

Abstract
Pyroptosis is a lytic type of cell death that is initiated by inflammatory caspases. These caspases are activated within multi‐protein inflammasome complexes that assemble in response to pathogens and endogenous danger signals. Pyroptotic cell death has been proposed to proceed via the formation of a plasma membrane pore, but the underlying molecular mechanism has remained unclear. Recently, gasdermin D (GSDMD), a member of the ill‐characterized gasdermin protein family, was identified as a caspase substrate and an essential mediator of pyroptosis. GSDMD is thus a candidate for pyroptotic pore formation. Here, we characterize GSDMD function in live cells and in vitro. We show that the N‐terminal fragment of caspase‐1‐cleaved GSDMD rapidly targets the membrane fraction of macrophages and that it induces the formation of a plasma membrane pore. In vitro, the N‐terminal fragment of caspase‐1‐cleaved recombinant GSDMD tightly binds liposomes and forms large permeability pores. Visualization of liposome‐inserted GSDMD at nanometer resolution by cryo‐electron and atomic force microscopy shows circular pores with variable ring diameters around 20 nm. Overall, these data demonstrate that GSDMD is the direct and final executor of pyroptotic cell death.
Keywords: atomic force microscopy, cell death, gasdermin, inflammasomes, inflammation, pyroptosis
Subject Categories: Autophagy & Cell Death, Immunology
Introduction
Inflammatory caspases (caspase‐1, human caspase‐4, human caspase‐5, and murine caspase‐11) are a group of cysteine‐dependent aspartate‐directed proteases that is essential for host innate immune defense. Caspase‐1 is activated within large multi‐protein complexes termed inflammasomes, which are assembled by the protein pyrin or members of the NOD‐like receptor (NLR) and PYHIN protein families (Latz et al, 2013; von Moltke et al, 2013). These proteins act as cytosolic pattern‐recognition receptors (PRRs) and detect a variety of pathogen‐associated molecular patterns (PAMPs) or endogenous danger signals (DAMPs). In contrast, the bacterial cell wall component lipopolysaccharide (LPS), one of the strongest immune‐system activators, leads to the assembly of “non‐canonical” inflammasomes through activation of caspase‐4, caspase‐5 or caspase‐11 (Kayagaki et al, 2011, 2013; Hagar et al, 2013; Shi et al, 2014).
The downstream signaling pathways that follow the activation of inflammatory caspases and how the active caspases initiate these events are still poorly understood (Lamkanfi, 2011). Initial work has identified the pro‐inflammatory cytokine interleukin (IL)‐1β as a key substrate of caspase‐1 (Thornberry et al, 1992). Subsequently, it was found that caspase‐1, as well as caspase‐11 and its human orthologs caspase‐4 and caspase‐5, induces a novel programmed cell death pathway that is characterized by cell swelling, lysis, and the release of cytoplasmic content (Fink & Cookson, 2007; Kayagaki et al, 2011; Shi et al, 2014), presumably as a result of the formation of membrane pores (Fink et al, 2008). Since this type of cell death is morphologically distinct from apoptosis and intrinsically pro‐inflammatory, it was named pyroptosis, from the Greek pyro (fire or fever) and ptosis (to fall) (Bergsbaken et al, 2009). The physiological function of pyroptosis is thought to be the prevention of intracellular pathogen replication and to re‐expose pathogens to extracellular killing mechanisms (Miao et al, 2010).
Several landmark studies have recently identified GSDMD (gasdermin D), a member of the gasdermin protein family, as an essential mediator of pyroptosis in human and murine cells (He et al, 2015; Kayagaki et al, 2015; Shi et al, 2015). GSDMD is required for pyroptosis induction after canonical and non‐canonical inflammasome activation and is processed by caspase‐1, caspase‐11, caspase‐4, and caspase‐5, but not by apoptotic caspases. The N‐terminal fragment of GSDMD (GSDMDNterm) was found sufficient to induce cell death with the morphological features of pyroptosis (Kayagaki et al, 2015; Shi et al, 2015), and overexpression of the C‐terminal domain GSDMDCterm was found to block GSDMDNterm‐dependent cell death (Shi et al, 2015). These results gave rise to the hypothesis that caspase‐dependent cleavage releases the N‐terminal domain from an inhibitory interaction with the C‐terminus, allowing GSDMDNterm to induce cell death by a yet undefined mechanism. Since pyroptosis had long been speculated to involve the formation of a plasma membrane pore, immediate destruction of the electrochemical gradient, and subsequent osmotic lysis of the host cell (Lamkanfi, 2011), it is likely that GSDMDNterm either promotes the formation of this pore or itself has pore‐forming activity (Broz, 2015). Here, we investigate the functional role of GSDMDNterm in live cells and in vitro. We demonstrate that after cleavage by caspase‐1, GSDMDNterm targets cellular membranes and that it induces the formation of a large permeability pore in the plasma membrane. In vitro experiments with purified recombinant GSDMD show that GSDMDNterm forms large pores in liposomes. We visualize these with cryo‐electron and atomic force microscopy. Overall, these results close the gaps in the pyroptotic signaling pathway by providing the proof that GSDMD is the final and direct executor of pyroptotic cell death.
Results
The N‐terminal GSDMD fragment induces the formation of a large plasma membrane pore
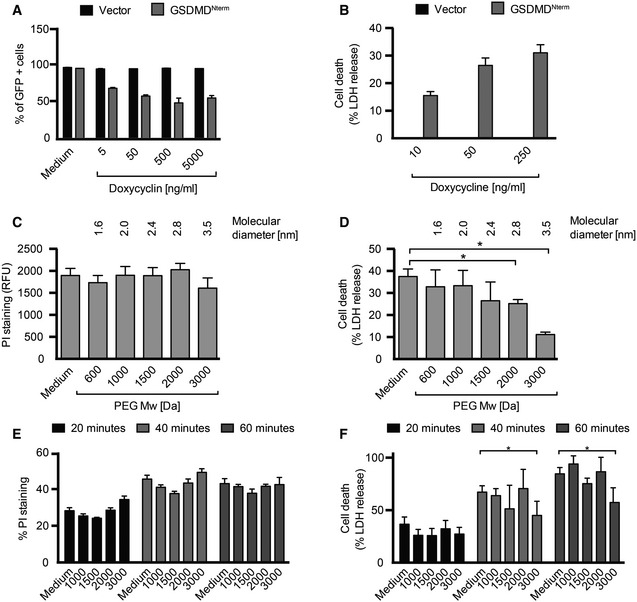
Pyroptotic cell death involves the formation of a plasma membrane pore, cell swelling, and rupture of the plasma membrane. To investigate whether GSDMD, a recently identified mediator of pyroptosis, mediates pore formation directly, we developed a doxycycline‐inducible system to express the N‐terminal fragment (GSDMDNterm) of mouse GSDMD in HEK293T cells. Doxycycline treatment caused cell death in HEK293T cells harboring the GSDMDNterm‐expressing plasmid, but not in cells harboring a vector control, in a concentration‐dependent manner (Fig 1A). To characterize whether GSDMDNterm expression induced a lytic type of cell death, characteristic for pyroptosis, we next measured the amount of LDH (lactate dehydrogenase) release after doxycycline‐induced expression of the GSDMDNterm (Fig 1B). Increasing levels of GSDMDNterm expression resulted in increased levels of LDH release, indicating that ectopic expression of the GSDMDNterm induced death through cell lysis. Microscopy analysis showed that GSDMDNterm‐induced death had the morphological features of pyroptosis, that is, cell swelling and nuclear condensation. To estimate the size of the GSDMDNterm‐induced plasma membrane pore, we next employed an osmoprotection assay based on the addition of polyethylene glycols (PEGs) of increasing molecular weight. Addition of these high‐molecular polymers can prevent water influx through pores and the resulting swelling and osmotic lysis, if the molecular diameter of the agent is larger than the diameter of the pore (Appendix Fig S1A). We induced GSDMDNterm expression by doxycycline addition in HEK293T cells in the presence of PEGs and measured LDH release as a readout for osmotic lysis and propidium iodide (PI) staining as a measure of plasma membrane pore formation (Fig 1C and D). Only the largest sized agent, PEG3000, was able to reduce LDH release partially, while smaller PEGs did not reduce cell lysis. Importantly, PEG3000 did not prevent PI influx, indicating that it does not block the pore directly, but functions as an osmoprotectant. PEGs in the range of 600–3,000 Da did not induce significant levels of cell death when added to cells (Appendix Fig S1B), but larger PEGs could not be used, since they proved to be cytotoxic.
Figure 1. GSDMD Nterm induces the formation of a large plasma membrane pore.
-
ACell viability as assessed by GFP expression in HEK293T cells transfected with the pRetroX TetOne3G‐eGFP plasmid only (vector) or pRetroX TetOne3G‐eGFP harboring the N‐terminal fragment of GSDMD. Cells were treated with the indicated concentrations of doxycycline 24 h post‐transfection, and the percentage of GFP‐positive cells was determined 16 h later by flow cytometry.
-
BLDH release from HEK293T cells transfected with the pRetroX TetOne3G‐eGFP plasmid only (vector) or pRetroX TetOne3G‐eGFP harboring the N‐terminal fragment of GSDMD. At 24 h post‐transfection, cells were treated with the indicated concentrations of doxycycline for 8 h and the percentage of LDH release was determined. Graphs show mean and s.d. of quadruplicate wells.
-
C, DPI staining of and LDH release from HEK293T cells transfected with pRetroX TetOne3G‐eGFP harboring the N‐terminal fragment of GSDMD in the presence of osmoprotectants. At 24 h post‐transfection, PEGs of the indicated molecular weights were added to a final concentration of 30 mM, cells were treated with 250 ng ml−1 doxycycline for 8 h, and the level of PI staining (C) or LDH release (D) was determined.
-
E, FPI staining of and LDH release from LPS‐primed primary BMDMs infected with log‐phase S. typhimurium for the indicated time points in the presence of PEGs of the indicated molecular weight (numbers on the x‐axis, 30 mM final concentration).
Infection of primary murine bone marrow‐derived macrophages (BMDMs) with Salmonella enterica serovar Typhimurium (S. typhimurium) activates the NLRC4 inflammasome (Mariathasan et al, 2004) and results in caspase‐1‐ and GSDMD‐dependent pyroptosis and cytokine release (Appendix Fig S1C and D). To estimate the size of the GSDMD‐dependent plasma membrane pore in BMDMs, we measured cell lysis as a function of time in the presence of PEGs of increasing size (Fig 1E and F). Consistent with the osmoprotection experiment done in HEK293T cells, we observed that only PEG3000 had a small protective effect, while all smaller PEGs did not prevent pyroptosis. PI influx was not affected by any of the osmoprotectants. IL‐1β release was also partially affected by PEG treatment (Appendix Fig S1E); consistent with the observation that pyroptosis is required for efficient release of the mature cytokine in BMDMs (Shi et al, 2015). Overall, these experiments suggest that GSDMD‐dependent pyroptosis involves the formation of a plasma membrane pore with an inner diameter of over 3.5 nm, the estimated molecular size of PEG3000 (Scherrer & Gerhardt, 1971).
The N‐terminal GSDMD fragment targets cellular membranes
GSDMDNterm might itself form a pore in the plasma membrane or alternatively initiate other events that result in pore formation (Broz, 2015). To define the fate of GSDMDNterm after caspase‐1‐dependent cleavage of GSDMD, we followed caspase‐1 activation, GSDMD processing, and cell death over time in immortalized wild‐type macrophages infected with S. typhimurium (Fig 2A, Appendix Fig S2A). The processed caspase‐1 p20 fragment, an indicator of caspase‐1 activation, appeared within 20 min after infection in the supernatant of macrophages. GSDMD processing correlated with caspase‐1 activation and was detectable in the cell lysate as well as in the cell supernatant. LDH release was also detectable at the same time points (Fig 1F and Appendix Fig S1C). Based on these data, we decided to determine the subcellular localization of full‐length GSDMD and GSDMDNterm in either uninfected cells or in cells infected with S. typhimurium for 10 and 20 min. Cells were harvested at each of these time points and subjected to subcellular fractionation as outlined (Fig 2B). GSDMD full length was exclusively found in the cytosolic fraction (S150) in uninfected cells (Fig 2C and Appendix Fig S2B), in line with the notion that it is a soluble, cytosolic protein. After infection, full‐length GSDMD, but very little GSDMDNterm was detected in the S150 fraction. Instead, the majority of GSDMDNterm was found in the P150 fraction and in the P10 fraction, correlating with the presence of the plasma membrane marker Na+K+ ATPase. The Na+K+ ATPase was also strongly present in the P10, presumably since it is secreted via the ER/Golgi pathway. The mitochondrial marker VDAC, a porin of the outer mitochondrial membrane, did not correlate with the GSDMDNterm and was mainly found in the P0.7 and P10 fractions, but not in the P150 fraction. Overall, these results suggest that the GSDMDNterm targets membranes after caspase‐1‐mediated cleavage. To characterize the interaction, we isolated plasma membrane fractions of BMDMs after S. typhimurium infection and subjected them to different treatments (Fig 2D). Conditions known to release membrane‐associated proteins or destabilize protein–protein interactions (Gatfield & Pieters, 2000), such as high salt (1 M NaCl) and sodium carbonate (pH 11) did not solubilize GSDMDNterm. Extraction of the plasma membrane with 0.02% digitonin, a cholesterol‐sequestering detergent, did not solubilize GSDMDNterm either. Only the disruption of the membrane with low concentrations of the detergent SDS (0.1%) was able to fully solubilize GSDMDNterm. Consistently, extraction of BMDMs membranes with 1% Triton was also able to partly solubilize GSDMDNterm (Appendix Fig S2C). Thus, GSDMDNterm integrates into cellular membranes in a cholesterol‐independent manner, and this integration is associated with formation of a pore and cell lysis.
Figure 2. GSDMD Nterm localizes to cellular membranes after inflammasome activation.
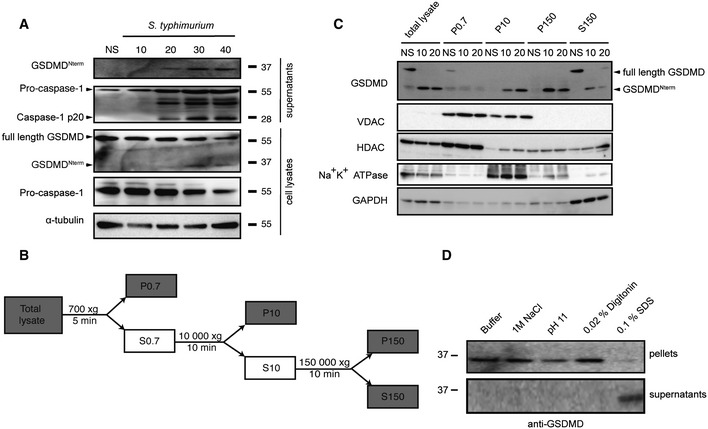
- Immunoblot analysis of cleaved GSDMD in culture supernatants and full‐length GSDMD, cleaved GSDMD and α‐tubulin in the cell lysates of immortalized LPS‐primed WT macrophages left uninfected (NS) or infected for 10–40 min with log‐phase S. typhimurium (MOI = 50).
- Schematic representation of the subcellular fractionation shown in (C).
- Fractionation and immunoblot analysis for GSDMD, Na+K+ ATPase, VDAC (voltage‐dependent anion channel), HDAC1 (histone deacetylase 1), and GAPDH (glycerinaldehyd‐3‐phosphate dehydrogenase) of WT macrophages infected for 10 and 20 min with log‐phase S. typhimurium (MOI = 50). Fractionation was carried out as described in the Materials and Methods section, and equivalent amount of protein was loaded per lane.
- Extraction of cleaved GSDMD from isolated membranes of WT macrophages infected for 10 min with log‐phase S. typhimurium (MOI = 50). Extraction was carried in variable conditions as described in the Materials and Methods section.
Source data are available online for this figure.
The N‐terminal fragment of GSDMD associates to liposomes in vitro
These findings encouraged us to attempt the reconstitution of a possible pore‐forming function of GSDMDNterm in vitro. We established recombinant expression for full‐length human GSDMD in E. coli BL21(DE3) expression cells. The protein expressed well with yields of 1.5 mg l−1 cell culture. Notably, following the same protocol, GSDMDNterm did not express in E. coli BL21(DE3) cells to detectable levels by SDS–PAGE, in agreement with the hypothesis that the protein might have a toxic effect on the host cells. Full‐length GSDMD was isolated and purified to homogeneity (Appendix Fig S3A). It elutes as a monodisperse, homogeneous elution peak from size exclusion chromatography, at the position of the expected monomeric species. Thermal denaturation showed a melting point of 43°C, indicating that the protein is folded and can be thermally denatured. Upon incubation with different human caspases, we confirmed that recombinant GSDMD is cleaved by caspase‐1, but not by the apoptotic caspase‐3 or caspase‐8 (Shi et al, 2015). Recombinant GSDMD is thus a functional substrate of its native enzyme. Then, we characterized the time dependence of GSDMD cleavage (Fig 3A). About 5 nM of caspase‐1 cleaves more than 50% of 2 μM of GSDMD in 40 min. GSDMD cleavage by caspase‐1 results in a 30‐kDa N‐terminal (GSDMDNterm) and a 22‐kDa C‐terminal (GSDMDCterm) fragment. In aqueous solution in the absence of a lipidic phase, GSDMDNterm is not soluble and forms aggregates, as demonstrated in a cross‐linking experiment (Fig 3B). After cleavage, the N‐terminus is highly cross‐linked by DSS (disuccinimidyl suberate), while the 22‐kDa GSDMDCterm remains soluble. To determine whether the poorly soluble GSDMDNterm associates with lipids, GSDMD was incubated in the presence or absence of active caspase‐1 with unilamellar liposomes made of either 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC) or from E. coli polar lipid extract. Ultracentrifugation allowed for separation of the liposomes from the soluble fraction. Whereas full‐length GSDMD did not associate with either the DMPC or the E. coli polar extract liposomes, the GSDMDNterm fully associated with either of the two membrane mimetics. GSDMDCterm did not associate with the liposomes (Fig 3C). Therefore, the soluble GSDMDCterm domain is acting in full‐length GSDMD not only as an inhibitor of GSDMDNterm but also as a solubility tag for the intrinsically insoluble and lipophilic GSDMDNterm, preventing it from aggregation and membrane association. Once cleaved, GSDMDNterm associates strongly to available membranes. DMPC liposomes are made from a chemically pure compound, showing that membrane association by GSDMDNterm does not require any specific receptors in the membrane.
Figure 3. GSDMD Nterm targets liposomes after caspase‐1 cleavage.
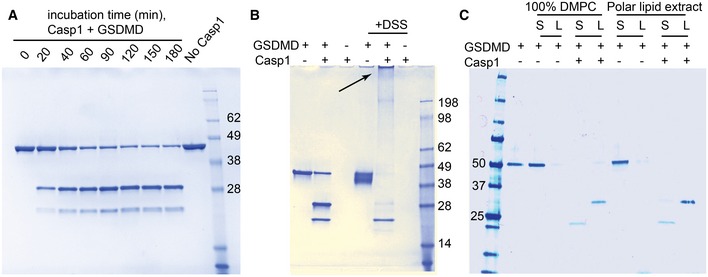
- Human GSDMD at a concentration of 2 μM was incubated at room temperature with 5 nM caspase‐1. The protein was cleaved in a time‐dependent manner into two bands of 31 kDa (GSDMD Nterm) and 22 kDa (GSDMDCterm).
- Cross‐linking experiment of full‐length and cleaved GSDMD. GSDMD at a concentration of 2 μM was incubated at room temperature with 5 nM caspase‐1. After enzymatic cleavage, GSDMDNterm is highly cross‐linked by DSS, resulting in the gel‐impenetrating species highlighted by the arrow. GSDMDCterm is not cross‐linked.
- GSDMD at a concentration of 1 μM was incubated at room temperature with 5 nM caspase‐1 and liposomes composed of 4 mM DMPC or polar lipid extract derived from E. coli. After 2 h, the lipid fraction (L) was separated from the supernatant (S) by ultracentrifugation at 4°C for 1 h at 120,000 g.
Source data are available online for this figure.
The N‐terminal fragment of GSDMD forms pores in liposomes
Next, we asked whether liposome‐associated GSDMDNterm forms transmembrane pores. Liposomes filled with a self‐quenching concentration of the fluorophore 6‐carboxyfluorescein were incubated with recombinant full‐length GSDMD in the presence or absence of active caspase‐1 (Fig 4). Release of the fluorophore from the liposome interior results in a strong reduction in the concentration‐dependent self‐quenching effect and consequently in an increase of the overall fluorescence signal, as demonstrated by chemical rupture of the liposomes with the detergent Triton X in a control experiment (Appendix Fig S5). Neither caspase‐1 nor full‐length GSDMD alone was able to release dye from the liposomes, but in the presence of both GSDMD and caspase‐1, dye release was observed, indicating the formation of permeability pores with open diameters of at least the molecular size of 6‐carboxyfluorescein (≈1 nm). The dye release reaction from liposomes includes at least three kinetic steps: the first step is the proteolytic cleavage of GSDMD by caspase‐1, the second step is the membrane association, and the third step is pore formation of GSDMDNterm. Whereas the first step follows classical Michaelis–Menten kinetics, the reaction mechanisms of the second and third steps may include additional oligomerization steps with non‐trivial concentration dependence. In an attempt to visualize the concentration dependence of the overall reaction, we measured the kinetics of dye release as a function of GSDMD concentration at a constant caspase‐1 concentration of 5 nM (Fig 4A). At a GSDMD concentration of 520 nM, the dye release is very efficient so that in 20 min, already more than 90% of the total fluorescence signal is observed. With decreasing GSDMD concentration, the overall reaction rate decreases, but the overall dye release nonetheless reaches 100%, showing that sufficient GSDMD is available to permeate all liposomes. This conclusion breaks down at a GSDMD concentration of 65 nM, where only about 50% of the liposomes are permeated at late time points.
Figure 4. GSDMD Nterm causes liposome permeability by pore formation.
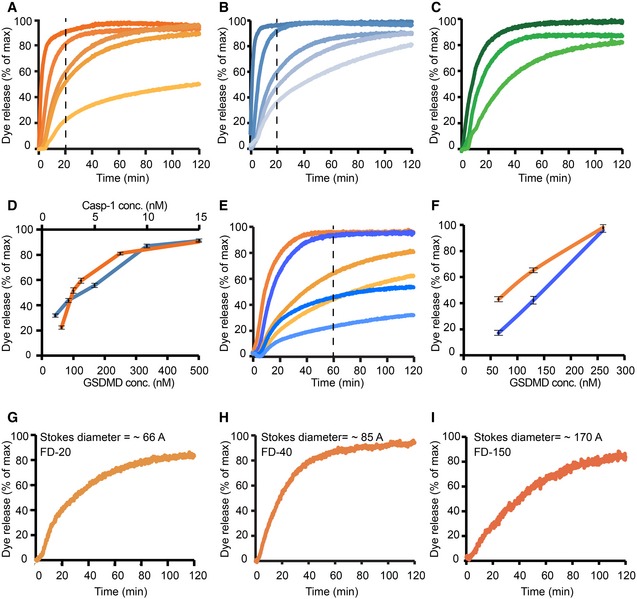
-
A–IDye release time courses from liposomes as a percentage of maximal release. (A) Five different reactions, where 5 nM caspase‐1 and 400 μM 6‐carboxyfluorescein‐loaded liposomes prepared with E. coli polar lipids were incubated with GSDMD concentrations of (nM): 520, 260, 130, 100, 65 (colored dark to light orange). The time point of 20 min is highlighted by a vertical dashed line. (B) Five different reactions, where 130 nM of GSDMD and 400 μM 6‐carboxyfluorescein‐loaded liposomes prepared with E. coli extract polar lipid, were incubated with caspase‐1 concentration of (nM): 15, 8, 5, 2.5, 1.2 (colored dark to light blue). The time point of 20 min is highlighted by a vertical dashed line. (C) Three different reactions, where 5 nM caspase‐1 and 400 μM 6‐carboxyfluorescein‐loaded liposomes prepared with porcine brain total lipid extract, were incubated with GSDMD concentrations of (nM): 520, 260, 100 (colored dark to light green). (D) Dye release at 20‐min reaction as a function of GSDMD (dark orange) and caspase‐1 (dark blue) concentrations. Error bars for three independent experiments are shown. (E) Two different sets of reactions, where wild‐type GSDMD (dark to light orange) and the mutant GSDMDI104N (dark to light blue) were independently incubated at the concentrations of 260, 130, and 65 nM with 5 nM caspase‐1 and 400 μM 6‐carboxyfluorescein‐loaded liposomes. The time point of 60 min is highlighted by a vertical dashed line. (F) Dye release at 60 min of reaction as a function of GSDMD wild‐type (dark orange) and GSDMDI104N (dark blue) concentration. Error bars for three independent experiments are shown. (G–I) Dye release from 400 μM liposomes loaded with the 6‐carboxyfluorescein derivates FD‐20, FD‐40, and FD‐150, with variable Stokes diameters, as indicated. 130 nM of GSDMD and 5 nM caspase‐1 were incubated with the liposomes. For each experiment, a representative from three independent experiments is shown. The corresponding raw data are shown in Appendix Figure S5.
We then measured the kinetics of dye release as a function of the caspase‐1 concentration in the range 1.2–15 nM, while keeping the GSDMD concentration constant at 130 nM (Fig 4B). In this experiment, by decreasing the concentration of caspase‐1, we expect a reduction of the availability of cleaved GSDMDNterm by the initial protease cleavage step, and consequently, we observe a deceleration of the dye release reaction. The total amount of GSDMD is always sufficient to permeate all liposomes in the setup, and consequently, we observe 100% dye release levels in all measurements. Importantly, GSDMD did not only permeate liposomes made of bacterial lipid extract (Fig 4A and B), but similarly also liposomes from a eukaryotic source (Fig 4C). In a next experiment, we examined the functionality of the I105N mutant of GSDMD. This mutant had played a key role in the discovery of GSDMD, since it had previously been identified as a loss‐of‐function mutant in mouse models (Kayagaki et al, 2015). We generated the analogous mutation I104N in GSDMD and expressed and purified the mutant protein with the same biochemical protocols as the wild‐type protein. GSDMDI104N is cleaved by caspase‐1 with kinetics indistinguishable from wild‐type GSDMD (Appendix Fig S4A and B). In the dye release assay, at high protein concentrations, GSDMDI104N is able to form functional pores with only minor differences to the wild‐type protein. At reduced proteins concentrations, however, it showed reduced activity compared to the wild‐type protein (Fig 4E and F). Consistent with these data and previously published work (Kayagaki et al, 2015), we found that GSDMDI104N could also induce cell death of HEK293T cells when expressed by a doxycycline‐inducible promoter, although significantly less than the WT protein (Appendix Fig S4D). Similarly, expression of the I104N mutant of human GSDMD in immortalized Gsdmd‐deficient mouse macrophages partially restored pyroptosis after Salmonella infection when compared to wild‐type human GSDMD (Appendix Fig S4C). The quantitative differences observed here may well translate into an effective loss‐of‐function effect in whole animals, and our experiments thus confirm the functional deficiency of the mouse I105N mutant. As an additional control experiment, we verified that uncleaved GSDMD full length does not form pores in liposomes (Appendix Fig S5E and F). Next, we addressed the size of the GSDMD pore in liposomes by using carbohydrate‐conjugated fluorophores as markers. These fluorophores have differently long carbohydrate chains attached, resulting in different overall Stokes radii. We selected three different fluorophores with Stokes radii of 33, 45, and 85 Å, but all of them were still released by the GSDMD pore (Fig 4G–I). This suggests that the pyroptosis pore formed by GSDMDNterm can reach diameters of at least 15 nm, in full agreement with the cellular experiments.
Visualization of GSDMD pores
We employed two methods to resolve pores of GSDMDNterm in liposomes at nanometer resolution. Cryo‐electron micrographs of untreated liposomes show intact spherical shapes (Appendix Fig S6A). Also the incubation with full‐length GSDMD in the absence of caspase‐1 did not result in visual distortions of the liposomes (Appendix Fig S6B). However, upon addition of caspase‐1 and subsequent incubation, large ruptures of the liposome structure were observed. In these preparations, the liposome surface features numerous large rings of dense material with inner diameters of ≈20 nm (Fig 5A–D and Appendix Fig S6C–E). These rings are formed by GSDMDNterm and although the liposomes appear completely covered by the assemblies, from the available contrast of the micrographs, it remains inconclusive, whether these rings are lipid‐filled protrusions or actual transmembrane pores. The lack of contrast also did not allow for a clear identification of the different shapes of GSDMD oligomers. To characterize the assembly of the transmembrane pores convincingly, we thus imaged GSDMD oligomers and pore formation on liposomes by atomic force microscopy (AFM). In an initial control experiment, we adsorbed either GSDMD alone, or caspase‐1 alone, or GSDMD and caspase‐1 together to freshly cleaved mica, which we used as sample support in our AFM studies (Appendix Fig S7A–D). GSDMD and caspase‐1 readily adsorbed to the negatively charged hydrophilic mica surface as monomers or small oligomers, but did not assemble into arcs or rings. After this, we incubated liposomes composed of E. coli polar lipids with GSDMD and caspase‐1 for 90 min at 37°C and adsorbed the sample onto freshly cleaved mica. The AFM topographs recorded in buffer solution showed that upon adsorption to mica, the liposomes opened as single‐layered membrane patches (Appendix Fig S7E). At higher resolution, the topographs showed GSDMD bound to the lipid membranes and forming arc‐, slit‐ and ring‐shaped oligomers (Fig 6A–E and Appendix Fig S7F). The height profile around the arc‐, slit‐ and ring‐like oligomers indicates that they protruded 4.3 ± 0.3 nm (average ± s.d.; n = 218) from the lipid surface and that each of them could form pores through the lipid membrane (Fig 6F and G). Occasionally, arcs and slits combined into ring‐shaped structures, which were not yet perfectly circular and only partially associated with the formation of membrane pores. Several pore‐forming toxins (PFTs) have shown the ability to bind to lipid membranes as oligomers forming arcs or slits that can dynamically rearrange on the membrane surface to form larger pores (Leung et al, 2014; Sonnen et al, 2014; Mulvihill et al, 2015; Podobnik et al, 2015). In agreement with this observation, GSDMD arcs and slits were found that presumably fused into perfect ring‐shaped GSDMDNterm oligomers of variable diameters, with an average value of 21 nm (Fig 6F–H). These AFM topographs thus clearly show a structural variability of the GSDMDNterm assembly, which is, however, determined to eventually lead to the assembly of ring‐like structures. These ring‐like structures correspond to the pyroptotic membrane pores.
Figure 5. Visualization of GSDMD Nterm pores in liposomes by cryo‐electron microscopy.
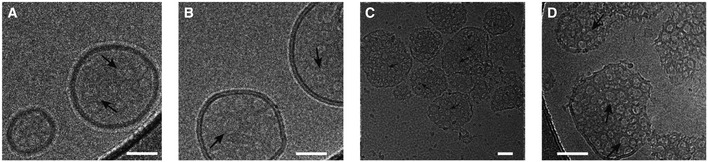
-
A–CCryo‐electron micrographs of GSDMDNterm pores in E. coli polar lipid liposomes. The micrographs were acquired at protein/lipid molar ratios of 1/1,000, 1/500, and 1/100, respectively. Black arrows indicate ring‐shaped structures corresponding to oligomeric GSDMDNterm pore forms. Scale bars = 80 nm.
-
DProteoliposome with protein/lipid molar ratio of 1/100 at higher magnification. Black arrows indicate ring‐shaped structures corresponding to oligomeric GSDMDNterm pore forms. Scale bar = 80 nm.
Figure 6. Characterization of GSDMD Nterm pores by atomic force microscopy.
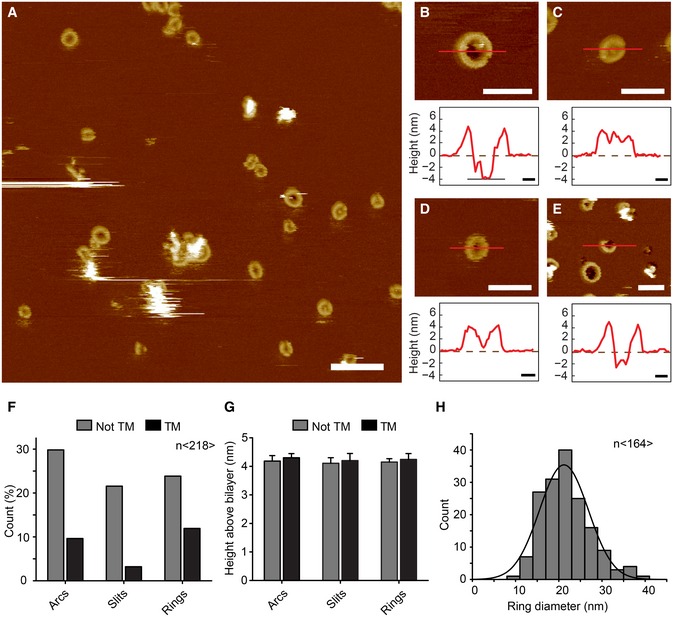
-
AAFM topograph of GSDMDNterm bound to lipid membranes of E. coli polar lipids. Overview topograph showing arc‐, slit‐, and ring‐like GSDMD oligomers. Scale bar corresponds to 100 nm.
-
B–EHigh‐resolution AFM topographs and height profiles of GSDMDNterm oligomers (red curves). Height profiles were measured along the red lines in the topographs. Dashed zero lines (0 nm) indicate the surface of the lipid membrane; heights of < −4 nm indicate that the oligomers formed transmembrane pores. Topographs were taken in buffer solution (150 NaCl, 20 mM Hepes, pH 7.8) at room temperature. The experiment was reproduced at least three times using independent liposome and GSDMD preparations. Scale bars of topographs correspond to 50 nm and of height profiles 10 nm. The full color range of the AFM topographs corresponds to a vertical scale (height) of 20 nm.
-
F–HAnalysis of GSDMDNterm oligomers bound to lipid membranes of E. coli polar lipids. (F) GSDMDNterm oligomers assembled into arcs, slits, and rings leading to transmembrane (TM) pores or not transmembrane aggregate (n = 218). (G) Height of GSDMDNterm oligomers protruding from the lipid membrane. Bars represent mean, and error bars represent s.e.m. (n = 218). (H) Distribution of the diameters of rings formed by GSDMDNterm oligomers. The average distribution was 21.2 ± 5.6 nm (n = 164; average ± s.d.), and the bin size was 3 nm.
Discussion
Pyroptotic death is a defining feature of canonical and non‐canonical inflammasome engagement and the final step associated with the activation of human and mouse caspase‐1, but also mouse caspase‐11 and human caspase‐4/‐5. Although the morphological features of pyroptotic cell death indicated the formation of a plasma membrane pore and subsequent lysis of the cell (Lamkanfi, 2011), the molecules involved in pyroptosis induction had remained elusive. The recent identification of GSDMD as an essential mediator of pyroptosis significantly expanded our understanding of pyroptotic cell death (He et al, 2015; Kayagaki et al, 2015; Shi et al, 2015), but whether after cleavage by caspases GSDMD executes pyroptosis directly, or by initiating other signaling events remained unclear. In this work, we have shown that the N‐terminal fragment of GSDMD, GSDMDNterm, can form pores with average diameters of 21 nm in artificial liposomes, as well as large pores in the plasma membrane of cells (Figs 5 and 6). GSDMD oligomers were found to form pre‐pore states consisting of arcs, slits, and rings. Each of these oligomeric states had a certain probability to also show a pore state, among which the GSDMD rings with their variable diameter formed the largest transmembrane pores. The variable pore size suggests that GSDMD possesses an intrinsic high structural variability and that GSDMD arcs and slits represent intermediates that presumably may fuse toward the ring‐shaped structures, similar to observations for several other pore‐forming toxins (PFTs) that dynamically rearrange on the membrane surface (Leung et al, 2014; Sonnen et al, 2014; Mulvihill et al, 2015; Podobnik et al, 2015). Importantly, our in vitro assays show that the formation of the GSDMD pore does not require other proteins as co‐factors or membrane receptors, demonstrating that GSDMDNterm is the sole and final executor of pyroptotic cell death (Fig 7). While this manuscript was in revision, other reports have been published that confirm our findings that the N‐terminal fragment of GSDMD causes pyroptosis through the formation of a plasma membrane pore and that demonstrate that the N‐terminal domains of other gasdermin family members share this pore‐forming activity (Aglietti et al, 2016; Ding et al, 2016).
Figure 7. Model for GSDMD‐mediated pyroptotic cell death.
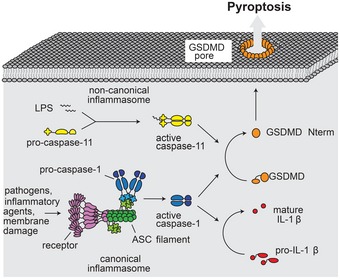
Canonical inflammasomes act as sensors for a variety of pathogens and cellular insults. Assembly of these types of inflammasomes involves multimerization of the receptor (purple), which initiates filament formation of the adaptor protein ASC (green). ASC filaments act as activation platforms for caspase‐1 (blue). In the noncanonical pathway, LPS is directly bound by caspase‐11 (yellow), resulting in its activation. Caspase‐1 processes interleukin IL‐1β (red). Both caspase‐1 and caspase‐11 process GSDMD (orange), which results in release of the GSDMDNterm fragment. GSDMDNterm forms a large pore in the plasma membrane. Pore formation results in rapid loss of membrane integrity, the dissipation of the electrochemical gradient, and ultimately in cell death.
The placement and function of GSDMD in the canonical and non‐canonical inflammasome pathway strongly resembles the placement and function of Bax (Bcl‐2‐associated X protein) in the apoptotic cell death pathways (Youle & Strasser, 2008). Bax is the final executor of apoptosis and forms a large pore in the mitochondrial outer membrane. Interestingly, whereas both proteins form large permeability pores in their respective target membranes, their mechanism of activation is entirely different. Whereas GSDMD is activated by proteolytic cleavage, Bax is activated by interactions within the Bcl‐2 family of proteins (Gavathiotis et al, 2008; Czabotar et al, 2013). A strong difference can also be expected for the atomic structure of the two proteins. Bax features a bundle of nine α‐helices (Suzuki et al, 2000), thereby burying the two presumed pore‐forming helices in its central core. The prediction of secondary structure elements shows that GSDMDNterm consists mainly of β‐strand secondary structure, whereas the soluble GSDMDCterm consists of α‐helical secondary structure (Appendix Fig S8). Thus, while Bax resembles the bacterial pore‐forming colicins, structural studies of the membrane‐inserted form of GSDMD will be of highest interest.
Another functionally related pathway is necroptotic cell death, which is for instance initiated by TNF (tumor necrosis factor)‐α or the engagement of Toll‐like receptor (TLRs) in absence of caspase‐8 activity (Pasparakis & Vandenabeele, 2015). Necroptosis involves the formation of the necrosome, a complex consisting of RIP (receptor serine–threonine–protein kinase)1, RIP3 and MLKL (mixed lineage kinase domain‐like) and similarly to pyroptosis it is characterized by cell lysis (Pasparakis & Vandenabeele, 2015). Recent work has demonstrated that recruitment of MLKL to RIP3 and MLKL phosphorylation results in the conformational change that leads to the exposure of its four‐helix bundle domain (FHBD) and oligomerization of MLKL (Murphy et al, 2013). Binding of the FHBD to negatively charged phosphatidylinositol phosphates has been proposed to recruit MLKL to the plasma membrane allowing it to directly permeabilize the membrane by forming a pore (Dondelinger et al, 2014; Su et al, 2014; Wang et al, 2014). Alternatively, it has been also proposed that plasma membrane‐bound MLKL recruits Ca2+ or Na+ ion channels to permeabilize the membrane (Cai et al, 2014; Chen et al, 2014). Thus, although the steps leading up to necroptosis are markedly different from pyroptosis, the execution of both types of lytic cell death involves the formation of plasma membrane pores.
The fact that GSDMD targets membranes after cleavage by caspases and forms permeability pores also sheds some light on the function of the regulatory C‐terminal domain. Cleavage of GSDMD by inflammatory caspases results in the release of the GSDMD C‐terminal fragment (Kayagaki et al, 2015; Shi et al, 2015), but overexpression of GSDMDCterm, which was found to bind GSDMDNterm, results in the inhibition of cell death (Shi et al, 2015). This gave rise to a model in which cleavage releases an intramolecular autoinhibition exerted by GSDMDCterm on the GSDMDNterm, thus preventing GSDMDNterm to unleash its cytotoxic activity. Interestingly, we have observed that GSDMDCterm is a soluble protein, while GSDMDNterm is insoluble and aggregates, when GSDMD is cleaved in the absence of a target membrane (Fig 3). This result supports a slightly different model in which GSDMDCterm does not only act as an inhibitory domain, but simultaneously also as a solubility tag for the N‐terminal domain, maybe by shielding certain hydrophobic or amphipathic segments required for membrane insertion. Based on these functions, the alternative names pore‐forming domain (PFD) and solubilizing inhibitory domain (SID) appear well suited for the GSDMDNterm and GSDMDCterm, respectively.
The identification of GSDMD as an essential mediator of pyroptosis has shed light on the possible functions of other gasdermin family members in programmed cell death. All gasdermins (GSDMA, GSDMB, GSDMC, and GSDMD in humans; GSDMA1‐3, GSDMC1‐4, and GSDMD in mice) are composed of a distinct N‐terminal and C‐terminal domain (Tanaka et al, 2013), a feature shared by the extended gasdermin family members DFNA5 and DFNB59 (deafness, autosomal‐dominant 5/autosomal‐recessive 59). Several lines of evidence suggest that the N‐terminal domain of other gasdermin family members is intrinsically cytotoxic (Saeki et al, 2007; Op de Beeck et al, 2011; Shi et al, 2015). Given the functional and sequence similarity between gasdermin N‐terminal domains, it is not surprising that all gasdermins induce membrane permeability pores and subsequent cell death (Ding et al, 2016). Structural studies of GSDMD and other gasdermin family members in their full‐length and membrane inserted forms as well as the stoichiometry of these pores will thus remain of high interest. Furthermore, it remains to be shown whether several gasdermins might cooperate in pore formation, given that caspase‐1‐dependent cell death does not solely rely on GSDMD (Kayagaki et al, 2015). Finally, to fully understand this emerging group of cell death effectors, it will be necessary to define how other gasdermin family members are activated and to identify the physiological context in which they induce cell death.
Materials and Methods
Cell culture and reagents
HEK293T were maintained in DMEM (Sigma), 10% FCS (Amimed), 1% penicillin/streptomycin (Amimed) and split, when they reached 90% confluency. Immortalized macrophages were obtained from R. Vance (UC Berkeley) and cultured in DMEM, 10% FCS, 1% PenStrep, 10 mM HEPES (Amimed), 1% non‐essential amino acids (NEAA, Amimed). Polyethylenimine was from Polysciences Inc, LDH detection kit from Takara, IL‐1β ELISA from eBiosciences, near IR live/dead marker from Thermo Scientific, propidium iodide from Santa Cruz Biotechnology, PEGs of different sizes from Merck. pRetroX TetONE3G‐Puro was obtained from Clontech. The puromycin resistance cassette was replaced by an EGFP by standard cloning procedures yielding pRetroX TetONE‐eGFP. GSDMDNterm was amplified from murine cDNA and cloned into pRetroX TetONE‐eGFP by standard cloning procedures. Anti‐GSDMD and anti‐GAPDH antibodies were from SCBT (both used at 1:250), anti‐caspase‐1 p20 from Adipogen (used at 1:4,000), anti‐alpha tubulin from Abcam (used at 1:2,500), anti‐VDAC, anti‐HDAC1, and anti‐Na+/K+ ATPase antibodies were from Cell Signaling Technologies (all at 1:1,000). HEK293T cells were seeded in 96‐well plates (30,000 cells per well) 1 day prior to transfection. Cells were subsequently transfected with 300 ng of plasmid DNA and 3× (wt/vol) linear PEI (polyethylenimine). Twenty‐four post‐transfection cells were treated as indicated in the respective figures.
Generation of stable cell lines
For complementation of Gsdmd KO cell lines (Dick et al, 2016), human Gsdmd was amplified from HeLa cell cDNA (Clontech) and cloned into pLJM1 (addgene #19319) where the puromycin resistance cassette was replaced with a hygromycin resistance cassette. The I104N mutant was generated using the Q5 site‐directed mutagenesis kit (NEB). To produce lentiviral particles, 1 × 106 HEK239T cells were transfected with the 2 μg lentiviral plasmid, 2 μg psPax2, and 0.4 μg VSV‐G for 6 hrs. Medium was exchanged, and lentiviral particles were collected 24 h later. Gsdmd‐deficient immortalized macrophages were spin‐infected with these particles, and transduced cells were selected with Hygromycin (500 μg/ml, invivogen) for 7 days. Selected cells were used for experiments.
Macrophage infection
Immortalized macrophages were seeded the day before the experiment (30,000 cells per well of a 96‐well plate, 1.5 × 106 cells per well of a 6‐well plate). S. typhimurium (SL1344) was grown overnight in Luria broth (LB) medium. On the day of experiment, bacteria were subcultured 1:40 in LB and grown for 4 h to logarithmic phase to induce SPI‐1 expression. Cells were washed once with PBS and infected at an MOI of 50 with S. typhimurium diluted in OptiMEM. LDH and IL‐1β release was determined as described in the respective manufacturer's instructions. Cell lysates and supernatants were prepared for Western blot and analyzed by SDS–PAGE as described previously (Broz et al, 2012).
Subcellular fractionation and membrane extraction
About 1.5 × 107 million immortalized macrophages were seeded in 10‐cm tissue culture‐treated dishes the day before the experiment. The next day, cells were left either uninfected or infected with log‐phase S. Typhimurium at a MOI of 50 for 10 and 20 min at 37°C. Cells were washed three times with homogenization buffer (10 mM Tris, 10 mM acetic acid, 1 mM EDTA, 7.5 mM MgCl2, 250 mM sucrose), scraped, and homogenized using a syringe equipped with a 25G needle (30 strokes) (Total lysate). Samples were centrifuged for 700 g for 5 min; supernatants were transferred to a new tube and centrifuged the same way again. Combined pellets yielded the P0.7 fraction, while the remaining supernatant yielded the S0.7 fraction. Pellets were washed twice with homogenization buffer and lysed in RIPA buffer. Supernatants (S0.7) were centrifuged 10,000 g for 15 min; supernatant was transferred to new tube and centrifuged the same way again. Pellets (P10) were combined, washed twice with homogenization buffer, and resuspended in SOL buffer (50 mM Tris–HCl pH 6.8, 1 mM EDTA, 1% Triton X‐100). Supernatants (S10) were centrifuged at 150,000 g for 30 min; supernatant was transferred to a new tube and centrifuged the same way again. Pellets were combined, washed again twice, and resuspended in SOL buffer (P150). The supernatants were analyzed as S150. Protein concentration of every sample was determined by BCA, and equal amounts were analyzed by SDS–PAGE. For extraction of membrane fractions, cells were infected for 15 min with log‐phase S. typhimurium, homogenized as described above, and membrane preparation and extraction were performed as described in Gatfield and Pieters (2000).
Cloning, expression, and purification of GSDMD and caspases
cDNA coding for the full‐length human GSDMD was cloned with an N‐terminal His6‐SUMO‐tag into a pET28a vector under control of a T7 promoter. Single amino acid point mutation I104N was generated by QuickChange site‐directed mutagenesis kit (Stratagene). All plasmids were verified by DNA sequencing. The protein construct was transformed in BL21 (DE3) E. coli strains, and the protein was expressed by growing the cultures at 37°C to an OD600 of 0.7 and by inducing with 0.5 mM IPTG overnight at 18°C. The cells were harvested by centrifugation and the pellet was resuspended in 20 mM Tris buffer pH 7.5, 50 mM NaCl, 5 mM imidazole, 20 mM MgCl2, 10 mM KCl, 0.5 mM TCEP, 0.1 mM protease inhibitor, and DNase I. The resuspended cells were disrupted by high‐pressure microfluidization and centrifuged at 30,000 g at 4°C for 45 min. The supernatant was incubated for 2 h at room temperature with pre‐equilibrated Ni‐NTA affinity resin (Thermo Scientific) and then passed through a column for gravity flow purification. The column was washed with 20 column volumes of resuspension buffer containing 15 mM imidazole, and the fusion protein was eluted with 3 column volumes of the same buffer with 250 mM imidazole. SUMO‐tag cleavage was achieved by addition of ULP1 protease to the solution and subsequent dialysis overnight at 4°C against 20 mM Tris buffer pH 7.5, 50 mM NaCl, 0.5 mM TCEP. GSDMD was eluted from a second round of purification through pre‐equilibrated Ni‐NTA affinity resin. The protein was further purified by Hi‐trapQ ion‐exchange and a Superdex 75 gel filtration column (GE Healthcare) pre‐equilibrated with 20 mM Tris buffer pH 7.5, 50 mM NaCl, 0.5 mM TCEP. Purified GSDMD was frozen in small aliquots in liquid N2. Human caspase‐1, caspase‐3, and caspase‐8 were cloned into pET22 expression vectors, expressed as inclusion bodies in E. coli, and refolded as previously described (Ramage et al, 1995). Briefly, inclusion bodies were solubilized in 8M urea, and protein was refolded by rapid dilution; during which the enzymes auto‐activated. A gel filtration column was run to purify the final product, and the concentration of active enzyme determined by active site titration.
Liposome preparation
The polar lipid extract derived from E. coli, total lipid extract derived from porcine brain, and DMPC were purchased from Avanti Polar Lipids, Inc. (Alabaster AL, USA). Chloroform lipid solutions at a concentration of 25 mg ml−1 were gently dried in a glass tube into a thin film under nitrogen flow and placed under vacuum overnight to further evaporate any residual solvent. The lipid layers were that rehydrated in 1 ml of 50 mM HEPES buffer, 150 mM NaCl, pH 7.5 under continuous shaking at 50°C for 2 h. The lipid dispersions were subjected to 10 freeze–thaw cycles, and the resultant liposomes were extruded 20 times through 100‐nm polycarbonate membranes to form large unilamellar vesicles (LUV). The mean size diameter of the liposomes was verified by dynamic light scattering (DLS). To prepare dye‐filled LUVs, the dry lipid film was hydrated with 0.5 ml of 50 mM HEPES buffer, 50 mM NaCl, 70 mM 6‐carboxyfluorescein, 5 mM TCEP, pH 7.5) or by adding 100 mg ml−1 of fluorescein isothiocyanate dextran FD‐20, FD‐40, and FD‐150. For the 6‐carboxyfluorescein containing liposome, the removal of extra‐vesicular dye was achieved by a purification step through a PD‐10 column (GE Healthcare) pre‐equilibrated with 50 mM HEPES buffer, 150 mM NaCl, 5 mM TCEP pH 7.5. The FD‐20, FD‐40, and FD‐150 dye‐filled liposomes were washed twice by ultracentrifugation at 4°C for 20 min at 100,000 g and then resuspended in 0.5 ml of the same isotonic solution. All liposomes were stored at 4°C and used within 24 h.
Liposome leakage assay
Membrane leakage experiments were performed using 6‐carboxyfluorescein, FD‐20, FD‐40, and FD‐150‐filled liposomes composed of polar lipid extract derived from E. coli and total lipid extract derived from porcine brain. The samples were initially prepared by diluting 40 mM total lipid concentration in 100 μl solution of 50 mM HEPES buffer, 150 mM NaCl, 5 mM TCEP, pH 7.5 supplemented with different aliquots of a 2.6 μM GSDMD stock solution. The NaCl concentration in the reaction buffer outside the LUV was optimized to preserve caspase‐1 activity and to minimize the dye release due to the osmotic pressure exerted by the dye on the membrane bilayer. To initiate the cleavage of GSDMD and the consequent pore formation reaction, different amounts of caspase‐1 from a 12 μM stock solution were added to 100 μl of premixed LUV–GSDMD solution. The membrane leakage was detected by measuring the time‐course increase in fluorescence resulting from the dye dilution and subsequent dequenching upon membrane pore formation. The experiments were carried out in a Corning 384‐well non‐binding surface plate, and the fluorescence was continuously recorded for 2 h at 10‐s intervals using a Biotek Synergy 2 plate reader with excitation and emission wavelengths at 492 and 520 nm, respectively. At each time point, the percentage of dye release was calculated as:
where I is the emission intensity of the sample, I 0 is the emission intensity from an experiment with the liposome solution only (negative control), and I max is the emission intensity from an experiment with added Triton X‐100 to the liposome solution (positive control).
Gel‐shift assays
Recombinant GSDMD was incubated with different caspases for the indicated time period in a reaction vessel in caspase activity buffer of 100 mM Hepes, pH 7.4, 0.5 mM EDTA, 50 mM NaCl, 0.1% CHAPS, 0.005% Novexin, and 5 mM TCEP. Final concentrations were 5 nM enzyme, 2 μM GSDMD. Reactions were quenched with 6xSDS loading buffer and analyzed by SDS–PAGE. For cross‐linking experiments, 2 mM DSS (ThermoFisher) were added to the reaction, and protein was cross‐linked for 30 min before quenching the reaction.
Thermofluor assay
To monitor GSDMD thermal stability, the protein was incubated with the fluorescent dye Sypro orange and the thermal shift assay was conducted in the CFX96 Real Time Detection System (Bio‐Rad, Hercules, CA). Solutions of 2 μl of 2 μM GSDMD, 8 μl of 5× Sypro orange, 10 μl solution of screened condition were loaded to a 96‐well plate. The plate was heated from 10 to 90°C with a heating rate of 0.5°C min−1. The fluorescence intensity was measured with excitation and emission wave lengths of 490 and 530 nm, respectively.
Cryo‐electron microscopy (cryo‐EM)
Cryo‐EM was used to visualize GSDMD‐mediated pore on liposome surfaces. The samples were prepared by 2‐h incubation of freshly prepared liposome (2 mM lipids) with 2.6 μM of GSDMD in the presence and the absence of catalytic amounts of caspase‐1. All samples were adsorbed for 10 s on glow‐discharged thin carbon film‐coated 300‐mesh lacey EM grids, blotted for 2 s, and plunge frozen in liquid ethane using a FEI Vitrobot MK4 (Vitrobot, Maastricht Instruments). Cryo‐EM micrographs were acquired with a Philips CM200FEG transmission electron microscope operated at 200 kV and a nominal magnification of 66,000×. Images were recorded with a TVIPS F416 CMOS camera.
Liposome Preparation for atomic force microscopy (AFM)
Unilamellar liposomes were prepared at room temperature (≈23°C) by hydration of lipid films and extrusion through polycarbonate filters with 0.1‐μm pore diameter (Nucleopore Polycarbonate, Whatman) according to the method described by Avanti Polar Lipids (www.avantilipids.com). The E. coli polar lipids and the extruding equipment used for liposome preparation were purchased from Avanti Polar Lipids. The liposomes were stored at −80°C in buffer solution (150 mM NaCl, 20 mM Hepes, pH 7.25). After incubation with 1 μM mM GSDMD and 0.2 μM caspase‐1 for 90 min at 37°C in buffer solution (50 mM NaCl, 100 mM Hepes, 5 mM TCEP, pH 7.4), the liposomes were adsorbed onto freshly cleaved mica in buffer solution (50 mM NaCl, 20 mM Hepes, pH 7.4) (Muller et al, 1997). After an adsorption time of 30 min, the sample was washed several times with the AFM imaging buffer (150 mM NaCl, 20 mM Hepes, pH 7.8) to remove weakly adsorbed protein. Buffer solutions were freshly made using nanopure water (18.2 MΩ cm−1) and pro‐analysis (> 98.5%) purity grade reagents from Sigma‐Aldrich and Merck. Each experimental condition characterized by AFM was reproduced at least three times. Liposomes made from E. coli polar lipids incubated in buffer solution but in the absence of GSDMD showed no arc‐, slit‐ or ring‐like structures when imaged by AFM (Mulvihill et al, 2015).
AFM
Force–distance curve‐based AFM (FD‐based AFM) (Dufrene et al, 2013) was performed using a Nanoscope Multimode 8 (Bruker, Santa Barbara, USA) operated in the PeakForce Tapping mode. The AFM was equipped with a 120‐μm piezoelectric scanner and fluid cell. AFM cantilevers used (BioLever mini BL‐AC40, Olympus Corporation, Tokyo, Japan) had a nominal spring constant of 0.1 N m−1, a resonance frequency of ≈110 kHz in liquid and sharpened silicon tip with a nominal radius of 8–10 nm. The FD‐based AFM topographs were recorded in AFM imaging buffer (150 mM NaCl, 20 mM Hepes, pH 7.8) and at room temperature as described (Pfreundschuh et al, 2014). The maximum force applied to image the samples was 70 pN, and the oscillation frequency and oscillation amplitude of the cantilever were set to 2 kHz and 40 nm, respectively. The AFM was placed inside a home‐built temperature controlled acoustic isolation box. For data analysis, we took unprocessed AFM topographs. Diameters of ring‐like GSDMD oligomers were measured from the highest protruding rim. Heights of GSDMD arcs, slits, and rings were measured from their highest protruding feature relative to the surface of the lipid membrane. GSDMD oligomers were classified to having formed transmembrane pores if the inside of the pore was at least 3.5 nm deeper compared to the surface of the surrounding lipid membrane.
Author contributions
LS, SR, PB, SH designed the study. LS, SR, EM, JP, RH, CJF carried out research experiments. All authors (including HS) analyzed data. LS, SR, DJM, PB, SH wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank Paul Erbel for help with the protein preparation. This work was supported by the European Research Council (FP7 contract MOMP 281764 to S.H.), the Swiss National Science Foundation (PP00P3_139120/1 to P.B.), and by the European Union Marie Curie Actions program through the ACRITAS Initial Training Network (FP7‐PEOPLE‐2012‐ITN, Project 317348 to D.J.M.).
The EMBO Journal (2016) 35: 1766–1778
References
- Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016) GsdmD p30 elicited by caspase‐11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA 113: 7858–7963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase‐11 increases susceptibility to Salmonella infection in the absence of caspase‐1. Nature 490: 288–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P (2015) Immunology: caspase target drives pyroptosis. Nature 526: 642–643 [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF‐induced necroptosis. Nat Cell Biol 16: 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J (2014) Translocation of mixed lineage kinase domain‐like protein to plasma membrane leads to necrotic cell death. Cell Res 24: 105–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152: 519–531 [DOI] [PubMed] [Google Scholar]
- Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P (2016) ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 7: 11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F (2016) Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116 [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7: 971–981 [DOI] [PubMed] [Google Scholar]
- Dufrene YF, Martinez‐Martin D, Medalsy I, Alsteens D, Muller DJ (2013) Multiparametric imaging of biological systems by force‐distance curve‐based AFM. Nat Methods 10: 847–854 [DOI] [PubMed] [Google Scholar]
- Fink SL, Cookson BT (2007) Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol 9: 2562–2570 [DOI] [PubMed] [Google Scholar]
- Fink SL, Bergsbaken T, Cookson BT (2008) Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase‐1‐dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA 105: 4312–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield J, Pieters J (2000) Essential role for cholesterol in entry of mycobacteria into macrophages. Science 288: 1647–1650 [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD (2008) BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 341: 1250–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J (2015) Gasdermin D is an executor of pyroptosis and required for interleukin‐1beta secretion. Cell Res 25: 1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose‐Girma M, Dixit VM (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: 1246–1249 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Lamkanfi M (2011) Emerging inflammasome effector mechanisms. Nat Rev Immunol 11: 213–220 [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung C, Dudkina NV, Lukoyanova N, Hodel AW, Farabella I, Pandurangan AP, Jahan N, Pires Damaso M, Osmanovic D, Reboul CF, Dunstone MA, Andrew PW, Lonnen R, Topf M, Saibil HR, Hoogenboom BW (2014) Stepwise visualization of membrane pore formation by suilysin, a bacterial cholesterol‐dependent cytolysin. elife 3: e04247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose‐Girma M, Erickson S, Dixit VM (2004) Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 430: 213–218 [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010) Caspase‐1‐induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ayres JS, Kofoed EM, Chavarria‐Smith J, Vance RE (2013) Recognition of bacteria by inflammasomes. Annu Rev Immunol 31: 73–106 [DOI] [PubMed] [Google Scholar]
- Muller DJ, Amrein M, Engel A (1997) Adsorption of biological molecules to a solid support for scanning probe microscopy. J Struct Biol 119: 172–188 [DOI] [PubMed] [Google Scholar]
- Mulvihill E, van Pee K, Mari SA, Muller DJ, Yildiz O (2015) Directly Observing the Lipid‐Dependent Self‐Assembly and Pore‐Forming Mechanism of the Cytolytic Toxin Listeriolysin O. Nano Lett 15: 6965–6973 [DOI] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez‐Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA et al (2013) The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453 [DOI] [PubMed] [Google Scholar]
- Op de Beeck K, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, Van Nassauw L, Van Tendeloo VF, Timmermans JP, Van Laer L (2011) The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis‐inducing protein. Eur J Hum Genet 19: 965–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517: 311–320 [DOI] [PubMed] [Google Scholar]
- Pfreundschuh M, Martinez‐Martin D, Mulvihill E, Wegmann S, Muller DJ (2014) Multiparametric high‐resolution imaging of native proteins by force‐distance curve‐based AFM. Nat Protoc 9: 1113–1130 [DOI] [PubMed] [Google Scholar]
- Podobnik M, Marchioretto M, Zanetti M, Bavdek A, Kisovec M, Cajnko MM, Lunelli L, Dalla Serra M, Anderluh G (2015) Plasticity of listeriolysin O pores and its regulation by pH and unique histidine. Sci Rep 5: 9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage P, Cheneval D, Chvei M, Graff P, Hemmig R, Heng R, Kocher HP, Mackenzie A, Memmert K, Revesz L, Wishart W (1995) Expression, refolding, and autocatalytic proteolytic processing of the interleukin‐1 beta‐converting enzyme precursor. J Biol Chem 270: 9378–9383 [DOI] [PubMed] [Google Scholar]
- Saeki N, Kim DH, Usui T, Aoyagi K, Tatsuta T, Aoki K, Yanagihara K, Tamura M, Mizushima H, Sakamoto H, Ogawa K, Ohki M, Shiroishi T, Yoshida T, Sasaki H (2007) GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF‐beta‐dependent apoptotic signalling. Oncogene 26: 6488–6498 [DOI] [PubMed] [Google Scholar]
- Scherrer R, Gerhardt P (1971) Molecular sieving by the Bacillus megaterium cell wall and protoplast. J Bacteriol 107: 718–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192 [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Sonnen AF, Plitzko JM, Gilbert RJ (2014) Incomplete pneumolysin oligomers form membrane pores. Open Biol 4: 140044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L, Quade B, Wang H, Sun L, Wang X, Rizo J (2014) A plug release mechanism for membrane permeation by MLKL. Structure 22: 1489–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654 [DOI] [PubMed] [Google Scholar]
- Tanaka S, Mizushina Y, Kato Y, Tamura M, Shiroishi T (2013) Functional conservation of Gsdma cluster genes specifically duplicated in the mouse genome. G3 (Bethesda) 3: 1843–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casano FJ, Chin J, Ding GJF, Egger LA, Gaffney EP, Limjuco G, Palyha OC, Raju SM et al (1992) A novel heterodimeric cysteine protease is required for interleukin‐1 beta processing in monocytes. Nature 356: 768–774 [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X (2014) Mixed lineage kinase domain‐like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133–146 [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A (2008) The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3