

Abstract
We report that phosphotyrosine–cholesterol conjugates effectively and selectively kill cancer cells, including platinum-resistant ovarian cancer cells. The conjugate increases the degree of noncovalent oligomerization upon enzymatic dephosphorylation in aqueous buffer. This enzymatic conversion also results in the assembly of the cholesterol conjugates inside and outside cells and leads to cell death. Preliminary mechanistic studies suggest that the formed assemblies of the conjugates not only interact with actin filaments and microtubules but also affect lipid rafts. As the first report of multifaceted supramolecular assemblies of cholesterol conjugates against cancer cells, this work illustrates the integration of enzyme catalysis and self-assembly of essential biological small molecules on and inside cancer cells as a promising strategy for developing multifunctional therapeutics to treat drug-resistant cancers.
We report enzymatic, in situ conversion of supramolecular assemblies of cholesterol conjugates for selectively killing cancer cells, including platinum-resistant1 ovarian cancer cells. Due to the emergence of resistance to platinum-based chemotherapy, little progress has been made in treating some types of cancer. For example, ovarian cancer, a common cancer worldwide, remains difficult to treat (the five-year survival rate has remained the same over recent decades).2 This challenge with anticancer-drug resistance demands innovative approaches for developing cancer therapy. Departing from the dogma of tight ligand–receptor interactions in molecular therapy, we3−5 and others6 are exploring enzyme catalysis and self-assembly of small molecules for developing new strategies for future cancer therapy, especially for cancers that respond poorly to immunotherapy.7 Recent results have supported the concept of enzyme-instructed self-assembly (EISA) of small molecules for selectively inhibiting cancer cells,4,5,8,9 which employ enzymatic reactions to generate assemblies of small molecules in situ either on the surface or inside of cancer cells. However, the inhibitory concentrations of those self-assembling molecules are still higher than for drugs in the clinic. Therefore, new strategies are needed for maximizing the efficacy of EISA so that its excellent selectivity can be leveraged for developing clinical medicines for cancer therapy.
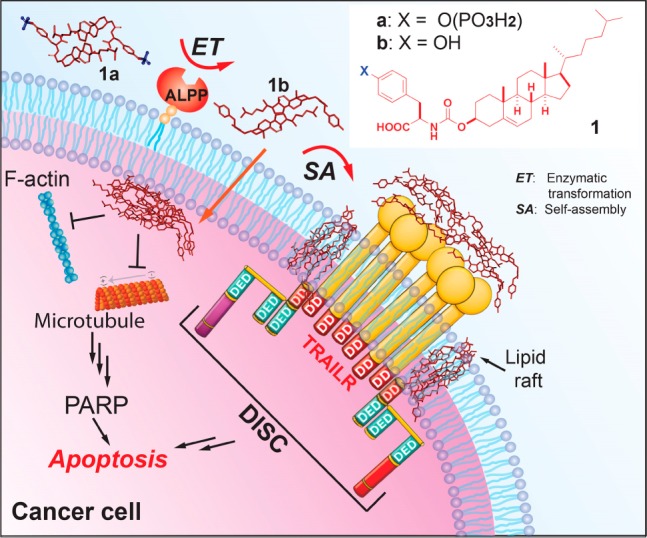
We hypothesize that simultaneously activating extrinsic and intrinsic cell death signaling10 by EISA would effectively kill cancer cells. Thus, we choose cholesterol, an evolutionarily optimized molecule known to be present both on the cell surface11 and inside cells,12 as a building block for designing EISA precursors. Further motivation for the use of cholesterol is that the interaction between proteins and cholesterol is critical for cellular functions.13 Additionally, cholesterol has been used as a motif to enable self-assembly to form various nanostructures.14 We covalently conjugate cholesterol with d-phosphotyrosine to generate a precursor (1a) for EISA. Our results reveal that (i) 1a, besides being orders of magnitude more potent than the previous reported precursors for EISA,4 is more potent than cisplatin for inhibiting platinum-resistant ovarian cancer cells; (ii) 1a inhibits cancer cells selectively because EISA generates assemblies of 1b in situ on or inside the cancer cells; (iii) the assemblies of 1b, indeed, are able to activate extrinsic and intrinsic cell death signaling simultaneously. Thus, this work illustrates EISA as a multistep process to generate multifaceted nanomedicine from small molecules, including the building blocks of life.15
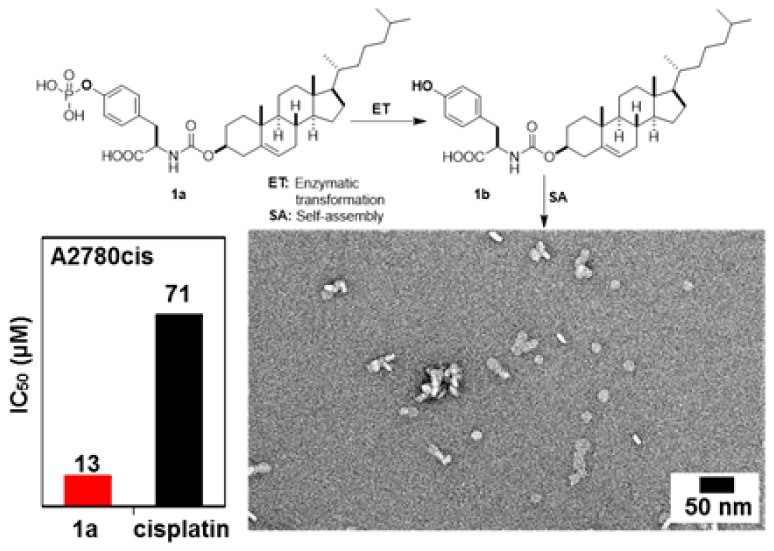
Although conjugates of cholesterol and amino acids have been reported,16 there is no report of the conjugate of tyrosine and cholesterol. After using a facile synthesis (Scheme S1) to generate 1a, we examine its activity against a platinum-resistant ovarian cancer cells (A2780cis).17 As shown in Figure 1, 1a inhibits A2780cis cells and is more potent than cisplatin, a clinical drug. The IC50 of 1a is 13 ± 1.3 μM (48 h), which is about five times lower than the IC50 of cisplatin against A2780cis, 71 ± 1.2 μM (48 h). Importantly, the dosage of 1a (8.7 ± 0.8 μg/mL) is much lower than that of cisplatin (21.2 ± 0.4 μg/mL) against A2780cis, promising an effective drug candidate. The dosage curve of 1a (Figure 1A) exhibits a threshold concentration, deviating from the conventional dosage curve and agreeing with molecular aggregation.18 To examine the effect of the chirality of the tyrosine, we use l-phosphotyrosine to replace the d-phosphotyrosine in 1a to generate 2a (Scheme S2 and Figure S1) and find that the IC50 of 2a against A2780cis is 22 μM (72 h), which coincides with 1b and 2b forming different-size assemblies (vide infra). We also test the inhibitory activity of 1b (or 2b) itself, the dephosphorylated product of 1a (or 2a), against A2780cis. The IC50 values (at 72 h) of 1b and 2b are 50 μM and 36 μM, respectively, considerably higher than those of 1a and 2a (11 and 22 μM). These results indicate that enzymatic dephosphorylation likely contributes to the higher activity of 1a (or 2a) relative to that of 1b (or 2b) against A2780cis cells.
Figure 1.
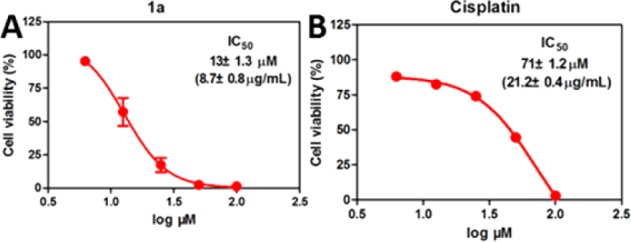
Dosage curves of (A) 1a and (B) cisplatin against A2780cis cells at 48 h.
As shown by transmission electron microscopy (TEM), in PBS, 1a (100 μM) formed particles with a mean size of 5.7 ± 0.37 nm (Figure 2A,C), indicating oligomerization of 1a. In contrast, bigger particles with a mean size of 13.7 ± 0.37 nm (Figure 2B,D) form after the addition of alkaline phosphatase (ALP) into the solution of 1a. The formation of larger oligomers of 1b likely originates from dephosphorylation of 1a, possibly due to the elimination of the repulsive phosphates groups. Without the use of ALP, the direct addition of 1b (100 μM) in PBS buffer results in much larger particles (bilayer-like with a diameter of 20 nm and some bigger ones around 100 nm) (Figure S3). We have also measured magic-angle-spinning (MAS) direct-polarization (DP) 31P NMR spectra of 1a (40 wt %) before and after the addition of ALPs (Figure S4). The spectra confirm that ∼44% of 1a is converted to 1b after treatment with ALP. Static 31P solid-state NMR results are in agreement with the formation of spherical nanoparticles (18 nm diameter) of 1b, consistent with TEM images of the NMR sample (Figure S5). Similar to 1a, adding 2a in PBS buffer results in oligomers as particles (diameters of 9 ± 2 nm). The addition of ALP into the solution of 2a results in clustering of bigger particles (diameter of 16 ± 2 nm). The direct addition of 2b provides an aqueous sample containing bilayer-like assemblies with a diameter of about 20–50 nm (Figure S3). As shown in Figure S6, the CMC of 1a is 1.3 μM in PBS buffer. There is little variation in CMCs of 2a and 2b, which are 2.1 and 4.5 μM, respectively, whereas the CMC of 1b is 22.3 μM. These observations, collectively, confirm that an enzymatic reaction modulates the morphology of the assemblies of the cholesterol conjugates, which likely contributes to the different inhibitory activities of 1a, 1b, 2a, and 2b. That is, being incubated with cancer cells, 1a (or 2a) turns into 1b (or 2b), a process catalyzed by the extra- or intracellular phosphatase of the cancer cells; the resulting 1b (or 2b) forms oligomers, which results in death of the cancer cells. Not being generated by the EISA process in situ, 1b or 2b shows less cytotoxicity than the 1a or 2a.
Figure 2.
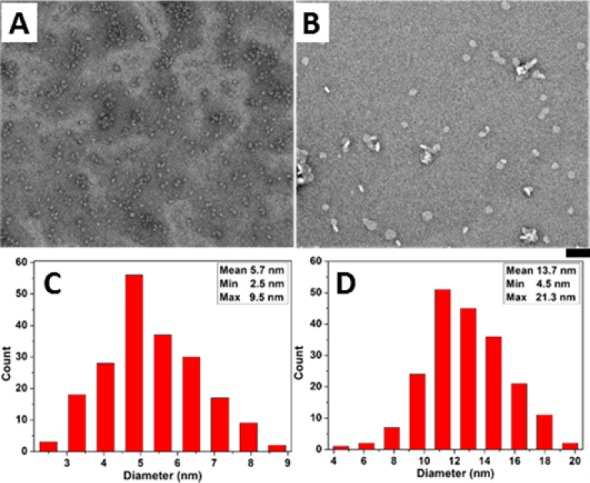
(A,B) TEM images of (A) 1a (100 μM); (B) 1a (100 μM) treated with ALP (1 U/mL) in PBS (pH7.4) after 24 h; bar is 50 nm. (C,D) Histogram of the size of oligomers (according to TEM, for 200 particles) in (C) A and (D) B.
Next, we use HS-5 cells (a stroma cell line expressing low level of ALP)9 to verify the selectivity of 1a against cancer cells. Figure 3A shows that 1a at 12.5 μM is innocuous to HS-5. Moreover, 1b hardly inhibits the proliferation of HS-5 cells. To confirm the selectivity of 1a in targeting cancer cells, we coculture HS-5 and A2780cis cells. Cell viability (Figure S7) indicates that 1a selectively kills the cancer cells, agreeing with the results from the separate cultures of A2780cis and HS-5 (Figure 3A). To further confirm that membrane-anchored ALP (i.e., as an ectophosphatase)19 contributes to the inhibitory activity of 1a, we incubate A2780cis with 1a and exogenous ALP (1 U/mL) and find that exogenous ALP, indeed, partially rescues the A2780cis cells (Figure 3B). The addition of l-phenylalanine (l-Phe) or levamisole, two types of inhibitors of ALP,20 also results in higher cell viability of A2780cis. These partial rescue effects validate the contribution of the membrane ALPs on the cell surface for cell death. The residual cytotoxicity implies that mechanisms other than EISA likely contribute to the activity of 1a.
Figure 3.
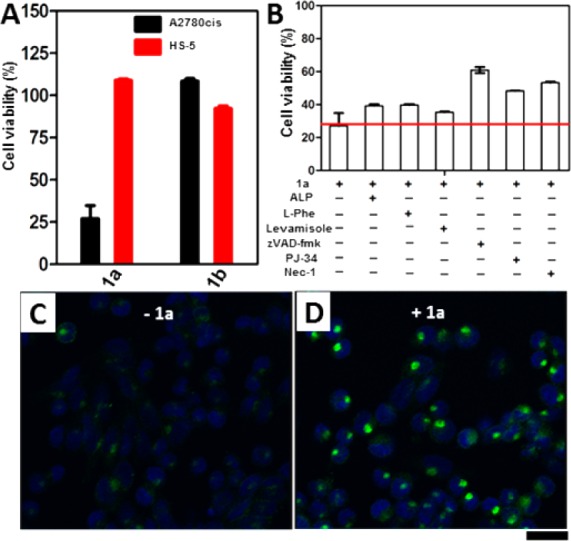
(A) Viability of A2780cis or HS-5 cell lines incubated with 12.5 μM of 1a or 1b for 48 h. (B) Viability of A2780cis treated by 1a (12.5 μM) in the presence of phosphatase inhibitors or cell death signaling inhibitors at 48h ([l-Phe] = [levamisole] = 1 mM, [ALP] = 1 U/mL, [zVAD-fmk] = 45 μM, [PJ34] = 1 μM, [Nec-1] = 50 μM). (C, D) Confocal images of A2780cis cells treated with anti-DR5 (C) without or (D) with the addition of 1a (12.5 μM) for 24 h. Scale bar = 30 μm.
We co-incubate A2780cis with 1a in the presence of a pan-caspase inhibitor (zVAD-fmk),21 a PARP-1 inhibitor (PJ34),22 or a necroptosis inhibitor (Nec-1).23 The addition of zVAD-fmk, PJ34, or Nec-1 all rescues the cells, but only partially. These results confirm that 1a induces cell death via multiple mechanisms, including apoptosis and regulated necrosis, by activating both extrinsic and intrinsic cell death signaling. After adding the primary antibodies of extrinsic cell-death receptors24 (i.e., anti-CD95, anti-DR3, anti-DR5, anti-TNFR1, and anti-TNFR2) to bind their corresponding receptors on the cells, we use fluorescent secondary antibodies to reveal the binding. The addition of 1a results in significantly clustering of DR5 (Figure 3D), while the clustering of DR3 or TNFR1 is rather moderate (Figure S9), and there is little clustering of CD95 or TNFR2 (Figure S9). These preliminary results suggest that, after ALP turns 1a to 1b, the assemblies of 1b likely promiscuously interact with DR5, DR3, and TNFR1 to result in cell death. Moreover, TRAIL, a ligand of DR5, increases the cytotoxicity in the presence of 1a (Figure S10), further suggesting that the assemblies of 1b enhance the clustering of the death receptors for TRAIL and lead to cell death. To better understand signaling molecules involved in survival and apoptosis pathways, we use PathScan apoptosis multitarget sandwich ELISA to detect their changes of expression,25 which suggests that assemblies of 1a/1b mainly initiate the activation of p53, which later activates the caspase cascade and downstream poly(ADP-ribose) polymerase to eventually induce apoptosis in A2780cis cells (Figure S11).
Cholesterol is a key component of lipid rafts, dynamic assemblies in the cell membrane.26 To examine the changes of lipid rafts in the presence of 1b assemblies (after EISA), we use laurdan as a fluorescence marker.27 In the absence of treatment with 1a, the fluorescence of laurdan disperses relatively evenly in the whole cell membrane (Figure 4). But the addition of 1a results in increased inhomogeneity of the fluorescence (Figure 4), indicating that the addition of 1a increases the microheterogeneity of the cell membrane.28 This result is in agreement with the notion that lipid rafts participate in cell death signaling.29
Figure 4.
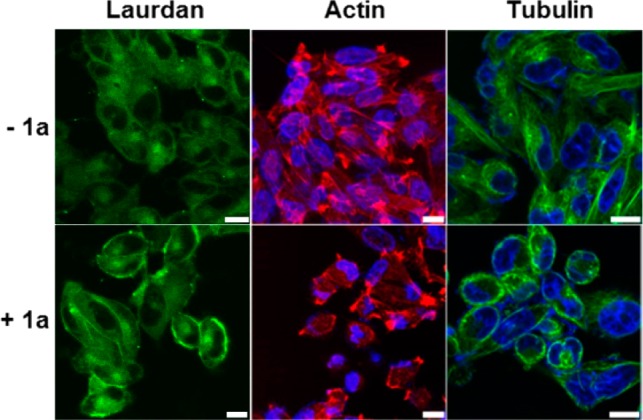
Confocal images of A2780cis cells stained with laurdan (10 μM), Alexa Fluor 633 phalloidin (F-actin, red), and Hoechst (nuclei, blue) or tubulin tracker (green) without or with the addition of 1a (12.5 μM) for 12 h (scale bar is 10 μm).
To examine whether the intracellular assemblies of 1b, formed after 1a enters the cell, alter the dynamics of cytoskeletons to cause cell death,8 we examine the changes of F-actin and microtubules. Compared with the control, A2780cis cells treated with 1a (12.5 μM) mainly exhibit short, ill-defined actin filaments (Figures 4 and S12), indicating that the assemblies of 1b interact with F-actin and disrupt the dynamics of F-actin. Unlike the microtubules extending through the entire control cells, microtubules in the A2780cis treated with 1a reorganize in the proximity of plasma membranes, suggesting that the assemblies of 1b promote the formation of apoptotic microtubule networks.30 These results are consistent with cytoskeleton proteins actively interacting with membrane rafts.31 Although the alteration of cytoskeletons can be the consequence of cell death, the observation that more than 80% of cells are viable after incubation with 1a for 24 h suggests that the interaction between the assemblies of 1b and F-actin and microtubules results in cell death (Scheme 1), agreeing with apoptosis caused by intracellular assemblies of small molecules.25
Scheme 1. EISA of the Tyrosine-Cholesterol Conjugate Activates Extrinsic and Intrinsic Cell Death Signaling.
In conclusion, we demonstrate that a simple conjugate of cholesterol and phosphotyrosine exhibits higher potency and higher selectivity than cisplatin against platinum-resistant ovarian cancer cells. Besides inhibiting HeLa cells with IC50 of 16 μM, 1a exhibits almost the same activity against A2780cis and A2780 cells (Table S1), implying that the mechanism of platinum resistance is ineffective toward the assemblies of 1b formed by EISA. The tyrosine residue is necessary because the conjugate of phenylalanine and cholesterol is innocuous to A2780cis and HeLa cells (Figures S1 and S13). The incorporation of d-Phe-d-Phe (Scheme S2 and Figure S13) between cholesterol and tyrosine significantly decreases the cytotoxicity of 4a or 4b, suggesting that tyrosine needs to connect to cholesterol directly. Most importantly, this work illustrates a new way to use one kind of molecule to regulate multiple biological targets, possibly including the dynamics of lipid rafts. Such a “one-to-many” targeting via supramolecular assemblies may be useful to counter anticancer drug resistance.
Acknowledgments
This work was partially supported by NIH (CA142746) and the W. M. Keck Foundation. Z.F. is grateful for a Dean’s fellowship, and J.Z. is a HHMI fellow.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06075.
Details of synthesis, methods, cell toxicity results, antibody staining, and other figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ozols R. F.; Corden B. J.; Jacob J.; Wesley M. N.; Ostchega Y.; Young R. C. Ann. Intern. Med. 1984, 100, 19. 10.7326/0003-4819-100-1-19. [DOI] [PubMed] [Google Scholar]
- Holmes D. Nature 2015, 527, S218. 10.1038/527S218a. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Xu K.; Guo Z.; Guo Z.; Xu B. Adv. Mater. 2007, 19, 3152. 10.1002/adma.200701971. [DOI] [Google Scholar]; Shi J.; Du X.; Yuan D.; Zhou J.; Zhou N.; Huang Y.; Xu B. Biomacromolecules 2014, 15, 3559. 10.1021/bm5010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Xu B. Bioconjugate Chem. 2015, 26, 987. 10.1021/acs.bioconjchem.5b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Y.; Shi J.; Li J.; Yuan D.; Alberti K. A.; Xu Q.; Xu B. Angew. Chem., Int. Ed. 2014, 53, 8104. 10.1002/anie.201402216. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhou J.; Du X. W.; Berciu C.; He H. G.; Shi J. F.; Nicastro D.; Xu B. Chem. 2016, 1, 246. 10.1016/j.chempr.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A.; Fukuoka Y.; Morimoto Y.; Honjo T.; Koda D.; Goto M.; Maruyama T. J. Am. Chem. Soc. 2015, 137, 770. 10.1021/ja510156v. [DOI] [PubMed] [Google Scholar]; Pires R. A.; Abul-Haija Y. M.; Costa D. S.; Novoa-Carballal R.; Reis R. L.; Ulijn R. V.; Pashkuleva I. J. J. Am. Chem. Soc. 2015, 137, 576. 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]
- Vaughan S.; Coward J. I.; Bast R. C.; Berchuck A.; Berek J. S.; Brenton J. D.; Coukos G.; Crum C. C.; Drapkin R.; Etemadmoghadam D.; Friedlander M.; Gabra H.; Kaye S. B.; Lord C. J.; Lengyel E.; Levine D. A.; McNeish I. A.; Menon U.; Mills G. B.; Nephew K. P.; Oza A. M.; Sood A. K.; Stronach E. A.; Walczak H.; Bowtell D. D.; Balkwill F. R. Nat. Rev. Cancer 2011, 11, 719. 10.1038/nrc3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Kuang Y.; Shi J. F.; Zhou J.; Medina J. E.; Zhou R.; Yuan D.; Yang C. H.; Wang H. M.; Yang Z. M.; Liu J. F.; Dinulescu D. M.; Xu B. Angew. Chem., Int. Ed. 2015, 54, 13307. 10.1002/anie.201507157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Du X.; Yamagata N.; Xu B. J. Am. Chem. Soc. 2016, 138, 3813. 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A.; Salvesen G. Annu. Rev. Cell Dev. Biol. 2014, 30, 337. 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- Heino S.; Lusa S.; Somerharju P.; Ehnholm C.; Olkkonen V. M.; Ikonen E. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 8375. 10.1073/pnas.140218797. [DOI] [PMC free article] [PubMed] [Google Scholar]; Slotte J. P.; Oram J. F.; Bierman E. L. J. Biol. Chem. 1987, 262, 12904. [PubMed] [Google Scholar]
- Neufeld E. B.; Cooney A. M.; Pitha J.; Dawidowicz E. A.; Dwyer N. K.; Pentchev P. G.; BlanchetteMackie E. J. J. Biol. Chem. 1996, 271, 21604. 10.1074/jbc.271.35.21604. [DOI] [PubMed] [Google Scholar]; Simons K.; Ikonen E. Science 2000, 290, 1721. 10.1126/science.290.5497.1721. [DOI] [PubMed] [Google Scholar]; Rajendran L.; Knoelker H.-J.; Simons K. Nat. Rev. Drug Discovery 2010, 9, 29. 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A.; Goldstein J. L.; McDonald J. G.; Brown M. S. Cell Metab. 2008, 8, 512. 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Raghavan S. R.; Terech P.; Weiss R. G. J. Am. Chem. Soc. 2006, 128, 15341. 10.1021/ja0657206. [DOI] [PubMed] [Google Scholar]; Majumdar S.; Hajduczki A.; Vithayathil R.; Olsen T. J.; Spitler R. M.; Mendez A. S.; Thompson T. D.; Weiss G. A. J. Am. Chem. Soc. 2011, 133, 9855. 10.1021/ja201792q. [DOI] [PMC free article] [PubMed] [Google Scholar]; Murata K.; Aoki M.; Suzuki T.; Harada T.; Kawabata H.; Komori T.; Ohseto F.; Ueda K.; Shinkai S. J. Am. Chem. Soc. 1994, 116, 6664. 10.1021/ja00094a023. [DOI] [Google Scholar]; Michinobu T.; Shinoda S.; Nakanishi T.; Hill J. P.; Fujii K.; Player T. N.; Tsukube H.; Ariga K. J. Am. Chem. Soc. 2006, 128, 14478. 10.1021/ja066429t. [DOI] [PubMed] [Google Scholar]
- Marth J. D. Nat. Cell Biol. 2008, 10, 1015. 10.1038/ncb0908-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphlett W. J.; Wilson C. V. J. Org. Chem. 1961, 26, 2511. 10.1021/jo01351a089. [DOI] [Google Scholar]
- Behrens B. C.; Hamilton T. C.; Masuda H.; Grotzinger K. R.; Whangpeng J.; Louie K. G.; Knutsen T.; McKoy W. M.; Young R. C.; Ozols R. F. Cancer Res. 1987, 47, 414. [PubMed] [Google Scholar]; Masuda H.; Ozols R. F.; Lai G. M.; Fojo A.; Rothenberg M.; Hamilton T. C. Cancer Res. 1988, 48, 5713. [PubMed] [Google Scholar]
- Irwin J. J.; Duan D.; Torosyan H.; Doak A. K.; Ziebart K. T.; Sterling T.; Tumanian G.; Shoichet B. K. J. Med. Chem. 2015, 58, 7076. 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sassano M. F.; Doak A. K.; Roth B. L.; Shoichet B. K. J. Med. Chem. 2013, 56, 2406. 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Owen S. C.; Doak A. K.; Wassam P.; Shoichet M. S.; Shoichet B. K. ACS Chem. Biol. 2012, 7, 1429. 10.1021/cb300189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino-Gomes D.; Meyer-Fernandes J. R. J. Bioenerg. Biomembr. 2011, 43, 89. 10.1007/s10863-011-9334-y. [DOI] [PubMed] [Google Scholar]
- Fernley H. N.; Walker P. G. Biochem. J. 1970, 116, 543. 10.1042/bj1160543. [DOI] [PMC free article] [PubMed] [Google Scholar]; Borgers M. J. Histochem. Cytochem. 1973, 21, 812. 10.1177/21.9.812. [DOI] [PubMed] [Google Scholar]
- Slee E. A.; Zhu H. J.; Chow S. C.; MacFarlane M.; Nicholson D. W.; Cohen G. M. Biochem. J. 1996, 315, 21. 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelkarim G. E.; Gertz K.; Harms C.; Katchanov J.; Dirnagl U.; Szabo C.; Endres M. Int. J. Mol. Med. 2001, 7, 255. 10.3892/ijmm.7.3.255. [DOI] [PubMed] [Google Scholar]
- Degterev A.; Huang Z. H.; Boyce M.; Li Y. Q.; Jagtap P.; Mizushima N.; Cuny G. D.; Mitchison T. J.; Moskowitz M. A.; Yuan J. Y. Nat. Chem. Biol. 2005, 1, 112. 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]; Degterev A.; Hitomi J.; Germscheid M.; Ch’en I. L.; Korkina O.; Teng X.; Abbott D.; Cuny G. D.; Yuan C.; Wagner G.; Hedrick S. M.; Gerber S. A.; Lugovskoy A.; Yuan J. Nat. Chem. Biol. 2008, 4, 313. 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair P.; Lu M.; Petersen S.; Ashkenazi A. Methods Enzymol. 2014, 544, 99. 10.1016/B978-0-12-417158-9.00005-4. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Long M. J.; Zhou J.; Shi J.; Gao Y.; Xu C.; Hedstrom L.; Xu B. J. Biol. Chem. 2014, 289, 29208. 10.1074/jbc.M114.600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Ikonen E. Nature 1997, 387, 569. 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Parasassi T.; De Stasio G.; Ravagnan G.; Rusch R. M.; Gratton E. Biophys. J. 1991, 60, 179. 10.1016/S0006-3495(91)82041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathy R.; Anilkumar A. A.; Polley A.; Singh P. P.; Yadav M.; Johnson C.; Suryawanshi S.; Saikam V.; Sawant S. D.; Panda A.; Guo Z.; Vishwakarma R. A.; Rao M.; Mayor S. Cell 2015, 161, 581. 10.1016/j.cell.2015.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Toomre D. Nat. Rev. Mol. Cell Biol. 2000, 1, 31. 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Sanchez-Alcazar J. A.; Rodriguez-Hernandez A.; Cordero M. D.; Fernandez-Ayala D. J. M.; Brea-Calvo G.; Garcia K.; Navas P. Apoptosis 2007, 12, 1195. 10.1007/s10495-006-0044-6. [DOI] [PubMed] [Google Scholar]
- Edidin M. Nat. Rev. Mol. Cell Biol. 2003, 4, 414. 10.1038/nrm1102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.