

Abstract
LINE-1 (long interspersed element-1) retroelements are the only active autonomous endogenous retroelements in human genomes. Their retrotransposition activity has created close to 50% of the current human genome. Due to the apparent costs of this proliferation, host genomes have evolved multiple mechanisms to curb LINE-1 retrotransposition. Here, we investigate the evolution and function of the LINE-1 restriction factor APOBEC3A, a member of the APOBEC3 cytidine deaminase gene family. We find that APOBEC3A genes have evolved rapidly under diversifying selection in primates, suggesting changes in APOBEC3A have been recurrently selected in a host–pathogen “arms race.” Nonetheless, in contrast to previous reports, we find that the LINE-1 restriction activity of APOBEC3A proteins has been strictly conserved throughout simian primate evolution in spite of its pervasive diversifying selection. Based on these results, we conclude that LINE-1s have not driven the rapid evolution of APOBEC3A in primates. In contrast to this conserved LINE-1 restriction, we find that a subset of primate APOBEC3A genes have enhanced antiviral restriction. We trace this gain of antiviral restriction in APOBEC3A to the common ancestor of a subset of Old World monkeys. Thus, APOBEC3A has not only maintained its LINE-1 restriction ability, but also evolved a gain of antiviral specificity against other pathogens. Our findings suggest that while APOBEC3A has evolved to restrict additional pathogens, only those adaptive amino acid changes that leave LINE-1 restriction unperturbed have been tolerated.
Keywords: retroelements, LINE-1, APOBEC3A, positive selection, restriction factor, innate immunity, primates.
Introduction
Perhaps no other genetic entity has had a more profound impact on the present-day human genome than the LINE-1 (long interspersed element-1) retroelement, which is the only known autonomous mobile element in the genome. Not only do LINE-1 copies make up 13% of the genome themselves (Lander et al. 2001), but their retrotransposition activity also mobilizes other nonautonomous elements that comprise a further 25% of the human genome (Richardson 2015). By some estimates, the products of retrotransposition have created as much as two-thirds of the human genome (Lander et al. 2001; Jurka et al. 2005; de Koning et al. 2011). This impressive contribution suggests an ancient and ongoing battle for the “real estate” of the human genome. Active LINE-1 elements encode an approximately 6.7 kb transcript, which contains the coding sequence for two proteins (Scott et al. 1987; Dombroski et al. 1991)—ORF1p, an RNA-binding and chaperone protein (Martin and Bushman 2001), and ORF2p, which encodes endonuclease (Feng et al. 1996) and reverse transcriptase activities (Mathias et al. 1991). While these components primarily mobilize LINE-1 sequences, their reverse transcriptase activity also mobilizes other nonautonomous elements (e.g., Alu, SVA elements), as well as cellular RNAs to generate processed pseudogenes (Richardson et al. 2015).
Due to their mutagenic retrotransposition activity, LINE-1 retroelements have a detrimental consequence on host fitness (Boissinot et al. 2001, 2006). For example, nearly a hundred cases of human diseases have been associated with novel LINE-1 or LINE-1-facilitated germline insertions (Hancks and Kazazian 2012). Furthermore, LINE-1 elements impose an increasing hazard of ectopic recombination among element copies that generates chromosomal rearrangements (Boissinot et al. 2001; Montgomery et al. 2007; Song and Boissinot 2007; Beck et al. 2011). These costs have likely driven the evolution of a suite of host restriction factors that limit LINE-1 retrotransposition, both pre- and post-transcription, as well as other forms of both endogenous and infectious mobile elements (Molaro and Malik 2016). Transcription-silencing modifications of DNA and chromatin provide a principal mechanism of LINE-1 restriction (Yoder et al. 1997; Ooi et al. 2009). For instance, KRAB-ZNF proteins specifically recruit the silencing machinery to LINE-1 elements via direct DNA binding (Castro-Diaz et al. 2014; Jacobs et al. 2014; Turelli et al. 2014). Recent findings have demonstrated that LINE-1 elements may evolve to evade these KRAB-ZNF factors in primate genomes (Jacobs et al. 2014). Post-transcriptional restriction factors form an additional layer of genome defense, targeting a variety of steps in the lifecycle of LINE-1. For example, the TREX1 nuclease acts as a post-transcriptional regulator of transposons by degrading cytoplasmic DNA (Crow et al. 2006; Stetson et al. 2008; Zhao et al. 2013), and SAMHD1 enhances sequestration of LINE-1 ribonucleoproteins to block LINE-1 replication (Zhao et al. 2013; Hu et al. 2015). The importance of these post-transcriptional regulatory mechanisms is evident in diseases like Aicardi Goutieres Syndrome (AGS), wherein loss of function mutations in TREX1, SAMHD1, and other AGS genes drive autoimmunity, likely through the activity of unrestricted retroelements, triggering the immune system akin to a chronic viral infection (Crow and Rehwinkel 2009; Volkman and Stetson 2014).
One of the most potent post-transcriptional restrictors of LINE-1 retroelements in cell culture models is the human APOBEC3A cytidine deaminase (referred to as A3A henceforth) (Bogerd, Wiegand, Hulme, et al. 2006; Chen et al. 2006). A3A belongs to the APOBEC3 gene family, which has undergone a series of expansions and contractions in mammals, resulting in seven paralogs in primates but only a single A3 gene in rodents (Conticello et al. 2005; Munk et al. 2012). APOBEC3G (A3G) was the first A3 gene discovered with antiviral activity against some HIV-1 strains (Sheehy et al. 2002). HIV-1 and other lentiviruses encode Vif, an antagonist that binds host A3G proteins and mediates their degradation (Sheehy et al. 2003). Vif antagonism of A3G thus sets up an “evolutionary arms race” between host and viral genomes, whereby host A3G genes change to evade Vif binding and viral Vif genes subsequently evolve to reestablish A3G binding. This recurrent selection for functional innovation produces a characteristic pattern of an excess of non-synonymous (or amino acid altering) mutations relative to synonymous mutations at specific sites in both A3G and Vif genes. Indeed, signatures of diversifying selection coincide, in part, with sites that are shown to affect binding affinity between Vif and A3G directly (Compton et al. 2012; Compton and Emerman 2013). These findings demonstrate that primate lentiviruses have shaped the evolution of A3G, and vice-versa (Compton et al. 2013). However, not all of the positive selection in A3G can be attributed to pressure to evade Vif (Compton et al. 2012; Compton and Emerman 2013). In fact, A3G has likely evolved in response to pressure from multiple pathogenic viruses.
In contrast to A3G, which only weakly restricts human LINE-1, A3B and A3F have been shown to inhibit LINE-1 activity independent of their encoded deaminase activity (Steinglein and Harris 2006; Bogerd, Wiegand, Doegle, et al. 2006). However, the most potent anti-LINE-1 restriction activity among the human A3 genes is encoded by A3A, which has been shown to restrict human LINE-1 as well as Alu elements using in vitro retrotransposition assays which measure the reverse transcriptase-mediated replication of these elements (Bogerd, Wiegand, Hulme, et al. 2006; Chen et al. 2006). A recent mechanistic study shows that A3A deaminates the single-stranded DNA transiently generated during the process of LINE-1 retrotransposition, suggesting retroelement restriction functions of A3A likely require deaminase activity (Richardson et al. 2014). A3A mRNA levels transiently increase during reprogramming of pluripotent stem cells (Friedli et al. 2014) coincident with increased expression and protein production from LINE-1 elements (Coufal et al. 2009; Wissing et al. 2012). Unlike infectious viruses, endogenous mobile elements like LINE-1 must replicate in the germline or early embryo in order to increase or maintain their copy number in the genome. The expression of A3A in reprogramming pluripotent stem cells (a similar reprogramming also occurs during development) is, therefore, consistent with an integral role of A3A in the germline to defend against endogenous elements. We reasoned that if the anti-LINE-1 activity were an important driver of A3A evolution, there might be considerable variation in the restrictive capabilities of various primate A3A proteins against human LINE-1 under the “arms-race” paradigm. Indeed, species-specific adaptive changes in host and pathogen proteins often result in species-specific functional variation of host–pathogen interactions (Daugherty and Malik 2012). We were therefore intrigued by recent studies, which reported that at least some A3A proteins (e.g., from the rhesus macaque Macaca mulatta) might have lost LINE-1 restriction but gained lentiviral restriction instead (Schmitt et al. 2011). The recent findings suggested that rhesus A3A might restrict lentiviruses via a novel mechanism (Schmitt et al. 2011, 2013; Katuwal et al. 2014). Thus, the simultaneous loss of LINE-1 activity might reflect an apparent evolutionary tradeoff between lentiviral and LINE-1 restriction.
To investigate the molecular basis of this apparent tradeoff and its potential evolutionary consequences, we undertook a detailed analysis of A3A function and evolution across simian primate evolution. Our study revealed that some of the previous findings of A3A gain and loss of function were confounded by the inclusion of incorrect sequences of A3A genes deposited in public databases. Many of these incorrect sequences arose from polymerase chain reaction (PCR)-generated chimeras between A3A and A3G genes that replaced one A3A exon with its paralogous A3G counterpart. Upon resolving these discrepancies, we find that although A3A has evolved rapidly under positive selection in primates, it has strictly conserved its ability to restrict LINE-1 throughout its evolution. Despite this strict conservation of one A3A function, we find that a gain of antiviral function occurred in the A3A of the common ancestor of all cercopithecine primates, at least 11 Ma. Our data suggests that A3A has maintained its LINE-1 restriction but evolved rapidly due to selective pressure from a distinct pathogen, somehow partitioning these functions to preserve LINE-1 restriction in the face of recurrent bouts of selection driven by another pathogen.
Results
Phylogenetic Incongruence Reveals PCR-Based Chimeras in Primate APOBEC3A Sequences
A recent study reported the apparent loss of LINE-1 restriction in some primate A3A proteins (Schmitt et al. 2011). We wished to understand the molecular basis for this loss and ascertain whether other parallel losses of LINE-1 restriction had occurred during A3A evolution in primates. We, therefore, extended and re-examined the sampling of primate A3A sequences. For the latter, we gathered all available A3A sequences from public sequence databases. We found two sources for these previously reported A3A sequences. The first set derived from annotations of whole-genome assemblies. The second set was derived by targeted exon-by-exon PCR and sequencing, in which primers were designed to anneal to the intronic regions just outside each A3A exon boundary (Henry et al. 2012). To confirm and expand this previously described set of A3A sequences, we performed additional sequencing of the A3A locus from hominoids, Old World monkeys, and New World monkeys (supplementary table S1, Supplementary Material online).
As previously reported, A3A shows striking sequence variation across simian primates (Henry et al. 2012) (see supplementary data set S1, Supplementary Material online, for alignment). Our examination of the second exon from Old World monkey A3A sequences revealed two distinct sets of sequences with a surprising phylogenetic distribution. For instance, rhesus macaque and olive baboon A3A gene sequences were found to encode “DLSVRGRHQ” (hereafter referred to as “type 2”) in exon 2. In contrast, mandrill, African green monkey (AGM), colobus, and Debrazza’s monkey encoded “KL(/P)WVSGQR(/H)E” (“type x”). This divergence between type 2 and type x A3A exons is unexpected. Indeed, an A3A phylogeny built using only exon 2 sequences is incongruent with the primate phylogeny (fig. 1A).
Fig. 1.

Primate A3A-A3G chimeras contain exon 5 of A3G in place of A3A exon 2. (A) We generated an unrooted maximum likelihood phylogeny using an alignment of A3A exon 2, and A3G exon 5 nucleotide sequences (see Materials and Methods for details). Branches leading to A3A, A3G, or chimeric sequences are shown in black, gray, and dotted lines, respectively. Bootstrap values represent percentages based on 1,000 iterations. The previously reported “alternative exon 2-containing” A3As from mandrill, AGM, colobus (marked *) group with A3Gs, whereas the newly sequenced, corrected A3A exon 2s group with the rest of the A3As. Although the phylogeny is not well resolved within the A3A and A3G clades, the bootstrap support for separation of these two clades is high, grouping the chimeric A3A* sequences with the A3Gs. (B) The chimeric A3A-A3Gs comprise A3G coding exon 5 swapped into A3A in place of the second coding exon of A3A. These chimeras resulted from sequence similarity between the introns on either side of these two exons.
Upon closer examination, we found that the primers used in the previous exon-by-exon amplification approach (Henry et al. 2012) also matched the intronic sequence on either side of exon 5 of APOBEC3G. Indeed, the previously reported type x sequence of exon 2 in AGM A3A was almost identical to that of A3G exon 5 from the same species. As a result, phylogenetic analyses group all A3A exon 2 sequences together, except for type x A3A exon 2 sequences, which instead cluster with A3G exon 5 sequences (fig. 1A). We also used long PCR to amplify a 4 kb portion of the Colobus guereza genome that spans exons 1–3 and intervening introns using primers that should only bind A3A and not other A3 paralogs. Sequencing of this PCR product revealed the anticipated sequences of exons 1 and 3, as well as an exon 2 sequence similar to that found in the hominoid A3As (“type 1”—“GIG(/R)R(/W)”). Whole-genome assemblies are not immune to these assembly errors in multigene families. For example, the A3A sequence from the proboscis monkey (Nasalis larvatus WGS JMHX01000683) also reports a type x sequence of A3A exon 2. However, our targeted sequencing of the A3A from this species confirmed that they instead have a type 2 A3A exon 2. A recent release of a genome assembly of another species of colobus (Colobus angolensis, BCM-HGSC, NCBI BioProject Accession PRJNA251421) supports our findings in colobus and proboscis monkey.
Together, our data suggest that all type x A3A sequences, previously reported for mandrill, AGM, colobus, and DeBrazza’s monkey, likely represent artifactual A3A-A3G chimeras, which were generated by an exon-by-exon PCR and assembly process that used primers that would amplify both A3A and A3G (fig. 1B). Our targeted resequencing produced sequences that fit parsimoniously with the rest of the primate A3A sequences (fig. 1A). Using this corrected set of A3A sequences, we next re-examined the evolutionary signatures and functional conservation of biochemical and restriction activities of primate A3A proteins.
Diversifying Selection of Primate APOBEC3A
Using the corrected A3A sequences from our PCR-based sequencing from genomic DNA as well as additional A3A sequences from publicly available databases that we screened for any sign of chimeric sequences, we created an alignment of 19 simian primate A3A orthologs. This alignment included A3A representatives from New World monkeys, Old World monkeys, and hominoids, species that together represent more than 40 My of evolution (Perelman et al. 2011). Our data show that all primate A3As conserve the catalytic motifs of the deaminase domain (His-X-Asp and Cys-X2-4-Cys) (fig. 2A, in red lettering above the exon diagram). However, we observed substantial variation in the coding sequences of these A3As outside of the highly conserved catalytic positions.
Fig. 2.

Sequence variation and positive selection in primate A3A. (A) We created an alignment of 19 A3A sequences spanning the hominoids, Old World monkeys, and New World monkeys using publicly available databases and sequencing. Several primates have lost the methionine at “position 1” and instead initiate translation using the methionine at “position 13”. Exon boundaries are denoted by black squiggly lines and the conserved deaminase motif is noted in red labels, top. The exon 2 sequence of A3A varies among primates, which we classify into three general “types.” We tested the signature of positive selection using PAML’s NSsites maximum likelihood method and observe a significant signature of gene-wide positive selection (PAML M8a vs. M8). We also identify 13 codons with statistically significant positive selection (PAML M8, BEB, P > 0.9, table 1), as indicated by shading in the protein alignment and orange teardrops in the exon diagram. (B) The positively selected positions are shown as orange space-filling spheres with positions numbered in black on the white ribbon diagram (A3A NMR structure, PDB ID 2M65 [Byeon et al. 2013]); active site residues are shown in red sticks. Positively selected positions cluster in secondary structure as well tertiary structure along the “polynucleotide accommodating groove” (Bulliard et al. 2011). Amino acid positions numbered in red were shown to increase restriction of LINE-1 by human A3G when swapped from A3A into A3G (Bulliard et al. 2011).
A3A sequences are quite variable in the N-terminus as a result of the recurrent loss of methionine at position 1 (M1) and use of an alternate methionine at position 13 (M13) as the start codon. Previous studies have noted these alternative start codons; both isoforms of human A3A are expressed in interferon-alpha-treated PBMCs (peripheral blood mononuclear cells) and function as active deaminases (Thielen et al. 2010). Several species encode A3A genes that have lost M1 (fig. 2A) and presumably use only M13 as the start codon, as supported by adequate Kozak context at these M13 codons (Henry et al. 2012). It is currently unclear whether all species that encode both M1 and M13 produce both isoforms of the A3A protein and what functional significance such dual isoforms hold.
In addition to the variable N-terminus, we found that the A3A proteins are also highly variable in a short region of exon 2 even after correcting for the A3A-A3G chimeras (fig. 2A). Within exon 2 of A3A, there are two major sequence variants within Catarrhine primates, that is, Old World monkeys and hominoids. We found that hominoids (humans, chimps, gorilla, orangutan, gibbons) and Colobinae (colobus and snub-nosed monkey) share a similar GIG(/R)R(/W) type 1 motif. In contrast, Cercopithecinae (the macaques, sooty mangabey, baboon, AGM) have a “DLSVR(/L)GR” type 2 motif instead. This dichotomy suggests that the type 2 exon 2 sequence arose in the common ancestor of Cercopithecinae. The New World monkeys encode a different sequence from either type 1 or type 2 in this region—“PG(/R/E)VS(/F)GL(/Q/R)” (“type 3,” fig. 2A).
We analyzed our revised and expanded data set of primate A3A sequences for evidence of diversifying selection. We used maximum likelihood methods (PAML NSsites [Nielsen and Yang 1998]) to test whether models that permit positive selection on individual codons (dN/dS > 1) are a better statistical fit than models that disallow positive selection. We find that A3A displays a significant signature of diversifying selection across the entire gene (PAML NSsites M8 vs. M8a, P < 8 × 10−8) (table 1). This signature results from a high fraction of codons evolving with a high dN/dS ratio (14.6%, average dN/dS = 3.2). Thirteen codon positions have a high posterior probability of having evolved under diversifying selection (PAML NSsites M8, BEB P > 0.9) (fig. 2A); several of these sites are corroborated by the FUBAR program implemented in the HyPhy suite of analyses (Murrell et al. 2013) (table 1). Six of these PAML-identified residues are clustered within coding exon 2, suggesting that this region has been a major determinant of adaptation in the evolution of A3A. Although the majority of the A3A gene aligns unambiguously, even with multiple alignment methods (see Materials and Methods), some of the indel positions in exon 2 are difficult to unambiguously align (fig. 2A). However, eliminating the seven codon positions with ambiguous alignment from our analysis did not affect our conclusion of positive selection (PAML NSsites M8 vs. M8a, P < 8 × 10−7). Nevertheless, such clustering of positively selected residues is highly suggestive of protein domains that have been repeatedly influenced by recurrent diversifying selection to potentially “gain recognition” of a pathogenic entity or “escape recognition” by a pathogen antagonist (Sawyer et al. 2005; Daugherty and Malik 2012; Mitchell et al. 2012).
Table 1.
Primate APOBEC3A is Evolving under Positive Selection.
| Model | 2Δlnλ | df | P value | Position | AA | PAML | FUBAR |
|---|---|---|---|---|---|---|---|
| M8a vs. M8 | 32.63 | 2 | 8.22 × 10−8 | 16 | H | 0.999 | 0.763 |
| 17 | I | 0.934 | 0.217 | ||||
| 20 | S | 0.929 | 0.893 | ||||
| 30 | — | 0.950 | 0.796 | ||||
| 32 | — | 0.981 | 0.609 | ||||
| 34 | R | 0.983 | 0.442 | ||||
| 63 | H | 0.715 | 0.926 | ||||
| 71 | C | 1.000 | 0.997 | ||||
| 73 | F | 0.907 | 0.774 | ||||
| 85 | L | 0.926 | 0.848 | ||||
| 110 | S | 0.839 | 0.910 | ||||
| 115 | G | 0.965 | 0.946 | ||||
| 141 | P | 0.941 | 0.825 | ||||
| 142 | L | 0.993 | 0.777 | ||||
| 166 | K | 0.978 | 0.932 | ||||
| 167 | H | 0.868 | 0.940 | ||||
| 175 | H | 0.623 | 0.912 |
Note.—Positions refer to the numbering in figure 2 and the included supplementary alignment. AA refers to the amino acids found in the chimpanzee sequence. A gap (“—”) indicates that that position is not present in chimpanzee, but is still under positive selection in the other species. Positions found to be significant in both PAML and FUBAR are indicated in underline.
The positively selected positions in A3A mapped to surface-exposed residues in the published A3A structures (Byeon et al. 2013; Bohn et al. 2015). Many of them clustered within the polynucleotide-accommodating groove, a region previously predicted to comprise the ssDNA binding groove and underlie the specificity differences between A3A and A3G (Bulliard et al. 2011) (fig. 2B). Previous work showed that swapping some of the residues in this groove from A3A into A3G increases the ability of A3G to restrict LINE-1 (Bulliard et al. 2011) (fig. 2B, residues 36 and 68). We therefore posit that the A3A positive selection may be driven by selection to alter target-binding specificity. We concluded that, like other A3 paralogs, A3A has evolved under diversifying selection in primates, suggestive of a role in host defense against pathogens.
All Primate APOBEC3A Proteins Restrict LINE-1 Retroelements
The positive selection we described suggested that diversifying selection altered A3A’s specificity of interaction with a pathogen. However, these data provided us with little information on the identity of the coevolving pathogen. One candidate genomic pathogen that might have driven the diversifying selection of A3A in primates is the LINE-1 retroelement. Human A3A has been shown to potently restrict both human LINE-1 as well as other non-long terminal repeat (non-LTR) retroelements (Bogerd, Wiegand, Doehle, et al. 2006; Bogerd, Wiegand, Hulme, et al. 2006; Chen et al. 2006). Under A3A restriction, any LINE-1 with mutations that allow it to escape A3A restriction would presumably have a fitness advantage. However, if “escaped” LINE-1 elements proliferate and impact host fitness, A3A should evolve to recognize and limit this new LINE-1 variant. If this hypothesis were correct, we might expect an evolutionary arms race between A3A and LINE-1 elements.
To test this hypothesis, we examined a diverse panel of primate A3A proteins for their ability to restrict LINE-1. If primate A3As have coevolved with their respective LINE-1, we would expect to see variation in the ability of one species’ A3A to restrict another species’ LINE-1; in this case, not all primate A3As would be expected to restrict human LINE-1. We synthesized codon-optimized versions of N-terminally HA-tagged A3A proteins from a representative panel of extant simian primates, including New World monkeys, Old World monkeys, and hominoids. We made sure to include Old World monkeys with both forms of exon 2 (type 1 and type 2). We tested the ability of this panel of primate A3A proteins to restrict either human LINE-1 (LRE3) (Brouha et al. 2002) or a codon-optimized mouse LINE-1 (ORFeus_Mm) (An et al. 2011). Both assays used an episomal LINE-1 with a reporter gene in the antisense orientation (relative to the LINE-1 coding sequence) and interrupted by an intron (Moran et al. 1996). Expression of the reporter required that the LINE-1 bearing the reporter gene be transcribed, processed (removing the intron), reverse transcribed, and integrated into the host genome. Only successful retrotransposition would create a version of the antisense reporter gene that can be expressed. The reporter constructs we employed produced either GFP (green fluorescent protein) (human LINE-1) (Ostertag et al. 2000) or firefly luciferase (mouse LINE-1), and transcribed the LINE-1 sequence under the control of an active promoter (CMV or CAG, respectively). We normalized each assay for transfection efficiency and toxicity, since high levels of A3A may be toxic to host cells and degrade transfected DNA (Stenglein et al. 2010; Richardson et al. 2014). This normalization was achieved by measuring the expression of either a cotransfected dsRed plasmid (human LINE-1) or Renilla luciferase expressed from the same plasmid (mouse LINE-1) (An et al. 2011) (supplementary fig. S1A, Supplementary Material online). The amount of plasmid we transfected for each A3A resulted in very little toxicity, and the raw data showed the same patterns as the displayed normalized data. We empirically determined the amount of plasmid encoding each species’ A3A that gives comparable expression by Western blot (fig. 3). We used the readout of the reporter gene, relative to transfection control, to measure the degree to which expressed A3A proteins restrict either of the two LINE-1 elements.
Fig. 3.

LINE-1 restriction is conserved across primate A3A genes. We synthesized 9 codon-optimized primate A3As and tested their ability to restrict mouse ORFeus_Mm (left bar graph) or human LRE3 LINE-1 (right bar graph) using in vitro retrotransposition assays in 293T cells (see Materials and Methods). The amount of transfected A3A plasmid was optimized for similar levels of protein production as measured by Western blot against each A3A’s HA-tag. The Western blot image, shown on the right, represents an independent transfection of 25,000 293T cells with the same mass of A3A-expressing or empty plasmid transfected for the mouse LINE-1 assay (for the human assay, the plasmid mass was scaled to transfect the same ng plasmid/cell, see Materials and Methods). Each A3A restricted mouse LINE-1 by around 100-fold and human LINE-1 by around 10-fold; data are shown on a log scale. The mouse LINE-1 restriction data points represent the mean ± standard error of at least eight independent transfected wells and the human LINE-1 restriction data points represent the mean ± standard error of at least three independent transfected wells.
We first tested the restrictive activities of the primate A3A proteins against the highly diverged (relative to human) and codon-optimized mouse LINE-1 (ORFeus_Mm). Using the dual luciferase retrotransposition assay, we found that the LINE-1 element produces reporter activity approximately 1,000-fold higher than a mutant LINE-1 containing mutations in ORF1 that abolish retrotransposition (Moran et al. 1996; unrestricted vs. inactive LINE-1, fig. 3, left bar graph). When we cotransfected human A3A and the LINE-1 reporter plasmid into 293T cells, we found that retrotransposition-dependent luciferase activity was reduced 100-fold compared with an empty vector control (fig. 3, left bar graph). Further, we observed a dose-dependent relationship between the amount of A3A transfected/A3A produced and the resulting LINE-1 activity (supplementary fig. S1B, Supplementary Material online). Extending our analysis to nonhuman A3A proteins, we found that all nine primate A3A orthologs tested restrict mouse LINE-1 (ORFeus_Mm) by at least 100-fold (fig. 3, left bar graph).
The ORFeus_Mm LINE-1 sequence used in the luciferase reporter assay was generated by decreasing the high A-T content of the genomic LINE-1 sequence while maintaining its amino acid sequence (An et al. 2006). Such recoding leads to increased transcription and retrotransposition. While this construct provided a sensitive experimental tool to test the restrictive capabilities of A3A proteins, coevolutionary dynamics might be occluded by the sequence divergence relative to primate LINE-1s. Furthermore, the increased %GC content of this construct could change the sensitivity of this LINE-1 to A3A restriction relative to its wild-type counterpart. We, therefore, tested our panel of A3A constructs against an active wild-type/un-recoded human LINE-1 in a GFP-retrotransposition assay (Ostertag et al. 2000). Using this assay, we observed potent restriction by all tested A3As (fig. 3, right bar graph).
In contrast to the previously reported lack of LINE-1 restriction in the rhesus macaque A3A, we observed potent restriction by this gene. The discrepancy in these data likely resulted from the use of the wild-type A3A nucleotide sequence in the previous reports, which has much lower expression (Schmitt et al. 2011) than our codon-optimized version of rhesus A3A. Indeed, when we performed a dose-response curve with the wild-type rhesus A3A sequence, we found that it did restrict LINE-1 when expressed at levels similar to the codon-optimized sequence (supplementary fig. S1B, Supplementary Material online). In addition, we found that the corrected colobus A3A potently restricted LINE-1, even though previously described colobus A3A/A3G chimera did not (supplementary fig. S2, Supplementary Material online). Our findings suggest that LINE-1 restriction activity has been conserved throughout the evolution of primate A3As in spite of the strong signature of diversifying selection. We saw no obvious species-specific differences in the ability of different A3A orthologs to inhibit mouse or human LINE-1 elements. We, therefore, conclude that LINE-1 retrotransposons did not drive the diversifying selection in primate A3A.
Cercopithecinae A3A Proteins Gained Viral Restriction
Our finding that divergent primate A3A proteins have conserved LINE-1 restriction activity suggests that the rapid evolution we observe in A3A occurred in response to some other pathogen. In addition to LINE-1, A3A has been shown to restrict diverse viruses (Wiegand et al. 2004; Rose et al. 2005; Bogerd, Wiegand, Doehle, et al. 2006; Bogerd, Wiegand, Hulme, et al. 2006; Chen et al. 2006; Kinomoto et al. 2007; Bogerd et al. 2008; Marin et al. 2008). However, human A3A does not potently restrict HIV-1 unless specifically expressed in the target cell, likely as a result of its exclusion from packaging in the egressing viral particle (Bishop et al. 2004; Goila-Gaur et al. 2007; Aguiar et al. 2008; Stavrou et al. 2014). Recently, some Old World monkey A3A genes were shown to effectively restrict the replication of lentiviruses including HIV-1 (strain NL4-3) and SHIV (strain KU-2MC4, a human macaque chimera) (Schmitt et al. 2011, 2013; Katuwal et al. 2014). We re-examined these findings in light of our discovery that some of the previously tested genes were in fact A3A/A3G chimeras. We therefore retested the codon-optimized rhesus and (corrected) colobus A3A genes for their ability to restrict HIV-1 in a single cycle replication assay. We carefully controlled for any effects of A3A expression on the transfected plasmids by measuring p24 gag levels in the supernatant of transfected producer cells. We found that transfection of human, colobus, rhesus, or AGM A3A did not affect the production of viral particles from producer cells (supplementary fig. S3, Supplementary Material online). When we measured the infectivity of the resulting viral particles, we found that the A3A from rhesus restricted HIV-1 by about 10-fold. Although this restriction activity is less potent than human A3G (fig. 4), it is nonetheless more potent than either human A3A or (corrected) colobus A3A. Our findings are consistent with previous results that show that human A3A does not potently restrict HIV-1 (Aguiar et al. 2008). We, therefore, conclude that the previously observed restriction of HIV-1 resulted from the use of an A3A/A3G chimeric sequence for colobus (supplementary fig. S3, Supplementary Material online), and that the natural colobus A3A does not potently restrict HIV-1 (fig. 4).
Fig. 4.
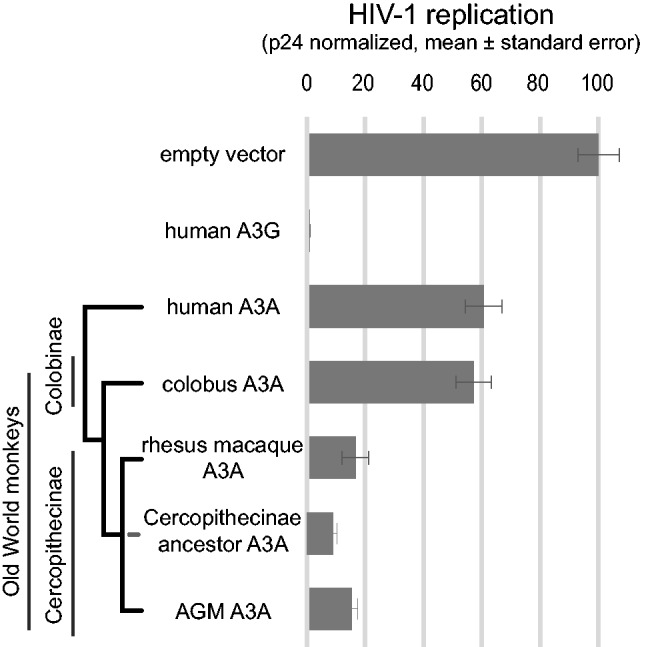
Some Old World monkey A3As restrict HIV-1. We tested the ability of selected A3A genes to restrict HIV-1 (pLai3ΔenvLuc2Δvif [OhAinle et al. 2006]) in single-cycle infectivity assays as detailed in the methods. Each bar represents the mean ± standard error of the luciferase measurement of at least three wells normalized to empty vector, each infected with the virus equivalent of 2 ng p24 gag. Human A3G restricts HIV-1 greater than 100-fold, whereas A3As from AGM, rhesus, and the reconstructed Cercopithecinae ancestor restrict approximately 10-fold compared with empty vector. In contrast, human and colobus A3A show less than 2-fold restriction.
Motivated by the finding that rhesus A3A restricted HIV-1, we next tested whether other Old World monkey A3As may also be capable of HIV-1 restriction. We found that AGM A3A also restricts HIV-1 approximately 10-fold (fig. 4). Our finding that rhesus and AGM A3A, but not colobus or human A3A, can restrict HIV-1 led us to hypothesize that gain of lentiviral restriction occurred in the common ancestor of Cercopithecinae, the subfamily of Old World monkeys which includes macaques and vervet monkeys such as AGM, after this lineage split from Colobinae, the subfamily which includes the colobus monkeys. We used maximum likelihood methods to reconstruct the A3A sequence of the most recent common ancestor of the Cercopithecinae (supplementary data set S1, Supplementary Material online). Using FastML we were able to reconstruct the identity of the ancestral A3A sequence with high confidence for 200/205 amino acids (posterior probabilities >0.9). We were not able to reconstruct five positions with high confidence; for these positions, we chose the most likely ancestral amino acid (AAs 62, 69, 114, 68, and 109; P= 0.62, 0.63, 0.78, 0.82, 0.83, respectively). When we tested this cercopithecine ancestor against HIV-1 and LINE-1, it restricted both pathogens (fig. 4 and supplementary fig. S4, Supplementary Material online). This suggests that the retroviral restriction ability of A3A was gained at least 11 Ma (Perelman et al. 2011) in the common ancestor of all cercopithecine primates, possibly in response to selection from an ancient virus.
Discussion
Our analysis of A3A evolution and function was partially motivated by the possibility of a functional tradeoff, whereby selection for gained retroviral restriction in colobus A3A occurred at the expense of losing LINE-1 restriction (Schmitt et al. 2011, 2013). Instead, our analysis of A3A evolution revealed inconsistencies between gene (exon 2) trees and species phylogeny. We discovered that this inconsistency was a result of a targeted exon-by-exon sequencing approach, which resulted in artifactual A3A-A3G chimeras (fig. 1B) (Henry et al. 2012). We conclude that the PCR-generated chimeras of A3A resulted from unintended priming of oligos due to the high sequence identity of the introns surrounding A3A coding exon 2 and A3G coding exon 5. This illustrates the potential perils of sequencing multicopy gene families, which arise from gene duplication and subsequent modification. Similar problems might exist in the databases for other A3s and other multiexon multigene families.
Our functional studies of A3A confirm that several previous findings on the unusual properties of the colobus A3A, including restriction of HIV-1 (Schmitt et al. 2011, 2013; Katuwal et al. 2014), are likely a result of the inadvertent use of an artifactual A3A/A3G chimera. While such a chimeric sequence has yet to be found in the primate genome sequences, the high sequence identity between recently duplicated multigene families increases the possibility of such exon shuffling that could alter A3 repertoires creating hybrid genes with novel functional properties. Indeed, one such recombination events led to the loss of the A3B gene in many human haplotypes (Kidd et al. 2007), whereas similar events led to the generation of novel functions in other antiviral gene families (Woelk et al. 2007; Mitchell et al. 2015).
The dynamic expansion and contraction of the A3 gene family across mammals over evolutionary time suggests they perform important but pliable roles in host defense. The variable number of A3 genes in each species may indicate significant idiosyncrasy in the function of both orthologs and paralogs among species (Munk et al. 2012). There is precedence for positive selection within A3 family members; this selection is ascribed to either escape from viral antagonists, as seen with A3G and Vif (Compton et al. 2012), modulation of target specificities (A3DE) (Duggal et al. 2011), or changes in expression level to modulate antiviral activity (mouse A3) (Takeda et al. 2008). In spite of this dynamism, we demonstrate in this study that at least one function—A3A restriction of the non-LTR retroelement LINE-1—has been strictly conserved over 40 My of primate evolution. This conservation is surprising for a couple of reasons. First, there are other A3 genes that can also restrict LINE-1 (Kinomoto et al. 2007; Arias et al. 2012); their presence could have relaxed selective pressure to maintain A3A restriction of LINE-1 over primate evolution. Second, interferon dramatically induces the expression of A3A (Stenglein et al. 2010; Thielen et al. 2010), suggesting A3A likely functions as a bona fide restriction factor for some virus. A3A has been reported to restrict several infectious and endogenous retroviruses including HIV-1, HTLV-1, and HERV-K (Goila-Gaur et al. 2007; Aguiar et al. 2008; Esnault et al. 2008; Schmitt et al. 2011, 2013; Ooms et al. 2012; Katuwal et al. 2014), as well as DNA viruses such as adeno-associated virus and human papillomavirus (Chen et al. 2006; Warren et al. 2015). The diverse restrictive capabilities of A3 genes can complicate the assignment of which selective agent(s) drove their rapid evolution, but suggests that multiple viruses may have driven the evolution of A3A. Yet, in spite of the genetic redundancy of restriction functions among A3s and the rapid evolution of A3A (likely in response to multiple viruses), LINE-1 restriction by A3A remains intact.
We consider two possibilities to explain this conservation of function. The first possibility is that LINE-1 restriction is an intrinsic property of A3A, that is, the ability of A3A to restrict LINE-1 is maintained regardless of what changes might occur to adapt to other pathogens. Under this model, the fact that A3A encodes a ssDNA-specific nucleus-localized cytidine deaminase is sufficient to explain its retention of LINE-1 restriction activity. An alternate possibility is that A3A has evolved under constant selection to maintain its LINE-1 restriction activity to ensure host fitness while also constantly adapting to some (as yet unknown) pathogens. Under this scenario, A3A restriction of LINE-1 might be non-redundant either because it is the most potent restrictor or because of its tissue-specific pattern of expression. An important presumption of this model is that loss of A3A entails a LINE-1-dependent fitness effect in the host; that is, A3A actually controls LINE-1 in vivo, not just in the context of in vitro retrotransposition assays. This has yet to be demonstrated, as mice deficient in APOBEC3 have no obvious impairment (Mikl et al. 2005), although fitness measurements in the lab may not truly reflect requirements in the wild. Moreover, the burden of LINE-1 elements on fitness might be higher in primates, which might partially explain the expansion of the A3 gene family in primates. Under this possibility, some subset of adaptive mutations would disrupt LINE-1 restriction, but diversifying selection in A3A is net beneficial only in the subset of residues that leave the LINE-1 restriction function intact. Other antiviral factors, especially those subject to pathogen mimicry, frequently undergo diversifying selection to selectively alter their binding preferences to some proteins while preserving affinity to others (Elde and Malik 2009; Elde et al. 2009).
If A3A plays an important role in the restriction of LINE-1, it would be advantageous to LINE-1 to evade A3A restriction. Yet, from human to mouse we saw no indication that LINE-1 has ever evaded restriction by A3A. This could be because A3A does not require any sequence-specific pattern to target LINE-1, so that LINE-1 has no available means to evolve to escape restriction. Indeed, the recently elucidated molecular mechanism of human A3A restriction of LINE-1 supports this scenario (Richardson et al. 2014). A3A restriction involves deamination of ssDNA overhangs generated during the process of target-primed reverse transcription and integration, which ultimately give rise to the target site duplications on either side of the LINE-1 integrant (Richardson et al. 2014). Variation in LINE-1 that could alter this structure in the lifecycle of the elements seems unlikely to occur. A3A action may thus be very difficult to evade by LINE-1 because there is no apparent “specificity” determinant to avoid.
If not LINE-1, what might have driven the diversifying selection on A3A? One possibility is that A3A diversification was driven by another primate retroelement (e.g., Alu, SVA, or ERVs). Alternatively, the rapid evolution could result from selection to restrict some infectious virus. We do find that restriction of one retrovirus appears to have been specifically gained by the A3A gene of the Cercopithecinae subfamily of the Old World monkeys. This gain of activity suggests that positive selection of A3A in response to some ancient virus has altered its properties over primate evolution. Whether directly selected for or not, this ancient adaptation may nevertheless influence the viral susceptibility of modern day cercopithecine primates. Future studies on the molecular mechanism of this gain of function may help identify the ancient driving pathogen.
We have demonstrated that the A3A gene of primates displays sequence variation consistent with a role in host defense against a changing pathogen or set of pathogens. Our data suggest that though A3A potently restricts LINE-1 retroelements, the rapid evolution in A3A was likely not driven by LINE-1, since we observe strict conservation of LINE-1 restriction across a diverse set of primate A3As. In contrast, we find that some Old World monkey A3As gained the ability to restrict an ancient virus, as evidenced in the restriction of HIV-1 by A3As from modern day cercopithecine primates. This suggests that A3A evolution may be directed by pressure to restrict new or evolving targets. However, despite this rapid change in sequence and function, the ability of A3A to restrict LINE-1 has remained intact. This system provides important insight into the evolution of multifunctional and multicopy gene families involved in host defense against both endogenous and infectious pathogens.
Materials and Methods
Plasmids and Antibodies
APOBEC3A genes were cloned into pCMV-HA (Clontech). Human APOBEC3G was cloned into the Xho/EcoRI sites of pCDNA3.1 with a primer-encoded 5′-HA tag (MYPYDVPDYA). LINE-1 plasmids were a gift from John Moran, JM101 99_gfp_LRE3_puro and JM111 99_gfp_LRE3_puro (ORF1 mutant) (Moran et al. 1996; Ostertag et al. 2000) and Wenfang An, pYX016 (CAG promoter—mouse ORFeus—Fluc—globin intron with renilla luciferase constitutive expression from the same plasmid) and pYX015 (same as pYX016 except containing JM111 ORF1 mutant) (Xie et al. 2011). A3A Western blots were carried out using Covance mouse HA.11 Clone 16B12 anti-HA monoclonal antibody.
A3A Sequence Collection and Alignment
APOBEC3A sequences were obtained from publicly available primate genome databases using PSI-BLAST (Altschul et al. 1997) against the NR database or TBLASTN (Altschul et al. 1997) against the HTGS database, with human APOBEC3A as a search seed. To sequence anew and “resequence” the Old World monkeys, exons were individually amplified from genomic DNA using NEB Q5 High-Fidelity DNA Polymerase and oligonucleotides designed against unique intergenic regions (as defined by an alignment of intronic sequence from A3A, A3B, and A3G) that also yield a single product in UCSC’s in silico PCR tool (http://genome.ucsc.edu/cgi-bin/hgPcr; last accessed March 1, 2016) (A3A exon 2 sense oligo: 5′ CAG AGA TTT TTC TAA TTC TGG TTA TGT CAG GAT GCA TAG TGA GG; A3A exon 2 sense oligo: 5′ GAA GCC CCC AGT TTC CAG CAC CAG AG). PCR products were sequenced using standard Sanger sequencing. A3A genes were codon optimized and synthesized by GENEWIZ or SGI (see supplementary data set S1 , Supplementary Material online, for sequences). Synthesized double stranded DNA was assembled using Gibson assembly (Gibson et al. 2009) into pCMV with an N-terminal HA tag. The corrected colobus complete A3A sequence and the corrected partial De Brazza’s monkey A3A sequence have been deposited at NCBI, accession numbers KU985237and KU985238. Supplementary table S1, Supplementary Material online lists all of the accession numbers and sources for the sequences used in this article.
Positive Selection Analysis
Primate nucleotide sequences were aligned using the ClustalW or MAFFT translation align function in Geneious Pro (Biomatters Ltd.). The alignment was refined manually to account for insertions and deletions in the exon 2 region. The alignment was analyzed with HyPhy SBP and GARD to ensure no signals of recombination were found (Kosakovsky Pond et al. 2006). This alignment and an established primate phylogeny (Perelman et al. 2011) or the gene tree (roughly equivalent to the species tree) were input into the CODEML sites model of PAML (Yang 1997) to detect positive selection at individual sites. The PAML P value was calculated using twice the difference in log-likelihood between models M8a and M8 and two degrees of freedom. PAML analysis was carried out using the F61 model of codon frequencies, but similar results were obtained for the F3x4 model and various initial omega values. Positively selected sites were classified as those sites with a M8 Bayes empirical Bayes posterior probability greater than 90%.
Ancestral Reconstruction
Reconstruction of the Cercopithecinae ancestor A3A sequences was performed with the FastML webserver (http://fastml.tau.ac.il/; last accessed March 1, 2016; Ashkenazy et al. 2012), to generate marginal reconstructions of both characters and indels using the 19 species primate nucleotide alignment (supplementary data set S1, Supplementary Material online).
Phylogenetic Inference
To construct the tree shown in figure 1A, we aligned A3A exon 2 and A3G exon 5 nucleotide sequences and generated a maximum likelihood phylogeny using PhyML (Guindon and Gascuel 2003) using the general time reversible evolutionary model with estimated proportion of invariable sites, one substitution rate category, optimization of topology/length/rate, and BEST topology search.
LINE-1 Retrotransposition Assays
LINE-1 retrotransposition assays were carried out as previously described (Ostertag et al. 2000; Xie et al. 2011). Briefly, for the mouse ORFeus luciferase assays 25,000 293T cells were seeded into each well of a 96-well, white-wall plate. 24 h later, each well was transfected with 200 ng pYX016 (CAG promoter driving mouse ORFeus LINE-1 with globin intron and luciferase reporter) or pYX015 (JM111 inactive human LINE-1 construct which contains loss-of-function mutations in ORF1p of LINE-1 [Moran et al. 1996]) and pCMV-HA-A3A or pCMV-HA-empty. 24 h post-transfection, transfected cells were selected with 2.5 μg/ml puromycin for 72 h. Cells were lysed and luciferase substrates provided using the Dual-Glo Luciferase Assay System (Promega E2920). Renilla and firefly luciferase activity were measured using the LUMIstar Omega luminometer. Retrotransposition is reported as firefly/Renilla activity to control for A3A toxicity. For the human LRE3 assays, 200,000 293T cells were seeded into each well of a six-well tissue culture plate. Twenty-four hours later, each well was transfected with 1 μg 99_GFP LRE3 or JM111_99_GFP LRE3 (which contains loss-of-function mutations in ORF1p of LINE-1 [Moran et al. 1996]) along with pCMV plasmid expressing an HA-tagged A3A or no A3A and 10 ng pDsRed-express vector (Matz et al. 1999). Each well was washed with phosphate-buffered saline (PBS), resuspended in 0.5 ml PBS, filtered through a 0.2 μm filter, and analyzed on a BD FACSAria II flow cytometer for GFP intensity. The gate for calling GFP-positive cells was set based upon the distribution of the JM111 nonfunctional LINE-1 construct.
Viral Infectivity Assays
Single-round HIV-1 infectivity assays were performed as described previously (Yamashita and Emerman 2004). To produce VSV-G-pseudotyped HIV-1, 50,000 293T cells were plated in a 24-well plate, and 24 h later, cotransfected with 0.3 μg lentiviral vector encoding luciferase in the place of the nef gene (pLai3ΔenvLuc2 [Yamashita and Emerman 2004], pLai3ΔenvLuc2Δvif [OhAinle et al. 2006]), 50 ng L-VSV-G, and 0.3 μg APOBEC or empty plasmid. All viruses were harvested 48 h after transfection and filtered through a 0.2-μm filter. p24 gag in viral supernatants was quantified using an HIV-1 p24 Antigen Capture Assay (ABL Inc.). Virus equivalents to two nanograms of p24 gag were used to infect 50,000 SupT1 cells per well in a 96-well plate in the presence of 20 μg/ml DEAE-dextran. Forty-eight hours after infection, cells from triplicate infections were lysed in 100 μl Bright-Glo luciferase assay reagent (Promega) and read on a LUMIstar Omega luminometer (BMG Labtech).
Supplementary Material
Supplementary figures S1–S4, table S1, and data set S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by a Howard Hughes Medical Institute postdoctoral fellowship of the Helen Hay Whitney Foundation (to R.N.M.), a National Institute of General Medical Sciences at the National Institutes of Health K99/R00 Pathway to Independence Award (grant number 1K99GM112941 to R.N.M.), a novel project grant from the Lupus Research Institute (to H.S.M.), a National Institute of General Medical Sciences at the National Institutes of Health P50 grant (grant number GM107632, PI: Jef Boeke, NYU; subaward: H.S.M.), a National Institute of Allergy and Infectious Diseases at the National Institutes of Health T32 STD/AIDS Training Grant Fellowship (grant number AI07140 to C.J.W.), and a National Institute of Allergy and Infectious Diseases at the National Institutes of Health R01 (grant number R01AI030927 to M.E.). H.S.M. is an Investigator of the Howard Hughes Medical Institute. The authors thank Janet Young, Matt Daugherty, and members of the Malik and Emerman labs for critical comments on the manuscript. The authors also thank John Moran for the LRE3-GFP plasmids, Wenfeng An for the LINE-1 luciferase plasmids, and Mario Santiago, Ed Stephens, and Kimberly Schmitt for APOBEC3 constructs. They also thank colleagues and genome sequencing centers for public release of genome sequences.
References
- Aguiar RS, Lovsin N, Tanuri A, Peterlin BM. 2008. Vpr.A3A chimera inhibits HIV replication. J Biol Chem. 283:2518–2525. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W, Dai L, Niewiadomska AM, Yetil A, O'Donnell KA, Han JS, Boeke JD. 2011. Characterization of a synthetic human LINE-1 retrotransposon ORFeus-Hs. Mob DNA. 2:2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W, Han JS, Wheelan SJ, Davis ES, Coombes CE, Ye P, Triplett C, Boeke JD. 2006. Active retrotransposition by a synthetic L1 element in mice. Proc Natl Acad Sci U S A. 103:18662–18667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias JF, Koyama T, Kinomoto M, Tokunaga K. 2012. Retroelements versus APOBEC3 family members: no great escape from the magnificent seven. Front Microbiol. 3:275.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Penn O, Doron-Faigenboim A, Cohen O, Cannarozzi G, Zomer O, Pupko T. 2012. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 40:W580–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Garcia-Perez JL, Badge RM, Moran JV. 2011. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 12:187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 14:1392–1396. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Tallmadge RL, Oaks JL, Carpenter S, Cullen BR. 2008. Equine infectious anemia virus resists the antiretroviral activity of equine APOBEC3 proteins through a packaging-independent mechanism. J Virol. 82:11889–11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. 2006. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 34:89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. 2006. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A. 103:8780–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn MF, Shandilya SM, Silvas TV, Nalivaika EA, Kouno T, Kelch BA, Ryder SP, Kurt-Yilmaz N, Somasundaran M, Schiffer CA. 2015. The ssDNA mutator APOBEC3A is regulated by cooperative dimerization. Structure 23:903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Davis J, Entezam A, Petrov D, Furano AV. 2006. Fitness cost of LINE-1 (L1) activity in humans. Proc Natl Acad Sci U S A. 103:9590–9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Furano AV. 2001. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol. 18:926–935. [DOI] [PubMed] [Google Scholar]
- Brouha B, Meischl C, Ostertag E, de Boer M, Zhang Y, Neijens H, Roos D, Kazazian HH., Jr. 2002. Evidence consistent with human L1 retrotransposition in maternal meiosis I. Am J Hum Genet. 71:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulliard Y, Narvaiza I, Bertero A, Peddi S, Rohrig UF, Ortiz M, Zoete V, Castro-Diaz N, Turelli P, Telenti A, et al. 2011. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J Virol. 85:1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon IJ, Ahn J, Mitra M, Byeon CH, Hercik K, Hritz J, Charlton LM, Levin JG, Gronenborn AM. 2013. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun. 4:1890.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Diaz N, Ecco G, Coluccio A, Kapopoulou A, Yazdanpanah B, Friedli M, Duc J, Jang SM, Turelli P, Trono D. 2014. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev. 28:1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. 2006. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 16:480–485. [DOI] [PubMed] [Google Scholar]
- Compton AA, Emerman M. 2013. Convergence and divergence in the evolution of the APOBEC3G-Vif interaction reveal ancient origins of simian immunodeficiency viruses. PLoS Pathog. 9:e1003135.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton AA, Hirsch VM, Emerman M. 2012. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe. 11:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton AA, Malik HS, Emerman M. 2013. Host gene evolution traces the evolutionary history of ancient primate lentiviruses. Philos Trans R Soc Lond B Biol Sci. 368:20120496.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. 2005. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol Biol Evol. 22:367–377. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O'Shea KS, Moran JV, Gage FH. 2009. L1 retrotransposition in human neural progenitor cells. Nature. 460:1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. 2006. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 38:917–920. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Rehwinkel J. 2009. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 18:R130–R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Malik HS. 2012. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet. 46:677–700. [DOI] [PubMed] [Google Scholar]
- de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. 2011. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 7:e1002384.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombroski BA, Mathias SL, Nanthakumar E, Scott AF, Kazazian HH., Jr. 1991. Isolation of an active human transposable element. Science 254:1805–1808. [DOI] [PubMed] [Google Scholar]
- Duggal NK, Malik HS, Emerman M. 2011. The breadth of antiviral activity of Apobec3DE in chimpanzees has been driven by positive selection. J Virol. 85:11361–11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Child SJ, Geballe AP, Malik HS. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457:485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Malik HS. 2009. The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol. 7:787–797. [DOI] [PubMed] [Google Scholar]
- Esnault C, Priet S, Ribet D, Heidmann O, Heidmann T. 2008. Restriction by APOBEC3 proteins of endogenous retroviruses with an extracellular life cycle: ex vivo effects and in vivo “traces” on the murine IAPE and human HERV-K elements. Retrovirology 5:75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87:905–916. [DOI] [PubMed] [Google Scholar]
- Friedli M, Turelli P, Kapopoulou A, Rauwel B, Castro-Diaz N, Rowe HM, Ecco G, Unzu C, Planet E, Lombardo A, et al. 2014. Loss of transcriptional control over endogenous retroelements during reprogramming to pluripotency. Genome Res. 24:1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. [DOI] [PubMed] [Google Scholar]
- Goila-Gaur R, Khan MA, Miyagi E, Kao S, Strebel K. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 4:61.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. [DOI] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH., Jr. 2012. Active human retrotransposons: variation and disease. Curr Opin Genet Dev. 22:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry M, Terzian C, Peeters M, Wain-Hobson S, Vartanian JP. 2012. Evolution of the primate APOBEC3A cytidine deaminase gene and identification of related coding regions. PLoS One 7:e30036.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Li J, Xu F, Mei S, Le Duff Y, Yin L, Pang X, Cen S, Jin Q, Liang C, et al. 2015. SAMHD1 inhibits LINE-1 retrotransposition by promoting stress granule formation. PLoS Genet. 11:e1005367.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs FM, Greenberg D, Nguyen N, Haeussler M, Ewing AD, Katzman S, Paten B, Salama SR, Haussler D. 2014. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516:242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110:462–467. [DOI] [PubMed] [Google Scholar]
- Katuwal M, Wang Y, Schmitt K, Guo K, Halemano K, Santiago ML, Stephens EB. 2014. Cellular HIV-1 inhibition by truncated old world primate APOBEC3A proteins lacking a complete deaminase domain. Virology 468-470:532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. 2007. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 3:e63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, Sata T, Tokunaga K. 2007. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 35:2955–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 23:1891–1901. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. [DOI] [PubMed] [Google Scholar]
- Marin M, Golem S, Rose KM, Kozak SL, Kabat D. 2008. Human immunodeficiency virus type 1 Vif functionally interacts with diverse APOBEC3 cytidine deaminases and moves with them between cytoplasmic sites of mRNA metabolism. J Virol. 82:987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SL, Bushman FD. 2001. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol Cell Biol. 21:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias SL, Scott AF, Kazazian HH, Jr, Boeke JD, Gabriel A. 1991. Reverse transcriptase encoded by a human transposable element. Science 254:1808–1810. [DOI] [PubMed] [Google Scholar]
- Matz MV, Fradkov AF, Labas YA, Savitsky AP, Zaraisky AG, Markelov ML, Lukyanov SA. 1999. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat Biotechnol. 17:969–973. [DOI] [PubMed] [Google Scholar]
- Mikl MC, Watt IN, Lu M, Reik W, Davies SL, Neuberger MS, Rada C. 2005. Mice deficient in APOBEC2 and APOBEC3. Mol Cell Biol. 25:7270–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Patzina C, Emerman M, Haller O, Malik HS, Kochs G. 2012. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe 12:598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Young JM, Emerman M, Malik HS. 2015. Evolutionary analyses suggest a function of MxB immunity proteins beyond lentivirus restriction. PLoS Pathog. 11:e1005304.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molaro A, Malik HS. 2016. Hide and seek: how chromatin-based pathways silence retroelements in the mammalian germline. Curr Opin Genet Dev. 37:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery E, Charlesworth B, Langley CH. 2007. A test for the role of natural selection in the stabilization of transposable element copy number in a population of Drosophila melanogaster. Genet Res. 89:435–445. [DOI] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr. 1996. High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927. [DOI] [PubMed] [Google Scholar]
- Munk C, Willemsen A, Bravo IG. 2012. An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol Biol. 12:71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 30:1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhAinle M, Kerns JA, Malik HS, Emerman M. 2006. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol. 80:3853–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SK, O'Donnell AH, Bestor TH. 2009. Mammalian cytosine methylation at a glance. J Cell Sci. 122:2787–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Krikoni A, Kress AK, Simon V, Munk C. 2012. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J Virol. 86:6097–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH., Jr. 2000. Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res. 28:1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y, et al. 2011. A molecular phylogeny of living primates. PLoS Genet. 7:e1001342.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. 2015. The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr. 3:MDNA3–M0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SDA, Kopera H, Moldovan J, Garcia-Perez J, Moran J. 2015. The Influence of LINE-1 and SINE Retrotransposons on Mammalian Genomes In: Craig NCM, Gellert M, Lambowitz A, Rice P, Sandmeyer S, editors. Mobile DNA III. Washington, DC: ASM Press; p. 1165–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Narvaiza I, Planegger RA, Weitzman MD, Moran JV. 2014. APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. Elife 3:e02008.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose KM, Marin M, Kozak SL, Kabat D. 2005. Regulated production and anti-HIV type 1 activities of cytidine deaminases APOBEC3B, 3F, and 3G. AIDS Res Hum Retroviruses 21:611–619. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A. 102:2832–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K, Guo K, Algaier M, Ruiz A, Cheng F, Qiu J, Wissing S, Santiago ML, Stephens EB. 2011. Differential virus restriction patterns of rhesus macaque and human APOBEC3A: implications for lentivirus evolution. Virology 419:24–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K, Guo K, Katuwal M, Wilson D, Prochnow C, Bransteitter R, Chen XS, Santiago ML, Stephens EB. 2013. Lentivirus restriction by diverse primate APOBEC3A proteins. Virology 442:82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AF, Schmeckpeper BJ, Abdelrazik M, Comey CT, O'Hara B, Rossiter JP, Cooley T, Heath P, Smith KD, Margolet L. 1987. Origin of the human L1 elements: proposed progenitor genes deduced from a consensus DNA sequence. Genomics 1:113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 9:1404–1407. [DOI] [PubMed] [Google Scholar]
- Steinglein MD, Harris RS. 2006. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 281:16837–16841. [DOI] [PubMed] [Google Scholar]
- Song M, Boissinot S. 2007. Selection against LINE-1 retrotransposons results principally from their ability to mediate ectopic recombination. Gene 390:206–213. [DOI] [PubMed] [Google Scholar]
- Stavrou S, Crawford D, Blouch K, Browne EP, Kohli RM, Ross SR. 2014. Different modes of retrovirus restriction by human APOBEC3A and APOBEC3G in vivo. PLoS Pathog. 10:e1004145.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. 2010. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol. 17:222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. 2008. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda E, Tsuji-Kawahara S, Sakamoto M, Langlois MA, Neuberger MS, Rada C, Miyazawa M. 2008. Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J Virol. 82:10998–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielen BK, McNevin JP, McElrath MJ, Hunt BV, Klein KC, Lingappa JR. 2010. Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J Biol Chem. 285:27753–27766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P, Castro-Diaz N, Marzetta F, Kapopoulou A, Raclot C, Duc J, Tieng V, Quenneville S, Trono D. 2014. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res. 24:1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Stetson DB. 2014. The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol. 15:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, Lambert PF, Santiago ML, Pyeon D. 2015. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol. 89:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 23:2451–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissing S, Munoz-Lopez M, Macia A, Yang Z, Montano M, Collins W, Garcia-Perez JL, Moran JV, Greene WC. 2012. Reprogramming somatic cells into iPS cells activates LINE-1 retroelement mobility. Hum Mol Genet 21:208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelk CH, Frost SD, Richman DD, Higley PE, Kosakovsky Pond SL. 2007. Evolution of the interferon alpha gene family in eutherian mammals. Gene 397:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Rosser JM, Thompson TL, Boeke JD, An W. 2011. Characterization of L1 retrotransposition with high-throughput dual-luciferase assays. Nucleic Acids Res. 39:e16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Emerman M. 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol. 78:5670–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 13:555–556. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13:335–340. [DOI] [PubMed] [Google Scholar]
- Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X, Wei W, Evans SL, Li L, Zhang W, et al. 2013. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell Rep. 4:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.