

Abstract
The enzyme 15-lipoxygenase-1 (15-LOX-1) plays a dual role in diseases with an inflammatory component. On one hand 15-LOX-1 plays a role in pro-inflammatory gene expression and on the other hand it has been shown to be involved in central nervous system (CNS) disorders by its ability to mediate oxidative stress and damage of mitochondrial membranes under hypoxic conditions. In order to further explore applications in the CNS, novel 15-LOX-1 inhibitors with favorable physicochemical properties need to be developed. Here, we present Substitution Oriented fragment Screening (SOS) in combination with Multi Component Chemistry (MCR) as an effective strategy to identify a diversely substituted small heterocyclic inhibitors for 15-LOX-1, denoted ThioLox, with physicochemical properties superior to previously identified inhibitors. Ex vivo biological evaluation in precision-cut lung slices (PCLS) showed inhibition of pro-inflammatory gene expression and in vitro studies on neuronal HT-22 cells showed a strong protection against glutamate toxicity for this 15-LOX-1 inhibitor. This provides a novel approach to identify novel small with favorable physicochemical properties for exploring 15-LOX-1 as a drug target in inflammatory diseases and neurodegeneration.
Keywords: 15-lipoxygenase, thiophene-based inhibitor, substitution oriented fragment screening, Multi Component Chemistry, inflammation, neurodegeneration
1. Introduction
Despite initial speculations that inflammation plays a minor role in central nervous system (CNS) disorders, most recent evidence shows the opposite. In various acute, chronic and psychiatric CNS disorders, pro-inflammatory mediators like cytokines, prostaglandins and leukotrienes, have been found to play pivotal roles.[1–4] In addition, elevated levels of IL-1, IL-6 and TNFα have been identified in brain tissue of patients with Alzheimer’s (AD)[1,5,6] and Parkinson’s (PD)[1,7] disease. Furthermore, several animal studies suggest a connection between IL-1 and stroke,[1] multiple sclerosis[1,6,8] and depression[1]. These findings have led to a search for novel therapeutic agents that can target inflammation in the CNS.[9]
One possible class of targets for anti-inflammatory therapy in the CNS are the Lipoxygenases (LOXs). LOXs are an important class of enzymes that play regulatory roles in the inflammatory process. Several lines of evidence suggest that 15-LOX-1 (also known as 12/15-LOX, leukocyte-type 12-LOX), could be a potential therapeutic target for various neurological diseases with an inflammatory component. This enzyme is widely expressed in the CNS, and its catalysis products are critical factors in the emergence of brain pathology.[10] Moreover, it has been shown that 15-LOX-1 is upregulated in mice following a stroke and the ensuing ischemia.[11] In addition, 15-LOX-1 has been linked to airway inflammation diseases[12–16] such as asthma, chronic obstructive pulmonary disease (COPD) and chronic bronchitis, and more recently in various CNS diseases[11,17–25] like Alzheimer’s and Parkinson’s diseases as well as stroke. Recent evidence shows that 15-LOX-1 plays conserved roles in the molecular mechanisms contributing to the pathophysiology of these diseases.
It is believed that 15-LOX-1 is involved in two different mechanistic pathways. In airway inflammation, it converts arachidonic acid into its corresponding lipid peroxides, which are further converted, in the presence of glutathione, to different inflammatory mediators (eoxins, 12-HETE and 15-HETE). Generation of the inflammatory mediators is followed by the production of pro-inflammatory cytokines such as IL-1, IL-6, IL-8 or IL-12.[26–31] While in the airway, 15-LOX-1 causes inflammation, in the CNS, it can trigger apoptosis. In CNS diseases, a similar mechanism appears to play a role. However, depletion of glutathione under oxidative stress conditions gives rise to another mechanism involving Bid activation that induces mitochondrion dependent cell death.[23] This provides a model for several inflammatory diseases in which 15-LOX-1 exhibits a key role in both the production of cytokines and the induction of apoptosis under oxidative stress conditions (Figure 1).
Figure 1.

The dual biological role of 15-LOX-1. In macrophages, after LPS/IFNγ stimulation, activation of cPLA2 yields the overproduction of AA. As a result, the conversion of AA from 15-LOX-1 leads to increased levels of lipid-peroxides. Then, the lipid-peroxides in presence of GSH, either can be reduce to 12- or 15-HETE or after a second conversion from 15-LOX-1 can follow the EX pathway. These formed metabolites cause inflammatory responses, while increased levels of pro-inflammatory cytokines such as IL-1, IL-6, IL-8 and IL-12 are also observed. (LPS, Lipopolysaccharide; IFNγ, Interferon gamma; cPLA2, Phospholipase A2; AA, arachidonic acid; GSH, glutathione; EX, eoxins). In adult neurons, glutamate promotes cell death by sustained activation of NMDA/AMPA receptors inducing excitotoxicity, while in immature and immortalized neurons high levels of extracellular glutamate initiate cell death via inhibition of the xCT antiporter. As a result, decreased GSH levels and the loss in GpX4 activity leads to enhanced 15-LOX-1 activity, and formation of reactive oxygen/nitrogen species and lipid peroxides. Downstream of ROS production, proteins such as Bid, Bax and Drp1 get activated and translocate to the mitochondria triggering mitochondrial dysfunction. Afterwards, mitochondrial release of apoptosis-inducing factor promotes large scale chromatin condensation, DNA fragmentation and subsequent cell death. (AIF, apoptosis-inducing factor; Bax, B-cell lymphoma protein-2-associated X protein; Drp1, dynamin-related protein; GSH, glutathione; LOX, lipoxygenenases; NMDA, N-Methyl-D-aspartate; ROS, reactive oxygen species; xCT, glutamate-cystine-antiporter).
Thus, 15-LOX-1 appears as an emerging drug target for the treatment of neurological diseases. Therefore, different 15-LOX-1 inhibitors, such as Baicalein or LOXBlock-1, have shown encouraging results in neuroprotective studies.[11,21–23] In addition, another 15-LOX-1 inhibitor, PD-146176, has been found to mitigate the effects of the AD phenotype.[19] However, many of these inhibitors suffer from low inhibitory potency and/or poor physicochemical properties, such as a high logP value, which limits their potential therapeutic value.
In particular for 15-LOX-1, it proved to be challenging to develop compact inhibitors with favorable physicochemical properties. A common drug discovery strategy is based on employing the screening of fragments, followed by hits optimization. Utilizing this approach for 15-LOX-1, yields very lipophilic molecules most likely due to the lipid character of the active site. Therefore, alternative approaches are needed to generate more compact and diversely substituted molecular scaffolds for this type of challenging drug targets. Towards this aim, we describe a substitution oriented screening (SOS) approach in which a focused compound collection of diversely substituted scaffolds is screened for inhibitors of a particular molecular target.[32] Application of this SOS approach on heterocycles that can be assembled by multicomponent reactions (MCR) enables the exploration of very compact molecules with a very diverse substitution pattern.
In this study, we report the application of our SOS approach with MCR chemistry for identification of novel thiophene-based inhibitors for 15-LOX-1 with the desired physicochemical properties for potential applications in CNS diseases with an inflammatory component. The most potent inhibitors demonstrated anti-inflammatory effects in precision-cut lung slices (PCLS) of mouse lung tissue, while it demonstrated neuroprotective effects in in vitro studies on glutamate-induced cytotoxicity of neuronal cells.
2. Results and Discussion
2.1. Initial screening and discovery of potent inhibitors
Having previously established a successful strategy based on SOS, we applied the same approach in combination with MCR chemistry for the discovery of unique substitution patterns on novel scaffolds (Figure 2). Sulfur containing heterocycles such as thiophenes, thiazoles and thiadiazoles appear to be good candidates for such an approach. In particular, thiophenes have been very successfully employed in the pharmaceutical industry for numerous targets against not only cancer, inflammation, immune system but also in CNS disorders (Figure S1). Hence, we screened a fragment collection consisting of organosulfur heterocycles, mostly thiophene derivatives with a diverse substitution pattern (Supplementary Library screening).
Figure 2.

The “discovery flow” of thiophene derivatives for 15-LOX-1 inhibition using the Substitution Oriented fragment Screening (SOS) approach. Screening of an organosulfur fragment collection enabled the identification of a Gewald multicomponent reaction product with a unique substitution pattern that was employed for further elucidation of Structure Activity Relationships (SAR).
The enzyme activity studies were performed, as previously described, using the UV absorbance of the 15-LOX-1 (234 nm) product after its enzymatic conversion from linoleic acid.[32–34] All the assays including the initial screening, IC50 measurements and enzyme kinetics, were done in a 96-well format. After the initial library screening, two hits were identified providing more than 70% inhibition of the enzyme activity at concentrations of 50 μM. The inhibitory potency of these hits, confirmed by determination of their IC50 values, proved to be 18.6 ± 6.0 μM and 20.6 ± 3.8 μM for compounds C2 and C6 respectively. The two hits are three-substituted thiophene derivatives with similar substitution patterns bearing a free amino group, an amide with different aliphatic chain and a phenyl ring on positions 2, 3 and 5 respectively. The specific substitution pattern can be accessed in a one single step by the Gewald three-component reaction.
2.2. Gewald thiophenes
These findings led us in a second screening of a specially designed Gewald thiophene library. A collection of about 40 2-amino thiophenes with diverse amide substitution patterns (aliphatic, aromatic or heterocyclic) on the 3-position as well as diverse substitution patterns on the 5-position was investigated. This enabled the identification of structure activity relationships for this novel scaffold. It was observed that the compounds (A9, B4, C2 and C3) with higher inhibitory potency at 50 μM have an amino group on the 2-position, an amide with an aliphatic chain on the 3-position and a phenyl group on the 5-position. Comparison of the compounds A9 with B8 or C6 with B6 clearly indicates the importance of the phenyl group on 5-position. In addition, the substitution of phenyl with benzyl group, causes a dramatic decrease in inhibitory potency, as can be recognized by comparing D9 and B4. Branched tails or (hetero)cycles in the 3-position are not present among the hits from this focused screening, supporting most likely the use of the aliphatic chain in that position. Inhibition around 60% was also observed from the compounds A2, C3 and D2. However, these compounds bearing into the 3-position an indole moiety which is possibly responsible for the inhibition, while it has been previously described in many 15-LOX inhibitors. The IC50’s were determined and compound A9 proved to be the most potent hit with an IC50 of 12.4 ± 0.9 μM. A9 exhibited almost 2-3 fold better inhibitory potency compared to the B4, C2 and C6 (IC50’s; 29.8 ± 3.68, 18.6 ± 6.0 and 20.6 ± 3.8 μM respectively).
2.3. Focused optimization
A focused optimization of the substitution pattern of the Gewald thiophene A9 was performed. The Gewald multicomponent reaction (Gewald-3CR) was employed, using different aldehydes or ketones (1), cyanoacetamides (2) and elementary sulfur as starting materials. The desired substituted 2-amino thiophenes were obtained after reflux overnight in EtOH with triethylamine (TEA) as base (Scheme 1). The 3-, 4- and 5-positions were varied with differently branched aliphatic and substituted aromatic substituents. Finally, variations of the 2-amino group were explored by acylation with different acyl chlorides.
Scheme 1.

General synthesis. Reagents and conditions: a) TEA, EtOH, reflux, overnight; b) TiCl4, TEA, CH2Cl2, 40°C, overnight; c) LiAlH4, THF, 0 °C to rt, overnight; d) DMP, CH2Cl2, rt, 2h; e) CuCN, Et2O, 0 °C to rt, overnight; f) TEA, EtOAc, reflux, 48h; g) Pyridine, acyl chloride, DMF, rt, overnight; h) DIPEA, CDI, DMF, reflux, 16h.
In our first series of experiments, in order to explore the lipophilic interactions, the tail length on the 3-position was varied keeping at the same time the other substituents constant. In our previous screening steps, only tails of 3 (C2 and C6) or 4 (A9) carbons were present in combination with a phenyl group in the 5-position. Cyanoacetamides with various tail lengths can conveniently be synthesized by methyl 2-cyanoacetate and a primary or secondary amine carrying the desired tail. Cyanoacetamides with the aliphatic tails of 5, 8 and 12 carbons as well as one with double tail of 4-carbon length were synthesized and then used in a Gewald-3CR.
IC50 measurements with the obtained products demonstrated a clear relationship between tail length and inhibitory potency (Table 1). As noted earlier, 3-carbon tails (C2 and C6) yielded less potent inhibitors than 4 carbon tails (A9). Results with longer tail length indicated that increasing the length is paralleled with decreasing inhibitory potency. Furthermore, introducing phenyl substituent in the aliphatic tail (3d) also caused an increase in IC50, as did by adding an additional 4-carbon tail to the amide (3e). In conclusion, this data indicate that a linear 4-carbon is the optimal substituent in the 3-position.
Table 1.
IC50 values of the Gewald products in which tail on the amide in the 3-position was varied.
| Compound | R1 | R2 | IC50 (µM) | |
|---|---|---|---|---|
 |
C6 |  |
H | 20.6 ± 3.8 |
| C2 |  |
H | 18.6 ± 6.0 | |
| A9 |  |
H | 12.4 ± 0.9 | |
| 3a |  |
H | 17.9 ± 5.6 | |
| 3b |  |
H | 41.5 ± 19.6 | |
| 3c |  |
H | 47.3 ± 14.8 | |
| 3d |  |
H | >50 | |
| 3e |  |
 |
>50 |
In the next series of experiments the substituent on the thiophene 5-position was varied, while keeping the 4 carbon tail in 3-position constant. Gewald compounds with various groups in 5-position were synthesized by choosing different aldehydes for the reaction. Lipophilic tails from 1 to 9 carbons, branched aliphatic tail as well as benzyl group were incorporated into the thiophene derivatives (Table 2). In addition, the phenyl group of A9 was halogen substituted with para-chloro (3l) and para-bromo (3m). The IC50 measurements of the compounds were subjected, showing again a clear structure-activity relationship between inhibitory potency and tail length. Compounds with small tails in 5-position were found to be very poor inhibitors while increasing of the tail length is paralleled with increasing the inhibitory potency with the compound 3k to be the most potent (IC50 = 15.2 ± 8.4 µM ). The compounds substituted in para position of the phenyl ring or benzyl group in thiophene 5-position were also found to be inactive (3l, 3m, 3n). Hence, the original substitution of A9 was found to be optimal while these finding also corroborate hypothesis that the phenyl is constrained in this direction when A9 binds in the active site.
Table 2.
IC50 values of the Gewald products with different substituent on the thiophene 5-position.
| Compound | R2 | IC50 (µM) | |
|---|---|---|---|
 |
3f |  |
>50 |
| 3g |  |
>50 | |
| 3h |  |
>50 | |
| 3i |  |
>50 | |
| 3j |  |
25.9 ± 7.3 | |
| 3k |  |
15.2 ± 8.4 | |
| A9 |  |
12.4 ± 0.9 | |
| 3l |  |
>50 | |
| 3m |  |
>50 | |
| 3n |  |
>50 |
In the last series of the optimization experiments, we explored the 4-position of the thiophene ring as well as the importance of the 2-amino group. Firstly, we introduced a methyl or butyl group in 4-position while keeping constant the rest optimized substitutions. Whereas the starting material for the methyl product (phenyl acetone) was commercially available, the ketone required for 4-n-butyl substitution was not. We utilized a reaction in which a cyanocuprate formed in situ from n-butyl lithium and cuprous cyanide, cleanly converts 2-phenylacetic acid into benzyl n-butyl ketone (Scheme 1). Next, three additional thiophenes were synthesized, using a two-step procedure including a Knoevenagel condensation followed by the Gewald reaction (Scheme 1). None of these compounds were able to achieve more than 20% reduction of enzymatic activity at 50 µM, meaning that their IC50 must lie above this concentration (Table 3). These results indicate that substitution of the 4-position is not a promising route for further exploration.
Table 3.
IC50 values of the compounds with different substituent on the thiophene 4- and 5-position.
| Compound | R2 | R3 | IC50 (µM) | |
|---|---|---|---|---|
 |
4a |  |
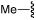 |
>50 |
| 4b |  |
 |
>50 | |
| 4c |  |
 |
>50 |
Secondly, in order to study the importance of the 2-amino group, acylation with different acyl chlorides was performed (Scheme 1). In three derivatives (5a, 5b and 5c) the amine group was acylated with linear tails of different lengths. Additionally, in compound 6 the amine group was acetylated using 1,1'-Carbonyldiimidazole (CDI), followed by ring closure at the 3N-position (Scheme 1). None of the compounds were better than A9, with the calculated IC50 to be for 5a and 5b around 20 µM, for the 5c to be 28,2 µM while for the compound 6 were found to be above 50 µM (Table 4). Although, there is no clear relationship between inhibitory potency and tail length in the thiophene 2-position, we can consider that elongation in the 2-position is not improving the potency.
Table 4.
IC50 values of the compounds with different aliphatic tails in amino group.
| Compound | R1 | R2 | IC50 (µM) | |
|---|---|---|---|---|
 |
5a |  |
H | 19.8 ± 5.0 |
| 5b |  |
H | 19.0 ± 6.8 | |
| 5c |  |
H | 28.2 ± 6.7 | |
 |
6 | - | - | >50 |
In brief, we can conclude that the optimum substitution pattern is present on the compound A9, which proved to be the most potent 15-LOX-1 inhibitor. In terms of inhibitory potency, the compounds 3a, 3k and A9 provided comparable IC50 values. However, the compound A9 has better physicochemical properties such as low molecular weight or logP value and so was chosen for further studies. We denote this inhibitor, ThioLox.
2.4. Enzyme kinetic analysis
The mechanism of 15-LOX-1 inhibition was established performing Michaelis-Menten enzyme kinetics analysis in presence of ThioLox (Figure 3A,B). The Lineweaver-Burk plot shows that the inhibitor causes an increase in the Km values, whereas the Vmax values remain constant (Table S2), indicating competitive inhibition. In addition, the binding affinities (Ki) of all the inhibitors can be calculated, using the Cheng-Prusoff equation. The Ki value of ThioLox was calculated to be 3.30 ± 0.24 µM.
Figure 3.

Steady-State kinetic characterization of human 15-lipoxygenase-1 (15-LOX-1) in the presence of different concentrations of compound ThioLox: A) Michaelis-Menten representation and B) Lineweaver-Burk representation.
2.5. Molecular modeling with ThioLox
In order to verify our findings and link the observed SAR to structural information, the competitive inhibitor ThioLox was docked in the active site of the enzyme. There is no crystal structure of human 15-LOX-1 available but as previously described due to the high sequence similarity (87%) rabbit 15-LOX (PDB: 1LOX) was used in the molecular modeling study.[32,33] The molecular modeling studies were performed in MOE software (2012.10) and highest scoring docking poses were chosen. The experiments were performed with rescoring model 1 London dG (refinement: forcefield) and rescoring 2: GBVI/WSA dG, followed by minimization energy (forcefield: MMFF94X; eps = r, cutoff {8,10}). This enabled the proposal of a binding configuration in which the sulfur and the 2-amino group interact with Glu357 forming two hydrogen bonds, one with the Glu357 backbone and one with its side chain. The carbonyl oxygen of the amide participates in a hydrogen bond with Gln548, while the phenyl group makes a π-π stacking interaction with His366 that is involved in the binding of the catalytic iron (Figure 4A,B).
Figure 4.

A) Binding of ThioLox in the active site of 15-LOX-1 after molecular modeling studies. B) 2D illustration of the interactions of ThioLox with the active site of the enzyme. C) Calculated properties (logP, PSA, MW, HBA and HBD) of 15-LOX-1 inhibitors. The grey area represents the desired properties for CNS-likeness and every line with different color represents a 15-LOX-1 inhibitor as mentioned in the legend. Our new inhibitor ThioLox, which is shown with the red line, fulfils all the criteria for CNS-likeness.
Further, the proposed binding of ThioLox is in line and can explain our SAR findings. Starting with the 5-position, the phenyl ring is highly constrained, explaining why para-substitution (3l, 3m) or exchange for a benzyl group (3n), cause loss of activity. Substitution of the phenyl for a short length linear tail up to 5-carbons (3f, 3g, 3h, 3i) was also observed that cause a loss of activity. This can be explained with the proposed binding pose, where the π-π stacking interaction of the phenyl His366 is lost. Longer tails in this position gave better IC50 values (3j, 3k), presumably because an increase in lipophilic interactions make up for the loss of π-π stacking (Figure S4). Next, the 3-position also seems to be constrained, explaining why longer tails (3a, 3b, 3c, 3d) or double tails (3e) were found to be less potent. The shorter tail like in case of C2 and C6 are not constrained, but cause loss of lipophilic interactions. Finally, the compounds 5a, 5b, 5c and 6 are found to be less potent probably because of the absence of one of the hydrogen bonds between the amine group and Glu357.
We note that 2-amino thiophenes frequently appear as hits in bioactivity screenings, which is considered to be disadvantageous.[35,36] Nevertheless, the 2-amino thiophene inhibitor ThioLox shows competitive inhibition thus indicating non-covalent interaction with the active site of the 15-LOX-1. In addition, a clear SAR profile has been identified that can be explained by molecular modeling. Altogether, this combined evidence clearly argues for ThioLox to be a valuable 15-LOX-1 inhibitor.
2.6. Ligand efficiency metrics and physicochemical properties
The SOS approach and subsequent optimization provided ThioLox as the most potent inhibitor for further investigation in biochemical studies towards applications in drug discovery projects. In drug discovery, the potential drugs should have suitable physicochemical properties in order to achieve an acceptable ADME-Tox (absorption, distribution, metabolism, excretion and toxicity) profile in vivo. Ligand efficiency metrics rendered into a generally accepted tool to estimate the value of lead compounds in this perspective.[37] Therefore, ligand efficiency metrics such as the ligand efficiency (LE) were calculated for ThioLox in comparison to previously described inhibitors (Table S3). The ligand efficiency metrics were calculated according to the Equation LE = (1.37/HA) x pKi, whereas HA represents the heavy atoms. Values above 0.3 kcal per mol are considered to be acceptable values for LE of drug candidates.[38] The LE value of ThioLox is 0.4 kcal per mol, which is among highest value reported together with our previously published inhibitor N247 and the known 15-LOX inhibitor PD-146176.
In addition to ligand efficiency metrics, criteria like the Lipinski’s ‘rule of five’ are important guidelines in drug discovery. The ‘rule of five’, which forms the basis for the concept of ‘drug-likeness’, makes allowance for the typical gain in size and lipophilicity during compound optimization.[39] However, especially for drugs being designed to target CNS diseases, the limits are even more rigorous due to the requirement to pass the blood-brain barrier (BBB). Towards this aim the putative properties have been described to be: i) logP bellow 3, ii) polar surface area (PSA) less than 90 Å2, iii) molecular weight (MW) bellow 450 Da, and iv) hydrogen bond acceptors (HBA) and hydrogen bond donors (HBD) less than 8 and 4, respectively.
To evaluate how these newly identified 15-LOX-1 inhibitors relate to previously described inhibitors, the calculated physicochemical properties were compared to values that should enable permeation of the BBB. ThioLox has a logP of 3, a MW of 274.38 Da, 3 HBA and HBD and PSA of 55.12 Å2, which are values that comply very well with the BBB permeability. The respective values for these physicochemical parameters were calculated for previously identified inhibitors such as N247, PD-146176, imidazole and pyrazole sulfamides and plotted in Figure 4C in comparison with the limits considered to enable activity in the CNS. Parameters indicating the lipophilicity, logP and the size, PSA and MW are in particular more favorable for ThioLox compared to previous identified inhibitors. These results show that our hit finding strategy provides a starting structure with favorable properties for applications in biochemical studies in drug discovery. The calculation of physicochemical properties for the inhibitors was performed using Marvin Sketch 15.6.29.
2.7. Gene expression
15-LOX-1 has been shown to play a key role in pro-inflammatory gene expression. To assess the influence of ThioLox on inflammatory signaling, an ex vivo model system for airway inflammation was applied. In this model, precision-cut lung slices (PCLS) were applied to provide a complex testing matrix with a relatively high level of similarity to the in vivo situation. The viability of PCLS upon ThioLox treatment was determined by measuring the release of lactate dehydrogenase (LDH) into the medium upon incubation with the inhibitor. The LDH release demonstrated that ThioLox is not affecting the PCLS viability at concentrations up to 50 μM (Figure S5). Upon inflammatory stimulation using LPS the effect of ThioLox on the expression of the pro-inflammatory gene IL-1β, IL-6, IL-8, IL-12b, TNFα and inducible nitric oxide synthase (iNOS) was investigated. Since the cellular concentration of free fatty acids (preferred substrate of mammalian LOXs) is rather low, we supplemented the culture medium with linoleic acid (C18:Δ2, n – 6) which is the most abundant polyenoic fatty acid in mammalian cells and serves as a natural substrate for 15-LOX-1. Linoleic acid itself did not alter the basal gene expression levels in PCLS (data not shown).
ThioLox provided almost 50% inhibition of the expression of the pro-inflammatory genes IL-1β, IL-6, IL-8, IL-12b, TNFα and iNOS at 50 μM (Figure 5). For the pro-inflammatory genes IL-6 and IL-12b, significant downregulation was observed at concentrations as low as 5 μM. This can be explained by the fact that these genes are direct related to 15-LOX-1 activity.[40–44] This is in line with a previous study reporting that 15-LOX-1 regulates the production of IL-12b in macrophages.[44]
Figure 5.

Effects of 15-LOX-1 inhibition on interleukin expression in precision-cut lung slices (PCLS). Stimulated PCLS were subjected to ThioLox in combination with 10 μM linoleic acid for 20 h and stimulated with 10 ng/ml LPS for the last 4 h. Subsequently, PCLS were lysed and gene expression was assessed by RT-qPCR and expressed as fold change compared to control (LPS/linoleic acid-treated) group. Data are presented as mean values ± SD of 4-6 independent experiments. * p < 0.05; *** p < 0.001 compared to control.
The efficiency of ThioLox as anti-inflammatory agent in ex vivo studies was investigated. The effect of ThioLox on the expression of the pro-inflammatory genes is remarkable. The importance of interleukin production has been previously highlighted in lung tissue of COPD patients after LPS stimulation.[45] In addition, it has been shown that 15-LOX plays an important role in the progression of inflammatory arthritis, while affecting the matrix metalloproteinase (MMP) expression and being involved in the inflammatory action induced by TNFα and IL-1β.[46] It has been also found that 15-LOX shows significant association with IL-6 expression and alters the expression of IL-1β and TNFα, cytokines both intimately associated with the acute inflammatory response.[40] Furthermore, it has been proven that 15-LOX regulates the production of IL-12 and it was established that 15-LOX is also a critical mediator of the chronic type 1 inflammatory response.[43,44] Taking together, the effects we observed for treatment with ThioLox are in agreement with the hypothesis that 15-LOX-1 inhibition provides an anti-inflammatory effect. In fact, there is a growing body of evidence that 15-LOX-1 plays an important role in regulation of these pro-inflammatory genes. This sets the stage for further investigation and optimization of ThioLox for in vivo applications towards the development of novel therapeutics for inflammatory lung diseases.
2.8. Neuroprotective activity
In CNS diseases, 15-LOX-1 plays a role in apoptosis of neuronal cells under hypoxic conditions. In view of its favorable physicochemical properties and its inhibitory effect on pro-inflammatory gene expression in PCLS, the neuroprotective properties of ThioLox were investigated in a model of cellular oxidative stress employing neuronal cells. In the CNS, activation of 15-LOX-1 mediates lipid peroxidation that subsequently triggers cell death in a variety of brain cell types. Besides cortical/hippocampal neurons, oligodendroglial and brain endothelial cells are also subject to 15-LOX-1 mediated toxicity.[47,48] To mimic these conditions, in our studies we used HT-22 immortalized hippocampal cells, and cell death was induced by glutamate exposure and subsequent 15-LOX-1 activation.
In HT-22 cells, lipid peroxidation rapidly accumulates in the cytosol within 6-8 h following glutamate challenge and to a much larger extent as a second more robust increase within 10-14 h after the glutamate exposure.[23] To examine in a cellular system the potential inhibitory properties of newly developed chemical compounds on the formation of lipid peroxidation, we tested the 15-LOX-1 inhibitors in our current model system of glutamate-induced lipid peroxidation. The analysis of the fluorescent dye BODIPY (4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid) by fluorescence-activated cell sorting (FACS) revealed that the newly developed 15-LOX-1 inhibitor ThioLox and the conventional 15-LOX-1inhibitor PD-146176 significantly attenuated the boost of lipid peroxides detected at 16 h after initiation of the glutamate challenge (Figure 6). Notably, as illustrated in Figure 6, a 10 µM concentration of ThioLox perfectly matched the inhibitory property of lipid peroxide formation in a cellular system and the enzyme inhibition studies in vitro.
Figure 6.

15-LOX-1inhibitors, ThioLox prevent lipid peroxidation and mitochondrial superoxide formation in neuronal cells. A) Representative scatter plots of BODIPY green fluorescence staining measured by FACS assay of neuronal HT-22 cells challenged with glutamate in the presence and absence of classical 15-LOX-1 inhibitor, PD-146176 (1 μM) and recently identified 15-LOX-1 inhibitor ThioLox (10 μM). B) Analysis of FACS measurements of BODIPY staining. C) Representative plots of MitoSOX red fluorescence staining of neuronal cells. D) Analysis of MitoSOX staining by FACS assay of neuronal HT-22 cells treated with glutamate and PD-146176 or ThioLox. Results are represented as mean ± SD of n=3, (3 independent experiments). *** p < 0.001; compared to control, ### p < 0.001 compared to glutamate-treated cells.
The cytotoxic effect of 15-LOX-1 has been associated with the oxidized polyunsaturated fatty acids 12- and 15-hydroxy-eicosatetraenoic acid (12- and 15-HETE) and 12- and 15-hydroperoxy-eicosatetraenoic acid (12- and 15-HPETE).[49] Besides 12/15-LOX metabolites, 12- and 15-H(P)ETE, 15-LOX-1 alone mediates direct damage at the level of mitochondria as demonstrated in red blood precursor cells, where mitochondria are eliminated during the physiological process of maturation.[50] Although 15-LOX-1 is required for lipid peroxide formation, 15-LOX-1 may also promote mitochondrial membrane depolarization and cytochrome c release in intact mitochondria isolated from HT-22 cells.[51] Therefore, in our studies we investigated whether the newly developed 15-LOX-1 inhibitor ThioLox attained the property of preserving mitochondrial function. As detected by the fluorescent dye MitoSOX and subsequent FACS analysis (Figure 6C,D), the glutamate-induced formation of mitochondrial superoxides was largely attenuated by the 15-LOX-1inhibitor, suggesting that ThioLox can prevent both lipid peroxidation and mitochondrial dysfunction.
Most importantly, ThioLox significantly prevented glutamate-induced cell death in a concentration-dependent manner (Figure 7A,B). Real-time recording of cell impedance by the xCELLigence system of HT-22 cells showed that cell death followed 9-10 h after the initiation of the glutamate challenge and it was concluded within additional 2-4 h (Figure 7).[52] Notably, ThioLox fully protected HT-22 cells against glutamate toxicity at a concentration of 5-10 µM and significantly reduced neuronal cell death (Figure 7A,B). Comparable results were obtained by using the conventional 15-LOX-1 inhibitor PD-146176 (Figure 7A,B). Next, we determined the protective time window of 15-LOX-1-dependent lethal oxidative stress in neuronal cell death and added ThioLox at different time points between 2 and 8 h after onset of the glutamate treatment. HT-22 cells were protected against glutamate toxicity even when ThioLox was added up to 8 h after the glutamate challenge (Figure 7C,D), indicating that beyond that time point glutamate-induced cell death proceeded too far for protection by attenuation of either lipid peroxidation or mitochondrial dysfunction. Our studies identified a therapeutic time window for the interference with 15-LOX-1 activation in neuronal cell death that may be relevant for therapeutic strategies in related neurological diseases. Previously, 15-LOX-1 has been proposed as a potential target for neuroprotective strategies in stroke treatment based on data on genetic 12/15-LOX deletion and on pharmacological inhibition of 15-LOX-1 with PD-146176 that significantly reduced the infarct size in a mouse model of transient cerebral ischemia.[11,23] Targeting 15-LOX-1 with our newly developed inhibitor in HT-22 cells, we suggest that ThioLox can prevent both lipid peroxidation and mitochondrial dysfunction. In conclusion, ThioLox was proved to be a key regulator in glutamate-induced oxidative stress, process highly relevant for neurodegenerative diseases and acute neurological disorders such as ischemic stroke or brain trauma.
Figure 7.

Inhibition of 15-LOX-1 prevents neuronal cell death. A) Real-time impedance measurements of neuronal HT-22 cells challenged with glutamate in the absence or presence of different concentrations (5, 10 and 20μM) of the novel 15-LOX-1 inhibitor ThioLox or classical 15-LOX-1 inhibitor PD-146176 (PD, 1μM). Normalized cell index was performed prior the glutamate application, as indicated at time=0 in the graph. B) MTT analysis of neuronal cell death initiated by glutamate challenge. Different concentrations of 15-LOX-1 inhibitor ThioLox were applied at the same time as glutamate. C) Real-time impedance measurements of neuronal cells challenged with glutamate and subsequent application (2, 4, 6 and 8h) of 15-LOX-1 inhibitor ThioLox at a concentration of 10μM. D) MTT analysis of neuronal cells damaged by toxic concentration of glutamate. 15-LOX-1 inhibitor ThioLox (5 and 10μM) was applied 2, 4, 6 and 8 h following glutamate challenge exposure. Data are represented as mean ± SD, n= 6. * p < 0.05; *** p < 0.001 compared to control, ### p < 0.001 compared to glutamate-treated cells.
3. Conclusions
There is an increasing interest in 15-LOX-1 as a therapeutic target for various inflammatory and neurological diseases with an inflammatory component. However, currently identified inhibitors have physicochemical properties that are unfavorable for applications in drug discovery for CNS diseases. In this study, we utilized a substitution oriented screening (SOS) approach with MCR chemistry to identify the thiophene-based 15-LOX-1 inhibitor, ThioLox. The enzyme kinetic analysis as well as the molecular modeling studies showed competitive inhibition. ThioLox was calculated to have a Ki value of 3.30 μM, very good ligand efficiency metric but also the disired physicochemical properties. This inhibitor was evaluated in ex-vivo studies in precision-cut lung slices (PCLS) of mouse lung tissue showing a strong anti-inflammatory effect. In addition, considering the acceptable physiochemical properties, neuroprotective studies were performed in HT-22 neuronal cells showing strong protection. Identification of ThioLox provides a starting point to target 15-LOX-1 with a competitive inhibitor with limited size and lipophilicity. These findings point out the way toward the development of therapeutic agents against diseases with an inflammatory component, such as asthma and COPD as well as neurological disorders like Alzheimer’s and Parkinson’s disease.
4. Experimental Section
4.1. Chemistry
4.1.1. General
The solvent and reagents were purchased from Sigma-Aldrich and Acros chemicals and were used without further purification unless otherwise noted. Reactions were monitored by thin layer chromatography (TLC). Merck silica gel 60 F254 plates were used and spots were detected under UV light or after staining with potassium permanganate for the non UV-active compounds. MP Ecochrom silica 32-63, 60Å was used for flash column chromatography. 1H NMR (500 MHz) and 13C NMR (126 MHz) spectra were recorded with a Bruker Avance 4-channel NMR Spectrometer with TXI probe. Chemical shifts were referenced to the residual proton and carbon signal of the deuterated solvent CDCl3: δ = 7.26 ppm (1H) and 77.05 ppm (13C). The following abbreviations were used for spin multiplicity: s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet, dd = double of doublets, ddd = double of doublet of doublets, m = multiplet. Fourier Transform Mass Spectrometry (FTMS) was recorded on an Orbitrap XL Hybrid Ion Trap-Orbitrap Mass Spectrometer to give high-resolution mass spectra (HRMS). All the compounds were analyzed by Waters Investigator Semi-prep 15 SFC-MS instrument confirming purity ≥ 95%.
The thiophene compounds A1 to A12, B1 to B12, C1 to C12 and D1 to D12 were previously fully characterized.[53]
4.1.2. Synthetic procedure 1: Gewald 3 Component Reaction
To a stirred solution of the cyanoacetamide (2.0 mmol) in EtOH (3 mL), the ketone or aldehyde (2.0 mmol) was added and dissolved. Then, triethylamine (Et3N) (2.0 mmol) and S8 (0.25 mmol) were added and the reaction mixture was refluxed overnight. Afterwards, it was diluted with EtOAc (25 mL) and washed with water (2 x 25 mL) and brine (2 x 25 mL). The combined organic layers were dried over MgSO4, filtrated and the solvent was removed under reduced pressure. Further purification by flash chromatography, with heptane:EtOAc 3:1 (v/v) as eluent, was performed when needed.
4.1.3. Synthetic procedure 2: Cyanoacetamide preparation
To a stirred solution solution of methyl 2-cyanoacetate (5.0 mmol) in EtOAc (3 mL), the amine (5.0 mmol) was added, followed by Et3N (5.0 mmol). The reaction mixture was refluxed for 48 h and then diluted with EtOAc (25 mL) and washed with water (2 x 50 mL) and brine (2 x 50 mL). The combined organic layers where dried over MgSO4, filtered and the solvent was removed under reduced pressure.
4.1.4. Synthetic procedure 3: Reduction of carboxylic acid with LiAlH4
In a 3-neck flask, LiAlH4 (8.0 mmol) was dissolved in anhydrous tetrahydrofuran (THF) (10 mL), under nitrogen atmosphere at 0 °C. A solution of phenyl acetic acid (4.0 mmol) in anhydrous THF (10 mL), was added dropwise to the reaction mixture over 20 min, followed by addition of 5 mL of anhydrous THF. The mixture was allowed to warm to rt, after which the reaction was quenched with EtOAc (50 mL) and water (50 mL). The mixture was washed with water (3 x 50 mL) and the organic layer was dried over MgSO4, filtered and the solvent was removed under reduced pressure.
4.1.5. Synthetic procedure 4: Primary alcohol oxidation with Dess-Martin reagent
To a stirred solution of the Dess-Martin reagent (DMP) (1.5 mmol) in CH2Cl2 (10 mL) under nitrogen, the alcohol (1.0 mmol) was added dropwise. The reaction mixture was stirred for 2 h and subsequently quenched with saturated aqueous sodium thiosulfate (20 mL). After 15 minutes of stirring the layers were separated, the organic layer was washed with water (50 mL) and brine (2 x 50 mL) and the aqueous layer was washed with EtOAc (2 x 25 mL). The combined organic layers were dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The thus obtained semisolid was triturated with 10% EtOAc in hexanes. The solid was removed by filtration. The filtrate was concentrated under reduced pressure to yield the final product.
4.1.6. Synthetic procedure 5: Knoevenagel Condensation with titanium activated carbonyl
The cyanoacetamide (1.0 mmol) and the benzyl ketone (1.0 mmol) were dissolved in THF (1 mL). To the stirred solution, TiCl4 (2 mL of 1 M in CH2Cl2, 2.0 mmol) was added dropwise, followed by addition of Et3N (0.3 mL). The reaction mixture was stirred overnight at 40 °C, followed by the addition of 1.2 N HCl (25 mL) and then extracted with EtOAc (3 x 20 mL). The combined organic layers were washed with 2 M NaOH (25 mL), dried over MgSO4, filtrated and the solvent was removed under reduced pressure.
4.1.7. Synthetic procedure 6: n-Butyl ketones using butyl lithium and copper (I) cyanide
To a dried flask, CuCN (10.0 mmol) and then Et2O (10 mL) were added under nitrogen atmosphere. The reaction mixture was cooled to 0 °C and n-BuLi (8 mL of 2.5 M in hexanes, 20.0 mmol) was added dropwise. The mixture was stirred for 5 minutes at 0 °C, after which the carboxylic acid (2.0 mmol) was added dropwise at approximately 1 mL/min. The reaction mixture was allowed to warm to rt and left stirring overnight. The mixture was quenched with an aqueous saturated NH4Cl (10 mL) solution, then diluted with CH2Cl2 and washed with water (2 x 20 mL) and brine (2 x 20 mL). The combined organic layers were dried over MgSO4, filtered and the solvent was removed under reduced pressure.
4.1.8. Synthetic procedure 7: Gewald reaction with the Knoevenagel condensation product
To a stirred solution of the cyanoacetamide (1.0 mmol) in EtOH (3 mL), S8 (1.0 mmol) and Et3N (2.0 mmol) were added and dissolved. The reaction mixture was refluxed overnight. Then, the mixture was diluted with EtOAc (25 mL) and washed with water (2 x 50 mL) and brine (2 x 50 mL). The organic layers were dried over MgSO4, filtrated and the solvent was removed under reduced pressure. The product was obtained after flash chromatography with heptane:EtOAc 3:1 (v/v) as eluent.
4.1.9. Synthetic procedure 8: Acylation of ThioLox
To a stirred solution of the thiophene (1.0 mmol) in dimethylformamide (DMF) (4 mL), acyl chloride (2.0 mmol) and pyridine (2 mmol) were added and dissolved. The reaction mixture was stirred overnight at rt. Then, the mixture was diluted with CH2Cl2 (20 mL) and was washed with 1N HCl (20 mL), NaHCO3 (20 mL), water (20 mL) and brine (2 x 20 mL). The organic layer was dried over MgSO4, filtrated and the solvent was removed under reduced pressure.
4.1.10. Synthetic procedure 9: Synthesis of compound 6
To a stirred solution of the thiophene (1.0 mmol) in DMF (10 mL), N,N-diisopropylethylamine (DIPEA) (5 mmol) were added and dissolved. After 10 min,1,1'-Carbonyldiimidazole (CDI) (3.0 mmol) was added and the reaction mixture was refluxed for 16h. Then, the solvent was removed under reduce pressure, EtOAc (25 mL) was added and washed with water (25 mL) and brine (25 mL). The organic layer was dried over MgSO4, filtrated and the solvent was removed under reduced pressure. The product was obtained after flash chromatography with heptane:EtOAc 3:1 (v/v) as eluent.
4.1.11. 2-amino-N-butyl-5-phenylthiophene-3-carboxamide (ThioLox)
The product was obtained using synthetic procedure 1. Brown solid, yield 64%. 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 7.3 Hz, 2H), 7.32 (t, J = 7.6 Hz, 2H), 7.20 (t, J = 7.4 Hz, 1H), 6.95 (s, 1H), 6.21 (br s, 2H), 5.78 (t, J = 5.8 Hz, 1H), 3.39 (q, J = 7.1 Hz, 2H), 1.61 - 1.55 (m, 2H), 1.44 - 1.37 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 160.2, 134.0, (2x)128.9, 126.6, 125.3, (2x)124.7, 118.1, 109.9, 39.1, 32.0, 20.2, 13.8. HRMS, calculated for C15H19N2OS [M+H]+ : 275.12126, found 275.12116.
4.1.12. 2-amino-N-pentyl-5-phenylthiophene-3-carboxamide (3a)
The product was obtained using synthetic procedure 1. Brown solid, yield 54%. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 7.6 Hz, 2H), 7.32 (t, J = 7.8 Hz, 2H), 7.20 (t, J = 7.4 Hz, 1H), 6.94 (s, 1H), 6.19 (br s, 2H), 5.72 (t, J = 6.1 Hz, 1H), 3.38 (q, J = 6.9 Hz, 2H), 1.63 - 1.57 (m, 2H), 1.36 (m, 4H), 0.92 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 160.2, 134.0, (2x)128.8, 128.6, 126.6, (2x)124.6, 118.1, 109.9, 39.3, 29.7, 29.2, 22.4, 14.0. HRMS, calculated for C16H20N2OS [M+H]+: 289.13691, found 289.13681.
4.1.13. 2-amino-N-octyl-5-phenylthiophene-3-carboxamide (3b)
The product was obtained using synthetic procedure 1. Brown solid, yield 79%. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 7.3 Hz, 2H), 7.33 (t, J = 7.8 Hz, 2H), 7.21 (t, J = 7.3 Hz, 1H), 6.93 (s, 1H), 5.71 (br s, 1H), 3.31 - 3.43 (m, 2H), 1.65 - 1.55 (m, 2H), 1.37 - 1.25 (m, 10H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 160.1, 134.0, (2x)128.9, 128.3, (2x)126.6, 124.7, 118.1, 109.9, 39.4, 31.8, 30.0, 29.4, 29.3, 27.1, 22.7, 14.1. HRMS, calculated for C19H26N2OS [M+H]+: 331.18386, found 331.18370.
4.1.14. 2-amino-N-dodecyl-5-phenylthiophene-3-carboxamide (3c)
The product was obtained from 2-cyano-N-dodecylacetamide using synthetic procedure 1. Brown solid, yield 73%. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 7.4 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 5.71 (s, 1H), 3.38 (t, J = 7.7 Hz, 4H), 1.56 - 1.60 (m, 2H), 1.34 - 1.21(m, 19H), 0.88 (t, J = 7.4 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 166.0, 160.4, 134.3, (2x)129.1, 126.9, 125.5, (2x)124.9, 118.2, 39.6, 32.1, 30.2, 29.9, 29.8, 29.6, 27.3, 22.9, 14.3. HRMS, calculated for C23H34N2OS [M+H]+: 387.23918, found 387.23920.
4.1.15. 2-amino-5-phenyl-N-(4-phenylbutyl)thiophene-3-carboxamide (3d)
The product was obtained using synthetic procedure 1 without column chromatography purification. Brown solid, yield 34%. 1H NMR (500 MHz, CDCl3) δ 7.42 (d, J = 7.3 Hz, 2H), 7.34 - 7.27 (m, 4H), 7.21 - 7.17 (m, 4H), 6.92 (s, 1H), 6.19 (s, 2H), 5.77 - 5.70 (m, 1H), 3.41 (q, J = 6.7 Hz, 2H), 2.66 (t, J = 7.4 Hz, 2H), 1.75 - 1.70 (m, 2H), 1.66 - 1.61 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.8, 160.2, 142.2, 134.0, (2x)128.8, (2x)128.4, 128.4, (2x)128.3, 126.6, 125.8, (2x)124.6, 118.1, 109.8, 39.1, 35.5, 29.5, 28.8. HRMS, calculated for C21H22N2OS [M+H]+: 351.15256, found 351.15243.
4.1.16. N,N-dibutyl-2-cyanoacetamide (HP191)
The product was obtained using synthetic procedure 2. Brown oil, yield 30%. 1H NMR (500 MHz, CDCl3) δ 3.47 (s, 2H), 3.32 (t, J = 7.7 Hz, 2H), 3.20 (t, J = 7.8 Hz, 2H), 1.56 - 1.51 (m, 4H), 1.35 - 1.31 (m, 4H), 0.9 - 0.89 (m, 6H).
4.1.17. 2-amino-N,N-dibutyl-5-phenylthiophene-3-carboxamide (3e)
The product was obtained from N,N-dibutyl-2-cyanoacetamide (HP191) using synthetic procedure 1. Brown solid, yield 40%. 1H NMR (500 MHz, CDCl3) δ 7.40 (d, J = 7.4 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.20 (t, J = 7.4 Hz, 1H), 6.93 (s, 1H), 3.43 (t, J = 7.7 Hz, 4H), 1.66 - 1.60 (m, 4H), 1.37 - 1.33 (m, 4H), 0.95 (t, J = 7.4 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 167.6, 158.0, 134.3, (2x)128.9, 126.4, 125.3, (2x)124.6, 120.7, 108.7, 33.4, 30.5, 20.2, 13.9. HRMS, calculated for C19H26N2OS [M+H]+: 331.18386, found 331.18389.
4.1.18. 2-amino-N-butyl-5-methylthiophene-3-carboxamide (3f)
The product was obtained using synthetic procedure 1. Brown solid, yield 20%. 1H NMR (500 MHz, CDCl3) δ 6.30 (s, 1H), 5.88 (s, 2H), 5.52 (s, 1H), 3.33 (q, J = 6.7 Hz, 2H), 2.25 (s, 3H), 1.55 - 1.51 (m, 2H), 1.39 - 1.34 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 158.9, 121.6, 119.6, 108.6, 38.9, 32.0, 20.2, 15.0, 13.8. HRMS, calculated for C10H16N2OS [M+H]+: 213.10561, found 213.10539.
4.1.19. 2-amino-N-butyl-5-ethylthiophene-3-carboxamide (3g)
The product was obtained using synthetic procedure 1. Brown solid, yield 30%. 1H NMR (500 MHz, CDCl3) δ 6.35 (s, 1H), 5.92 (s, 2H), 5.63 (s, 1H), 3.34 (q, J = 6.7 Hz, 2H), 2.61 (q, J = 7.5 Hz, 2H), 1.57 - 1.53 (m, 2H), 1.42 - 1.32 (m, 2H), 1.21 (t, J = 7.5 Hz, 3H), 0.93 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.9, 158.7, 129.2, 117.7, 108.3, 38.9, 32.1, 23.2, 20.2, 15.5, 13.8. HRMS, calculated for C11H18N2OS [M+H]+: 227.12126, found 227.1213
4.1.20. 2-amino-N-butyl-5-propylthiophene-3-carboxamide (3h)
The product was obtained using synthetic procedure 1. Brown solid, yield 29%. 1H NMR (500 MHz, CDCl3) δ 6.34 (s, 1H), 5.91 (s, 2H), 5.57 (s, 1H), 3.35 (q, J = 6.7 Hz, 2H), 2.56 (t, J = 7.5, 2H), 1.61 - 1.54 (m, 4H), 1.42 - 1.35 (m, 2H), 0.94 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 165.86, 158.8, 127.5, 118.6, 108.4, 38.9, 32.1, 31.9, 24.5, 20.2, 13.9, 13.6. HRMS, calculated for C12H20N2OS [M+H]+: 241.13691, found 241.13689.
4.1.21. 2-amino-N-butyl-5-pentylthiophene-3-carboxamide (3i)
The product was obtained using synthetic procedure 1. Brown solid, yield 30%. 1H NMR (500 MHz, CDCl3) δ 6.33 (s, 1H), 5.91 (s, 2H), 5.56 (s, 1H), 3.35 (q, J = 6.7 Hz, 2H), 2.58 (t, J = 7.5 Hz, 2H), 1.61 - 1.56 (m, 4H), 1.40 - 1.32 (m, 6H), 0.94 (t, J = 7.3 Hz, 3H), 0.89 (t, J = 6.9, 3H). 13C NMR (126 MHz, CDCl3) δ 165.9, 158.8, 127.8, 118.5, 108.4, 38.9, 32.1, 31.2, 30.9, 29.9, 22.4, 20.2, 14.0, 13.8. HRMS, calculated for C14H24N2OS [M+H]+: 269.16821, found 269.16821.
4.1.22. 2-amino-N-butyl-5-(6-methylhept-5-en-2-yl)thiophene-3-carboxamide (3j)
The product was obtained using synthetic procedure 1. Brown oil, yield 26%. 1H NMR (500 MHz, CDCl3) δ 6.33 (s, 1H), 5.92 (s, 2H), 5.58 (s, 1H), 5.08 (t, J = 7.0 Hz, 1H), 3.35, (q, J = 6.7 Hz, 2H), 2.80 - 2.72 (m, 1H), 1.96 (q, J = 7.6 Hz, 2H), 1.68 (s, 3H), 1.57 (s, 3H), 1.56 - 1.50 (m, 4H), 1.42 - 1.36 (m, 2H), 1.22 (d, J = 6.8 Hz, 3H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.9, 158.5, 133.8, 131.9, 124.0, 117.2, 108.1, 38.9, 38.7, 34.9, 32.1, 25.8, 25.8, 22.7, 20.2, 17.8, 13.9. HRMS, calculated for C17H28N2OS [M+H]+: 309.19951, found 309.19949.
4.1.23. 2-amino-N-butyl-5-(non-8-en-1-yl)thiophene-3-carboxamide (3k)
The product was obtained using synthetic procedure 1. Brown solid, yield 30%. 1H NMR (500 MHz, CDCl3) δ 6.33 (s, 1H), 5.91 (s, 2H), 5.80 (ddt, J = 17.2, 10.3, 6.7 Hz, 1H), 5.58 (t, J = 5.9 Hz, 1H), 4.99 (dd, J = 1.4, 17.1 Hz, 1H), 4.93 (dd, J = 1.3, 10.2 Hz, 1H), 3.35 (q, J = 6.8, 2H), 2.57 (t, J = 7.5 Hz, 2H), 2.04 (q, J = 7.1, 2H), 1.58 - 1.52 (m, 4H), 1.43 - 1.21 (m, 10H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.9, 158.8, 139.1, 127.7, (2x)118.5, (2x)114.2, 108.4, 38.9, 33.8, 32.1, 31.2, 29.9, 29.2, 29.0, 28.9, 28.9, 20.2, 13.8. HRMS, calculated for C18H30N2OS [M+H]+ : 323.21516 found 323.21515.
4.1.24. 2-(4-chlorophenyl)ethan-1-ol (HP129)
The product was obtained from 2-(4-chlorophenyl)acetic acid using synthetic procedure 3. Yellow oil, yield 80%. 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 3.85 (t, J = 6.5 Hz, 2H), 2.84 (t, J = 6.5 Hz, 2H), 1.46 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 137.0, 132.3, 130.4, 128.7, 63.5, 38.5.
4.1.25. 2-(4-chlorophenyl)acetaldehyde (HP151)
The product was obtained from 2-(4-chlorophenyl)ethan-1-ol (HP129) using synthetic procedure 4. Yellow solid, yield 87%. 1H NMR (500 MHz, CDCl3) δ 9.75 (t, J = 2.2 Hz, 1H), 7.34 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 3.69 (d, J = 2.2 Hz, 2H).[54]
4.1.26. 2-amino-N-butyl-5-(4-chlorophenyl)thiophene-3-carboxamide (3l)
The product was obtained from 2-(4-chlorophenyl)acetaldehyde (HP151) using synthetic procedure 1. Yellow solid, yield 31%. 1H NMR (500 MHz, CDCl3) δ 7.34 (d, J = 8.6 Hz, 2H), 7.29 (d, J = 8.7 Hz, 2H), 6.91 (s, 1H), 5.69 (br s, 1H), 3.39 (t, J = 7.2 Hz, 3H), 1.62 - 1.55 (m, 2H), 1.45 - 1.38 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.7, 160.2, 132.6, 132.2, 131.9, (2x)129.0, 126.1, (2x)125.8, 118.5, 39.1, 32.0, 20.2, 13.8. HRMS, calculated for C15H17ClN2OS [M+H]+: 309.08229, found 309.08222.
4.1.27. 2-(4-bromophenyl)ethan-1-ol (HP149)
The product was obtained from 2-(4-bromophenyl)acetic acid using synthetic procedure 3. Yellow oil, yield 78%. 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 8.2 Hz, 2H), 7.11 (d, J = 8.3 Hz, 2H), 3.85 (t, J = 6.5 Hz, 2H), 2.83 (t, J = 6.5 Hz, 2H), 1.46 (s, 1H).[55]
4.1.28. 2-(4-bromophenyl)acetaldehyde (HP145)
The product was obtained from 2-(4-bromophenyl)ethan-1-ol (HP149) using synthetic procedure 4. Yellow solid, yield 71%. 1H NMR (500 MHz, CDCl3) δ 9.74 (t, J = 2.1 Hz, 1H), 7.50 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.3 Hz, 2H), 3.67 (d, J = 2.2 Hz, 2H).[54]
4.1.29. 2-amino-5-(4-bromophenyl)-N-butylthiophene-3-carboxamide (3m)
The product was obtained from 2-(4-bromophenyl)acetaldehyde (HP145) using synthetic procedure 1. Brown solid, yield 23%. 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 8.6 Hz, 2H), 7.31 (d, J = 8.6 Hz, 2H), 6.95 (s, 1H), 5.71 (s, 1H), 3.42 (t, J = 7.2 Hz, 2H), 1.64 - 1.57 (m, 2H), 1.46 -1.42 (m, 2H), 0.99 (t, J = 7.4, 3H). 13C NMR (126 MHz, CDCl3) δ 165.7, 160.1, 133.0, (2x)131.9, 128.9, (2x)126.1, 124.7, 120.2, 118.7, 39.1, 32.0, 20.2, 13.8. HRMS, calculated for C15H17BrN2OS [M+H]+: 353.03177, found 353.03169.
4.1.30. 2-amino-5-benzyl-N-butylthiophene-3-carboxamide (3n)
The product was obtained using synthetic procedure 1 without column chromatography purification. Brown solid, yield 74%. 1H NMR (500 MHz, CDCl3) δ 7.34 - 7.28 (m, 2H), 7.24 - 7.21 (m, 3H), 6.36 (s, 1H), 5.95 (br s, 2H), 5.56 (s, 1H), 3.91 (s, 2H), 3.33 (q, J = 5.8 Hz, 2H), 1.56 - 1.50 (m, 2H), 1.40 - 1.35 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.8, 159.7, 139.8, (2x)128.6, (2x)128.5, 126.6, 125.8, 119.9, 108.3, 38.9, 36.0, 32.0, 20.2, 13.8. HRMS, calculated for C16H20N2OS [M+H]+: 289.13691, found 289.13692.
4.1.31. N-butyl-2-cyano-3-methyl-4-phenylbut-2-enamide (HP175)
Using synthetic procedure 5, the product was obtained as a mixture of E:Z-isomers (2:1). Brown oil, yield 56%. major isomer: 1H NMR (500 MHz, CDCl3) δ 7.35 - 7.27 (m, 3H), 7.23 (m, 2H), 6.20 (s, 1H), 4.17 (s, 2H), 3.37 (q, J = 6.7, Hz, 2H), 2.13 (s, 3H), 1.60 - 1.52 (m, 2H), 1.41 - 1.36 (m, 2H) , 0.95 (t, J = 7.4 Hz, 3H).
4.1.32. 2-amino-N-butyl-4-methyl-5-phenylthiophene-3-carboxamide (4a)
The product was obtained from N-butyl-2-cyano-3-methyl-4-phenylbut-2-enamide (HP175) using synthetic procedure 7. Brown oil, yield 64%. 1H NMR (500 MHz, CDCl3) δ 7.40 - 7.27 (m, 5H), 5.81 (br s, 1H), 3.42 (q, J = 6.7 Hz, 2H), 2.34 (s, 3H), 1.62 - 1.56 (m, 2H), 1.45 - 1.39 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.7, 159.2, 134.2, (2x)129.7, (2x)128.5, 127.4, 127.0, 121.1, 111.8, 39.1, 31.9, 20.3, 16.2, 13.8. HRMS, calculated for C16H20N2OS [M+H]+ : 289.13691 , found 289.13688.
4.1.33. 1-(4-chlorophenyl)hexan-2-one (HP179)
The product was obtained using synthetic procedure 6. Green oil, yield 54%. 1H NMR (500 MHz, CDCl3) δ 7.29 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 3.65 (s, 2H), 2.44 (t, J = 7.4 Hz, 2H), 1.57 - 1.50 (m, 2H), 1.29 - 1.25 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H).[56]
4.1.34. N-butyl-3-(4-chlorobenzyl)-2-cyanohept-2-enamide (HP181)
The product was obtained from 1-(4-chlorophenyl)hexan-2-one (HP179) as a mixture of E:Z-isomers (70:30) using synthetic procedure 5. Brown oil, yield 65%. 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 6.20 (s, 1H), 4.15 (s, 2H), 3.33 (m, 2H), 2.38 (t, J = 7.9 Hz, 2H), 1.56 - 1.48 (m, 4H), 1.37 - 1.35 (m, 4H), 0.93 - 0.89 (m, 6H).
4.1.35. 2-amino-N,4-dibutyl-5-(4-chlorophenyl)thiophene-3-carboxamide (4b)
The product was obtained from N-butyl-3-(4-chlorobenzyl)-2-cyanohept-2-enamide (HP181) using synthetic procedure 7. Brown solid, yield 32%. 1H NMR (500 MHz, CDCl3) δ 7.34 (d, J = 8.5 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 5.83 (br s, 1H), 3.42 (q, J = 6.7 Hz, 2H), 2.61 (t, J = 8.0 Hz, 2H), 1.61 - 1.54 (m, 2H), 1.51 - 1.46 (m, 2H), 1.43 - 1.39 (m, 2H), 1.23 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.5, 158.6, 133.9, 133.1, 132.9, (2x)131.2, (2x)128.6, 120.0, 111.7, 39.1, 32.8, 31.8, 28.7, 22.6, 20.3, 13.8, 13.7. HRMS, calculated for C19H25ClN2OS [M+H]+: 365.14489, found 365.14478.
4.1.36. 1-phenylhexan-2-one (HP183)
The product was obtained using synthetic procedure 6 (4.0 mmol scale). Yellow oil, yield 47%. 1H NMR (500 MHz, CDCl3) δ 7.35 - 7.27 (m, 3H), 7.21 - 7.19 (m, 2H), 3.68 (s, 2H), 2.44 (t, J = 7.4 Hz, 2H), 1.56 - 1.53 (m, 2H), 1.30 - 1.23 (m 2H), 0.86 (t, J = 7.4 Hz, 3H).[57]
4.1.37. 3-benzyl-N-butyl-2-cyanohept-2-enamide (HP195)
The product was obtained from 1-phenylhexan-2-one (HP183) using synthetic procedure 5 as a mixture of E:Z-isomers (70:30). Brown oil, yield 90%. 1H NMR (500 MHz, CDCl3) δ 7.31 - 7.27 (m, 3H), 7.24 - 7.20 (m, 2H), 6.22 (s, 1H), 4.18 (s, 2H), 3.38 - 3.33 (m, 2H), 2.40 (t, J = 7.9 Hz, 2H), 1.55 - 1.49 (m, 4H), 1.39 - 1.33 (m, 4H), 0.95 - 0.88 (m, 6H).
4.1.38. 2-amino-N,4-dibutyl-5-phenylthiophene-3-carboxamide (4c)
The product was obtained from 3-benzyl-N-butyl-2-cyanohept-2-enamide (HP195) using synthetic procedure 7. Brown solid, yield 36%. 1H NMR (500 MHz, CDCl3) δ 7.40 - 7.27 (m, 5H), 5.87 (s, 1H), 3.43 (q, J = 6.7 Hz, 2H), 2.64 (t, J = 8.0 Hz, 2H), 1.62 - 1.54 (m, 2H), 1.54 - 1.47 (m, 2H), 1.45 - 1.39 (m, 2H), 1.26 - 1.22 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 166.6, 158.5, 134.4, 133.4, (2x)130.0, (2x)128.5, 127.2, 121.6, 111.6, 39.1, 32.9, 31.8, 28.8, 22.6, 20.3, 13.8, 13.7. HRMS, calculated for C19H26N2OS [M+H]+: 331.18386, found 331.18364.
4.1.39. 2-acetamido-N-butyl-5-phenylthiophene-3-carboxamide (5a)
The product was obtained using synthetic procedure 8. Brown solid, yield 75%. 1H NMR (500 MHz, CDCl3) δ 11.95 (s, 1H), 7.56 (d, J = 7.3 Hz, 2H), 7.37 (t, J = 7.6 Hz, 2H), 7.28 (d, J = 7.3 Hz, 1H), 7.03 (s, 1H), 3.44 (q, J = 7.1 Hz, 2H), 2.28 (s, 1H), 1.66 - 1.60 (m, 2H), 1.47 – 1.40 (m, 2H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 167.1, 165.3, 145.8, 134.0, 133.6, 128.8, 127.3, 125.2, 116.1, 115.3, 39.3, 31.7, 23.4, 20.1, 13.7. HRMS, calculated for C17H21N2O2S [M+H]+: 317.13183, found 317.13165
4.1.40. N-butyl-2-butyramido-5-phenylthiophene-3-carboxamide (5b)
The product was obtained using synthetic procedure 8. Brown solid, yield 74%. 1H NMR (500 MHz, CDCl3) δ 12.01 (s, 1H), 7.49 (d, J = 7.3 Hz, 2H), 7.30 (t, J = 7.6 Hz, 2H), 7.22 (m, 1H), 6.45 (s, 1H), 3.41 (q, J = 6.8 Hz, 2H), 2.44 (t, J = 7.3 Hz, 2H), 1.80 - 1.73 (m, 2H), 1.60 – 1.57 (m, 2H), 1.43 – 1.36 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.4, 165.4, 145.9, 134.0, 133.7, 128.9, 127.3, 125.3, 116.1, 115.3, 39.3, 38.6, 31.7, 20.1, 18.8, 13.7, 13.7. HRMS, calculated for C19H25N2O2S [M+H]+: 345.16313, found 345.16299
4.1.41. N-butyl-2-octanamido-5-phenylthiophene-3-carboxamide (5c)
The product was obtained using synthetic procedure 8. Brown solid, yield 76%. 1H NMR (500 MHz, CDCl3) δ 12.02 (s, 1H), 7.50 (d, J = 7.3 Hz, 2H), 7.31 (t, J = 7.6 Hz, 2H), 7.27 (s, 1H), 7.22 (t, J = 7.8 Hz, 2H), 6.61 (t, J = 6.3 Hz, 1H), 3.44 (q, J = 7.22 Hz, 2H), 2.47 (t, J = 7.6 Hz, 2H), 2.35 (t, J = 7.6 Hz, 2H), 1.77 - 1.71 (m, 2H), 1.65 – 1.61 (m, 2H), 1.45 – 1.39 (m, 2H), 1.29 – 1.23 (m, 8H), 0.95 (t, J = 7.5 Hz, 3H), 0.89 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 170.6, 165.5, 145.9, 133.9, 133.7, 128.8, 127.3, 125.2, 116.2, 115.3, 39.4, 36.7, 31.6, 29.1, 29.0, 28.9, 25.3, 24.7, 22.5, 20.1, 14.0, 13.7. HRMS, calculated for C23H33N2O2S [M+H]+: 401.22573, found 401.22552
4.1.42. 3-butyl-6-phenylthieno[2,3-d]pyrimidine-2,4(1H,3H)-dione (6)
The product was obtained using synthetic procedure 9. Brown solid, yield 70%. 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 7.7 Hz, 2H), 7.49 (s, 1H), 7.40 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.2 Hz, 1H), 4.04 (t, J = 7.6 Hz, 2H), 1.72 - 1.66 (m, 2H), 1.47 – 1.40 (m, 2H), 0.98 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 159.1, 151.1, 149.7, 134.2, 133.0, 128.9, 127.7, 125.2, 117.7, 116.1, 40.4, 29.8, 20.1, 13.7. HRMS, calculated for C16H17N2O2S [M+H]+: 301.10053, found 301.10037
4.2. Gene expression
4.2.1. Animals
C57bl/6 male mice (weight 20-25 g; age 8-10 weeks) were purchased from Harlan (Zeist, the Netherlands). Animals were maintained on mouse chow and tap water ad libitum in a humidity- and temperature-controlled room at 24°C with a 12 h light/dark cycle. All experiments were performed according to national guidelines and upon approval of the experimental procedures by the local Animal Care and Use committee of Groningen University, DEC number 6962A. Mice were randomly assigned to the experiments.
4.2.2. Precision-cut lung slices
Mouse precision-cut lung slices (PCLS) were prepared as previously described for guinea pig with the following modifications [58]. Male mice were anesthetized by subcutaneous injection of ketamin (75 mg/kg, Alfasan, Woerden, the Netherlands) and dexdomitor (0.5 mg/kg, Orion Pharma, Mechelen, Belgium). Subsequently, the trachea was cannulated and the animal was exsanguinated by cutting the jugular vein, after which the lungs were filled trough the cannula with 1.5 ml low melting-point agarose solution (1.5% final concentration (Gerbu Biotechnik GmbH, Wieblingen, Germany) in CaCl2 (0.9 mM), MgSO4 (0.4 mM), KCl (2.7 mM), NaCl (58.2 mM), NaH2PO4 (0.6 mM), glucose (8.4 mM), NaHCO3 (13 mM), Hepes (12.6 mM, Gibco® by Life Technologies, Bleiswijk, the Netherlands), sodium pyruvate (0.5 mM, GE Healthcare Life Sciences, Eindhoven, the Netherlands), glutamine (1 mM, Gibco® by Life Technologies), MEM-amino acid mixture (1:50, Gibco® by Life Technologies) and MEM-vitamins mixture (1:100, Gibco® by Life Technologies), pH 7.2). The lungs were placed on ice for 15 min to solidify the agarose for slicing. The lobes were separated and tissue cores were prepared of the individual lobes, after which the lobes were sliced at a thickness of 250 μm in medium composed of CaCl2 (1.8 mM), MgSO4 (0.8 mM), KCl (5.4 mM), NaCl (116.4 mM), NaH2PO4 (1.2 mM), glucose (16.7 mM), NaHCO3 (26.1 mM), Hepes (25.2 mM), pH 7.2, using a tissue slicer (Compresstome™ VF-300 microtome, Precisionary Instruments, San Jose, CA, USA). Tissue slices were incubated at 37 °C in a humid atmosphere under 5% CO2/95% air. In order to remove the agarose and cell debris from the tissue, slices were washed every 30 min (four times in total) in medium composed of CaCl2 (1.8 mM), MgSO4 (0.8 mM), KCl (5.4 mM), NaCl (116.4 mM), NaH2PO4 (1.2 mM), glucose (16.7 mM), NaHCO3 (26.1 mM), Hepes (25.2 mM), sodium pyruvate (1 mM), glutamine (2 mM), MEM-amino acid mixture (1:50), MEM-vitamins mixture (1:100), penicillin (100 U/ml, Gibco® by Life Technologies) and streptomycin (100 μg/ml, Gibco® by Life Technologies), pH 7.2. Chemicals to prepare the media described above were obtained from Sigma-Aldrich (Zwijndrecht, the Netherlands) unless stated otherwise, and were of analytical grade.
4.2.3. Treatment of lung slices
PCLS were incubated in Dulbecco’s Modification of Eagle’s Medium (DMEM, Gibco® by Life Technologies) supplemented with sodium pyruvate (1 mM), MEM non-essential amino acid mixture (1:100, Gibco® by Life Technologies), gentamycin (45 μg/ml, Gibco® by Life Technologies), penicillin (100 U/ml), streptomycin (100 μg/ml) and amphotericin B (1.5 μg/ml, Gibco® by Life Technologies). Slices were cultured at 37 °C in a humidified atmosphere under 5% CO2/95% air in 12-well tissue culture plates (Costar Europe, Badhoevedorp, the Netherlands), using 3 slices per well. Slices were treated with the LOX inhibitor (LOXi) ThioLox at a final concentration of 1-500 nM for 20 h, and were co-incubated with 10 μM Linoleic acid (L1376; Sigma Aldrich). The last 4 h of the experiments tissue slices were stimulated with 10 ng/ml lipopolysaccharide (LPS, Escherichia coli, serotype 0111:B4; Sigma-Aldrich) and 10 ng/ml interferon gamma (IFNγ, cat.#315-05; PeproTech, Hamburg, Germany).
4.2.4. Assessment of tissue viability using lactate dehydrogenase
To assess the viability of the PCLS subjected to increasing concentrations ThioLox, the amount of lactate dehydrogenase (LDH) released from the tissue slices into the incubation medium was analyzed. Maximal LDH release was determined by lysing 3 slices with 1% Triton X-100 for 30 min at 37 °C at the start of the experiments. Supernatants were stored at -80 °C. LDH release was determined using an assay form Roche Diagnostics (Mannheim, Germany), and was measured using a Hitachi automatic analyzer (Modular Analytics, Roche Diagnostics). LDH release from the PCLS into the incubation medium was plotted relative to maximal LDH release.
4.2.5. Gene expression analysis by RT-qPCR
PCLS were washed twice with ice-cold DPBS (Gibco® by Life Technologies) and total RNA was isolated from PCLS using the Maxwell® 16 LEV simplyRNA Tissue Kit (Promega), according to the protocol of the manufacturer. RNA integrity was determined by 28S/18S ratio detection on an agarose gel, which was consistently found intact. For gene expression analysis, RNA was reverse transcribed using a reverse transcription kit (Promega). Subsequently, 10 ng of cDNA was applied for each real-time PCR, which was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Nieuwerkerk a/d IJssel, the Netherlands). The primers for TNFa (Mm00443258_m1), iNOS (Mm00440502_m1), IL-1β (Mm00434228_m1), IL-6 (Mm00446190_m1), IL-8 (Mm00441263_m1), IL-12 (Mm00434174_m1) and GAPDH (Mm99999915_g1) were purchased as Assay-on-Demand (Applied Biosystems). For each sample, the real-time PCR reactions were performed in duplicate or triplicate and the averages of the obtained Ct values were used for further calculations. Gene expression levels were normalized to the expression of the reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was not influenced by the experimental conditions resulting in the ΔCt value. Gene expression levels were calculated by the comparative Ct method (2-ΔΔCt).[59]
4.2.6. Statistical analysis
Statistical analysis of the results was performed by a two-tailed unpaired Student's t-test, assuming equal variances to compare two replicate groups. Analysis of differences between multiple replicate groups was analyzed with one-way ANOVA followed by Tukey post hoc analysis. p values <0.05 were considered to be significant. Data were analyzed with GraphPad Prism (GraphPad software 5.00, San Diego, CA, USA).
4.3. Neuroprotective studies
4.3.1. Cell viability measurement
HT-22 cells were cultivated in Dulbecco’s modified Eagle Medium (DMEM; PAA, Cölbe, Germany) supplemented with 10% fetal calf serum (FCS; PAA Cölbe, Germany), 100U/mL penicillin, 100µg/mL streptomycin and 2mM glutamine (D10; Invitrogen, Karlsruhe, Germany) at 37°C and 5% CO2. HT-22 cells were seeded into a 96well plate (8x103 cells per well) and incubated overnight. On the next day, cells were treated with D10 (control) or D10 containing 4mM glutamate in the presence or absence of 5/10µM ThioLox or 1µM PD146176. For post-treatment, 5µM or 10µM ThioLox were added to the corresponding wells 2h, 4h, 6h and 8h after initiating cell damage with glutamate. To generate a dose response curve, HT-22 cells were treated with different ThioLox concentrations (1µM, 2µM, 5µM, 10µM, 20µM). After 14-16h of incubation, cell viability was assessed by staining with 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at a final concentration of 0.5mg/mL for 1h at 37°C. Afterwards, the medium was removed and the plate was stored at -80°C for a minimum of 1h. Then, the resulting formazan was dissolved in 70µL of DMSO for 1h at 37°C. Absorbance was measured at 570nm using the FluoStar OPTIMA (BMG Labtech, Offenbach, Germany) with background subtraction at 630nm. Cell viability was calculated relative to control cells. Three independent experiments were performed with 6-8 replicates per condition.
4.3.2. Real-time cell viability measurement
HT-22 cells were seeded into a 96well plate containing 8x103 cells per well. After overnight incubation, cells were treated with D10 (control) or D10 containing 4mM glutamate in the presence or absence of different ThioLox concentrations (1µM, 2µM, 5µM, 10µM, 20µM) or 1µM PD146176. For post-treatment, 10µM ThioLox was added to the corresponding wells 2h, 4h, 6h and 8h after initiating cell damage with glutamate. Cell viability was analyzed in real-time for 16-20h using the xCELLigence impedance system (Roche, Munich, Germany).
4.3.3. Lipid peroxidation
HT-22 cells were seeded into a 24well plate containing 5x104 cells per well. After overnight incubation, cells were treated with D10 (control) or D10 containing 4mM glutamate in the presence or absence of 10µM ThioLox or 1µM PD146176 for 6-7h or 14-16h, respectively. Lipid peroxidation was detected by staining with BODIPY 581/591 C11 (Invitrogen, Karlsruhe, Germany) at a final concentration of 2µM for 1h at 37°C. The shift in fluorescence from red to green was analyzed by fluorescence-activated cell sorting (FACS) using the Guava Easy Cite 6-2L system (Merck Millipore, Darmstadt, Germany) by excitation at 488nm. At least three independent experiments were performed with three replicates per condition.
4.3.4. Mitochondrial superoxide (ROS) formation
HT-22 cells were seeded into a 24well plate containing 5x104 cells per well. After overnight incubation, cells were treated with D10 (control) or D10 containing 4mM glutamate in the presence or absence of 10µM ThioLox or 1µM PD146176 for 14-16h. Mitochondrial superoxides were detected by staining with MitoSOX (Invitrogen, Karlsruhe, Germany) at a final concentration of 2.5µM for 30min at 37°C. Fluorescence intensity was analyzed by fluorescence-activated cell sorting (FACS) using the Guava Easy Cite 6-2L system (Merck Millipore, Darmstadt, Germany) by excitation at 488nm. At least three independent experiments were performed with three replicates per condition.
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version, at…… NMR full spectra of the compound, enzyme kinetic and inhibition as well as molecular modeling data were reported.
Acknowledgments
We acknowledge the European Research Counsel for providing an ERC starting grant (309782) and the Netherlands Organisation for Scientific Research (NWO) for providing a VIDI grant (723.012.005) to F.J.D. We acknowledge the COST action ‘biomimetic radical chemistry’ CM1201. The work (C.N and A.D) was financially supported from the NIH (1R01GM097082-01) and by Innovative Medicines Initiative (grant agreement n° 115489). We acknowledge Prof. dr. Reinoud Gosens (Department of Molecular Pharmacology, University of Groningen) and Petra E. van der Wouden for their support with ex vivo experiments. We also acknowledge T. Holman (University of California, Santa Cruz) for providing the human 15-LOX-1 plasmid.
Abbreviations
- DMF
dimethylformamide
- EtOAc
ethyl acetate
- EtOH
ethanol
- Et2O
diethyl ether
- TEA
trimethylamine
- rt
room temperature
- DMP
Dess-Martin reagent
- DIPEA
N,N-Diisopropylethylamine
- CDI
1,1'-Carbonyldiimidazole
Footnotes
Author Contributions
F.J.D. designed the experiments and supervised the studies. N.E. and H.P. designed and performed the synthesis, enzyme inhibition and kinetic studies as well as the molecular modelling. C.G.N. and A.D. designed and contributed to the synthesis. N.G.J.L. performed the ex vivo studies. B.H. and A.D. performed the in vitro neuroprotective studies. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- [1].Lucas S-M, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2009;147:S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Phillis JW, Horrocks La, Farooqui Aa. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- [3].McMahon E, Bailey S, Miller S. CNS dendritic cells: Critical participants in CNS inflammation? Neurochem Int. 2006;49:195–203. doi: 10.1016/j.neuint.2006.04.004. [DOI] [PubMed] [Google Scholar]
- [4].Ubogu EE, Cossoy MB, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27:48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- [5].Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Weiner HL, Selkoe DJ. Inflammation and therapeutic vaccination in CNS diseases. Nature. 2002;420:879–884. doi: 10.1038/nature01325. [DOI] [PubMed] [Google Scholar]
- [7].Nagatsu T, Sawada M. Inflammatory Process in Parkinson’s Disease: Role for Cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- [8].Herz J, Zipp F, Siffrin V. Neurodegeneration in autoimmune CNS inflammation. Exp Neurol. 2010;225:9–17. doi: 10.1016/j.expneurol.2009.11.019. [DOI] [PubMed] [Google Scholar]
- [9].Pangalos MN, Schechter LE, Hurko O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat Rev Drug Discov. 2007;6:521–532. doi: 10.1038/nrd2094. [DOI] [PubMed] [Google Scholar]
- [10].Xu J, Zhang Y, Xiao Y, Ma S, Liu Q, Dang S, Jin M, Shi Y, Wan B. Inhibition of 12/15-lipoxygenase by baicalein induces microglia PPARβ/δ: a potential therapeutic role for CNS autoimmune disease. Cell Death Dis. 2013;4:e569. doi: 10.1038/cddis.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].van Leyen K, Kim HY, Lee S-R, Jin G, Arai K, Lo EH. Baicalein and 12/15-Lipoxygenase in the Ischemic Brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- [12].Zhao J, O’Donnell VB, Balzar S, St. Croix CM, Trudeau JB, Wenzel SE. 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc Natl Acad Sci U S A. 2011;108:14246–14251. doi: 10.1073/pnas.1018075108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mabalirajan U, Rehman R, Ahmad T, Kumar S, Leishangthem GD, Singh S, Dinda AK, Biswal S, Agrawal A, Ghosh B. 12/15-Lipoxygenase Expressed in Non-Epithelial Cells Causes Airway Epithelial Injury in Asthma. Sci Rep. 2013;3:1540. doi: 10.1038/srep01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mabalirajan U, Rehman R, Ahmad T, Kumar S, Singh S, Leishangthem GD, Aich J, Kumar M, Khanna K, Singh VP, Dinda AK, et al. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Sci Rep. 2013;3:1349. doi: 10.1038/srep01349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lindley AR, Crapster-Pregont M, Liu Y, Kuperman DA. 12/15-Lipoxygenase Is an Interleukin-13 and Interferon-γ Counterregulated-Mediator of Allergic Airway Inflammation. Mediators Inflamm. 2010;2010:727305. doi: 10.1155/2010/727305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Haeggström JZ, Funk CD. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- [17].van Leyen K. Lipoxygenase: An Emerging Target for Stroke Therapy. CNS Neurol Disord Drug Targets. 2013 doi: 10.2174/18715273112119990053. http://www.ncbi.nlm.nih.gov/pubmed/23394536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Giannopoulos PF, Joshi YB, Chu J, Praticò D. The 12-15-lipoxygenase is a modulator of Alzheimer’s-related tau pathology in vivo. Aging Cell. 2013;12:1082–1090. doi: 10.1111/acel.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Joshi YB, Giannopoulos PF, Praticò D. The 12/15-lipoxygenase as an emerging therapeutic target for Alzheimer’s disease. Trends Pharmacol Sci. 2015;36:181–186. doi: 10.1016/j.tips.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Praticò D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson Ja, Trojanowski JQ, Lee VM-Y. 12/15-Lipoxygenase Is Increased in Alzheimer’s Disease. Am J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fiedorowicz A, Car H, Prokopiuk S, Sacharzewska E, Żendzian M, Kowal K. Inhibition of 12/15-LOX activity and ceramide pattern in the murine brain. Prog Heal Sci. 2013;3:33–38. [Google Scholar]
- [22].van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg Pa, Holman TR, Lo EH. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res. 2008;86:904–909. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tobaben S, Grohm J, Seiler A, Conrad M, Plesnila N, Culmsee C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011;18:282–292. doi: 10.1038/cdd.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Succol F, Praticò D. A role for 12/15 lipoxygenase in the amyloid beta precursor protein metabolism. J Neurochem. 2007;103:380–387. doi: 10.1111/j.1471-4159.2007.04742.x. [DOI] [PubMed] [Google Scholar]
- [25].Chu J, Li J-G, Giannopoulos PF, Blass BE, Childers W, Abou-Gharbia M, Praticò D. Pharmacologic blockade of 12/15-lipoxygenase ameliorates memory deficits, Aβ and tau neuropathology in the triple-transgenic mice. Mol Psychiatry. 2015:1–10. doi: 10.1038/mp.2014.170. [DOI] [PubMed] [Google Scholar]
- [26].Feltenmark S, Gautam N, Brunnstrom A, Griffiths W, Backman L, Edenius C, Lindbom L, Bjorkholm M, Claesson H-E. Eoxins are proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase-1 pathway in human eosinophils and mast cells. Proc Natl Acad Sci. 2008;105:680–685. doi: 10.1073/pnas.0710127105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sachs-Olsen C, Sanak M, Lang AM, Gielicz A, Mowinckel P, Lødrup Carlsen KC, Carlsen K-H, Szczeklik A. Eoxins: A new inflammatory pathway in childhood asthma. J Allergy Clin Immunol. 2010;126:859–867.e9. doi: 10.1016/j.jaci.2010.07.015. [DOI] [PubMed] [Google Scholar]
- [28].Zhao J, Maskrey B, Balzar S, Chibana K, Mustovich A, Hu H, Trudeau JB, O’Donnell V, Wenzel SE. Interleukin-13–induced MUC5AC Is Regulated by 15-Lipoxygenase 1 Pathway in Human Bronchial Epithelial Cells. Am J Respir Crit Care Med. 2009;179:782–790. doi: 10.1164/rccm.200811-1744OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Profita M, Sala A, Riccobono L, Paternò A, Mirabella A, Bonanno A, Guerrera D, Pace E, Bonsignore G, Bousquet J, Vignola AM. 15-Lipoxygenase expression and 15(S)-hydroxyeicoisatetraenoic acid release and reincorporation in induced sputum of asthmatic subjects. J Allergy Clin Immunol. 2000;105:711–716. doi: 10.1067/mai.2000.105122. [DOI] [PubMed] [Google Scholar]
- [30].Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, Wenzel SE. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. 2002;32:1558–1565. doi: 10.1046/j.1365-2222.2002.01477.x. [DOI] [PubMed] [Google Scholar]
- [31].Zarbock A, DiStasi MR, Smith E, Sanders JM, Kronke G, Harry BL, von Vietinghoff S, Buscher K, Nadler JL, Ley K. Improved Survival and Reduced Vascular Permeability by Eliminating or Blocking 12/15-Lipoxygenase in Mouse Models of Acute Lung Injury (ALI) J Immunol. 2009;183:4715–4722. doi: 10.4049/jimmunol.0802592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Eleftheriadis N, Neochoritis CG, Leus NGJ, van der Wouden PE, Dömling A, Dekker FJ. Rational Development of a Potent 15-Lipoxygenase-1 Inhibitor with in Vitro and ex Vivo Anti-inflammatory Properties. J Med Chem. 2015;58:7850–7862. doi: 10.1021/acs.jmedchem.5b01121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Eleftheriadis N, Thee S, te Biesebeek J, van der Wouden P, Baas B-J, Dekker FJ. Identification of 6-benzyloxysalicylates as a novel class of inhibitors of 15-lipoxygenase-1. Eur J Med Chem. 2015;94:265–275. doi: 10.1016/j.ejmech.2015.03.007. [DOI] [PubMed] [Google Scholar]
- [34].Traven K, Eleftheriadis N, Seršen S, Kljun J, Bezenšek J, Stanovnik B, Turel I, Dekker FJ. Ruthenium complexes as inhibitors of 15-lipoxygenase-1. Polyhedron. 2015;101:306–313. doi: 10.1016/j.poly.2015.09.019. [DOI] [Google Scholar]
- [35].Huth JR, Mendoza R, Olejniczak ET, Johnson RW, Cothron DA, Liu Y, Lerner CG, Chen J, Hajduk PJ. ALARM NMR: A rapid and robust experimental method to detect reactive false positives in biochemical screens. J Am Chem Soc. 2005;127:217–224. doi: 10.1021/ja0455547. [DOI] [PubMed] [Google Scholar]
- [36].Devine SM, Mulcair MD, Debono CO, Leung EWW, Nissink JWM, Lim SS, Chandrashekaran IR, Vazirani M, Mohanty B, Simpson JS, Baell JB, et al. Promiscuous 2-aminothiazoles (PrATs): A frequent hitting scaffold. J Med Chem. 2015;58:1205–1214. doi: 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- [37].Abad-Zapatero C, Metz J. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10 doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- [38].Hopkins AL, Keserü GM, Leeson PD, Rees DC, Reynolds CH. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov. 2014;13:105–21. doi: 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- [39].Reichel A. The role of blood-brain barrier studies in the pharmaceutical industry. Curr Drug Metab. 2006;7:183–203. doi: 10.2174/138920006775541525. [DOI] [PubMed] [Google Scholar]
- [40].Fairfax BP, Vannberg FO, Radhakrishnan J, Hakonarson H, Keating BJ, Hill AVS, Knight JC. An integrated expression phenotype mapping approach defines common variants in LEP, ALOX15 and CAPNS1 associated with induction of IL-6. Hum Mol Genet. 2009;19:720–730. doi: 10.1093/hmg/ddp530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ibrahim AS, Tawfik AM, Hussein KA, Elshafey S, Markand S, Rizk N, Duh EJ, Smith SB, Al-Shabrawey M. Pigment epithelium-derived factor inhibits retinal microvascular dysfunction induced by 12/15-lipoxygenase-derived eicosanoids. Biochim Biophys Acta. 2015;1851:290–298. doi: 10.1016/j.bbalip.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mao F, Xu M, Zuo X, Yu J, Xu W, Moussalli MJ, Elias E, Li HS, Watowich SS, Shureiqi I. 15-Lipoxygenase-1 suppression of colitis-associated colon cancer through inhibition of the IL-6/STAT3 signaling pathway. FASEB J. 2015;29:2359–2370. doi: 10.1096/fj.14-264515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Middleton MK, Zukas AM, Rubinstein T, Kinder M, Wilson EH, Zhu P, Blair Ia, Hunter Ca, Puré E. 12/15-Lipoxygenase-Dependent Myeloid Production of Interleukin-12 Is Essential for Resistance To Chronic Toxoplasmosis. Infect Immun. 2009;77:5690–700. doi: 10.1128/IAI.00560-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Middleton MK, Rubinstein T, Pure E. Cellular and Molecular Mechanisms of the Selective Regulation of IL-12 Production by 12/15-Lipoxygenase. J Immunol. 2006;176:265–274. doi: 10.4049/jimmunol.176.1.265. [DOI] [PubMed] [Google Scholar]
- [45].Hackett TL, Holloway R, Holgate ST, Warner JA. Dynamics of pro-inflammatory and anti-inflammatory cytokine release during acute inflammation in chronic obstructive pulmonary disease: an ex vivo study. Respir Res. 2008;9:47. doi: 10.1186/1465-9921-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wu M-Y, Lin T-H, Chiu Y-C, Liou H-C, Yang R-S, Fu W-M. Involvement of 15-lipoxygenase in the inflammatory arthritis. J Cell Biochem. 2012;113:2279–2289. doi: 10.1002/jcb.24098. [DOI] [PubMed] [Google Scholar]
- [47].Wang H, Li J, Follett PL, Zhang Y, Cotanche Da, Jensen FE, Volpe JJ, Rosenberg Pa. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. 2004;20:2049–2058. doi: 10.1111/j.1460-9568.2004.03650.x. [DOI] [PubMed] [Google Scholar]
- [48].Jin G, Arai K, Murata Y, Wang S, Stins MF, Lo EH, Van Leyen K. Protecting against cerebrovascular injury: Contributions of 12/15-lipoxygenase to edema formation after transient focal ischemia. Stroke. 2008;39:2538–2543. doi: 10.1161/STROKEAHA.108.514927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Loscalzo J. Membrane Redox State and Apoptosis: Death by Peroxide. Cell Metab. 2008;8:182–183. doi: 10.1016/j.cmet.2008.08.004. [DOI] [PubMed] [Google Scholar]
- [50].van Leyen K, Duvoisin RM, Engelhardt H, Wiedmann M. A function for lipoxygenase in programmed organelle degradation. Nature. 1998;395:392–395. doi: 10.1038/26500. [DOI] [PubMed] [Google Scholar]
- [51].Pallast S, Arai K, Wang X, Lo EH, Van Leyen K. 12/15-Lipoxygenase targets neuronal mitochondria under oxidative stress. J Neurochem. 2009;111:882–889. doi: 10.1111/j.1471-4159.2009.06379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Diemert S, Dolga AM, Tobaben S, Grohm J, Pfeifer S, Oexler E, Culmsee C. Impedance measurement for real time detection of neuronal cell death. J Neurosci Methods. 2012;203:69. doi: 10.1016/j.jneumeth.2011.09.012. [DOI] [PubMed] [Google Scholar]
- [53].Wang K, Kim D, D#x00F6;mling A. Cyanoacetamide MCR (III): Three-Component Gewald Reactions Revisited. J Comb Chem. 2010;12:111–118. doi: 10.1021/cc9001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chernyak N, Buchwald SL. Continuous-Flow Synthesis of Monoarylated Acetaldehydes Using Aryldiazonium Salts. J Am Chem Soc. 2012;134:12466–12469. doi: 10.1021/ja305660a. [DOI] [PubMed] [Google Scholar]
- [55].Kovalenko OO, Adolfsson H. Highly Efficient and Chemoselective Zinc-Catalyzed Hydrosilylation of Esters under Mild Conditions. Chem – A Eur J. 2015;21:2785–2788. doi: 10.1002/chem.201406176. [DOI] [PubMed] [Google Scholar]
- [56].Michel J, et al. Bull Soc Chim Fr. 1968:4898–4901. No Title. [Google Scholar]
- [57].Genna DT, Posner GH. Cyanocuprates Convert Carboxylic Acids Directly into Ketones. Org Lett. 2011;13:5358–5361. doi: 10.1021/ol202237j. [DOI] [PubMed] [Google Scholar]
- [58].Oenema TA, Maarsingh H, Smit M, Groothuis GMM, Meurs H, Gosens R. Bronchoconstriction Induces TGF-β Release and Airway Remodelling in Guinea Pig Lung Slices. PLoS One. 2013;8:e65580. doi: 10.1371/journal.pone.0065580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.