive changes in the component interactions comprising such combinatorial control could ultimately result in a regulatory handoff between rewiring TFs across evolutionary time (Tuch et al. 2008). For example, the cis-element rewiring between RAP1 and TBF1 in RP genes was accompanied by a change in the protein domain of a common cofactor that they interact with (a heterodimer containing IFH1 and FHL). Specifically, correlated with the transition to the RAP1-regulated circuit in Scer, the Sc-IFH1 acquired an RAP1 interaction domain (Mallick and Whiteway 2013) that is not present in the Ca-IFH1 protein. To assess this possibility, we tested 1) if rewired TF pairs possess a common interacting TF more often than other TF pairs and 2) if the commonly interacting TF is more likely to bind to the target gene’s promoter when compared with other promoters. Although the first test only shows mild support (although not statistically significant) for the expected trend (fig. 7B; odds ratio = 1.2, Fisher’s P = 0.112), the second test showed a highly significant trend (fig. 7C; odds ratio = 14.1, Fisher’s p = 1 × 10−4). Overall, this suggests that cooperative binding of rewiring TFs to a common factor is perhaps one of the potential mechanisms facilitating regulatory rewiring.
Fig. 7.

Functional analyses of rewired TFs in regulons. Each panel represents Fisher’s test of a specific hypothesis. In each case, rewired TF pairs and all other TF pairs are binned into two classes based on a given functional criteria (except panel C, where the bins are regulon genes and all other genes), and compared using Fisher’s exact test. (A) Direct physical interaction: Based on BioGRID database, the plot shows the fraction of TF pairs that do (light gray) and do not (dark gray) physically interact. (B) Physical interaction with a common cofactor TF: Based on BioGRID database, the plot shows the fraction of TF pairs that do (light gray) and do not (dark gray) possess a common cofactor TF. (C) Cofactor binding at target regulons: This plot shows the fraction of cofactor TFs that do (light gray) and do not (dark gray) bind strongly at gene promoters (≥0.75 vs. <0.75 binding scores). (D) Structural similarity: This plot shows the fraction of TF pairs that do (light gray) and do not (dark gray) belong to same structural family. (E) Common KEGG pathways: This plot shows the fraction of TF pairs that do (light gray) and do not (dark gray) belong to the same KEGG pathway. (F) TF hierarchy: This plot shows the fraction of TF pairs that do (light gray) and do not (dark gray) belongs to lowest and middle hierarchies. (G) Common upstream regulator: This plot shows the fraction of TF pairs whose distance to a common upstream regulator is ≤4 (light gray) or >4 (dark gray).
Structural Similarity
Second, we gathered the structural family annotations of the TFs (Pfam; Finn et al. 2014), and tested if rewired TF pairs belong to the same family more or less often than background expectation. We observed that the rewired TFs in fact belong to the same TF family less often than the random nonrewired TF pairs (fig. 7D; odds ratio = 0.67, Fisher’s P = 0.001). Although the reasons for depletion of cofamily TFs among rewiring TF pairs is not entirely clear, we suspect it may partly be due to functional divergence of paralogous genes, consistent with our other results showing a greater functional similarity between the rewired TFs.
Common Pathways
Third, we hypothesized that the co-option by a group of genes of an alternate regulator may be influenced by whether or not the rewired TFs are already functioning in the same pathways. Based on KEGG (Kyoto Encyclopedia of Genes and Genomes; Kanehisa et al. 2014) pathway annotations, we assessed if TFs implicated in the same pathway are more likely to rewire than those involved in different pathways. TF annotations in KEGG were limited to cell cycle, signaling pathways, and meiosis, which substantially reduced the number of pairs we could test. Nevertheless, we observed greater likelihood for TFs of common pathways to rewire than that expected by random chance (fig. 7E; odds ratio = 3.4, Fisher’s P = 0.001).
Regulatory Hierarchy
Previous studies of the effects of network rewiring events (insertion or deletion of connections) in a broadly constructed regulatory hierarchy of transcriptional factors in yeast suggest that rewiring affecting upper levels of such a hierarchy is much less tolerated and result in cell proliferation and survival defects, when compared with those affecting lower levels of the hierarchy (Bhardwaj et al. 2010). Also, these upper level regulators were found to exhibit fewer functionally redundant copies across species. In light of these characteristics, we expect that the TFs in the upper level of the hierarchy should be less prone to rewiring. Using data on regulator hierarchy across 90 TFs (Bhardwaj et al. 2010), we indeed observe that there is significant depletion of rewiring events involving TFs belonging to the highest level of regulation when compared with lower and middle level TFs (fig. 7F; odds ratio = 1.67, Fisher’s P = 0.004).
Common Upstream Regulator
Next, we assessed the possibility that member TFs of a rewired TF pair are regulated by a common upstream regulator (UpR), and that this differential regulation between the species of a lineage and the ancestral species has enabled rewiring of the two factors. In general, however, this UpR may not be directly regulating either of the rewired TFs, but may instead exist further upstream in the regulatory network, at a point from which two alternative paths leading to the two rewired TFs originate. This possibility can be tested using species-specific TF–TF regulatory networks and checking if the two rewired TFs lead up to a common UpR in their respective species-specific networks. We generated TF–TF regulatory networks for Scer and Calb independently (see Methods). Next, for every TF pair, we checked if the members of the pair link to a common UpR in their respective species, such that the sum of the shortest path lengths to that UpR is smaller than that expected by random chance (i.e., shortest path length to common UpR for random TF pairs). We use shortest path length from the TF pair to the UpR as a proxy for the presence or absence of the UpR, that is, the smaller the metric is, the greater the chance that the common UpR exists. Similar to the analyses above, we binned TF pairs into whether or not they are close to a common UpR and whether or not they rewire across species (2 × 2 contingency table). Using a Fisher’s test, we conclude that rewired TF pairs do in fact possess a common UpR, more often than random TF pairs (fig. 7G; odds ratio = 1.28, Fisher’s P = 1 × 10−4). To remove possible confounding effects in the computation of shortest paths due to widely connected master regulators, we removed the top 5% TFs with the greatest degree before computing shortest path between nodes. This however does not affect our conclusion (supplementary fig. S8, Supplementary Material online). This notion of a nearby common UpR is further supported by a prior study in yeast (Ucar et al. 2009) that recovered MSN2 and FKH2 in a TF–TF interaction pathway (in oxidative stress conditions) that they generated by combining ChIP-chip, motif binding sites, nucleosome occupancy, and mRNA expression data sets in a probabilistic framework. Similarly, they recovered RTG1/3 and GCN4 from a network generated for AAS conditions.
Taken together, our results suggest that regulon rewiring under conserved target expression is limited to the lower level TFs in a given pathway, such that they might not necessarily interact with each other, but be implicated in the same process/pathway in a broader context.
Gene-Level Assessment of Rewiring Using Rotation Test
The application of the RS function to each orthogroup and TF pair triplet across 6 different evolutionary lineages resulted in ∼650 million individual RSs (∼110 million per branch). Thus, a major challenge was to devise a stringent control to assess significance of each individual RS. Therefore, we employed a rotation test (Langsrud 2005) based FDR approach whereby a background distribution of RSs using controlled permutations of TF binding probabilities across species is generated, and compared with the distribution of observed RSs to get an FDR for each data point in the set. We expect binding probabilities for a TF at an orthogene in sister species to be very similar due to expected sequence similarity. Traditional permutation of these binding probabilities would sample from each variable (binding probability in a given species) independently, despite the fact that there is an inherent constraint in the range of values each variable can adopt due to their mutual relationship, thus leading to overestimation of significance. The controlled permutation method (called rotation test) essentially has the effect of permuting the binding probabilities of a given TF across species while preserving the inherent phylogenetic relationships between species to simulate neutral evolution.
This is essentially equivalent to sampling from binding probabilities across species while maintaining a fixed covariance structure that represents the constraints of phylogeny; we derive this covariance matrix from the concatenated binding probabilities for all TFs at all gene loci in each species, which serves as a suitable proxy for phylogeny. Thus, binding probabilities for each TF across 23 species were permuted as above, and the “rotated” binding probability profiles of each TF were subsequently used to compute background RSs. The details of the rotation test are provided in the Methods section. We estimated the FDR of every RS. The background generation and FDR calculation was done independently for each of the six lineages mentioned above. At 0.1 FDR we identify 1,446 significant gene-level rewiring events. Although the total number of detected events is much smaller compared with regulon-level rewiring (which is expected due to our use of a highly stringent control), most of these are along branch #19 and #20 (fig. 1B) that best separate C. albicans from S. cerevisiae, consistent with our results from regulon-level findings.
Similar to regulon rewiring events, we assessed whether the expression correlation between the TF (say X) and the predicted target in a species is higher than that for replaced TF (say Y) and the same target. To this end, using the entire set of predicted (X,Y,g) events, we collected four sets of pooled correlations between 1) X and g in Scer, 2) Y and g in Scer, 3) X and g in Calb, and 4) Y and g in Calb across all branches. As shown in figure 8, rewiring events are strongly supported by coexpression as in the case of regulon-level rewiring (Wilcoxon P in Scer = 1 × 10−5, Wilcoxon P in Calb = 1 × 10−3). Most of the coexpression support in this case is also driven by branch #11, similar to what we observe for regulon rewiring (supplementary fig. S9, Supplementary Material online).
Fig. 8.
Rewiring at the individual gene level. (A) Number of significant rewiring events: Table summarizes the detected events per branch after applying a highly stringent screen controlling for phylogenetic relationships between TF binding profiles across species at FDR < 0.1. The corresponding maximum rewiring score of detected events is also shown here (“Score Threshold”), which varies between branches because each controlled permutation was carried out specific to a branch. (B) Species-specific TF-target coexpression analysis: In each panel, the predicted TF-target expression distribution is shown for the TF predicted to be active in a species (gray) and for the TF predicted not be active in the species (white); the distribution is based on pooled correlations across all significant rewiring events.
Next, we investigated the functional characteristics of rewired TF pairs represented in individual gene rewiring events (fig. 9). Akin to our regulon-level analyses, we found that the rewired TF pairs 1) do not necessarily interact physically with each other (fig. 9A; odds ratio = 1.09, Fisher’s P = 0.9) but yet show mild potential (although not statistically significant) for direct interaction with a common cofactor (fig. 9B; odds ratio = 1.2, Fisher’s P = 0.2) that co-operatively regulates its target genes (fig. 9C; odds ratio = 16.1, Fisher’s P = 1 × 10−4); 2) have lower than expected structural similarity (fig. 9D; odds ratio = 0.77, Fisher’s P = 0.001); 3) are enriched in common pathways, although to a lesser extent than rewired TFs in regulons (fig. 9E; odds ratio = 2.15, Fisher’s P = 0.01); and (4) are enriched in TFs belonging to the lower or middle hierarchies (fig. 9F; odds ratio = 2.02, Fisher’s P = 0.001). However, interestingly, we observed that TF pairs found to rewire in the context of regulating individual genes are “less” likely to possess a common UpR than random expectation (fig. 9G; odds ratio = 0.8, Fisher’s P < 0.01). This difference between TFs regulating genes with conserved expression patterns (i.e., at the regulon level), and TFs regulating genes with possibly divergent downstream expression (at the gene level) across species, could be an important distinguishing property of the mechanism of rewiring that leads to these alternate scenarios. To test this, we divided the detected gene level targets into groups with conserved and diverged expression patterns, and assessed the potential of rewired TFs in each group to possess a common UpR (supplementary fig. S10, Supplementary Material online). We found that although TFs regulating genes with conserved expression patterns showed no trend (Fisher’s P = 0.24), TFs regulating genes with divergent expression patterns were indeed less likely to possess a common UpR than expectation (Fisher’s P < 0.03). Note that because this analysis was carried out on few high-confidence conserved and diverged expression target genes, their small sizes limit our ability to conclude meaningful functional trends.
Fig. 9.

Functional analyses of rewired TFs in individual genes. See figure 7 legend for details.
Discussion
Prior studies have characterized a few cases of regulatory rewiring of specific genes/gene sets in great depth (Martchenko et al. 2007; Hogues et al. 2008; Lavoie et al. 2010). These previous works provide important insights into aspects of rewiring. For example, Mallick and Whiteway (2013) showed how regulatory connections local to rewired TFs can change to preserve gene target expression patterns (e.g., recruitment of IFH1-FHL1 to ribosomal gene targets is maintained in both systems). Yet, there are several aspects of regulatory rewiring that are poorly understood: 1) How widespread is a wholesale shift in transcriptional regulation of a regulon?, 2) what are the features of target genes that make them amenable to rewiring?, 3) what characterizes rewired TFs, and so on. By gathering more candidate rewiring events and collectively analyzing their trends, we can potentially answer these questions and gain further insights into conditions conducive to rewiring, as well as enable discovery of clade/species-specific instances of regulatory innovation.
A genome-wide screen for TF rewiring has not been reported thus far. Here, we present the first scalable probabilistic approach to detect rewiring. Its application to 23 yeast species has successfully recapitulated known rewiring events (in ribosomal genes, sugar metabolism genes, etc.), and also has generated specific testable hypotheses of rewiring in many genes, as well as regulons. Our results indicate that rewiring is pervasive; it is further likely that analysis of regulons with divergent expression across species will reveal many more rewiring events that have the potential to shed light on the divergence of functionally related genes’ expression mediated by rewiring. Although some of the detected events have functional consequences, it is very likely that a lot of these are manifestations of high cis-regulatory plasticity.
Similar to previous related works (Habib et al. 2012; Roy et al. 2013), ours is based on estimated TF binding probabilities and not in vivo binding. This is not a limitation of the approach but that of the availability of functional binding information such as ChIP-seq across 100+ TFs or high-resolution DNAse footprinting in all 23 species of yeast. Furthermore, reliance on such experimental data is limited due to their condition specificity, that an in silico approach avoids (Roy et al. 2013). On the other hand, in silico motif-based prediction of cis-regulation can be noisy. We attempted to reduce the noise by using experimentally measured nucleosome occupancy data (Tsankov et al. 2010, 2011) from 13 yeast species to gather additional support for functional binding. We have described this analysis in supplementary figure S11, Supplementary Material online, which suggests that incorporating nucleosome occupancy is not likely to improve the sensitivity of our approach. This is generally expected due to a poor association between nucleosome free regions (NFRs) and TF binding in yeast; Ozonov and van Nimwegen (2013) showed that only 10–20 of 158 TFs contribute to inducing NFRs and that nucleosome positioning is mainly determined by intrinsic sequence. Another study by Thompson et al. (2013) showed that TF binding sites are depleted from NFRs in most post-WGD species. Thus in our assessment, integrating nucleosome occupancy data in our analyses decreases statistical power without necessarily decreasing noise. Additionally, the presence of several high-quality positional weight matric (PWM) motif matches for a certain TF in a gene promoter would increase the confidence in the corresponding TF-gene regulation. As shown in supplementary figure S12, Supplementary Material online, we found that among the detected rewiring events, regulons possess a significantly higher number of motif hits for their true regulator compared with those for the replaced regulator across all species, thus providing support for our approach.
Our RS is based on the partition of the species on a defined lineage, and utilizes the binding probabilities in all extant species. Although our significance assessment does control for the phylogeny, in principle, the inherent phylogenetic relationships between species would be better exploited if ancestral sequences could be inferred at various internal nodes of the tree, and RS was computed based on the inferred ancestral sequences. However, ancestral sequence reconstruction (ASR) (for which we used FastML from Ashkenazy et al. 2012) relies critically on the quality of multiple sequence alignment (MSA), which is a major concern because the promoters of extant yeast orthologs are highly diverged with a potentially large amount of binding site turnover. We therefore first assessed the quality of the MSA, generated using two methods: M-Coffee (generates alignment consensus from multiple progressive and iterative methods; Wallace et al. 2006) and PRANK (a phylogeny aware method for sequence alignment; Löytynoja and Goldman 2008). We found that, as suspected, the length of the ancestral sequence produced by both methods were on average twice the length of the longest individual promoter (supplementary fig. S13, Supplementary Material online), suggesting a very poor alignment with several gaps. Furthermore, an IC calculation based on posterior probabilities of nucleotides at each position of the resulting ancestral sequences revealed that on average the IC is ∼0.3 (min = 0, max = 2), which is extremely low and inappropriate for ASR, thus ruling out the suitability of ASR approach to assess rewiring.
We observe many more cases of rewiring at the regulon level, as opposed to the gene level, while in reality one would expect the opposite. There are at least two possible reasons for this outcome. The first has to do with an extremely stringent control imposed by the rotation test in gene-level testing, as discussed further in the Methods section. The second potential reason is increased statistical power in regulon-level testing, that is, even if the individual gene rewiring events are not strongly evidenced by loss/gain of TF sites as supported by all species (relative to a stringent rotation test), it is easier to detect them when they occur in multiple functionally related genes of a regulon (such as in RP genes). Furthermore, these regulon-level events spanning rewiring at multiple gene loci are likely to have gone through a gradual switch in regulation across species. For instance, some RP genes in Sklu, Cgla, and Kwal contain strong binding sites for both RAP1 and TBF1 (Tanay et al. 2005). These RP genes, therefore, are not detectable in our gene-level analysis, despite retaining a rather strong signal for rewiring at the regulon level (fig. 3).
In the extreme case, rewiring posits that in any given species, all genes of a regulon will be regulated by exactly one of the two TFs in question. However, in reality, a gradual evolutionary transition in the regulation of a regulon’s member genes is expected. Such a transitionary stage is characterized by an ancestral species where the regulon genes are bound by both TFs without a clear winner. Moreover, such transitionary stage may be maintained in some of the extant species. To assess the extent of such a transitionary species, for each regulon detected to have undergone rewiring, we estimated the fraction of species (out of 23) that display an intermediate level of rewiring. For a rewiring event involving TFs X and Y, we defined a species to be transitionary if the fraction of gene promoters (in the particular regulon) more likely to be bound by X than by Y is between 0.4 and 0.6, that is, not extreme. We found that, on average across all detected events, ∼8 species (of 23) can be considered transitionary, that is, with regulons potentially regulated by both TFs.
In summary, our probabilistic approach, while recapitulating the well-established cases, implicates specific regulators involved in suspected cases of rewiring, for which the implied regulators are not known. A genome-wide unbiased screen suggests that evolutionary cis-regulatory rewiring is relatively frequent and may be a significant mechanism of introducing regulatory innovations and adaptations to changing environments. The detected rewiring events are well supported by regulator-target species-specific coexpression. Our observation that the rewired TF pairs tend to function in similar biological processes compliments a previous observation that evolutionarily diverged targets of a TF nevertheless possess common functions (Habib et al. 2012). Taken together, these two observations suggest high plasticity in regulatory networks. We also found that rewired TFs are generally controlled by a common UpR, and in general the rewired TFs occupy lower levels in regulatory hierarchy. It is likely that regulatory rewiring at the individual gene level is frequent and without strong selection (as also noted in Habib et al. 2012), while repeated rewiring at multiple loci consisting of functionally related genes may partly be due to directional selection. In the future, knowledge from deeper phylogeny could be used to infer the temporal ordering of specific events in cis-element evolution (e.g., the events within a regulon) which may help distinguish potential seeding events from the ones that follow, likely under selection.
Materials and Methods
Gene Orthology Groups, Annotations, and Sequences
An orthogroup comprises orthologs across a set of species. Gene orthogroup assignments for all predicted protein-coding genes across 23 Ascomycete fungal genomes were obtained from the Fungal Orthogroups Repository (Wapinski et al. 2007a) maintained by the Broad Institute (broadinstitute.org/regev/orthogroups: last accessed January 2014). For our analysis, we only considered the 3,844 orthogroups (Wapinski et al. 2007b) that had mappable orthologs across at least 14 or more species as a compromise between number of genes included and loss of power due to information across fewer species. The genome sequences and gene annotations were obtained from a variety of databases and studies summarized in “Data Sources” at the above link. Gene promoter sequences were defined as 600 bases upstream of the ATG codon and truncated when neighboring open reading frames (ORFs) overlapped with this region (also obtained from Wapinski et al. 2007a). All promoters of length <50 were excluded. Mean and standard deviation of lengths of retained promoters were 472.5 and 164.2 bp.
Probabilistic Rewiring Score
We demonstrate here a toy example of the framework used in this analysis. The sample tree in figure 1A shows four species (s1, s2, s3, s4) partitioned at a select internal branch b to produce the equivalent of two species in the left clade (s1, s2 Є S) and two species (s3, s4 Є T) in the right clade. Gene locus g represents the orthologous group of genes across all the four species (g1, g2, g3, g4 Є G) that hypothetically exhibits differential usage of regulating TFs X and Y, where X is used by species in the left clade and Y is used by species in the right clade, and not vice versa.
The function that tests if TF Y is predominantly used by genes in T, but was replaced by TF X in the S in a lineage specific manner is as follows:
where terms of the form P(TF, gene, species) represent the computed probability of a TF binding to gene’s promoter in the species (see below). The denominators in the right hand side of both equations represent the size of the left and right clades, respectively. Generalizing the same, we get
where Lb and Rb denote the sizes of the left and right clades resulting from a partition at branch b, respectively.
PWM-based TF Binding Probability
A list of 176 PWMs for S. cerevisiae TFs was obtained from TRANSFAC (Matys et al. 2006). A single PWM may map to one or more TFs, and vice versa. To compute the probability of a TF binding to a gene’s promoter in a given species, that is, P(TF, gene, species), we scan the gene’s promoter using PWMSCAN (Levy and Hannenhalli 2002) which provides a P value for each putative site based on a species-specific background of sequence composition. We note the lowest P value obtained in the promoter and transform that into a promoter-wide probability score based on a previously used approach (Chen et al. 2007) as follows:
P(TF, gene, species) = (1 – P)(L–w+1), where L is the length of the promoter and w is the length of the motif.
In (rare) cases where an orthogroup included multiple genes (paralogs) for the same species, we used the average binding probability for all such genes to obtain a species-specific binding probability for the orthogroup. Additionally, for orthogroups missing a gene in a given species, we imputed the value of P(TF, gene, species) by averaging the binding probabilities of all sibling species with detectable orthologs. This essentially has the effect of deriving binding potential from related species, when it cannot be directly estimated by binding scores, thereby providing a suitable proxy.
Species Tree and Selected Lineages to Assess Rewiring
The species tree showing the relationships between 23 related Ascomycota fungi in figure 1B (obtained from Wapinski et al. 2007b) was used to determine the lineages (partitions) in the phylogeny. The six branches were chosen (highlighted in bold in fig. 1B) such that the resulting partitions reflect some clade-specific differences in the biology of these species, namely, sensu-stricto versus non–sensu-stricto (branch #7), pre-WGD versus post-WGD (branch #11), mostly pathogenic versus mostly nonpathogenic species (branch #20), and so on.
Expression Data
Expression profiles of Scer comprised data for 6,206 genes across 1,011 conditions, and Calb comprised data for 6,167 genes across 198 conditions (Ihmels et al. 2005). Tab-delimited text files containing the log2 ratios are obtained from weizmann.ac.il/home/barkai/Rewiring (last accessed January 2014).
Regulon Discovery
We used expression data in Scer and Calb to identify conserved regulons—a set of genes with similar function that are coordinately expressed both within and across these two species. These two species have diverged sometime between 160 and 800 My, representing a long evolutionary time. Starting from 1,982 manually curated functionally related groups of genes (Field et al. 2009), we generated coexpression networks for each group in Scer and for its mappable orthologous genes in Calb. The nodes in the network are the component genes and edge weights between them are |ρ|, where ρ represents the Spearman correlation between the expression vectors of those genes. The individual networks in the two species were then merged, such that each merged network consists of nodes representing conserved orthologs and the edge weights are the product of the corresponding edge weights in the Scer and Calb networks. This unit of edge weight is a proxy for a combined measure of distance based on conserved coexpression (i.e., lesser the distance between nodes, the more likely they are to have conserved coexpression in both species).
Next, each network was subjected to unsupervised clustering to isolate dense subgraphs that are representative of regulons, as per the above definition. We used MCL, a Markov Cluster Algorithm (Van Dongen and Abreu-Goodger 2012), to identify these subnetworks using a setting of medium granularity in resolving clusters (“-I 2” option). Because these algorithms are not robust to large graphs with too many edges (despite using edge weights), we removed those edges with low combined measures of coexpression (<= 0.06). This cutoff provides a reasonably good proxy that preserves edges reflecting high correlation, while cutting out the noise significantly (supplementary fig. S14A, Supplementary Material online). The application of MCL resulted in several subgraphs of functionally related genes with high coexpression (regulons). We excluded regulons that were larger than 100 genes, as well those with 6 or fewer genes (except the galactose regulon), thus identifying 1,713 regulons. Although some overlap of genes across regulons of different functional processes is expected, it is relatively small (mean Jaccard index = 0.003) to be of any concern (supplementary fig. S14B, Supplementary Material online).
Note
For cases where a given species possessed multiple genes belonging to the same orthologous group, the expression profiles of the member genes were averaged before computing pairwise correlations with other ortholog groups within that species.
Generation of Species-Specific TF–TF Networks
We used PWMSCAN (Levy and Hannenhalli 2002) to scan the promoter sequences of TF-encoding genes in Scer and Calb. For all hits detected with a motif-match score of 0.95 (using a species-specific background of nucleotide composition), we assigned a directional edge between the corresponding TF pairs. Using the igraph package (Csárdi and Nepusz 2006) in R, we generated a species-specific network using data from the above, which was then used to compute shortest paths between pairs of TFs in a species-specific fashion.
Phylogeny-Preserving Rotation Test to Assess Significance of Rewiring Score at Gene Level
The overall aim of the rotation test is to enable sampling of related variables from a null distribution such that inherent covariance structure, i.e., the relationships between variables (TF-binding probability profile across species in our case) is preserved (Langsrud 2005). We first derive the species-by-species (23 × 23) covariance matrix ∑ based on concatenated binding probabilities for all TFs at all gene loci in each species (estimating covariance matrix for each TF separately does not influence the overall results). Next, for each TF, we obtain the 23-dimensional vector µ of TF-specific mean binding probabilities of all orthogroups in each of the 23 species. Finally, for a TF, given TF-specific vector µ and the general covariance matrix ∑, we randomly sample from a multivariate normal distribution (x ∼ N(µ, ∑)), which is analogous to sampling from the matrix of the TF’s binding probabilities in all 3,844 orthogroups across all 23 species, while preserving the covariance structure. We considered this a stringent control as the covariance matrix directly captures the relationship of TF binding probabilities across species (as required by the rotation test).
Upon generating these rotated TF binding probabilities for the same number of synthetic orthologous loci (and inverting these distributions back to the probability scale of (0, 1)), we applied the RS function to generate a background distribution of RSs. This enabled computation of an FDR value for every observed RS, as summarized for different thresholds in supplementary figure S15, Supplementary Material online.
Although in principle covariance matrix derived from the known phylogeny of the 23 species should be similar to the one based on all TF binding probabilities, we found that a phylogeny inferred from TF binding probabilities differs from the known species phylogeny (supplementary fig. S16, Supplementary Material online). This suggests that evolution of TF binding probability does not strictly follow the neutral expectation. Thus by directly controlling for overall TF binding probability relationships, our criteria for detecting gene-level rewiring should be considered highly stringent.
Nucleosome Occupancy Data
Genome-wide nucleosome occupancy data for 12 species, namely, Kluyveromyces waltii, Saccharomyces bayanus, Saccharomyces cerevisiae, Yarrowia lipolytica, Debaryomyces hansenii, Candida albicans, Candida glabrata, Saccharomyces castellii, Saccharomyces paradoxus, Kluyveromyces lactis, Saccharomyces mikatae, and Saccharomyces kluyveri was obtained from GSE22211 (Tsankov et al. 2010); and for Saccharomyces pombe from GSE28839 (Tsankov et al. 2011).
Supplementary Material
Supplementary figures S1–S16 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We would like to thank Avinash Das and Justin Malin for their useful comments and suggestions. This work was supported by NIH grant R01GM100335 to S.H.
References
- Ashkenazy H, Penn O, Doron-Faigenboim A, Cohen O, Cannarozzi G, Zomer O, Pupko T. 2012. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 40:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew C, Sellam A, Epp E, Hogues H, Mullick A, Nantel A, Whiteway M. 2009. Transcriptional regulation of carbohydrate metabolism in the human pathogen Candida albicans. PLoS Pathog. 5:e1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CR, Tuch BB, Johnson AD. 2011. Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proc Natl Acad Sci U S A. 108:7493–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M, Thompson D. 2008. UME6, a novel filament-specific regulator of Candida albicans hyphal extension and virulence. Mol Biol Cell. 19:1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew CR, Suzuki T, Du Z, Backues SK, Jin M, Lynch-Day MA, Umekawa M, Kamath A, Zhao M, Xie Z, et al. 2012. Ume6 transcription factor is part of a signaling cascade that regulates autophagy. Proc Natl Acad Sci U S A. 109:11206–11210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. 2005. Reverse engineering of regulatory networks in human B cells. Nat Genet. 37:382–390. [DOI] [PubMed] [Google Scholar]
- Bensen ES, Filler SG, Berman J. 2002. A forkhead transcription factor is important for true hyphal as well as yeast morphogenesis in Candida albicans. Eukaryot Cell. 1:787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N, Kim PM, Gerstein MB. 2010. Rewiring of transcriptional regulatory networks: hierarchy, rather than connectivity, better reflects the importance of regulators. Sci Signal. 3:ra79. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Warner JR. 2008. Tbf1 or not Tbf1? Mol Cell. 29:537–538. [DOI] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, Nixon J, Ramage L, Kolas N, O’Donnell L, et al. 2015. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 43:470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Jensen ST, Stoeckert CJ. 2007. Clustering of genes into regulons using integrated modeling-COGRIM. Genome Biol. 8:R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly CF, Wakefield J, Akey JM. 2014. Evolution and genetic architecture of chromatin accessibility and function in yeast. PLoS Genet. 10:e1004427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo J, Powers T. 2002. The TOR-controlled transcription activators GLN3, RTG1, and RTG3 are regulated in response to intracellular levels of glutamine. Proc Natl Acad Sci U S A. 99:6784–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JL, Hall MN. 2002. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 66:579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csárdi G, Nepusz T. 2006. The igraph software package for complex network research. InterJournal Complex Syst. 1695:1695. [Google Scholar]
- Elfving N, Chereji RV, Bharatula V, Björklund S, Morozov AV, Broach JR. 2014. A dynamic interplay of nucleosome and Msn2 binding regulates kinetics of gene activation and repression following stress. Nucleic Acids Res. 42:5468–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field Y, Fondufe-Mittendorf Y, Moore IK, Mieczkowski P, Kaplan N, Lubling Y, Lieb JD, Widom J, Segal E. 2009. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat Genet. 41:438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, Von Mering C, et al. 2013. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio S, Liu Z, Thornton J, Butow RA. 2005. Retrograde response to mitochondrial dysfunction is separable from TOR1/2 regulation of retrograde gene expression. J Biol Chem. 280:42528–42535. [DOI] [PubMed] [Google Scholar]
- Ha M, Kim ED, Chen ZJ. 2009. Duplicate genes increase expression diversity in closely related species and allopolyploids. Proc Natl Acad Sci U S A. 106:2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Wapinski I, Margalit H, Regev A, Friedman N. 2012. A functional selection model explains evolutionary robustness despite plasticity in regulatory networks. Mol Syst Biol. 8:619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol. 59:407–450. [DOI] [PubMed] [Google Scholar]
- Hogues H, Lavoie H, Sellam A, Mangos M, Roemer T, Purisima E, Nantel A, Whiteway M. 2008. Transcription factor substitution during the evolution of fungal ribosome regulation. Mol Cell. 29:552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann OR, Dea J, Noble SM, Johnson AD. 2009. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet. 5:e1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihmels J, Bergmann S, Gerami-Nejad M, Yanai I, McClellan M, Berman J, Barkai N. 2005. Rewiring of the yeast transcriptional network through the evolution of motif usage. Science 309:938–940. [DOI] [PubMed] [Google Scholar]
- Isalan M, Lemerle C, Michalodimitrakis K, Horn C, Beltrao P, Raineri E, Garriga-Canut M, Serrano L. 2008. Evolvability and hierarchy in rewired bacterial gene networks. Nature 452:840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42:D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M, Wilson A. 1975. Evolution at two levels in humans and chimpanzees. Science 188:107–116. [DOI] [PubMed] [Google Scholar]
- Langsrud Ø. 2005. Rotation tests. Stat Comput. 15:53–60. [Google Scholar]
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M. 2010. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol. 8:e1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Whiteway M. 2009. Rearrangements of the transcriptional regulatory networks of metabolic pathways in fungi. Curr Opin Microbiol. 12:655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Hannenhalli S. 2002. Identification of transcription factor binding sites in the human genome sequence. Mamm Genome. 13:510–514. [DOI] [PubMed] [Google Scholar]
- Li JT, Hou GY, Kong XF, Li CY, Zeng JM, Li HD, Xiao GB, Li XM, Sun XW. 2015. The fate of recent duplicated genes following a fourth-round whole genome duplication in a tetraploid fish, common carp (Cyprinus carpio). Sci Rep. 5:8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. 2008. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320:1632–1635. [DOI] [PubMed] [Google Scholar]
- Mallick J, Whiteway M. 2013. The evolutionary rewiring of the ribosomal protein transcription pathway modifies the interaction of transcription factor heteromer Ifh1-Fhl1 (interacts with forkhead 1-forkhead-like 1) with the DNA-binding specificity element. J Biol Chem. 288:17508–17519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martchenko M, Levitin A, Hogues H, Nantel A, Whiteway M. 2007. Transcriptional rewiring of fungal galactose-metabolism circuitry. Curr Biol. 17:1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34:D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis N, Kuziora MA, McGinnis W. 1990. Human Hox-4.2 and Drosophila deformed encode similar regulatory specificities in Drosophila embryos and larvae. Cell 63:969–976. [DOI] [PubMed] [Google Scholar]
- Nicholls S, Straffon M, Enjalbert B, Nantel A, Macaskill S, Whiteway M, Brown AJP. 2004. Msn2- and Msn4-like transcription factors play no obvious roles in the stress responses of the fungal pathogen Candida albicans. Eukaryot Cell. 3:1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozonov EA, van Nimwegen E. 2013. Nucleosome free regions in yeast promoters result from competitive binding of transcription factors that interact with chromatin modifiers. PLoS Comput Biol. 9:e1003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud’homme B, Gompel N, Carroll SB. 2007. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 104(Suppl. 1):8605–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Hittinger CT. 2007. Transcriptional rewiring: the proof is in the eating. Curr Biol. 17:R626–R628. [DOI] [PubMed] [Google Scholar]
- Romano LA, Wray GA. 2003. Conservation of Endo16 expression in sea urchins despite evolutionary divergence in both cis and trans-acting components of transcriptional regulation. Development 130:4187–4199. [DOI] [PubMed] [Google Scholar]
- Roy S, Wapinski I, Pfiffner J, French C. 2013. Arboretum: reconstruction and analysis of the evolutionary history of condition-specific transcriptional modules. Genome Res. 23:1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, Friedman N. 2003. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 34:166–176. [DOI] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Sandstrom R, Haugen E, Reynolds AP, Zhang M, Byron R, Canfield T, Stelhing-Sun S, Lee K, et al. 2014. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. 1995. A direct approach to false discovery rates. J R Stat Soc B. 64:479–498. [Google Scholar]
- Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. 2012. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8:e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Regev A, Shamir R. 2005. Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci U S A. 102:7203–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Roy S, Chan M, Styczynsky MP, Pfiffner J, French C, Socha A, Thielke A, Napolitano S, Muller P, et al. 2013. Evolutionary principles of modular gene regulation in yeasts. Elife 2:e00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov A, Yanagisawa Y, Rhind N, Regev A, Rando OJ. 2011. Evolutionary divergence of intrinsic and trans-regulated nucleosome positioning sequences reveals plastic rules for chromatin organization. Genome Res. 21:1851–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov AM, Thompson DA, Socha A, Regev A, Rando OJ. 2010. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol. 8:e100414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Li H, Johnson AD. 2008. Evolution of eukaryotic transcription circuits. Science 319:1797–1800. [DOI] [PubMed] [Google Scholar]
- Ucar D, Beyer A, Parthasarathy S, Workman CT. 2009. Predicting functionality of protein-DNA interactions by integrating diverse evidence. Bioinformatics 25:i137–i144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen S, Abreu-Goodger C. 2012. Using MCL to extract clusters from networks. Methods Mol Biol. 804:281–295. [DOI] [PubMed] [Google Scholar]
- Wallace IM, O’Sullivan O, Higgins DG, Notredame C. 2006. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34:1692–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A. 2007a. Natural history and evolutionary principles of gene duplication in fungi. Nature 449:54–61. [DOI] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A. 2007b. Automatic genome-wide reconstruction of phylogenetic gene trees. Bioinformatics 23:i549–i558. [DOI] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. 2010. Conserved expression without conserved regulatory sequence: the more things change, the more they stay the same. Trends Genet. 26:66–74. [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G. 2011. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet. 13:59–69. [DOI] [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 8:206–216 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.