

Abstract
By definition, pseudogenes are relics of former genes that no longer possess biological functions. Operationally, they are identified based on disruptions of open reading frames (ORFs) or presumed losses of promoter activities. Intriguingly, a recent human proteomic study reported peptides encoded by 107 pseudogenes. These peptides may play currently unrecognized physiological roles. Alternatively, they may have resulted from accidental translations of pseudogene transcripts and possess no function. Comparing between human and macaque orthologs, we show that the nonsynonymous to synonymous substitution rate ratio (ω) is significantly smaller for translated pseudogenes than other pseudogenes. In particular, five of 34 translated pseudogenes amenable to evolutionary analysis have ω values significantly lower than 1, indicative of the action of purifying selection. This and other findings demonstrate that some but not all translated pseudogenes have selected functions at the protein level. Hence, neither ORF disruption nor presence of protein product disproves or proves gene functionality at the protein level.
Keywords: nonsynonymous substitution rate, synonymous substitution rate, purifying selection, macaque, translation, transcription
Pseudogenes are defined as gene relics that no longer encode functional products. Most pseudogenes originate from duplicate copies of functional genes. They are referred to as unprocessed or processed pseudogenes, depending on whether the duplication is DNA mediated or RNA mediated (Podlaha and Zhang 2010). A functional gene may also become a pseudogene without duplication, if its function no longer confers a fitness advantage to the organism due to a change in the environment or genetic background. Such pseudogenes are called unitary pseudogenes (Zhang et al. 2010; Marques et al. 2012). Because it is difficult to prove the lack of biological function for a segment of DNA, a pseudogene is operationally defined by its homology to a functional gene yet the presence of signs of nonfunctionality. The most obvious sign of nonfunctionality is a disruption of the canonical open reading frame (ORF) that exists in a homologous functional gene. Because RNA-mediated gene duplication only copies the transcribed region of a gene, the duplicate lacks the original promoter and is most likely “dead-on-arrival” (Podlaha and Zhang 2010). Thus, RNA-mediated duplicates, which typically lack introns that exist in their parental genes, are generally considered processed pseudogenes. Occasionally, an RNA-mediated duplicate may retain the canonical ORF and has evidence for transcription. In such a case, it is annotated as a retrogene rather than a processed pseudogene (Pei et al. 2012). Based on these operational criteria, numerous pseudogenes have been annotated in sequenced genomes (Karro et al. 2007; Podlaha and Zhang 2010; Sisu et al. 2014).
Because the operational definition of pseudogene does not require proof of nonfunctionality, claims of functionality have been made a number of times for operationally defined pseudogenes especially when they are transcribed (Balakirev and Ayala 2003; Pink et al. 2011; Marques et al. 2012; Poliseno 2012). In particular, several pseudogenes have been shown to be involved in tumorigenesis (Pink et al. 2011; Poliseno 2012). For example, human PTENP1 is a highly transcribed pseudogene originating from a duplicate of the tumor suppressor gene PTEN. PTENP1 competes with PTEN for the microRNAs that suppress PTEN expression, and it was reported that PTENP1 tends to be lost in cancer patients compared with healthy controls (Poliseno et al. 2010). But because biochemical activities may have no fitness benefit, proof of a true biological function requires the demonstration that the activity or the pseudogene is under natural selection (Doolittle 2013; Graur et al. 2013; Doolittle et al. 2014). No such proof has been provided in the case of PTENP1. In an example unrelated to cancer, the transcript of the mouse pseudogene Makorin1-p1 was shown to regulate its parental gene (Hirotsune et al. 2003) and be under purifying selection (Podlaha and Zhang 2004). But subsequent studies questioned the validities of both the functional data (Gray et al. 2006) and evolutionary data (Kaneko et al. 2006). More recently, an evolutionary genomic analysis of human transcribed pseudogenes that have macaque orthologs detected a small yet significant decrease in human–macaque sequence divergence in transcribed pseudogene regions, compared with corresponding flanking regions, suggesting that some transcribed pseudogenes are under purifying selection (Khachane and Harrison 2009). But it is unknown how many transcribed pseudogenes have selected functions and by what means their transcripts function.
Very recently, a human proteomic study reported peptides encoded by 107 pseudogenes (Kim et al. 2014). These peptides may signal pseudogene function at the protein level, a rarely considered possibility. Alternatively, they may have resulted from spurious translations with no protein function. We here distinguish between these two hypotheses by comparing the nonsynonymous/synonymous substitution rate ratio (ω) between translated pseudogenes and other pseudogenes based on human–macaque orthologs. We chose macaque (Macaca mulatta) for comparison, because the divergence between human and macaque is high enough to offer reasonable statistical power in testing natural selection yet low enough to permit the identification of many one-to-one orthologous pseudogenes.
Evolutionary analysis of pseudogenes is more tedious and error-prone than that of functional genes, because pseudogenes typically evolve rapidly in sequence and are frequently lost in evolution due to the loss of functional constraints. We designed a protocol detailed in Materials and Methods that is relatively conservative in claiming functional pseudogenes but allows fair comparisons among different groups of pseudogenes. Briefly, we subjected 15,352 human pseudogenes annotated in Ensembl (version 78) to a bioinformatic pipeline to acquire a set of 34 human–macaque orthologous pseudogenes that encode peptides on the basis of human proteomic data and are sufficiently long for ω analysis. For comparison, we acquired a set of 656 human–macaque orthologous pseudogenes that are transcribed (but have no proteomic hit) in humans and a set of 1,464 human–macaque orthologous pseudogenes that are not transcribed (and have no proteomic hit) in humans. We estimated ω for the ORF region of each human–macaque orthologous pseudogene alignment. The median ω of the translated pseudogenes is 0.68, significantly lower than that (0.91) of the transcribed pseudogenes (P = 0.045, Mann–Whitney U test; fig. 1) and that (0.88) of nontranscribed pseudogenes (P = 0.027; fig. 1), whereas the latter two groups have similar ω (P = 0.305; fig. 1).
Fig. 1.
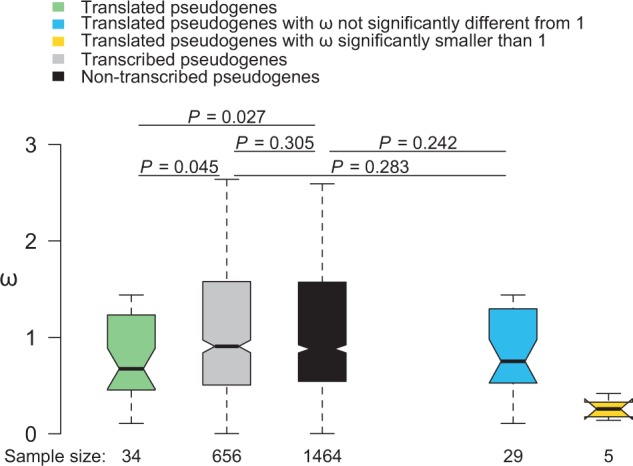
Comparison of nonsynonymous to synonymous substitution rate ratio (ω) among translated, transcribed, and nontranscribed human pseudogenes. In this bar plot, the notch indicates the median and the bar corresponds to the interquartile range (IQR), covering from the first quartile to the third quartile of the sample. The two whiskers of the bar show the minimum value not smaller than the first quartile minus 1.5 times IQR and the maximum value not greater than the third quartile plus 1.5 times IQR, respectively. The numbers of pseudogenes examined are provided beneath the bars. P values are from Mann–Whitney U tests.
For 19 of the 34 translated pseudogenes, an ortholog was also found in marmoset (Callithrix jacchus) by the same bioinformatic pipeline. Using marmoset as the outgroup, we tested whether ω is significantly different between the human and macaque lineages. Only two of these 19 pseudogenes showed a significant difference in ω between the two lineages at the nominal P value of 0.05 (i.e., without correction for multiple testing), with q values of 0.11 and 0.33, respectively. Furthermore, the median ω of the 19 pseudogenes is not significantly different between the human and macaque lineages. Thus, there is no evidence for different ω between human and macaque for the translated pseudogenes analyzed.
We found the median ORF length of the translated pseudogenes to be 458 nt, significantly greater than that (333) of the transcribed (but not translated) pseudogenes (P = 0.007), supporting the notion that the coding capacity is selectively maintained in at least some translated pseudogenes.
Perhaps not surprisingly, the median ω of the translated pseudogenes is substantially higher than that (0.12) of their parental genes (P < 3 × 10−7). This higher median ω may be because the translated pseudogenes are subject to weaker purifying selection and/or only a subset of them is subject to purifying selection. We found that only five (or 14.7%) of the 34 translated pseudogenes have ω values significantly lower than 1 (nominal P < 0.05, likelihood ratio test of the null hypothesis of ω = 1). Based on the computed q values (Storey et al. 2015), the false discovery rate (FDR) in the above finding is less than 8%. Therefore, all of the above five significant cases are likely genuine. These five translated pseudogenes have a median ω of 0.26. The remaining 29 translated pseudogenes have a median ω of 0.75, which is not significantly different from that of transcribed pseudogenes (P = 0.283; fig. 1) or nontranscribed pseudogenes (P = 0.242), suggesting that most of the translated pseudogenes are probably not under purifying selection. In other words, pseudogene translation does not indicate selected functionality in most cases.
When using the same FDR = 8% as the cutoff, we found 4.9% of transcribed pseudogenes and 0.49% of nontranscribed pseudogenes to have ω values significantly lower than 1. These results suggest that a small percentage of transcribed pseudogenes may have protein-level functions but their protein products have yet to be identified. Also, a tiny fraction of nontranscribed pseudogenes may have protein-level functions but their transcripts and peptides are still undetected. We also tested for positive selection by the criterion of having a ω value that is significantly greater than 1. Zero translated pseudogene, 3.7% of transcribed pseudogenes, and 3.6% of nontranscribed pseudogenes satisfy this criterion at the nominal P value of 0.05. Because none of these percentages exceed the expected false positive rate of 5%, we conclude that there is no evidence for positive selection acting on the pseudogenes.
All five translated pseudogenes with ω significantly lower than 1 (table 1) are processed pseudogenes, reminiscent of a number of reported cases of new genes that arose from retroduplicates initially thought to be pseudogenes (Long and Langley 1993; Jones et al. 2005; Kaessmann et al. 2009). Three of the above five pseudogenes preserve parental ORFs, whereas the other two (FUNDC2P2 and TUBB4AP1) have the original ORFs disrupted by premature stop codons. We found that the latter two pseudogenes exploit other in-frame start codons to circumvent the premature stop codons. For example, one of the cases involves FUNDC2P2, the pseudogene of a duplicate of FUNDC2 (FUN14 domain containing 2). In the pseudogene transcript, a premature stop codon appears downstream of the original start codon, which would result in a truncated peptide of 24 residues (fig. 2). Interestingly, a peptide identified in the proteomic data is uniquely mapped to the transcript sequence after the premature stop codon. An alternative ORF that starts with an in-frame ATG closely following the premature stop codon could code for a protein that contains the identified peptide (fig. 2). Thus, this in-frame ATG is likely the alternative start codon for the transcript. The protein encoded by the alternative ORF is 81% the length of the parental protein and contains the complete FUN14 domain of the parental protein, suggesting that it carries a similar molecular function. In addition to macaque, we also searched for the orthologs of FUNDC2P2 in chimpanzee, gorilla, orangutan, marmoset, and mouse. We were able to find its orthologs that include potential coding regions in chimpanzee, gorilla, and orangutan (table 1), suggesting that FUNDC2P2 has been maintained by natural selection in Old World primate evolution.
Table 1.
Five Translated Human Pseudogenes with ω Significantly Smaller Than 1.
| Pseudogene Symbol | Ensembl ID | ω | Protein Tissue Expression | Phylogenetic Distribution | Parental Gene |
|
|---|---|---|---|---|---|---|
| Ensembl ID | Description | |||||
| FUNDC2P2 | ENSG00000182814 | 0.327 | Testis | Human, chimpanzee, gorilla, orangutan, and macaque | ENSG00000165775 | FUN14 domain containing 2 |
| RP11‐34P1.2 | ENSG00000254373 | 0.257 | Frontal cortex | Human, chimpanzee, gorilla, orangutan, macaque, and mouse | ENSG00000156467 | Ubiquinol-cytochrome c reductase binding protein |
| TCEB2P2 | ENSG00000255262 | 0.418 | Fetal ovary | Human, chimpanzee, gorilla, orangutan, macaque, marmoset, and mouse | ENSG00000103363 | Transcription elongation factor B, polypeptide 2 |
| TUBB4AP1 | ENSG00000228466 | 0.177 | Frontal cortex | Human, chimpanzee, gorilla, orangutan, and macaque | ENSG00000173213 | Tubulin beta-8 chain-like protein |
| UBE2L5P | ENSG00000236444 | 0.139 | Testis | Human, chimpanzee, gorilla, orangutan, macaque, and marmoset | ENSG00000185651 | Ubiquitin-conjugating enzyme E2L 3 |
Fig. 2.

An example of a human translated pseudogene using an alternative start codon to circumvent a premature stop codon. Human FUNDC2 (FUN14 domain containing 2) is the parental functional gene, while FUNDC2P2 is the pseudogene. The coding DNA sequence of FUNDC2 is aligned with the homologous sequence of FUNDC2P2. The encoded amino acids are presented above or below the corresponding codons. In the pseudogene, the codon highlighted in blue is the original start codon, and the codon highlighted in green is the alternative start codon. The early stop codon in the pseudogene, created by a 1-nt insertion, is highlighted in yellow. The amino acids in red consist of the FUN14 domain in FUNDC2.
We found that the 34 translated pseudogenes have peptides identified from on average two out of 30 tissues (including cell lines) surveyed in the human proteomic data. The corresponding number (28) is much larger for their parental genes. Furthermore, the protein expression tissues of each translated pseudogene are a subset of those of its parental gene. The translated pseudogenes appear in 64 tissues in total (a tissue is counted as many times as the number of pseudogenes found translated in the tissue), including 6 times in testis and 58 times in other tissues. This ratio of 6/58 = 0.1 is greater than the corresponding ratio (0.04) for their parental genes with marginal significance (P = 0.07, Fisher’s exact test). A ratio of 0.7 was found among the translated pseudogenes with ω significantly smaller than 1. The preferred translation of pseudogenes in testis may be explained by the hyper-transcription hypothesis, which states that, in haploid germ cells of the testis, an overall permissive chromatin and abundant RNA polymerase II complexes promote widespread gene expression (Schmidt 1996; Soumillon et al. 2013).
The detection of purifying selection acting on five translated pseudogenes of humans raises the question of whether these pseudogenes were misannotated. Strictly speaking, the answer is yes, because by definition they are not pseudogenes if they are subject to purifying selection. A more practical question is whether their annotations as pseudogenes followed the commonly used guideline. The answer is also yes, because there was no evidence for their mRNA or protein expression at the time of annotation. Given that they are transcribed and translated and are under purifying selection, they should be reannotated as genes. These cases illustrate the point that neither ORF disruption nor presumed loss of promoter activity upon retroposition proves that a gene is nonfunctional.
In summary, our evolutionary analysis showed that human translated pseudogenes have significantly lower ω values than transcribed or nontranscribed pseudogenes. About 15% of translated pseudogenes have ω values significantly smaller than 1, suggesting that they possess selected functions at the protein level. But the rest of translated pseudogenes have ω values similar to transcribed or nontranscribed pseudogenes, suggesting that the majority of them likely possess no selected function at the protein level. We conclude that, while a small fraction of translated pseudogenes have selected functions, translation per se is not a guarantee of functionality.
Materials and Methods
Genome, Transcriptome, and Proteome Data
Human (hg38), macaque (rhsMac3), and marmoset (calJac3) genome sequences and exon coordinates were obtained from the UCSC genome browser (Rosenbloom et al. 2015). RNA sequencing (RNA-seq) data of all human genes in 16 tissues were downloaded from the human body map (Petryszak et al. 2014). Human pseudogenes and their peptides identified by mass spectrometry were from the human proteome draft generated by Kim et al. (2014). To reduce false discoveries, the authors manually curated the pseudogene peptides based on their mass spectra. They further excluded the peptides that may be explained by known alleles of human functional genes or alternative isoforms (Kim et al. 2014). Because the peptides generated in the study were short, because pseudogenes and their parental genes tend to be similar in sequence, and because pseudogene translation is expected to be rare, such data filtering were necessary to guard against false positives. Another recently published human proteome draft also reported translated pseudogenes (Wilhelm et al. 2014), but the data lacked such filtering and thus were not used in our analysis.
Orthologous Pseudogene Identification, Sequence Alignment, and ω Estimation
Human pseudogenes were obtained from Ensembl version 78 (Cunningham et al. 2015), including gene coordinates and pseudogene transcripts, which were annotated but not necessarily transcribed. From 15,352 annotated human pseudogenes, we removed 69 polymorphic and 226 immunity-related pseudogenes. The polymorphic pseudogenes have intact alleles in some human individuals and therefore were excluded. We removed immunity-related (i.e., immunoglobulin or T-cell receptor) pseudogenes because they may be subject to positive rather than negative selection when functional. For each human pseudogene, its syntenic region in macaque was identified in the LiftOver Browser (Kent et al. 2003). In parallel, the human pseudogene transcript was searched against the macaque genome using BLASTN (Altschul et al. 1990). The resulting high-scoring segment pairs that overlap the macaque syntenic region were considered orthologous exons. These macaque exons were tilted up to the human transcript following the BLAST alignment. A total of 8,070 human pseudogenes were found to have macaque orthologs.
We first aligned human and macaque orthologous pseudogene transcripts using ClustalW (Larkin et al. 2007). If the human transcript had peptide hits in the proteomic data, the longest ORF that codes for the peptide was identified as the coding ORF. If there was no peptide hit, the longest ORF was chosen as the potential coding ORF. In the coding ORF alignment, stop codons and codons with gaps were considered interruptive codons. The aligned codons between the human start codon and the first interruptive codon in the alignment were regarded as the coding region for the pseudogene. The likelihood-based CODEML program (Yang 2007) with default parameters was used to calculate ω for this region. For the parental genes of the pseudogenes concerned, we obtained from Ensembl the CODEML-derived estimates of ω based on human and macaque orthologs. The parental gene of a human pseudogene was defined as the human functional gene with the lowest E-value to the human pseudogene by BLAST.
Data Sets of Translated, Transcribed, and Nontranscribed Pseudogenes
Kim et al. (2014) identified peptides encoded by 107 human pseudogenes annotated in Ensembl version 78. We found that sometimes a peptide was perfectly mapped to a pseudogene transcript but the corresponding reading frame in the transcript has no start codon. Sometimes, a peptide was mapped to multiple pseudogenes. These peptides were removed, resulting in the final dataset of 75 unique translated pseudogenes. Sixty-eight of these human pseudogenes have macaque orthologs. Human–macaque alignments with 100% sequence identity or with fewer than 30 codons were removed because ω could not be estimated reliably. Occasionally, a pseudogene may have multiple transcripts and thus multiple alignments. The longest alignment was chosen for analysis. The above analyses resulted in 34 translated pseudogenes with qualified alignments.
Because pseudogene transcription may be tissue-specific, we used the human body map data (Petryszak et al. 2014) to identify transcribed pseudogenes. We followed the literature to use FPKM (fragments per kilobase of exon per million fragments mapped) ≥ 1 as a criterion for expression (Blazie et al. 2015). We found that 1,000 of the 8,070 human–macaque shared pseudogenes had FPKM ≥1 in at least one of the 16 tissues in the data but lacked peptide hits in the human proteomic data. These pseudogenes are referred to as transcribed pseudogenes. We regarded the longest ORF as the coding ORF in each pseudogene transcript, and applied the same procedure used for translated pseudogenes to generate codon alignments for these pseudogenes between human and macaque. This resulted in 656 transcribed pseudogenes with codon alignments.
To generate nontranscribed pseudogenes, we identified 2,274 human–macaque shared pseudogenes that have 0 FPKM in each of the 16 tissues and no peptide hit in the human proteome. From these pseudogenes, 1,464 had qualified codon alignments and were subject to further analyses.
The translated (34), transcribed (656), and nontranscribed (1,464) pseudogenes used for the comparison of ω , sequence alignments of the five translated pseudogenes under purifying selection, and the ω estimates of each branch of the tree of aligned sequences for the five pseudogenes are accessible from http://www.umich.edu/∼zhanglab/download/Jinrui_2015b/index.htm (last accessed November 26, 2015).
Acknowledgments
The authors thank Soochin Cho, Wei-Chin Ho, and Bryan Moyers for valuable comments. This work was supported in part by U.S. National Institutes of Health grant R01GM103232 to J.Z.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. 2003. Pseudogenes: are they “junk” or functional DNA? Annu Rev Genet. 37:123–151. [DOI] [PubMed] [Google Scholar]
- Blazie SM, Babb C, Wilky H, Rawls A, Park JG, Mangone M. 2015. Comparative RNA-Seq analysis reveals pervasive tissue-specific alternative polyadenylation in Caenorhabditis elegans intestine and muscles. BMC Biol. 13:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2015. Ensembl 2015. Nucleic Acids Res. 43:D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF. 2013. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A. 110:5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Brunet TD, Linquist S, Gregory TR. 2014. Distinguishing between “function” and “effect” in genome biology. Genome Biol Evol. 6:1234–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, Elhaik E. 2013. On the immortality of television sets: “function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol Evol. 5:578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray TA, Wilson A, Fortin PJ, Nicholls RD. 2006. The putatively functional Mkrn1-p1 pseudogene is neither expressed nor imprinted, nor does it regulate its source gene in trans. Proc Natl Acad Sci U S A. 103:12039–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S, Yoshida N, Chen A, Garrett L, Sugiyama F, Takahashi S, Yagami K, Wynshaw-Boris A, Yoshiki A. 2003. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature 423:91–96. [DOI] [PubMed] [Google Scholar]
- Jones CD, Custer AW, Begun DJ. 2005. Origin and evolution of a chimeric fusion gene in Drosophila subobscura, D. madeirensis and D. guanche. Genetics 170:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Vinckenbosch N, Long M. 2009. RNA-based gene duplication: mechanistic and evolutionary insights. Nat Rev Genet. 10:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Aki I, Tsuda K, Mekada K, Moriwaki K, Takahata N, Satta Y. 2006. Origin and evolution of processed pseudogenes that stabilize functional Makorin1 mRNAs in mice, primates and other mammals. Genetics 172:2421–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karro JE, Yan Y, Zheng D, Zhang Z, Carriero N, Cayting P, Harrrison P, Gerstein M. 2007. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 35:D55–D60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. 2003. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci U S A. 100:11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachane AN, Harrison PM. 2009. Assessing the genomic evidence for conserved transcribed pseudogenes under selection. BMC Genomics 10:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. 2014. A draft map of the human proteome. Nature 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- Long M, Langley CH. 1993. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science 260:91–95. [DOI] [PubMed] [Google Scholar]
- Marques AC, Tan J, Lee S, Kong L, Heger A, Ponting CP. 2012. Evidence for conserved post-transcriptional roles of unitary pseudogenes and for frequent bifunctionality of mRNAs. Genome Biol. 13:R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei B, Sisu C, Frankish A, Howald C, Habegger L, Mu XJ, Harte R, Balasubramanian S, Tanzer A, Diekhans M, et al. 2012. The GENCODE pseudogene resource. Genome Biol. 13:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E, Huber W, Jupp S, Keays M, Kryvych N, et al. 2014. Expression Atlas update–a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 42:D926–D932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. 2011. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA 17:792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlaha O, Zhang J. 2004. Nonneutral evolution of the transcribed pseudogene Makorin1-p1 in mice. Mol Biol Evol. 21:2202–2209. [DOI] [PubMed] [Google Scholar]
- Podlaha O, Zhang J. 2010. Pseudogenes and their evolution. In: Encyclopedia of Life Sciences. Chichester, UK: John Wiley & Sons; p. 1–8. [Google Scholar]
- Poliseno L. 2012. Pseudogenes: newly discovered players in human cancer. Sci Signal. 5:re5. [DOI] [PubMed] [Google Scholar]
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. 2010. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465:1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. 2015. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43:D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EE. 1996. Transcriptional promiscuity in testes. Curr Biol. 6:768–769. [DOI] [PubMed] [Google Scholar]
- Sisu C, Pei B, Leng J, Frankish A, Zhang Y, Balasubramanian S, Harte R, Wang D, Rutenberg-Schoenberg M, Clark W, et al. 2014. Comparative analysis of pseudogenes across three phyla. Proc Natl Acad Sci U S A. 111:13361–13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al. 2013. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 3:2179–2190. [DOI] [PubMed] [Google Scholar]
- Storey J, Bass A, Dabney A, Robinson D. 2015. Q-value estimation for false discovery rate control. Available from: http://bioconductor.org/packages/qvalue/. [Google Scholar]
- Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, et al. 2014. Mass-spectrometry-based draft of the human proteome. Nature 509:582–587. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhang ZD, Frankish A, Hunt T, Harrow J, Gerstein M. 2010. Identification and analysis of unitary pseudogenes: historic and contemporary gene losses in humans and other primates. Genome Biol. 11:R26. [DOI] [PMC free article] [PubMed] [Google Scholar]