

A dual role of the C9ORF72/SMCR8-containing complex in autophagy initiation and autophagic flux.
Keywords: C9ORF72, SMCR8, autophagy induction, ULK1, autophagic flux
Abstract
The intronic GGGGCC hexanucleotide repeat expansion in chromosome 9 open reading frame 72 (C9ORF72) is a prevalent genetic abnormality identified in both frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). Smith-Magenis syndrome chromosomal region candidate gene 8 (SMCR8) is a protein with unclear functions. We report that C9ORF72 is a component of a multiprotein complex containing SMCR8, WDR41, and ATG101 (an important regulator of autophagy). The C9ORF72 complex displays guanosine triphosphatase (GTPase) activity and acts as a guanosine diphosphate–guanosine 5′-triphosphate (GDP-GTP) exchange factor (GEF) for RAB39B. We created Smcr8 knockout mice and found that Smcr8 mutant cells exhibit impaired autophagy induction, which is similarly observed in C9orf72 knockdown cells. Mechanistically, SMCR8/C9ORF72 interacts with the key autophagy initiation ULK1 complex and regulates expression and activity of ULK1. The complex has an additional role in regulating later stages of autophagy. Whereas autophagic flux is enhanced in C9orf72 knockdown cells, depletion of Smcr8 results in a reduced flux with an abnormal expression of lysosomal enzymes. Thus, C9ORF72 and SMCR8 have similar functions in modulating autophagy induction by regulating ULK1 and play distinct roles in regulating autophagic flux.
INTRODUCTION
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative disorders leading to dementia and loss of motor coordination (1, 2). The aberrant regulation of RNA and protein metabolism has been suggested to underlie the pathogenesis of these diseases (3). An expanded hexanucleotide repeat (GGGGCC) in a noncoding region of chromosome 9 open reading frame 72 (C9ORF72) has been identified as the most common cause of familial FTD and ALS (4–7). Proposed mechanisms of disease onset include RNA toxicity due to accumulation of transcripts containing the GGGGCC repeat, accumulation of aberrantly expressed peptides, and C9ORF72 loss of function (8–17). Knockout of C9orf72 in mice does not result in neurodegeneration but instead causes defects in macrophage and microglial function (18–20), suggesting that loss of function of C9orf72 alone is not sufficient to cause FTD-ALS. However, C9ORF72 mRNA levels are reduced in FTD-ALS patients, and microglial dysfunction is tightly connected with FTD-ALS pathogenesis (4, 6, 18, 21, 22). These studies raise the possibility that haploinsufficiency of C9ORF72 could contribute to FTD-ALS diseases. It is important to understand the cellular functions of C9ORF72 and its mechanisms of action.
Autophagy is an evolutionarily conserved process characterized by engulfing cytoplasmic proteins or organelles into double-membrane vesicles called autophagosomes, which fuse with lysosomes to form autolysosomes primed for degradation (23–25). Autophagy is essential for cellular homeostasis because it removes aggregates of misfolded proteins and/or defective organelles, provides energy, and recycles cellular components (25). Autophagy is a highly regulated and multistep process (24, 25), and its deregulation has been implicated in a variety of neurodegenerative diseases including FTD, ALS, Parkinson’s, Alzheimer’s, and Huntington’s disease (26, 27). ULK1, a mammalian homolog of autophagy-related 1 (Atg1), forms a protein complex with FIP200, ATG13, and ATG101 to control autophagy initiation. After the initiation, Atg proteins are recruited to a membrane structure called the phagophore, which expands and fuses upon itself to form the autophagosome. The autophagosome is directed to and fuses with a lysosome, where the enclosed materials are degraded. Subsequently, the nutrients are released back into the cytosol for reuse. Accumulating evidence suggests that autophagy impairment at distinct regulatory steps may have different consequences for the pathogenesis of neurodegenerative diseases (26, 27). Therefore, it is crucial to understand the nature of defects in autophagy in individual neurodegenerative diseases.
Rab guanosine triphosphatases (GTPases) encode information about the state of membrane domains to control specific membrane trafficking events (28). Rabs are activated by specific guanine nucleotide exchange factors (GEFs). One of the emerging families of GEFs contains DENN (differentially expressed in normal and neoplastic cell) domains (29, 30). DENN domain–containing GEFs catalyze the dissociation of guanosine diphosphate (GDP) from the Rab GTPase followed by guanosine 5′-triphosphate (GTP) exchange. The GTP-bound Rab is then activated and recruits its effectors to regulate membrane trafficking. Bioinformatics studies predict that C9ORF72 contains DENN domains (31, 32), suggesting its potential functions in membrane trafficking as a GEF factor. Indeed, it has been reported that C9ORF72 is associated with RAB1, RAB5, RAB7, and RAB11, and is involved in endocytosis and autophagy (33). A recent study shows that C9ORF72 functions as a GEF for RAB8A and RAB39B, and its depletion has a partial deleterious effect on autophagy (34). Despite these progresses, the specific steps C9ORF72 regulates in autophagy and the molecular mechanisms by which C9ORF72 confers this regulation remain unclear.
To gain insights into the biology of the C9ORF72 protein, we isolated C9ORF72-containing complexes from human cells and characterized their functions. We found that C9ORF72 forms a protein complex with Smith-Magenis syndrome chromosomal region candidate gene 8 (SMCR8). Although it has been implicated in autophagy (35), the experimental evidence of SMCR8 functions is lacking. SMCR8 is also predicted to contain DENN domains and exhibits some homology with C9ORF72 (31, 32). We created Smcr8 knockout mice and found that mutant cells exhibit impaired autophagy induction, which is likewise observed in C9orf72 knockdown cells. Our mechanistic studies show that C9ORF72/SMCR8 interacts with the key autophagy initiation ULK1 complex, and the interaction is enhanced under starvation conditions. C9ORF72/SMCR8 regulates the expression and activity of ULK1. Furthermore, we identified unique roles for this complex at the later stage of autophagy. Whereas autophagic flux is enhanced in C9orf72 knockdown cells, depletion of Smcr8 leads to a reduced flux with an abnormal expression of lysosomal enzymes. Thus, C9ORF72 and SMCR8 have similar functions in modulating autophagy induction by regulating ULK1 and play distinct roles in regulating autophagic flux.
RESULTS
C9ORF72 forms a protein complex with SMCR8, WDR41, and ATG101
We developed a human embryonic kidney (HEK) 293 cell–derived stable cell line expressing Flag-C9ORF72 protein. The cytoplasmic extract enriched for the Flag-C9ORF72 protein was affinity-purified using Flag resin to examine C9ORF72-associated proteins. A Flag–green fluorescent protein (GFP) HEK293 stable cell line was used as a control. Flag-C9ORF72 affinity eluate was subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE) followed by silver staining (Fig. 1A). The C9ORF72 eluate was enriched for several proteins in association with C9ORF72. The individual bands were extracted from the gel and subjected to mass spectrometry. This analysis revealed the identification of SMCR8, WDR41, and ATG101 proteins with similar peptide coverage (~60%) to that of C9ORF72, reflecting a near-stoichiometric presence (Fig. 1A and table S1). The ULK1/FIP200/ATG13/ATG101 protein complex controls autophagy initiation (36–38). ATG101 is a binding partner of ATG13, interacts with ULK1, and regulates autophagy (39, 40). Mass spectrometry analysis of C9ORF72-associated proteins identified ATG13 and ULK1 with the coverage of ~26 and ~32%, respectively (table S1). Proteins at 68, 55, and 50 kD were respectively identified as SKB1, α-tubulin, and MEP50, which are common contaminants of Flag affinity purification (Fig. 1A, Flag-GFP, shown as asterisks). Together, these results suggest that C9ORF72, SMCR8, WDR41, and ATG101 form a protein complex, which is associated with the ULK1 complex and is potentially involved in autophagy.
Fig. 1. Isolation of C9ORF72-associated proteins.
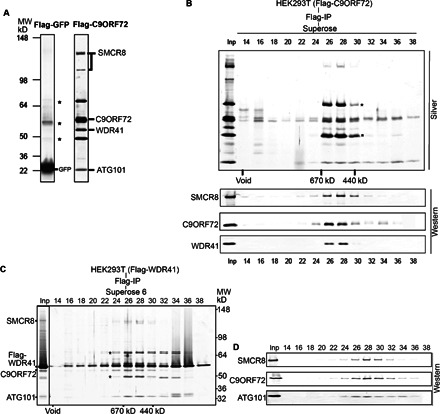
(A) Silver staining analysis of Flag affinity–purified fractions from cytoplasmic extracts of Flag-GFP and Flag-C9ORF72 HEK293 cell lines. Asterisks indicate common contaminants of Flag purification (SKB1, α-tubulin, and MEP50). Flag-C9ORF72–associated proteins as identified by mass spectrometry are indicated. MW, molecular weight; IP, immunoprecipitation. (B) Superose 6 gel filtration fractions from C9ORF72 cytoplasmic Flag affinity purification. Fractions were resolved by 4 to 12% SDS-PAGE and analyzed by silver staining (top) and Western blot with corresponding antibodies (bottom). The gel filtration purification scheme and fraction numbers are indicated on the top. (C and D) Purification of the WDR41-associated proteins from a HEK293 stable cell line expressing Flag-WDR41. Gel filtration fractions and Flag eluates were resolved on a 4 to 12% SDS-PAGE gel, followed with silver staining (left) and Western blot with corresponding antibodies (right). The gel filtration purification scheme and fraction numbers are indicated on the top. WDR41-associated proteins and molecular markers are indicated.
Next, we subjected the cytoplasmic Flag-C9ORF72 affinity eluate to size exclusion chromatography. After analysis of column fractions using silver stain and Western blot analysis, C9ORF72 protein coeluted with SMCR8, WDR41, and ATG101 in fractions 26 to 30, reflecting a complex of ~600 kD (Fig. 1B). A fraction of Flag-C9ORF72 also eluted at a lower molecular mass; SMCR8 and WDR41 did not appear to comigrate with this fraction of C9ORF72 (Fig. 1B). To further confirm the presence of C9ORF72 protein in a large protein complex in the cytoplasm, we developed HEK293-derived stable cell lines expressing Flag-WDR41 or Flag-SMCR8 protein. Flag affinity purification from cytoplasmic fractions followed by gel filtration chromatography revealed the presence of a similar-sized protein complex containing Flag-WDR41, SMCR8, C9ORF72, and ATG101 in fractions 26 through 30, corresponding to a complex of ~600 kD (Fig. 1C). Western blot analysis confirmed the coexistence of SMCR8, C9ORF72, and ATG101 in fractions 26 to 30 (Fig. 1D). A similar protein complex was also identified using a HEK293 cell line expressing the Flag-SMCR8 protein (fig. S1). Thus, our biochemical purification studies show that C9ORF72 forms a protein complex with SMCR8, WDR41, and ATG101.
The C9ORF72 complex exhibits a GTPase activity and acts as a GEF for RAB39B
Previous bioinformatics analyses of protein motifs in C9ORF72 and SMCR8 identified a conserved DENN domain region in both proteins (31, 32). DENN domain–containing proteins are suggested to function as GEFs to activate Rab GTPases and regulate membrane trafficking (28–30). We reasoned that a Rab GTPase might loosely associate with the C9ORF72 protein complex. To test this hypothesis, we decided to use the protein complex from the fractions of the Flag-WDR41 gel filtration. WDR41 contains a WD-repeat domain, which often functions as a scaffold for protein interaction (41). Because of this, we reasoned that Flag-WDR41 could be more effective to pull the associated GTPase activity compared to using Flag-C9ORF72 or Flag-SMCR8 fractions. We subjected the fractions of the Flag-WDR41 gel filtration to a GTPase assay (Fig. 2A). This study showed a peak of GTPase activity eluting in fractions 26 and 28 (Fig. 2A), coincident with the peak of the C9ORF72/SMCR8-containing complex (Fig. 1, B to D). We also found GTPase activity in a smaller molecular weight complex (fraction 34), reflecting an interaction between WDR41 protein and a putative Rab GTPase in a lower molecular range (Fig. 2A). To further confirm the GTPase activities associated with C9ORF72 protein complex, we generated stable Flag cell lines for ATG101 in HEK293 cells. Analysis of Flag affinity eluates from these stable cell lines similarly showed the presence of a specific and robust GTPase activity associating with C9ORF72-containing complexes (Fig. 2B). These data suggest that the C9ORF72 complex displays a GTPase activity.
Fig. 2. The C9ORF72 complex displays a GTPase activity and acts as a GEF for RAB39B.
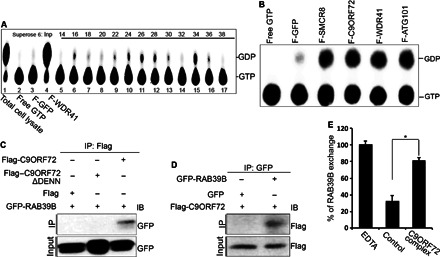
(A) GTPase assays were performed using WDR41 gel filtration fractions with [α-32P]GTP. The guanine nucleotides were separated by thin-layer chromatography plate. The positions of [α-32P]GTP and [α-32P]GDP are indicated on the right. (B) GTPase assay with Flag affinity–purified eluates from different Flag-tagged proteins in the C9ORF72 complex. Free [α-32P]GTP and Flag-GFP elutes were treated as controls. [α-32P]GTP and [α-32P]GDP are indicated on the right. (C and D) GFP-RAB39B, Flag-C9ORF72, or DENN domain–depleted C9ORF72ΔDENN was transfected into N2A cells. C9ORF72 proteins or RAB39B proteins were immunoprecipitated with M2 beads (anti-Flag) (C) or anti-GFP beads (D) followed by Western blot analyses using antibodies as listed. IB, immunoblot. (E) GEF assay of C9ORF72 protein complex and RAB39B. Purified His-tagged RAB39B proteins were preloaded with fluorescence-labeled BODIPY-GDP followed by addition of control or C9ORF72 protein complex. Fractions without C9ORF72 complex serve as the negative control. C9ORF72 complex promotes the release of GDP from RAB39B, suggesting its GEF activity against RAB39B.
To identify potential Rab GTPases associated with the C9ORF72 complex, we performed co-IP studies. Among the six candidate GTPases examined, including RAB33, RAB35, RAB39A, RAB39B, RAB31, and RAB24, we found that RAB31, RAB33, and RAB39B exhibit interactions with C9ORF72 (fig. S2A). Because mutations in RAB39B cause intellectual disability and early-onset Parkinson’s diseases (42, 43), we decided to focus on RAB39B for further investigations. Reciprocal co-IP analyses confirmed that C9ORF72 interacts with RAB39B (Fig. 2, C and D). In contrast, we failed to detect the interaction between SMCR8 and RAB39B under the same experimental conditions (fig. S2B). Emerging evidence suggests that DENN domains directly interact with Rab GTPases (29). To test whether the DENN domain of C9ORF72 is required for its interactions with RAB39B, we deleted the DENN domain and found that the interaction between C9ORF72 and RAB39B is abolished (Fig. 2C). These studies suggest that C9ORF72 interacts with RAB39B in a DENN domain–dependent manner. To determine whether the C9ORF72/SMCR8 complex functions as a GEF for RAB39B, we performed an in vitro activity assay (44). The His-tagged RAB39B protein was purified and preloaded with fluorescence-labeled BODIPY-GDP. Measurement of the release rate of BODIPY-GDP shows that the C9ORF72 protein complex strongly promotes GDP release from RAB39B (Fig. 2E), indicating that the C9ORF72/SMCR8 complex functions as a GEF for RAB39B.
C9ORF72 interacts with SMCR8 in a DENN domain–dependent manner
To examine whether C9ORF72 interacts with SMCR8 and WDR41 in cells other than HEK293, we performed co-IP studies in N2A cells, a mouse neuroblastoma cell line. Experimental results confirmed the interaction between C9ORF72 and SMCR8 or WDR41 in N2A cells (Fig. 3, A and B). Because DENN domains directly interact with Rab GTPases, in addition to serving as GEF enzymes (29), we examined the importance of DENN domains in protein interactions within the C9ORF72 complex. Deletion of the DENN domain in SMCR8 abolished its interaction with C9ORF72, but not with WDR41 (Fig. 3C). In contrast, both SMCR8’s and WDR41’s interactions with C9ORF72 are lost upon the deletion of C9ORF72 DENN domain (Fig. 3D). It is possible that DENN-depleted C9ORF72 or SMCR8 cannot fold correctly. These results suggest that the DENN domains are essential for the interaction between C9ORF72 and SMCR8.
Fig. 3. C9ORF72 interacts with SMCR8 in a DENN domain–dependent manner.
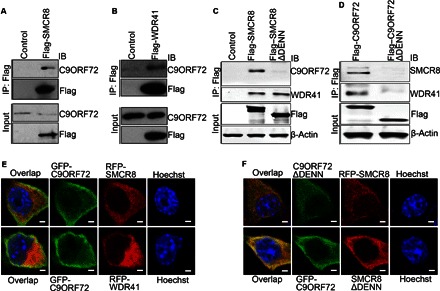
(A and B) Flag-tagged SMCR8 or WDR41 was transfected into N2A cells. SMCR8 or WDR41 proteins were immunoprecipitated with M2 beads (anti-Flag) followed by Western blot analysis using antibodies against endogenous C9ORF72. (C) Flag-tagged full-length or DENN domain–deleted SMCR8 was transfected into HEK293 cells. SMCR8 proteins were immunoprecipitated with M2 beads (anti-Flag) followed by Western blot analysis using antibodies against C9ORF72 or WDR41. (D) Flag-tagged full-length or DENN domain–deleted C9ORF72 constructs were transfected into HEK293 cells. C9ORF72 proteins were immunoprecipitated with M2 beads (anti-Flag) followed by Western blot analysis using antibodies against SMCR8 or WDR41. (E) GFP-tagged C9ORF72 was cotransfected with red fluorescent protein (RFP)–tagged SMCR8 or WDR41 into N2A cells. Confocal micrographs of N2A cells stained with antibodies against GFP (C9ORF72; green) and RFP (SMCR8 or WDR41; red). Hoechst stains the nuclei (blue). Scale bars, 10 μm. (F) GFP-tagged DENN domain–depleted C9ORF72 was cotransfected with RFP-tagged SMCR8 into N2A cells (upper panels). GFP-tagged C9ORF72 was cotransfected with RFP-tagged DENN domain–depleted SMCR8 into N2A cells (lower panels). Confocal micrographs of N2A cells stained with antibodies against GFP (C9ORF72ΔDENN or C9ORF72; green) and RFP (SMCR8 or SMCR8ΔDENN; red). Hoechst stains the nuclei (blue). Scale bars, 10 μm.
Next, we examined whether C9orf72 colocalizes with Smcr8 or Wdr41. Because commercially available antibodies failed to detect the endogenous localization of C9orf72 and Smcr8 in mouse cells, we constructed GFP-tagged C9ORF72 as well as RFP-tagged SMCR8 and WDR41. In addition to the nucleus, C9ORF72 is predominantly detected in the cytoplasm of N2A cells (Fig. 3E). Immunostaining results show that C9ORF72 is colocalized with both SMCR8 and WDR41 in the cytoplasm of N2A cells (Fig. 3E). To determine whether loss of the interaction between C9ORF72 and SMCR8 in the co-IP studies above is due to the aberrant localization of DENN domain–depleted C9ORF72 or SMCR8, we examined cellular localization of C9ORF72ΔDENN and SMCR8ΔDENN. We found that DENN domain depletion does not significantly change their cellular distributions (Fig. 3F). These studies suggest that C9ORF72 colocalizes with SMCR8 and WDR41, and C9ORF72 interacts with them in a DENN domain–dependent manner.
Autophagy induction is compromised in Smcr8-deficient cells
Autophagy is known to be the basic mechanism of delivering cytoplasmic contents to the lysosomes for degradation (24, 25). The ULK1/FIP200/ATG13 protein complex controls autophagy initiation (36–38). Because C9ORF72/SMCR8 is associated with ULK1/ATG13 in our mass spectrometry analysis of C9ORF72-associated proteins (table S1), we examined Smcr8’s potential involvement in autophagy. We generated two SMCR8 lentiviral short hairpin RNA (shRNA) constructs. Western blot and reverse transcription polymerase chain reaction (RT-PCR) analyses confirmed that SMCR8 shRNA1 and shRNA2 display ~85 to 90% knockdown efficiency with high specificities (fig. S3, A to C). Microtubule-associated protein 1 light chain 3 (LC3) protein expression has been widely used to monitor autophagy. LC3-I is a proteolytically processed form and is finally modified into the phosphatidylethanolamine (PE)–conjugated form, LC3-II (45). Knockdown of SMCR8 results in a significant increase in LC3-II protein levels relative to actin (Fig. 4, A and B). LC3 or GFP-tagged LC3 has been used to monitor autophagy at cellular levels by examining LC3-positive puncta (45). We examined autophagosome formation by staining GFP-LC3–positive puncta and found that there is a significant increase in puncta numbers per cell in SMCR8 knockdown cells compared to controls (Fig. 4, C and D). These data suggest that SMCR8 is involved in autophagy.
Fig. 4. Autophagy induction is compromised in Smcr8-deficient cells.
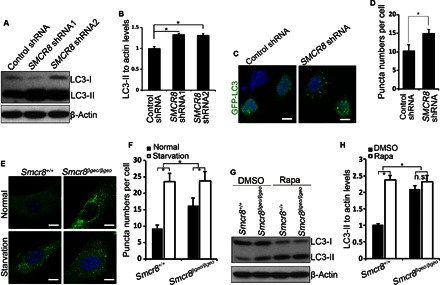
(A) HEK293 cells infected with lentiviruses expressing control shRNA, SMCR8 shRNA1, or SMCR8 shRNA2 were cultured 72 hours before protein lysate collection. The cell lysates were subjected to Western blot analyses using antibodies as indicated. (B) Quantification of LC3-II/actin ratio. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). (C) Confocal imaging of GFP-LC3 expression in SMCR8 knockdown HEK293 cells using antibodies against GFP. Hoechst stains the nuclei (blue). Scale bars, 10 μm. (D) Quantification of GFP-LC3–positive puncta per cell in (C). Error bars represent SEM of three independent experiments; ~100 GFP-positive cells were randomly selected for each experiment. *P < 0.05 (Student’s t test). (E) Confocal microscope images of wild-type and Smcr8 mutant MEFs stained with antibodies against LC3 (green) under normal or starvation conditions. Hoechst stains the nuclei (blue). Scale bars, 10 μm. (F) Quantification of LC3-positive puncta per cell in (E). Error bars represent SEM of three independent experiments; ~100 cells were randomly selected for each experiment. *P < 0.05 (Student’s t test). Two-way analysis of variance (ANOVA) detects a significant decrease in the magnitude of up-regulation of LC3-positive puncta after starvation in Smcr8 mutant MEFs compared to that in wild-type MEFs (P < 0.05). (G) Western blot analysis of LC3 expression. Wild-type or Smcr8 mutant MEFs were treated with dimethyl sulfoxide (DMSO) or rapamycin (0.1 μM) for 3.5 hours before protein lysate collection. β-Actin serves as the loading control. (H) Quantification of LC3-II/actin ratio from (G). Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). n.s. represents no significant difference in LC3-II expression between DMSO and rapamycin treatment in Smcr8 mutant MEFs. Two-way ANOVA detects a significant decrease in the magnitude of LC3-II up-regulation after rapamycin treatment in the Smcr8 mutant MEFs compared to wild-type MEFs (P < 0.05).
To further understand Smcr8’s functions, we created a mouse model using embryonic stem (ES) cells in which the Smcr8 gene has been disrupted by the β-geo reporter gene. We isolated mouse embryonic fibroblasts (MEFs) from Smcr8 mutant embryos. RT-PCR and Western blot analyses confirmed the depletion of Smcr8 at both mRNA and protein levels. Increased LC3-II levels in Smcr8-deficient cells could be due to increased autophagy induction or a block of autophagsome maturation. Amino acid starvation blocks mammalian target of rapamycin (mTOR) signaling and therefore induces autophagy by releasing mTOR signaling–mediated inhibition (25, 46). To determine whether autophagy induction is enhanced in Smcr8 mutant cells, we performed amino acid starvation experiments in MEFs. Immunostaining results show that LC3-positive puncta numbers are increased in Smcr8 mutant cells compared to controls (Fig. 4, E and F), which is consistent with SMCR8 shRNA knockdown studies (Fig. 4, C and D). Whereas puncta numbers per cell are increased in both wild-type and mutant MEFs under starvation, the magnitude of increase is significantly less in Smcr8 mutant MEFs compared to that in wild-type MEFs (Fig. 4, E and F), suggesting that autophagy induction is compromised in Smcr8 mutant cells. To further examine autophagy induction, we used rapamycin treatment to induce autophagy followed by Western blot analysis of LC3 expression. Rapamycin treatment results in autophagy induction reflected by induced expression of LC3-II in wild-type MEFs (Fig. 4, G and H). However, LC3-II levels are not significantly increased in Smcr8 mutant MEFs after rapamycin treatment (Fig. 4, G and H). Together, these results suggest that autophagy induction is compromised in Smcr8 mutant cells.
C9orf72 knockdown disrupts autophagy induction
Previous studies have implicated C9ORF72 in autophagy, but which step it regulates in autophagy remains unclear (33). To determine whether C9orf72 performs similar functions as Smcr8 in regulating autophagy induction, we generated two lentiviral C9orf72 shRNA constructs, which both effectively knock down C9orf72 expression (fig. S3D). We did not observe functional differences between these two shRNAs; therefore, we used C9orf72 shRNA1 in our studies. To determine how C9orf72 regulates autophagy, we performed Western blot analyses in MEFs. We found that the LC3-II/actin level is significantly reduced in C9orf72 knockdown cells (Fig. 5, A and B), suggesting a reduced number of autophagsomes. Reduced autophagsomes could be due to a decrease in autophagy induction or an enhanced autophagic flux. To determine whether C9orf72 regulates autophagy initiation, we used rapamycin to induce autophagy in MEFs. Rapamycin treatment results in a significant increase in LC3-II levels in control cells but not in C9orf72 knockdown cells (Fig. 5, A and B). Next, we examined LC3-positive puncta numbers. Control MEFs exhibit a significant increase in puncta numbers per cell under starvation conditions (Fig. 5, C and D). In contrast, C9orf72 knockdown MEFs exhibit normal puncta numbers under starvation compared to normal culture conditions (Fig. 5, C and D). Together, these results suggest that autophagy induction is impaired in C9orf72 knockdown cells. Last, we examined autophagy induction in both C9orf72- and Smcr8-deficient cells. Rapamycin treatment failed to promote autophagy induction in C9orf72, Smcr8, or C9orf72/Smcr8 double-deficient MEFs, although autophagy is significantly induced in wild-type MEFs (Fig. 5, E and F). Together, these studies suggest that autophagy induction is compromised in C9orf72, Smcr8, and C9orf72/Smcr8 double-depleted cells.
Fig. 5. C9orf72 knockdown disrupts autophagy induction.
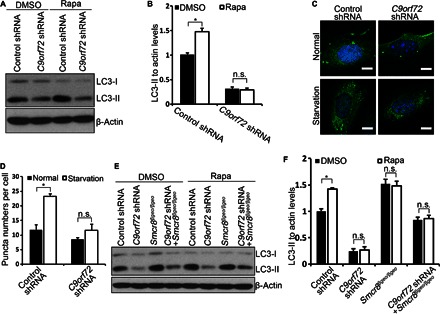
(A) Western blot analysis of LC3 expression. MEF cells infected with lentiviruses expressing control or C9orf72 shRNA were treated with DMSO or rapamycin (0.1 μM) for 3.5 hours before protein lysate collection. β-Actin serves as the loading control. (B) Quantification of LC3-II/actin ratio. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). n.s. represents no significant difference in LC3-II expression between DMSO and rapamycin treatment in C9orf72 knockdown MEFs. Two-way ANOVA detects a significant decrease in the magnitude of LC3-II up-regulation after rapamycin treatment in the C9orf72 knockdown MEFs compared to that in wild-type MEFs (P < 0.05). (C) Confocal imaging of MEFs stained with antibodies against LC3 (green). MEF cells were infected with lentiviruses expressing control or C9orf72 shRNA followed by starvation. Hoechst stains the nuclei (blue). Scale bars, 10 μm. (D) Quantification of LC3-positive puncta per cell in (C). Error bars represent SEM of three independent experiments; ~100 cells were randomly selected for each experiment. *P < 0.05 (Student’s t test). (E) Western blot analysis of LC3 expression. Wild-type or Smcr8 mutant MEF cells were infected with lentiviruses expressing control or C9orf72 shRNA as indicated followed by DMSO or rapamycin (0.1 μM) treatment for 3.5 hours before protein lysate collection. β-Actin serves as the loading control. (F) Quantification of LC3-II/actin ratio. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). n.s. represents no significant difference in LC3-II expression between DMSO and rapamycin treatment. Two-way ANOVA detects a significant decrease in the magnitude of LC3-II up-regulation after rapamycin treatment in the C9orf72 knockdown, Smcr8 mutant, and C9orf72/Smcr8 double depletion MEFs compared that in to wild-type MEFs (P < 0.05).
The C9ORF72/SMCR8 complex regulates ULK1
In mammalian cells, ULK1 forms a protein complex with FIP200/ATG13 regardless of nutrient status (36). During normal nutrient conditions, mTOR complex 1 phosphorylates Ser757 on ULK1, which results in repressing autophagy initiation (37, 38, 47). Under starvation, mTOR-mediated inhibition of autophagy is released, resulting in autophagy induction. Because our mass spectrometry data show that C9ORF72/SMCR8 is associated with ULK1 (table S1), we hypothesized that C9ORF72/SMCR8 regulates autophagy induction by affecting the expression or activity of ULK1. To test this hypothesis, we attempted to confirm the interaction between the C9ORF72/SMCR8 complex and ULK1/ATG13. We expressed C9ORF72 or SMCR8 alone or in combination in HEK293 cells. Endogenous ULK1 and ATG13 are coimmunoprecipitated with C9ORF72 when it is expressed together with SMCR8, but this co-IP is drastically reduced when C9ORF72 is expressed alone (Fig. 6A). Furthermore, the interaction between C9ORF72/SMCR8 and ULK1/ATG13 is enhanced under amino acid starvation conditions (Fig. 6A). Together, these results suggest that C9ORF72/SMCR8 interacts with the ULK1/ATG13 complex, which is facilitated by starvation conditions.
Fig. 6. The C9ORF72/SMCR8 complex regulates ULK1.
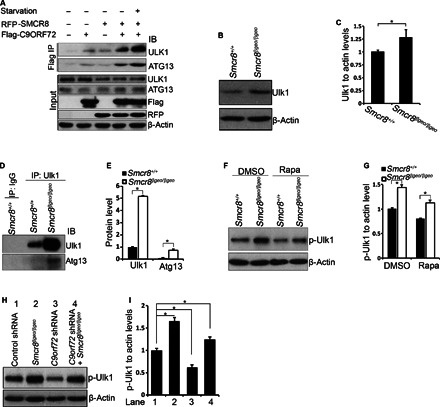
(A) The interaction between C9ORF72/SMCR8 complex with ULK1 is enhanced under starvation conditions. Flag-C9ORF72 and RFP-SMCR8 were transfected into HEK293 cells with or without amino acid starvation for 1 hour before protein lysate collection. C9ORF72 protein was immunoprecipitated with M2 beads (anti-Flag) followed by Western blot analyses using antibodies as listed. (B) Western blot analysis of Ulk1 expression in wild-type or Smcr8 mutant MEFs. β-Actin serves as the loading control. (C) Quantification of Ulk1 expression relative to actin. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). (D) Ulk1 protein in wild-type or Smcr8 mutant MEFs was immunoprecipitated with Ulk1 antibodies followed by Western blot analyses. IgG, immunoglobulin G. (E) Quantification of relative Ulk1 and Atg13 protein levels. Error bars represent SEM of three measurements from three independent experiments; *P < 0.01 (Student’s t test). (F) Western blot analysis of phospho-Ulk1 (p-Ser757) expression. Wild-type or Smcr8 mutant MEFs were treated with DMSO or rapamycin (0.1 μM) for 3.5 hours before protein lysate collection. (G) Quantification of phospo-Ulk1 (p-Ulk1)/actin ratio. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). (H) Western blot analysis of phospho-Ulk1 (p-Ser757) expression. Wild-type or Smcr8 mutant MEFs were infected with lentiviruses expressing control or C9orf72 shRNA constructs. β-Actin serves as the loading control. (I) Quantification of relative levels of phospho-Ulk1. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test).
To determine whether Ulk1 protein level is altered in Smcr8 mutant cells, we performed Western blot analyses and found that Ulk1 protein is significantly enhanced in mutant MEFs (Fig. 6, B and C). IP of endogenous Ulk1 in Smcr8 mutant cells shows markedly enhanced protein levels of Ulk1 and Atg13 (Fig. 6, D and E). It is possible that Smcr8 masks the epitope that Ulk1 antibody recognizes; therefore, Smcr8 depletion results in a more efficient IP of Ulk1. Together, these results suggest that Smcr8 is required for normal levels of Ulk1 and Atg13. Phosphorylation of Ser757 on Ulk1 controls autophagy initiation (37, 38, 47). The increased protein level of Ulk1 and defective autophagy induction in Smcr8 mutant cells prompted us to test whether Ulk1 phosphorylation is also altered in mutant MEFs. Indeed, phosphorylated Ulk1 (phospho-Ulk1) is significantly enhanced in Smcr8 mutant MEFs (Fig. 6, F and G). In rapamycin-treated cells, phospho-Ulk1 levels are also significantly increased (Fig. 6, F and G), which is consistent with the impaired autophagy induction in Smcr8 mutant cells (Fig. 4, E to H). Next, we examined phospho-Ulk1 expression in C9orf72 knockdown cells. In contrast to the up-regulation of phospho-Ulk1 in Smcr8 mutant MEFs, C9orf72 knockdown results in a decrease in phospho-Ulk1; C9orf72/Smcr8 double depletion leads to an intermediate level of phospho-Ulk1 in MEFs (Fig. 6, H and I). Together, these results suggest that the C9ORF72 and SMCR8 regulate autophagy induction by modulating ULK1 through distinct mechanisms.
Autophagic flux is defective in Smcr8-deficient cells
Autophagy induction is reduced (Fig. 4, E to H), but the autophagsome numbers are increased in Smcr8 mutant cells reflected by increased LC3-II levels (Figs. 4, G and H, and 5, E and F), suggesting that autophagsome maturation is defective in Smcr8-deficient cells. Autophagosome accumulation could be due to a blockage of autophagic flux (24, 25). We used double-tagged LC3 proteins (mCherry-GFP-LC3) to monitor autophagic flux. Simultaneous expression of mCherry and GFP results in yellow signals under physiological pH, whereas an acidic environment in the autolysosome quenches the GFP signal and results in exclusively red signals (24, 45). We found that the yellow dot/total red dot ratio is 50.1 ± 2.8% in control cells, whereas the yellow dot/total red dot ratio is higher at 68.0 ± 4.2% in SMCR8 knockdown cells (Fig. 7A), suggesting that relatively less GFP signal is quenched in knockdown cells. These results indicate that autophagsome maturation is compromised in Smcr8-deficient cells, which is consistent with LC3-II accumulation in Smcr8-deficient cells (Figs. 4, A, B, G, and H, and 7, D and E). Next, we examined a well-defined autophagy substrate, p62 (48). Consistent with an impaired degradation function of autophagsome, p62 is significantly accumulated in Smcr8-deficient cells (Fig. 7, B, C, F, and G). Together, these results suggest that Smcr8 depletion results in a defective autophagic flux.
Fig. 7. Autophagic flux is defective in Smcr8-deficient cells.
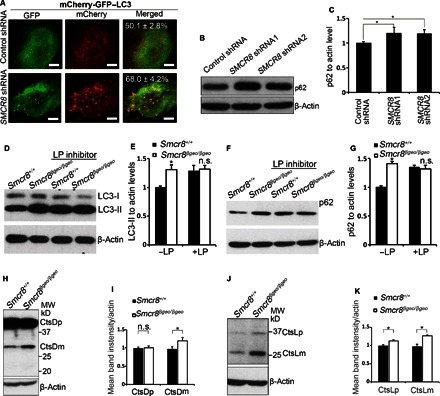
(A) Confocal imaging of HEK293 cells infected by lentiviruses expressing control or SMCR8 shRNA. Double-tagged LC3 (mCherry-GFP-LC3) protein was used to indicate autophagic flux. The percentage of yellow dots, which maintains both GFP and mCherry signal, out of total red dots is listed in the merged figures. Values represent SEM of three independent experiments; ~50 cells were randomly selected for each statistical analysis. *P < 0.05 (Student’s t test). (B) Western blot analysis of p62 expression. β-Actin serves as the loading control. (C) Quantification of p62 protein levels relative to actin. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). (D and F) Western blot analyses of LC3 (D) or p62 (F) expression in MEFs. MEFs were cultured in normal conditions or treated with lysosome inhibitors LP (100 μM each) for 3.5 hours before protein lysate collection. β-Actin serves as the loading control. (E and G) Quantification of LC3-II levels relative to actin (E) and p62 levels relative to actin (G). Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). n.s. represents no significant difference of LC3-II or p62 expression. Two-way ANOVA detects a significant decrease in the magnitude of LC3-II or p62 up-regulation after LP treatment in Smcr8 mutant MEFs compared to that in wild-type MEFs (P < 0.05). (H and J) Western blot analysis of cathepsin D (H) or cathepsin L (J) expression. β-Actin serves as the loading control. CtsDp, procathepsin D; CtsDm, mature cathepsin D; CtsLp, procathepsin L; CtsLm, mature cathepsin L. (I and K) Quantification of cathepsins D or L expression. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test); n.s. represents no significant difference detected.
Lysosomal degradation is mainly mediated by cysteine and aspartyl protease, which are inhibited by leupeptin and pepstatin, respectively. If an increased LC3-II level is due to the enhancement of autophagy induction, blocking lysosomal degradation using leupeptin and pepstatin A (LP) should result in a further increase of LC3-II levels. However, if an increased LC3-II level is because of defective autophagsome maturation, blocking lysosomal degradation using LP will not further increase LC3-II levels (45). To confirm that increased LC3-II levels are due to defective autophagsome maturation and not autophagy induction enhancement in Smcr8 mutant cells, we treated MEFs for 3.5 hours with lysosomal inhibitors (LP). This treatment leads to a significant accumulation of endogenous LC3-II, reflecting blockage of LC3-II degradation after lysosomal inhibitor (LP) treatment in control cells (Fig. 7D). However, there is no significant further increase of LC3-II levels in Smcr8 mutant cells (Fig. 7, D and E). Consistent with a blockage of autophagsome maturation, LP treatment fails to cause further accumulation of p62 in Smcr8 mutant cells (Fig. 7, F and G). Together, these results suggest that Smcr8 depletion results in defective autophagsome maturation and impaired autophagosome degradation.
The defective autophagsome maturation could be due to a block in autophagsome-lysosome fusion or lysosomal degradation. We failed to detect a significant alteration in autophagosome-lysosome fusions in Smcr8 mutant MEFs. Therefore, we examined lysosomal degradation. Cathepsins D and L are widely used as reporter molecules for trafficking and maturation of lysosomal hydrolases (49, 50). Cathepsins D and L are synthesized as preprocathepsins D and L, which are converted into procathepsins D and L in the endoplasmic reticulum (ER) after signaling peptides are removed. Procathepsin D (52 kD) and procathepsin L (36 kD) are generated in the ER transit from the trans-Golgi and are transported to late endosomes and lysosomes. Encountering the acidic environment in lysosomes, procathepsin D undergoes further proteolytic processing into a 44-kD form and finally into the 32-kD mature form. Procathepsin L is processed into a 24-kD mature form (49, 50). In wild-type MEFs, the procathepsin D 52-kD band was barely visible, and the dominant signals were from 44-kD procathepsin D and 32-kD mature cathepsin D bands (Fig. 7H). In Smcr8 mutant MEFs, the mature cathepsin D level is significantly increased (Fig. 7, H and I). For cathepsin L, both procathepsin L and mature cathepsin L levels were significantly increased in Smcr8 mutant MEFs (Fig. 7, J and K). The increased amounts of cathepsins D and L in Smcr8 mutant cells indicate either an increase in their synthesis or a decrease in their degradation, which is often observed when lysosomal function is disrupted (51–53). Together, the defective autophagic flux could be due to a block of lysosomal degradation in Smcr8 mutant cells.
C9orf72 knockdown results in an increase in autophagic flux
Because C9ORF72 and SMCR8 are within a protein complex, we asked whether autophagic flux is also compromised in C9orf72 knockdown cells. We used double-tagged LC3 proteins (mCherry-GFP-LC3) to monitor autophagic flux (45). Surprisingly, C9orf72 knockdown results in a significant decrease in the ratio of yellow to red puncta (Fig. 8, A and B). These results suggest that autophagic flux is enhanced in C9orf72 knockdown cells, which is opposite to the reduced autophagic flux in Smcr8-depleted cells (Fig. 7A). Next, we measured p62 expression and found that p62 is not significantly changed in C9orf72 knockdown cells compared to controls (Fig. 8, C and D). Whereas increased autophagic flux may lead to more efficient degradation of autophagy substrate p62, autophagy induction is compromised in C9orf72 knockdown cells (Fig. 5), which leads to more p62 accumulation. The combined alterations in autophagy induction and autophagic flux may result in normal p62 expression levels in C9orf72 knockdown cells. The protein level of p62 is slightly increased in C9orf72/Smcr8 double-depleted cells (Fig. 8, C and D). Together, these results suggest that autophagic flux is enhanced in C9orf72-deficient cells.
Fig. 8. C9orf72 knockdown results in an increase in autophagic flux.
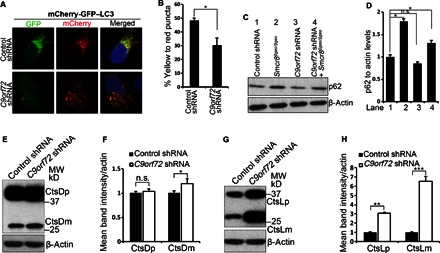
(A) Confocal imaging of GFP-mCherry expression in N2A cells infected with lentiviruses expressing control or C9orf72 shRNA. (B) Quantification of the percentage of yellow dots, which maintains both GFP and mCherry signal, out of total red dots. Values represent SEM of three independent experiments; ~50 cells were randomly selected for each statistical analysis. *P < 0.05 (Student’s t test). (C) Western blot analyses of p62 expression. Wild-type or Smcr8 mutant MEFs were infected with lentiviruses expressing control or C9orf72 shRNA followed by 72 hours of culture before protein lysate collection. β-Actin serves as the loading control. (D) Quantification of p62 levels relative to actin. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05 (Student’s t test). n.s. represents no significant difference detected. (E and G) Western blot analysis of cathepsin D (E) or cathepsin L (G) expression in the control or C9orf72 knockdown MEFs. β-Actin serves as the loading control. (F and H) Quantification of cathepsins D or L expression. Error bars represent SEM of three measurements from three independent experiments; *P < 0.05, ***P < 0.001 (Student’s t test); n.s. represents no significant difference detected.
Altered autophagic flux and degradation could be due to changes in lysosomal integrity. Next, we examined lysosomal integrity by monitoring the expression of cathepsins D and L. In C9orf72 knockdown MEFs, the mature cathepsin D level is slightly increased, whereas its precursor level appears normal (Fig. 8, E and F). In contrast, both procathepsin L and mature cathepsin L levels were significantly increased in C9orf72 knockdown MEFs (Fig. 8, G and H). Together, these results suggest that enhanced autophagic flux could be due to an increased lysosomal degradation in C9orf72 knockdown cells.
DISCUSSION
To gain insights into the biology of the C9ORF72 protein, we isolated C9ORF72-containing complexes from human cells and characterized their functions. We found that C9ORF72 forms a protein complex with SMCR8, WDR41, and ATG101. Here, we investigated C9ORF72/SMCR8’s functions in regulating autophagy and its action mechanisms.
Our studies provide novel insights into C9ORF72/SMCR8’s functions and its action mechanisms. First, in addition to forming a protein complex as reported (34, 54), we found that C9ORF72, SMCR8, WDR41, and ATG101 proteins exhibit similar peptide coverage (~60%) based on mass spectrometry analysis, reflecting a near-stoichiometric presence. In addition, C9ORF72 interacts with SMCR8 and WDR41 in a DENN domain–dependent manner. Starvation condition facilitates the interaction between C9ORF72/SMCR8 and ULK1. Our gel filtration identifies ATG101 as a component within the C9ORF72 complex, which could further aid in understanding this protein complex’s functions. ULK1/ATG13/FIP200/ATG101 complex acts as a key upstream factor to initiate autophagy (37, 38). It has been reported that Atg101 is essential for autophagy initiation; Atg101 binds to Atg13 and is required for its protein stabilization; Atg101 is responsible for recruiting downstream factors to the autophagosome formation sites (39, 40, 55). Future studies should reveal if and how the C9ORF72/SMCR8 complex regulates ATG101/ATG13 protein stabilization and ATG101’s functions in recruiting downstream autophagy factors. Second, the C9ORF72/SMCR8 complex has dual functions in autophagy by modulating both autophagy initiation and autophagic flux. Autophagy induction is compromised in Smcr8 mutant cells. SMCR8 is associated with and regulates the autophagy initiation factor ULK1. These data suggest that SMCR8 has a direct role in regulating autophagy initiation. However, it remains possible that a high baseline in autophagsome number is due to defective lysosomal degradation, which ultimately results in aberrant autophagy induction. Third, C9ORF72 and SMCR8 have overlapping and distinct functions, although they are within the same protein complex. C9orf72 or Smcr8 depletion results in impaired autophagy induction. However, phospho-Ulk1 expression is enhanced in Smcr8 mutant MEFs and is decreased in C9orf72 knockdown MEFs, suggesting that they have different mechanisms to regulate autophagy initiation. In addition, studies using double-tagged LC3 protein show that autophagic flux is enhanced in C9orf72 knockdown cells and is reduced in Smcr8 mutant cells. One potential explanation as to why C9ORF72 and SMCR8 have distinct functions in autophagy is because they interact with and activate different downstream Rab GTPases. Indeed, we found that C9ORF72 interacts with and activates RAB39B, but SMCR8 does not interact with RAB39B under the same experimental conditions. Future studies should further reveal the mechanisms underlying these overlapping and distinct functions of C9ORF72/SMCR8.
Our studies have important implications for C9ORF72-related neurodegeneration diseases. It has been reported that C9orf72 depletion alone in mice is not sufficient to cause motor neuron diseases (13, 19). The fact that C9ORF72 and SMCR8 form a protein complex and have overlapping functions in autophagy raises the possibility that Smcr8 has compensatory effects on C9orf72 loss of function. Future studies should examine whether double knockout of C9orf72 and Smcr8 causes autophagy defects in neurons. Given the importance of autophagy in the central nervous system (25, 56, 57), it is also important to determine whether double-knockout C9orf72/Smcr8 causes neurodegeneration due to defective autophagy in neurons. Multiple independent studies show that C9orf72 ablation in mice results in defective immune functions. It will be interesting to determine whether Smcr8 mutant mice also exhibit dysregulation of immune functions, which could be exacerbated in the C9orf72/Smcr8 double-knockout background. Because neurodegeneration is influenced by innate immune system and inflammation (58, 59), future studies should reveal if and how dysregulation of C9ORF72/SMCR8-mediated immune functions contributes to neurodegenerative diseases.
There are several reports about the C9ORF72 protein complex and its involvement in autophagy (34, 54, 60, 61). Our studies have revealed novel information about C9ORF72/SMCR8 complex functions compared to these recent studies. We found that ATG101 is a major component of C9ORF72 protein complex, which leads to the finding that C9ORF72 positively regulates ULK1 and autophagy initiation. Furthermore, we showed that C9orf72 plays a role in autophagic flux. Most notably, our studies using MEFs from Smcr8 knockout mice demonstrate that Smcr8 has dual roles in autophagy initiation and autophagic flux. Overall, our studies suggest that C9ORF72 and SMCR8 have distinct and overlapping functions at different steps of autophagy. It has been reported that a long form of C9ORF72 (C9-L) and not the short form of C9ORF72 (C9-S) interacts with SMCR8 and WDR41 (61, 62). C9-L, but not C9-S, is decreased in the frontal cortex of patients with FTD (61). Our gel filtration results show that a longer, but not shorter, form of C9ORF72 comigrates with SMCR8, WDR41, and ATG101 (Fig. 1B), suggesting that the longer form of C9ORF72 regulates autophagy. Future studies should determine whether C9-L and C9-S have different cellular functions relevant to the FTD diseases. Our co-IP experiments failed to detect the interaction between SMCR8 and RAB39B. However, Sellier et al. (34) showed an interaction between SMCR8 and RAB39B, and the presence of SMCR8 is essential for the complex to interact with RAB39B. These discrepancies could be because we performed studies in the N2A cells, whereas their co-IP was done in the HEK293 cells. Future studies should determine whether C9ORF72 and SMCR8 have differential interactions with other Rab GTPases and their functional importance.
MATERIALS AND METHODS
Affinity purification of Flag-C9ORF72, Flag-SMCR8, and Flag-WDR41
Expression constructs for Flag-C9ORF72, Flag-SMCR8, and Flag-WDR41 were individually cotransfected with a puromycin resistance plasmid into HEK293 cells. Protein complexes were purified (50 to 150 mg) from cytoplasmic extract (S100) with anti-Flag M2 affinity gel (Sigma). Two washes were performed using buffer A [20 mM tris-HCl (pH 7.9), 0.5 M KCl, 10% glycerol, 1 mM EDTA, 5 mM dithiothreitol (DTT), 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 0.5% NP-40] followed by one wash with buffer B [20 mM tris-HCl (pH 7.9), 0.1 M KCl, 10% glycerol, 1 mM EDTA, 5 mM DTT, 0.2 mM PMSF]. Then, the affinity column was eluted with Flag peptide (400 μg/ml). Proteins were further separated with Superose 6 gel filtration chromatography. Fractions were examined by SDS-PAGE analysis, and fractions containing C9ORF72/SMCR8 were combined and concentrated with Millipore Amicon Ultra (100K). Purified proteins were snap-frozen in liquid nitrogen and stored at −80°C.
Generation of Smcr8βgeo/βgeo mutant mice
The Smcr8tm1(KOMP)Vlcg ES cells were obtained from the University of California, Davis, Knockout Mouse Project Repository. Smcr8 locus is partially replaced by a cassette containing lacZ-polyA followed by a loxP-flanked hUbCpro-neo-polyA sequence. The Mouse Genetic Core Facility at National Jewish Health at Denver, CO, performed the ES cell injections into C57BL/6N blastocysts. The chimeric offsprings were mated to 129S1/SvImJ mice for germline transmission; germline-transmitted heterozygous females were crossed with CMV-Cre males to remove the Neo cassette. The PCR primers used for genotyping are as follows: LacF, acttgctttaaaaaacctcccaca; Smcr8-R, tgaacgaagactgctgtgtttaccc; Smcr8-wtR, gtcagtgttttccactccgaagtcc; and Smcr8-wtF, agaagcctatgcggataatgagggg. All animals were handled according to protocols approved by the Institutional Animal Care and Use Committee at the University of Georgia, Athens.
Constructs
For RFP-SMCR8, the SMCR8 gene was cloned into the pTag-RFP-N vector using the primers ggctcgagatgatcagcgcccctgac and ccgaattcggattttatacaaaaagct. For RFP-WDR41, the WDR41 gene was cloned into the pTag-RFP-N vector using the primers ggctcgagatgttgcgatggctgatc and ccgaattcggacagcaaggtataagtc. For the GFP-C9ORF72, the C9ORF72 gene was cloned into the pCAG–enhanced GFP vector using the primers gaattcatgtcgactctttgccca and ggtaccgtaaaagtcattagaacatc. For His-RAB39B, the RAB39B gene was cloned into the His vector using the primers aactcgaggaggccatctggctgtacca and ttaagcttctagcacaaacatctcctct. C9orf72 shRNA constructs were derived from the pLKO.1 vector using the targeting sequence gtgcagagaaagtaaataa and ggcctacactctttcatct. SMCR8 shRNA constructs were derived from the pLKO.1 vector using the targeting sequences ttacttctctttgcggattat and caagagctctcggccgaattt.
Antibodies
The following antibodies were used: rabbit anti-C9ORF72 (sc-138763, Santa Cruz Biotechnology), rabbit anti-SMCR8 (ab121682, Abcam), rabbit anti-WDR41 (sc-137922, Santa Cruz Biotechnology), rabbit anti-ATG101 (SAB4200175, Sigma), rabbit anti-ATG13 (13468, Cell Signaling), rabbit anti-ULK1 (8054, Cell Signaling), rabbit anti–phospho-ULK1 (6888, Cell Signaling), rabbit anti-p62 (5114, Cell Signaling), rabbit anti-RFP (R10367, Invitrogen), mouse anti-LC3 (0231-100/LC3-5F10, Nanotools), rabbit anti-LC3 (PM036, MBL), mouse anti–cathepsin D (AF1029, R&D Systems), rat anti–cathepsin L (MAB9521, R&D Systems).
GEF assay
For loading with GDP, 1 μM His-RAB39B protein in 110 mM NaCl, 50 mM tris-HCl (pH 8.0), 1 mM EDTA, 0.8 mM DTT, and 0.005% Triton X-100 was incubated with 50 μM BODIPY-GDP (Invitrogen) for 60 min at 30°C. Then, 0.1 μM preloaded RAB39B proteins were incubated with 7.5 nM C9ORF72 complex or control without C9ORF72 complex in the buffer containing 110 mM NaCl, 50 mM tris-HCl (pH 8.0), 12 mM MgCl2, 0.8 mM DTT, and 2 mM GDP solution for 7 min. As a positive control, preloaded RAB39B was incubated with 20 mM EDTA in the above solution. The fluorescence intensity was recorded with Synergy H4 Hybrid Multi-Mode Microplate Reader. The fluorescence intensity after equilibration with excess EDTA was defined as 100% exchange.
MEF isolation
MEFs were isolated from E15.5 wild-type and Smcr8 mutant embryos, as described (63), and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 15% fetal bovine serum (FBS) and penicillin/streptomycin (50 μg/ml).
Cell culture and lentivirus infection
To knock down SMCR8 in HEK293 cells or C9orf72 in N2A cells, cells were cultured in DMEM with 10% FBS plus penicillin/streptomycin (50 μg/ml). Around 60% confluent, cells were infected by lentiviruses expressing SMCR8 or C9orf72 shRNA followed by puromycin (2 μg/ml) selection for 72 hours before experiments. To examine autophagsome numbers or autophagic flux, GFP-LC3 or mCherry-GFP-LC3 plasmids were transfected into HEK293 or N2A cells using jetPRIME transfection reagents after puromycin (2 μg/ml) selection for 48 hours. Twenty-four hours after plasmid transfection, cells were fixed with 4% paraformaldehyde followed by immunostaining using antibodies against GFP or mCherry. For starvation, ~80% confluent, MEFs were infected by lentiviruses expressing C9orf72 shRNA followed by puromycin (2 μg/ml) selection for 72 hours before experiments. Smcr8 mutant or C9orf72 knockdown MEFs were cultured in DMEM with 15% FBS, washed with PBS, and then cultured in amino acid–free DMEM (United States Biological) for 3.5 hours before immunostaining using antibodies against LC3 (PM036, MBL). To induce autophagy with rapamycin, Smcr8 mutant or C9orf72 knockdown MEFs cultured in DMEM with 15% FBS were treated with rapamycin (0.1 μM) for 3.5 hours before protein lysate collection. To monitor the effects of blocking lysosomal degradation, Smcr8 mutant or C9orf72 knockdown MEFs were treated with lysosome inhibitors (LP) (100 μM each) for 3.5 hours before protein lysate collection.
Immunoprecipitation
HEK293 or N2A cells were transfected with plasmids, as indicated in the figures, and cultured for 36 hours before protein lysate collection. Cells were lysed in lysis buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 1 tablet protease inhibitor (Roche) per 10 ml]. Cell debris was pelleted at 12,500 rpm for 10 min at 4°C, and the supernatant was incubated with primary antibodies overnight at 4°C. The lysates with antibodies were incubated with M2 or anti-GFP beads for 2 hours, followed by washing of the immunoprecipitates three times with lysis buffer and elution of bound proteins in SDS-PAGE sampling buffer at 100°C for 10 min. Western blots were performed using the antibodies described above.
Supplementary Material
Acknowledgments
We thank J.-F.C.’s laboratory colleagues for the stimulating discussions. We thank J. Eggenschwiler, N. Manley, B. Condie, D. Menke, and J. Lauderdale for the use of their equipment. We also thank the Oncogenomics Core Facility at the Sylvester Comprehensive Cancer Center for their services. Funding: This work was supported by funds from the Office of the Vice President for Research at the University of Georgia, the University of Miami Miller School of Medicine, and the Sylvester Comprehensive Cancer Center, and by grants R00HD073269 (J.-F.C.), R01NS096176 (J.-F.C.), R01 GM078455 (R.S.), and R01 GM105754 (R.S.) from the NIH. Author contributions: M.Y., C.L., K.S., F.L., and J.-F.C. conceived and performed all the experiments. S.H. helped with the manuscript writing. J-F.C., R.S., and F.L. designed and interpreted the experiments and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1601167/DC1
fig. S1. Isolation of SMCR8-associated proteins.
fig. S2. Identifying C9orf72- or SMCR8-associated Rab GTPases.
fig. S3. Characterization of SMCR8 and C9orf72 shRNA constructs.
table S1. Polypeptide composition through Flag-C9ORF72 immunopurification by mass spectrometry.
REFERENCES AND NOTES
- 1.Bang J., Spina S., Miller B. L., Frontotemporal dementia. Lancet 386, 1672–1682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rademakers R., Neumann M., Mackenzie I. R., Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ling S.-C., Polymenidou M., Cleveland D. W., Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 79, 416–438 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gijselinck I., Van Langenhove T., van der Zee J., Sleegers K., Philtjens S., Kleinberger G., Janssens J., Bettens K., Van Cauwenberghe C., Pereson S., Engelborghs S., Sieben A., De Jonghe P., Vandenberghe R., Santens P., De Bleecker J., Maes G., Bäumer V., Dillen L., Joris G., Cuijt I., Corsmit E., Elinck E., Van Dongen J., Vermeulen S., Van den Broeck M., Vaerenberg C., Mattheijssens M., Peeters K., Robberecht W., Cras P., Martin J.-J., De Deyn P. P., Cruts M., Van Broeckhoven C., A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 11, 54–65 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., Scholz S. W., Duckworth J., Ding J., Harmer D. W., Hernandez D. G., Johnson J. O., Mok K., Ryten M., Trabzuni D., Guerreiro R. J., Orrell R. W., Neal J., Murray A., Pearson J., Jansen I. E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J. B., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M. A., Peuralinna T., Jansson L., Isoviita V.-M., Kaivorinne A.-L., Hölttä-Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chiò A., Restagno G., Borghero G., Sabatelli M.; ITALSGEN Consortium, Heckerman D., Rogaeva E., Zinman L., Rothstein J. D., Sendtner M., Drepper C., Eichler E. E., Alkan C., Abdullaev Z., Pack S. D., Dutra A., Pak E., Hardy J., Singleton A., Williams N. M., Heutink P., Pickering-Brown S., Morris H. R., Tienari P. J., Traynor B. J., A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.-Y. R., Karydas A., Seeley W. W., Josephs K. A., Coppola G., Geschwind D. H., Wszolek Z. K., Feldman H., Knopman D. S., Petersen R. C., Miller B. L., Dickson D. W., Boylan K. B., Graff-Radford N. R., Rademakers R., Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Majounie E., Renton A. E., Mok K., Dopper E. G. P., Waite A., Rollinson S., Chiò A., Restagno G., Nicolaou N., Simon-Sanchez J., van Swieten J. C., Abramzon Y., Johnson J. O., Sendtner M., Pamphlett R., Orrell R. W., Mead S., Sidle K. C., Houlden H., Rohrer J. D., Morrison K. E., Pall H., Talbot K., Ansorge O.; Chromosome 9-ALS/FTD Consortium; French research network on FTLD/FTLD/ALS; ITALSGEN Consortium, Hernandez D. G., Arepalli S., Sabatelli M., Mora G., Corbo M., Giannini F., Calvo A., Englund E., Borghero G., Floris G. L., Remes A. M., Laaksovirta H., McCluskey L., Trojanowski J. Q., Van Deerlin V. M., Schellenberg G. D., Nalls M. A., Drory V. E., Lu C.-S., Yeh T.-H., Ishiura H., Takahashi Y., Tsuji S., Le Ber I., Brice A., Drepper C., Williams N., Kirby J., Shaw P., Hardy J., Tienari P. J., Heutink P., Morris H. R., Pickering-Brown S., Traynor B. J., Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 11, 323–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z., Poidevin M., Li X., Li Y., Shu L., Nelson D. L., Li H., Hales C. M., Gearing M., Wingo T. S., Jin P., Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 7778–7783 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donnelly C. J., Zhang P.-W., Pham J. T., Haeusler A. R., Mistry N. A., Vidensky S., Daley E. L., Poth E. M., Hoover B., Fines D. M., Maragakis N., Tienari P. J., Petrucelli L., Traynor B. J., Wang J., Rigo F., Bennett C. F., Blackshaw S., Sattler R., Rothstein J. D., RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao Z., Wang H., Xia Q., Li K., Li K., Jiang X., Xu G., Wang G., Ying Z., Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 24, 2426–2441 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Mori K., Weng S.-M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H. A., Cruts M., Van Broeckhoven C., Haass C., Edbauer D., The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Zu T., Liu Y., Bañez-Coronel M., Reid T., Pletnikova O., Lewis J., Miller T. M., Harms M. B., Falchook A. E., Subramony S. H., Ostrow L. W., Rothstein J. D., Troncoso J. C., Ranum L. P. W., RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. U.S.A. 110, E4968–E4977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagier-Tourenne C., Baughn M., Rigo F., Sun S., Liu P., Li H.-R., Jiang J., Watt A. T., Chun S., Katz M., Qiu J., Sun Y., Ling S.-C., Zhu Q., Polymenidou M., Drenner K., Artates J. W., McAlonis-Downes M., Markmiller S., Hutt K. R., Pizzo D. P., Cady J., Harms M. B., Baloh R. H., Vandenberg S. R., Yeo G. W., Fu X.-D., Bennett C. F., Cleveland D. W., Ravits J., Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, E4530–E4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ash P. E. A., Bieniek K. F., Gendron T. F., Caulfield T., Lin W.-L., DeJesus-Hernandez M., van Blitterswijk M. M., Jansen-West K., Paul J. W. III, Rademakers R., Boylan K. B., Dickson D. W., Petrucelli L., Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mizielinska S., Grönke S., Niccoli T., Ridler C. E., Clayton E. L., Devoy A., Moens T., Norona F. E., Woollacott I. O. C., Pietrzyk J., Cleverley K., Nicoll A. J., Pickering-Brown S., Dols J., Cabecinha M., Hendrich O., Fratta P., Fisher E. M. C., Partridge L., Isaacs A. M., C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen X., Tan W., Westergard T., Krishnamurthy K., Markandaiah S. S., Shi Y., Lin S., Shneider N. A., Monaghan J., Pandey U. B., Pasinelli P., Ichida J. K., Trotti D., Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwon I., Xiang S., Kato M., Wu L., Theodoropoulos P., Wang T., Kim J., Yun J., Xie Y., McKnight S. L., Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Rourke J. G., Bogdanik L., Yáñez A., Lall D., Wolf A. J., Muhammad A. K. M. G., Ho R., Carmona S., Vit J. P., Zarrow J., Kim K. J., Bell S., Harms M. B., Miller T. M., Dangler C. A., Underhill D. M., Goodridge H. S., Lutz C. M., Baloh R. H., C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koppers M., Blokhuis A. M., Westeneng H.-J., Terpstra M. L., Zundel C. A. C., Vieira de Sá R., Schellevis R. D., Waite A. J., Blake D. J., Veldink J. H., van den Berg L. H., Pasterkamp R. J., C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atanasio A., Decman V., White D., Ramos M., Ikiz B., Lee H.-C., Siao C.-J., Brydges S., LaRosa E., Bai Y., Fury W., Burfeind P., Zamfirova R., Warshaw G., Orengo J., Oyejide A., Fralish M., Auerbach W., Poueymirou W., Freudenberg J., Gong G., Zambrowicz B., Valenzuela D., Yancopoulos G., Murphy A., Thurston G., Lai K.-M. V., C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 6, 23204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizielinska S., Lashley T., Norona F. E., Clayton E. L., Ridler C. E., Fratta P., Isaacs A. M., C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waite A. J., Bäumer D., East S., Neal J., Morris H. R., Ansorge O., Blake D. J., Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35, 1779.e5–1779.e13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ktistakis N. T., Tooze S. A., Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 26, 624–635 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Mizushima N., Levine B., Cuervo A. M., Klionsky D. J., Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N., Komatsu M., Autophagy: Renovation of cells and tissues. Cell 147, 728–741 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Vidal R. L., Matus S., Bargsted L., Hetz C., Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 35, 583–591 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Wong E., Cuervo A. M., Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stenmark H., Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Marat A. L., Dokainish H., McPherson P. S., DENN domain proteins: Regulators of Rab GTPases. J. Biol. Chem. 286, 13791–13800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshimura S.-i., Gerondopoulos A., Linford A., Rigden D. J., Barr F. A., Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J. Cell Biol. 191, 367–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine T. P., Daniels R. D., Gatta A. T., Wong L. H., Hayes M. J., The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang D., Iyer L. M., He F., Aravind L., Discovery of novel DENN proteins: Implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 3, 283 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farg M. A., Sundaramoorthy V., Sultana J. M., Yang S., Atkinson R. A. K., Levina V., Halloran M. A., Gleeson P. A., Blair I. P., Soo K. Y., King A. E., Atkin J. D., C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sellier C., Campanari M.-L., Julie Corbier C., Gaucherot A., Kolb-Cheynel I., Oulad-Abdelghani M., Ruffenach F., Page A., Ciura S., Kabashi E., Charlet-Berguerand N., Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behrends C., Sowa M. E., Gygi S. P., Harper J. W., Network organization of the human autophagy system. Nature 466, 68–76 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., Iemura S.-i., Natsume T., Takehana K., Yamada N., Guan J.-L., Oshiro N., Mizushima N., Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J., Kundu M., Viollet B., Guan K.-L., AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang L., Chen S., Du F., Li S., Zhao L., Wang X., Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hosokawa N., Sasaki T., Iemura S.-i., Natsume T., Hara T., Mizushima N., Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Mercer C. A., Kaliappan A., Dennis P. B., A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 5, 649–662 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Li D., Roberts R., WD-repeat proteins: Structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 58, 2085–2097 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson G. R., Sim J. C. H., McLean C., Giannandrea M., Galea C. A., Riseley J. R., Stephenson S. E. M., Fitzpatrick E., Haas S. A., Pope K., Hogan K. J., Gregg R. G., Bromhead C. J., Wargowski D. S., Lawrence C. H., James P. A., Churchyard A., Gao Y., Phelan D. G., Gillies G., Salce N., Stanford L., Marsh A. P. L., Mignogna M. L., Hayflick S. J., Leventer R. J., Delatycki M. B., Mellick G. D., Kalscheuer V. M., D’Adamo P., Bahlo M., Amor D. J., Lockhart P. J., Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am. J. Hum. Genet. 95, 729–735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giannandrea M., Bianchi V., Mignogna M. L., Sirri A., Carrabino S., D’Elia E., Vecellio M., Russo S., Cogliati F., Larizza L., Ropers H.-H., Tzschach A., Kalscheuer V., Oehl-Jaschkowitz B., Skinner C., Schwartz C. E., Gecz J., Van Esch H., Raynaud M., Chelly J., de Brouwer A. P. M., Toniolo D., D’Adamo P., Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am. J. Hum. Genet. 86, 185–195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong B., Bayat V., Jaiswal M., Zhang K., Sandoval H., Charng W.-L., Li T., David G., Duraine L., Lin Y.-Q., Neely G. G., Yamamoto S., Bellen H. J., Crag is a GEF for Rab11 required for rhodopsin trafficking and maintenance of adult photoreceptor cells. PLOS Biol. 10, e1001438 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klionsky D. J., Abdelmohsen K., Abe A., Abedin M. J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C. M., Adams P. D., Adeli K., Adhihetty P. J., Adler S. G., Agam G., Agarwal R., Aghi M. K., Agnello M., Agostinis P., Aguilar P. V., Aguirre-Ghiso J., Airoldi E. M., Ait-Si-Ali S., Akematsu T., Akporiaye E. T., Al-Rubeai M., Albaiceta G. M., Albanese C., Albani D., Albert M. L., Aldudo J., Algül H., Alirezaei M., Alloza I., Almasan A., Almonte-Beceril M., Alnemri E. S., Alonso C., Altan-Bonnet N., Altieri D. C., Alvarez S., Alvarez-Erviti L., Alves S., Amadoro G., Amano A., Amantini C., Ambrosio S., Amelio I., Amer A. O., Amessou M., Amon A., An Z., Anania F. A., Andersen S. U., Andley U. P., Andreadi C. K., Andrieu-Abadie N., Anel A., Ann D. K., Anoopkumar-Dukie S., Antonioli M., Aoki H., Apostolova N., Aquila S., Aquilano K., Araki K., Arama E., Aranda A., Araya J., Arcaro A., Arias E., Arimoto H., Ariosa A. R., Armstrong J. L., Arnould T., Arsov I., Asanuma K., Askanas V., Asselin E., Atarashi R., Atherton S. S., Atkin J. D., Attardi L. D., Auberger P., Auburger G., Aurelian L., Autelli R., Avagliano L., Avantaggiati M. L., Avrahami L., Awale S., Azad N., Bachetti T., Backer J. M., Bae D.-H., Bae J.-s., Bae O.-N., Bae S. H., Baehrecke E. H., Baek S.-H., Baghdiguian S., Bagniewska-Zadworna A., Bai H., Bai J., Bai X.-Y., Bailly Y., Balaji K. N., Balduini W., Ballabio A., Balzan R., Banerjee R., Bánhegyi G., Bao H., Barbeau B., Barrachina M. D., Barreiro E., Bartel B., Bartolomé A., Bassham D. C., Bassi M. T., Bast R. C. Jr, Basu A., Batista M. T., Batoko H., Battino M., Bauckman K., Baumgarner B. L., Bayer K. U., Beale R., Beaulieu J.-F., Beck G. R. Jr, Becker C., Beckham J. D., Bédard P.-A., Bednarski P. J., Begley T. J., Behl C., Behrends C., Behrens G. M. N., Behrns K. E., Bejarano E., Belaid A., Belleudi F., Bénard G., Berchem G., Bergamaschi D., Bergami M., Berkhout B., Berliocchi L., Bernard A., Bernard M., Bernassola F., Bertolotti A., Bess A. S., Besteiro S., Bettuzzi S., Bhalla S., Bhattacharyya S., Bhutia S. K., Biagosch C., Bianchi M. W., Biard-Piechaczyk M., Billes V., Bincoletto C., Bingol B., Bird S. W., Bitoun M., Bjedov I., Blackstone C., Blanc L., Blanco G. A., Blomhoff H. K., Boada-Romero E., Böckler S., Boes M., Boesze-Battaglia K., Boise L. H., Bolino A., Boman A., Bonaldo P., Bordi M., Bosch J., Botana L. M., Botti J., Bou G., Bouché M., Bouchecareilh M., Boucher M.-J., Boulton M. E., Bouret S. G., Boya P., Boyer-Guittaut M., Bozhkov P. V., Brady N., Braga V. M. M., Brancolini C., Braus G. H., Bravo-San Pedro J. M., Brennan L. A., Bresnick E. H., Brest P., Bridges D., Bringer M.-A., Brini M., Brito G. C., Brodin B., Brookes P. S., Brown E. J., Brown K., Broxmeyer H. E., Bruhat A., Brum P. C., Brumell J. H., Brunetti-Pierri N., Bryson-Richardson R. J., Buch S., Buchan A. M., Budak H., Bulavin D. V., Bultman S. J., Bultynck G., Bumbasirevic V., Burelle Y., Burke R. E., Burmeister M., Bütikofer P., Caberlotto L., Cadwell K., Cahova M., Cai D., Cai J., Cai Q., Calatayud S., Camougrand N., Campanella M., Campbell G. R., Campbell M., Campello S., Candau R., Caniggia I., Cantoni L., Cao L., Caplan A. B., Caraglia M., Cardinali C., Cardoso S. M., Carew J. S., Carleton L. A., Carlin C. R., Carloni S., Carlsson S. R., Carmona-Gutierrez D., Carneiro L. A. M., Carnevali O., Carra S., Carrier A., Carroll B., Casas C., Casas J., Cassinelli G., Castets P., Castro-Obregon S., Cavallini G., Ceccherini I., Cecconi F., Cederbaum A. I., Ceña V., Cenci S., Cerella C., Cervia D., Cetrullo S., Chaachouay H., Chae H.-J., Chagin A. S., Chai C.-Y., Chakrabarti G., Chamilos G., Chan E. Y. W., Chan M. T. V., Chandra D., Chandra P., Chang C.-P., Chang R. C.-C., Chang T. Y., Chatham J. C., Chatterjee S., Chauhan S., Che Y., Cheetham M. E., Cheluvappa R., Chen C.-J., Chen G., Chen G.-C., Chen G., Chen H., Chen J. W., Chen J.-K., Chen M., Chen M., Chen P., Chen Q., Chen Q., Chen S.-D., Chen S., Chen S. S.-L., Chen W., Chen W.-J., Chen W. Q., Chen W., Chen X., Chen Y.-H., Chen Y.-G., Chen Y., Chen Y., Chen Y., Chen Y.-J., Chen Y.-Q., Chen Y., Chen Z., Chen Z., Cheng A., Cheng C. H. K., Cheng H., Cheong H., Cherry S., Chesney J., Cheung C. H. A., Chevet E., Chi H. C., Chi S.-G., Chiacchiera F., Chiang H.-L., Chiarelli R., Chiariello M., Chieppa M., Chin L.-S., Chiong M., Chiu G. N. C., Cho D.-H., Cho S.-G., Cho W. C., Cho Y.-Y., Cho Y.-S., Choi A. M. K., Choi E.-J., Choi E.-K., Choi J., Choi M. E., Choi S.-I., Chou T.-F., Chouaib S., Choubey D., Choubey V., Chow K.-C., Chowdhury K., Chu C. T., Chuang T.-H., Chun T., Chung H., Chung T., Chung Y.-L., Chwae Y.-J., Cianfanelli V., Ciarcia R., Ciechomska I. A., Ciriolo M. R., Cirone M., Claerhout S., Clague M. J., Clària J., Clarke P. G. H., Clarke R., Clementi E., Cleyrat C., Cnop M., Coccia E. M., Cocco T., Codogno P., Coers J., Cohen E. E. W., Colecchia D., Coletto L., Coll N. S., Colucci-Guyon E., Comincini S., Condello M., Cook K. L., Coombs G. H., Cooper C. D., Cooper J. M., Coppens I., Corasaniti M. T., Corazzari M., Corbalan R., Corcelle-Termeau E., Cordero M. D., Corral-Ramos C., Corti O., Cossarizza A., Costelli P., Costes S., Cotman S. L., Coto-Montes A., Cottet S., Couve E., Covey L. R., Cowart L. A., Cox J. S., Coxon F. P., Coyne C. B., Cragg M. S., Craven R. J., Crepaldi T., Crespo J. L., Criollo A., Crippa V., Cruz M. T., Cuervo A. M., Cuezva J. M., Cui T., Cutillas P. R., Czaja M. J., Czyzyk-Krzeska M. F., Dagda R. K., Dahmen U., Dai C., Dai W., Dai Y., Dalby K. N., Dalla Valle L., Dalmasso G., D’Amelio M., Damme M., Darfeuille-Michaud A., Dargemont C., Darley-Usmar V. M., Dasarathy S., Dasgupta B., Dash S., Dass C. R., Davey H. M., Davids L. M., Dávila D., Davis R. J., Dawson T. M., Dawson V. L., Daza P., de Belleroche J., de Figueiredo P., de Figueiredo R. C. B. Q., de la Fuente J., De Martino L., De Matteis A., De Meyer G. R. Y., De Milito A., De Santi M., de Souza W., De Tata V., De Zio D., Debnath J., Dechant R., Decuypere J.-P., Deegan S., Dehay B., Del Bello B., Del Re D. P., Delage-Mourroux R., Delbridge L. M. D., Deldicque L., Delorme-Axford E., Deng Y., Dengjel J., Denizot M., Dent P., Der C. J., Deretic V., Derrien B., Deutsch E., Devarenne T. P., Devenish R. J., Di Bartolomeo S., Di Daniele N., Di Domenico F., Di Nardo A., Di Paola S., Di Pietro A., Di Renzo L., DiAntonio A., Díaz-Araya G., Díaz-Laviada I., Diaz-Meco M. T., Diaz-Nido J., Dickey C. A., Dickson R. C., Diederich M., Digard P., Dikic I., Dinesh-Kumar S. P., Ding C., Ding W.-X., Ding Z., Dini L., Distler J. H. W., Diwan A., Djavaheri-Mergny M., Dmytruk K., Dobson R. C. J., Doetsch V., Dokladny K., Dokudovskaya S., Donadelli M., Dong X. C., Dong X., Dong Z., Donohue T. M. Jr, Doran K. S., D’Orazi G., Dorn G. W. II, Dosenko V., Dridi S., Drucker L., Du J., Du L.-L., Du L., du Toit A., Dua P., Duan L., Duann P., Dubey V. K., Duchen M. R., Duchosal M. A., Duez H., Dugail I., Dumit V. I., Duncan M. C., Dunlop E. A., Dunn W. A. Jr, Dupont N., Dupuis L., Durán R. V., Durcan T. M., Duvezin-Caubet S., Duvvuri U., Eapen V., Ebrahimi-Fakhari D., Echard A., Eckhart L., Edelstein C. L., Edinger A. L., Eichinger L., Eisenberg T., Eisenberg-Lerner A., Eissa N. T., El-Deiry W. S., El-Khoury V., Elazar Z., Eldar-Finkelman H., Elliott C. J. H., Emanuele E., Emmenegger U., Engedal N., Engelbrecht A.-M., Engelender S., Enserink J. M., Erdmann R., Erenpreisa J., Eri R., Eriksen J. L., Erman A., Escalante R., Eskelinen E.-L., Espert L., Esteban-Martínez L., Evans T. J., Fabri M., Fabrias G., Fabrizi C., Facchiano A., Færgeman N. J., Faggioni A., Fairlie W. D., Fan C., Fan D., Fan J., Fang S., Fanto M., Fanzani A., Farkas T., Faure M., Favier F. B., Fearnhead H., Federici M., Fei E., Felizardo T. C., Feng H., Feng Y., Feng Y., Ferguson T. A., Fernández Á. F., Fernandez-Barrena M. G., Fernandez-Checa J. C., Fernández-López A., Fernandez-Zapico M. E., Feron O., Ferraro E., Ferreira-Halder C. V., Fesus L., Feuer R., Fiesel F. C., Filippi-Chiela E. C., Filomeni G., Fimia G. M., Fingert J. H., Finkbeiner S., Finkel T., Fiorito F., Fisher P. B., Flajolet M., Flamigni F., Florey O., Florio S., Floto R. A., Folini M., Follo C., Fon E. A., Fornai F., Fortunato F., Fraldi A., Franco R., Francois A., François A., Frankel L. B., Fraser I. D. C., Frey N., Freyssenet D. G., Frezza C., Friedman S. L., Frigo D. E., Fu D., Fuentes J. M., Fueyo J., Fujitani Y., Fujiwara Y., Fujiya M., Fukuda M., Fulda S., Fusco C., Gabryel B., Gaestel M., Gailly P., Gajewska M., Galadari S., Galili G., Galindo I., Galindo M. F., Galliciotti G., Galluzzi L., Galluzzi L., Galy V., Gammoh N., Gandy S., Ganesan A. K., Ganesan S., Ganley I. G., Gannagé M., Gao F.-B., Gao F., Gao J.-X., Nannig L. G., García Véscovi E., Garcia-Macía M., Garcia-Ruiz C., Garg A. D., Garg P. K., Gargini R., Gassen N. C., Gatica D., Gatti E., Gavard J., Gavathiotis E., Ge L., Ge P., Ge S., Gean P.-W., Gelmetti V., Genazzani A. A., Geng J., Genschik P., Gerner L., Gestwicki J. E., Gewirtz D. A., Ghavami S., Ghigo E., Ghosh D., Giammarioli A. M., Giampieri F., Giampietri C., Giatromanolaki A., Gibbings D. J., Gibellini L., Gibson S. B., Ginet V., Giordano A., Giorgini F., Giovannetti E., Girardin S. E., Gispert S., Giuliano S., Gladson C. L., Glavic A., Gleave M., Godefroy N., Gogal R. M. Jr, Gokulan K., Goldman G. H., Goletti D., Goligorsky M. S., Gomes A. V., Gomes L. C., Gomez H., Gomez-Manzano C., Gómez-Sánchez R., Gonçalves D. A. P., Goncu E., Gong Q., Gongora C., Gonzalez C. B., Gonzalez-Alegre P., Gonzalez-Cabo P., González-Polo R. A., Goping I. S., Gorbea C., Gorbunov N. V., Goring D. R., Gorman A. M., Gorski S. M., Goruppi S., Goto-Yamada S., Gotor C., Gottlieb R. A., Gozes I., Gozuacik D., Graba Y., Graef M., Granato G. E., Grant G. D., Grant S., Gravina G. L., Green D. R., Greenhough A., Greenwood M. T., Grimaldi B., Gros F., Grose C., Groulx J.-F., Gruber F., Grumati P., Grune T., Guan J.-L., Guan K.-L., Guerra B., Guillen C., Gulshan K., Gunst J., Guo C., Guo L., Guo M., Guo W., Guo X.-G., Gust A. A., Gustafsson Å. B., Gutierrez E., Gutierrez M. G., Gwak H.-S., Haas A., Haber J. E., Hadano S., Hagedorn M., Hahn D. R., Halayko A. J., Hamacher-Brady A., Hamada K., Hamai A., Hamann A., Hamasaki M., Hamer I., Hamid Q., Hammond E. M., Han F., Han W., Handa J. T., Hanover J. A., Hansen M., Harada M., Harhaji-Trajkovic L., Harper J. W., Harrath A. H., Harris A. L., Harris J., Hasler U., Hasselblatt P., Hasui K., Hawley R. G., Hawley T. S., He C., He C. Y., He F., He G., He R.-R., He X.-H., He Y.-W., He Y.-Y., Heath J. K., Hébert M.-J., Heinzen R. A., Helgason G. V., Hensel M., Henske E. P., Her C., Herman P. K., Hernández A., Hernandez C., Hernández-Tiedra S., Hetz C., Hiesinger P. R., Higaki K., Hilfiker S., Hill B. G., Hill J. A., Hill W. D., Hino K., Hofius D., Hofman P., Höglinger G. U., Höhfeld J., Holz M. K., Hong Y., Hood D. A., Hoozemans J. J. M., Hoppe T., Hsu C., Hsu C.-Y., Hsu L.-C., Hu D., Hu G., Hu H.-M., Hu H., Hu M. C., Hu Y.-C., Hu Z.-W., Hua F., Hua Y., Huang C., Huang H.-L., Huang K.-H., Huang K.-Y., Huang S., Huang S., Huang W.-P., Huang Y.-R., Huang Y., Huang Y., Huber T. B., Huebbe P., Huh W.-K., Hulmi J. J., Hur G. M., Hurley J. H., Husak Z., Hussain S. N. A., Hussain S., Hwang J. J., Hwang S., Hwang T. I. S., Ichihara A., Imai Y., Imbriano C., Inomata M., Into T., Iovane V., Iovanna J. L., Iozzo R. V., Ip N. Y., Irazoqui J. E., Iribarren P., Isaka Y., Isakovic A. J., Ischiropoulos H., Isenberg J. S., Ishaq M., Ishida H., Ishii I., Ishmael J. E., Isidoro C., Isobe K.-i., Isono E., Issazadeh-Navikas S., Itahana K., Itakura E., Ivanov A. I., Iyer A. K. V., Izquierdo J. M., Izumi Y., Izzo V., Jäättelä M., Jaber N., Jackson D. J., Jackson W. T., Jacob T. G., Jacques T. S., Jagannath C., Jain A., Jana N. R., Jang B. K., Jani A., Janji B., Jannig P. R., Jansson P. J., Jean S., Jendrach M., Jeon J.-H., Jessen N., Jeung E.-B., Jia K., Jia L., Jiang H., Jiang H., Jiang L., Jiang T., Jiang X., Jiang X., Jiang X., Jiang Y., Jiang Y., Jiménez A., Jin C., Jin H., Jin L., Jin M., Jin S., Jinwal U. K., Jo E.-K., Johansen T., Johnson D. E., Johnson G. V. W., Johnson J. D., Jonasch E., Jones C., Joosten L. A. B., Jordan J., Joseph A.-M., Joseph B., Joubert A. M., Ju D., Ju J., Juan H.-F., Juenemann K., Juhász G., Seung Jung H., Jung J. U., Jung Y.-K., Jungbluth H., Justice M. J., Jutten B., Kaakoush N. O., Kaarniranta K., Kaasik A., Kabuta T., Kaeffer B., Kågedal K., Kahana A., Kajimura S., Kakhlon O., Kalia M., Kalvakolanu D. V., Kamada Y., Kambas K., Kaminskyy V. O., Kampinga H. H., Kandouz M., Kang C., Kang R., Kang T.-C., Kanki T., Kanneganti T.-D., Kanno H., Kanthasamy A. G., Kantorow M., Kaparakis-Liaskos M., Kapuy O., Karantza V., Razaul Karim M., Karmakar P., Kaser A., Kaushik S., Kawula T., Kaynar A. M., Ke P.-Y., Ke Z.-J., Kehrl J. H., Keller K. E., Kemper J. K., Kenworthy A. K., Kepp O., Kern A., Kesari S., Kessel D., Ketteler R., Kettelhut I. d. C., Khambu B., Khan M. M., Khandelwal V. K. M., Khare S., Kiang J. G., Kiger A. A., Kihara A., Kim A. L., Kim C. H., Kim D. R., Kim D.-H., Kim E. K., Kim H. Y., Kim H.-R., Kim J.-S., Kim J. H., Kim J. C., Kim J. H., Kim K. W., Kim M. D., Kim M.-M., Kim P. K., Kim S. W., Kim S.-Y., Kim Y.-S., Kim Y., Kimchi A., Kimmelman A. C., Kimura T., King J. S., Kirkegaard K., Kirkin V., Kirshenbaum L. A., Kishi S., Kitajima Y., Kitamoto K., Kitaoka Y., Kitazato K., Kley R. A., Klimecki W. T., Klinkenberg M., Klucken J., Knævelsrud H., Knecht E., Knuppertz L., Ko J.-L., Kobayashi S., Koch J. C., Koechlin-Ramonatxo C., Koenig U., Koh Y. H., Köhler K., Kohlwein S. D., Koike M., Komatsu M., Kominami E., Kong D., Kong H. J., Konstantakou E. G., Kopp B. T., Korcsmaros T., Korhonen L., Korolchuk V. I., Koshkina N. V., Kou Y., Koukourakis M. I., Koumenis C., Kovács A. L., Kovács T., Kovacs W. J., Koya D., Kraft C., Krainc D., Kramer H., Kravic-Stevovic T., Krek W., Kretz-Remy C., Krick R., Krishnamurthy M., Kriston-Vizi J., Kroemer G., Kruer M. C., Kruger R., Ktistakis N. T., Kuchitsu K., Kuhn C., Pratap Kumar A., Kumar A., Kumar A., Kumar D., Kumar D., Kumar R., Kumar S., Kundu M., Kung H.-J., Kuno A., Kuo S.-H., Kuret J., Kurz T., Kwok T., Kyu Kwon T., Tae Kwon Y., Kyrmizi I., La Spada A. R., Lafont F., Lahm T., Lakkaraju A., Lam T., Lamark T., Lancel S., Landowski T. H., Lane D. J. R., Lane J. D., Lanzi C., Lapaquette P., Lapierre L. R., Laporte J., Laukkarinen J., Laurie G. W., Lavandero S., Lavie L., LaVoie M. J., Law B. Y. K., Law H. K.-w., Law K. B., Layfield R., Lazo P. A., Le Cam L., Le Roch K. G., Le Stunff H., Leardkamolkarn V., Lecuit M., Lee B.-H., Lee C.-H., Lee E. F., Lee G. M., Lee H.-J., Lee H., Lee J. K., Lee J., Lee J.-h., Lee J. H., Lee M., Lee M.-S., Lee P. J., Lee S. W., Lee S.-J., Lee S.-J., Lee S. Y., Lee S. H., Lee S. S., Lee S.-J., Lee S., Lee Y.-R., Lee Y. J., Lee Y. H., Leeuwenburgh C., Lefort S., Legouis R., Lei J., Lei Q.-Y., Leib D. A., Leibowitz G., Lekli I., Lemaire S. D., Lemasters J. J., Lemberg M. K., Lemoine A., Leng S., Lenz G., Lenzi P., Lerman L. O., Lettieri Barbato D., Leu J. I.-J., Leung H. Y., Levine B., Lewis P. A., Lezoualc’h F., Li C., Li F., Li F.-J., Li J., Li K., Li L., Li M., Li M., Li Q., Li R., Li S., Li W., Li W., Li X., Li Y., Lian J., Liang C., Liang Q., Liao Y., Liberal J., Liberski P. P., Lie P., Lieberman A. P., Lim H. J., Lim K.-L., Lim K., Lima R. T., Lin C.-S., Lin C.-F., Lin F., Lin F., Lin F.-C., Lin K., Lin K.-H., Lin P.-H., Lin T., Lin W.-W., Lin Y.-S., Lin Y., Linden R., Lindholm D., Lindqvist L. M., Lingor P., Linkermann A., Liotta L. A., Lipinski M. M., Lira V. A., Lisanti M. P., Liton P. B., Liu B., Liu C., Liu C.-F., Liu F., Liu H.-J., Liu J., Liu J.-J., Liu J.-L., Liu K., Liu L., Liu L., Liu Q., Liu R.-Y., Liu S., Liu S., Liu W., Liu X.-D., Liu X., Liu X.-H., Liu X., Liu X., Liu X., Liu Y., Liu Y., Liu Z., Liu Z., Liuzzi J. P., Lizard G., Ljujic M., Lodhi I. J., Logue S. E., Lokeshwar B. L., Long Y. C., Lonial S., Loos B., López-Otín C., López-Vicario C., Lorente M., Lorenzi P. L., Lõrincz P., Los M., Lotze M. T., Lovat P. E., Lu B., Lu B., Lu J., Lu Q., Lu S.-M., Lu S., Lu Y., Luciano F., Luckhart S., Lucocq J. M., Ludovico P., Lugea A., Lukacs N. W., Lum J. J., Lund A. H., Luo H., Luo J., Luo S., Luparello C., Lyons T., Ma J., Ma Y., Ma Y., Ma Z., Machado J., Machado-Santelli G. M., Macian F., MacIntosh G. C., MacKeigan J. P., Macleod K. F., MacMicking J. D., MacMillan-Crow L. A., Madeo F., Madesh M., Madrigal-Matute J., Maeda A., Maeda T., Maegawa G., Maellaro E., Maes H., Magariños M., Maiese K., Maiti T. K., Maiuri L., Maiuri M. C., Maki C. G., Malli R., Malorni W., Maloyan A., Mami-Chouaib F., Man N., Mancias J. D., Mandelkow E.-M., Mandell M. A., Manfredi A. A., Manié S. N., Manzoni C., Mao K., Mao Z., Mao Z.-W., Marambaud P., Marconi A. M., Marelja Z., Marfe G., Margeta M., Margittai E., Mari M., Mariani F. V., Marin C., Marinelli S., Mariño G., Markovic I., Marquez R., Martelli A. M., Martens S., Martin K. R., Martin S. J., Martin S., Martin-Acebes M. A., Martín-Sanz P., Martinand-Mari C., Martinet W., Martinez J., Martinez-Lopez N., Martinez-Outschoorn U., Martínez-Velázquez M., Martinez-Vicente M., Martins W. K., Mashima H., Mastrianni J. A., Matarese G., Matarrese P., Mateo R., Matoba S., Matsumoto N., Matsushita T., Matsuura A., Matsuzawa T., Mattson M. P., Matus S., Maugeri N., Mauvezin C., Mayer A., Maysinger D., Mazzolini G. D., McBrayer M. K., McCall K., McCormick C., McInerney G. M., McIver S. C., McKenna S., McMahon J. J., McNeish I. A., Mechta-Grigoriou F., Medema J. P., Medina D. L., Megyeri K., Mehrpour M., Mehta J. L., Mei Y., Meier U.-C., Meijer A. J., Meléndez A., Melino G., Melino S., de Melo E. J. T., Mena M. A., Meneghini M. D., Menendez J. A., Menezes R., Meng L., Meng L.-h., Meng S., Menghini R., Menko A. S., Menna-Barreto R. F. S., Menon M. B., Meraz-Ríos M. A., Merla G., Merlini L., Merlot A. M., Meryk A., Meschini S., Meyer J. N., Mi M.-t., Miao C.-Y., Micale L., Michaeli S., Michiels C., Migliaccio A. R., Mihailidou A. S., Mijaljica D., Mikoshiba K., Milan E., Miller-Fleming L., Mills G. B., Mills I. G., Minakaki G., Minassian B. A., Ming X.-F., Minibayeva F., Minina E. A., Mintern J. D., Minucci S., Miranda-Vizuete A., Mitchell C. H., Miyamoto S., Miyazawa K., Mizushima N., Mnich K., Mograbi B., Mohseni S., Moita L. F., Molinari M., Molinari M., Møller A. B., Mollereau B., Mollinedo F., Mongillo M., Monick M. M., Montagnaro S., Montell C., Moore D. J., Moore M. N., Mora-Rodriguez R., Moreira P. I., Morel E., Beatrice Morelli M., Moreno S., Morgan M. J., Moris A., Moriyasu Y., Morrison J. L., Morrison L. A., Morselli E., Moscat J., Moseley P. L., Mostowy S., Motori E., Mottet D., Mottram J. C., Moussa C. E.-H., Mpakou V. E., Mukhtar H., Mulcahy Levy J. M., Muller S., Muñoz-Moreno R., Muñoz-Pinedo C., Münz C., Murphy M. E., Murray J. T., Murthy A., Mysorekar I. U., Nabi I. R., Nabissi M., Nader G. A., Nagahara Y., Nagai Y., Nagata K., Nagelkerke A., Nagy P., Naidu S. R., Nair S., Nakano H., Nakatogawa H., Nanjundan M., Napolitano G., Naqvi N. I., Nardacci R., Narendra D. P., Narita M., Nascimbeni A. C., Natarajan R., Navegantes L. C., Nawrocki S. T., Nazarko T. Y., Nazarko V. Y., Neill T., Neri L. M., Netea M. G., Netea-Maier R. T., Neves B. M., Ney P. A., Nezis I. P., Nguyen H. T. T., Nguyen H. P., Nicot A.-S., Nilsen H., Nilsson P., Nishimura M., Nishino I., Niso-Santano M., Niu H., Nixon R. A., Njar V. C. O., Noda T., Noegel A. A., Nolte E. M., Norberg E., Norga K. K., Kazemi Noureini S., Notomi S., Notterpek L., Nowikovsky K., Nukina N., Nürnberger T., O’Donnell V. B., O’Donovan T., O’Dwyer P. J., Oehme I., Oeste C. L., Ogawa M., Ogretmen B., Ogura Y., Oh Y. J., Ohmuraya M., Ohshima T., Ojha R., Okamoto K., Okazaki T., Oliver F. J., Ollinger K., Olsson S., Orban D. P., Ordonez P., Orhon I., Orosz L., O’Rourke E. J., Orozco H., Ortega A. L., Ortona E., Osellame L. D., Oshima J., Oshima S., Osiewacz H. D., Otomo T., Otsu K., Ou J.-h. J., Outeiro T. F., Ouyang D.-y., Ouyang H., Overholtzer M., Ozbun M. A., Ozdinler P. H., Ozpolat B., Pacelli C., Paganetti P., Page G., Pages G., Pagnini U., Pajak B., Pak S. C., Pakos-Zebrucka K., Pakpour N., Palková Z., Palladino F., Pallauf K., Pallet N., Palmieri M., Paludan S. R., Palumbo C., Palumbo S., Pampliega O., Pan H., Pan W., Panaretakis T., Pandey A., Pantazopoulou A., Papackova Z., Papademetrio D. L., Papassideri I., Papini A., Parajuli N., Pardo J., Parekh V. V., Parenti G., Park J.-I., Park J., Park O. K., Parker R., Parlato R., Parys J. B., Parzych K. R., Pasquet J.-M., Pasquier B., Pasumarthi K. B. S., Patschan D., Patterson C., Pattingre S., Pattison S., Pause A., Pavenstädt H., Pavone F., Pedrozo Z., Peña F. J., Peñalva M. A., Pende M., Peng J., Penna F., Penninger J. M., Pensalfini A., Pepe S., Pereira G. J. S., Pereira P. C., Pérez-de la Cruz V., Pérez-Pérez M. E., Pérez-Rodríguez D., Pérez-Sala D., Perier C., Perl A., Perlmutter D. H., Perrotta I., Pervaiz S., Pesonen M., Pessin J. E., Peters G. J., Petersen M., Petrache I., Petrof B. J., Petrovski G., Phang J. M., Piacentini M., Pierdominici M., Pierre P., Pierrefite-Carle V., Pietrocola F., Pimentel-Muiños F. X., Pinar M., Pineda B., Pinkas-Kramarski R., Pinti M., Pinton P., Piperdi B., Piret J. M., Platanias L. C., Platta H. W., Plowey E. D., Pöggeler S., Poirot M., Polčic P., Poletti A., Poon A. H., Popelka H., Popova B., Poprawa I., Poulose S. M., Poulton J., Powers S. K., Powers T., Pozuelo-Rubio M., Prak K., Prange R., Prescott M., Priault M., Prince S., Proia R. L., Proikas-Cezanne T., Prokisch H., Promponas V. J., Przyklenk K., Puertollano R., Pugazhenthi S., Puglielli L., Pujol A., Puyal J., Pyeon D., Qi X., Qian W.-b., Qin Z.-H., Qiu Y., Qu Z., Quadrilatero J., Quinn F., Raben N., Rabinowich H., Radogna F., Ragusa M. J., Rahmani M., Raina K., Ramanadham S., Ramesh R., Rami A., Randall-Demllo S., Randow F., Rao H., Rao V. A., Rasmussen B. B., Rasse T. M., Ratovitski E. A., Rautou P.-E., Ray S. K., Razani B., Reed B. H., Reggiori F., Rehm M., Reichert A. S., Rein T., Reiner D. J., Reits E., Ren J., Ren X., Renna M., Reusch J. E. B., Revuelta J. L., Reyes L., Rezaie A. R., Richards R. I., Richardson D. R., Richetta C., Riehle M. A., Rihn B. H., Rikihisa Y., Riley B. E., Rimbach G., Rippo M. R., Ritis K., Rizzi F., Rizzo E., Roach P. J., Robbins J., Roberge M., Roca G., Roccheri M. C., Rocha S., Rodrigues C. M. P., Rodríguez C. I., Rodriguezde Cordoba S., Rodriguez-Muela N., Roelofs J., Rogov V. V., Rohn T. T., Rohrer B., Romanelli D., Romani L., Romano P. S., Roncero M. I. G., Rosa J. L., Rosello A., Rosen K. V., Rosenstiel P., Rost-Roszkowska M., Roth K. A., Roué G., Rouis M., Rouschop K. M., Ruan D. T., Ruano D., Rubinsztein D. C., Rucker E. B. III, Rudich A., Rudolf E., Rudolf R., Ruegg M. A., Ruiz-Roldan C., Ruparelia A. A., Rusmini P., Russ D. W., Russo G. L., Russo G., Russo R., Rusten T. E., Ryabovol V., Ryan K. M., Ryter S. W., Sabatini D. M., Sacher M., Sachse C., Sack M. N., Sadoshima J., Saftig P., Sagi-Eisenberg R., Sahni S., Saikumar P., Saito T., Saitoh T., Sakakura K., Sakoh-Nakatogawa M., Sakuraba Y., Salazar-Roa M., Salomoni P., Saluja A. K., Salvaterra P. M., Salvioli R., Samali A., Sanchez A. M. J., Sánchez-Alcázar J. A., Sanchez-Prieto R., Sandri M., Sanjuan M. A., Santaguida S., Santambrogio L., Santoni G., Dos Santos C. N., Saran S., Sardiello M., Sargent G., Sarkar P., Sarkar S., Rosa Sarrias M., Sarwal M. M., Sasakawa C., Sasaki M., Sass M., Sato K., Sato M., Satriano J., Savaraj N., Saveljeva S., Schaefer L., Schaible U. E., Scharl M., Schatzl H. M., Schekman R., Scheper W., Schiavi A., Schipper H. M., Schmeisser H., Schmidt J., Schmitz I., Schneider B. E., Schneider E. M., Schneider J. L., Schon E. A., Schönenberger M. J., Schönthal A. H., Schorderet D. F., Schröder B., Schuck S., Schulze R. J., Schwarten M., Schwarz T. L., Sciarretta S., Scotto K., Scovassi A. I., Screaton R. A., Screen M., Seca H., Sedej S., Segatori L., Segev N., Seglen P. O., Seguí-Simarro J. M., Segura-Aguilar J., Seki E., Seiliez I., Sell C., Semenkovich C. F., Semenza G. L., Sen U., Serra A. L., Serrano-Puebla A., Sesaki H., Setoguchi T., Settembre C., Shacka J. J., Shajahan-Haq A. N., Shapiro I. M., Sharma S., She H., Shen C.-K. J., Shen C.-C., Shen H.-M., Shen S., Shen W., Sheng R., Sheng X., Sheng Z.-H., Shepherd T. G., Shi J., Shi Q., Shi Q., Shi Y., Shibutani S., Shibuya K., Shidoji Y., Shieh J.-J., Shih C.-M., Shimada Y., Shimizu S., Shin D. W., Shinohara M. L., Shintani M., Shintani T., Shioi T., Shirabe K., Shiri-Sverdlov R., Shirihai O., Shore G. C., Shu C.-W., Shukla D., Sibirny A. A., Sica V., Sigurdson C. J., Sigurdsson E. M., Sijwali P. S., Sikorska B., Silveira W. A., Silvente-Poirot S., Silverman G. A., Simak J., Simmet T., Simon A. K., Simon H.-U., Simone C., Simons M., Simonsen A., Singh R., Singh S. V., Singh S. K., Sinha D., Sinha S., Sinicrope F. A., Sirko A., Sirohi K., Sishi B. J. N., Sittler A., Siu P. M., Sivridis E., Skwarska A., Slack R., Slaninová I., Slavov N., Smaili S. S., Smalley K. S. M., Smith D. R., Soenen S. J., Soleimanpour S. A., Solhaug A., Somasundaram K., Son J. H., Sonawane A., Song C., Song F., Song H. K., Song J.-X., Song W., Soo K. Y., Sood A. K., Soong T. W., Soontornniyomkij V., Sorice M., Sotgia F., Soto-Pantoja D. R., Sotthibundhu A., Sousa M. J., Spaink H. P., Span P. N., Spang A., Sparks J. D., Speck P. G., Spector S. A., Spies C. D., Springer W., St Clair D., Stacchiotti A., Staels B., Stang M. T., Starczynowski D. T., Starokadomskyy P., Steegborn C., Steele J. W., Stefanis L., Steffan J., Stellrecht C. M., Stenmark H., Stepkowski T. M., Stern S. T., Stevens C., Stockwell B. R., Stoka V., Storchova Z., Stork B., Stratoulias V., Stravopodis D. J., Strnad P., Marie Strohecker A., Ström A.-L., Stromhaug P., Stulik J., Su Y.-X., Su Z., Subauste C. S., Subramaniam S., Sue C. M., Suh S. W., Sui X., Sukseree S., Sulzer D., Sun F.-L., Sun J., Sun J., Sun S.-Y., Sun Y., Sun Y., Sun Y., Sundaramoorthy V., Sung J., Suzuki H., Suzuki K., Suzuki N., Suzuki T., Suzuki Y. J., Swanson M. S., Swanton C., Swärd K., Swarup G., Sweeney S. T., Sylvester P. W., Szatmari Z., Szegezdi E., Szlosarek P. W., Taegtmeyer H., Tafani M., Taillebourg E., Tait S. W. G., Takacs-Vellai K., Takahashi Y., Takáts S., Takemura G., Takigawa N., Talbot N. J., Tamagno E., Tamburini J., Tan C.-P., Tan L., Tan M. L., Tan M., Tan Y.-J., Tanaka K., Tanaka M., Tang D., Tang D., Tang G., Tanida I., Tanji K., Tannous B. A., Tapia J. A., Tasset-Cuevas I., Tatar M., Tavassoly I., Tavernarakis N., Taylor A., Taylor G. S., Taylor G. A., Taylor J. P., Taylor M. J., Tchetina E. V., Tee A. R., Teixeira-Clerc F., Telang S., Tencomnao T., Teng B.-B., Teng R.-J., Terro F., Tettamanti G., Theiss A. L., Theron A. E., Thomas K. J., Thomé M. P., Thomes P. G., Thorburn A., Thorner J., Thum T., Thumm M., Thurston T. L. M., Tian L., Till A., Ting J. P.-y., Titorenko V. I., Toker L., Toldo S., Tooze S. A., Topisirovic I., Torgersen M. L., Torosantucci L., Torriglia A., Torrisi M. R., Tournier C., Towns R., Trajkovic V., Travassos L. H., Triola G., Tripathi D. N., Trisciuoglio D., Troncoso R., Trougakos I. P., Truttmann A. C., Tsai K.-J., Tschan M. P., Tseng Y.-H., Tsukuba T., Tsung A., Tsvetkov A. S., Tu S., Tuan H.-Y., Tucci M., Tumbarello D. A., Turk B., Turk V., Turner R. F. B., Tveita A. A., Tyagi S. C., Ubukata M., Uchiyama Y., Udelnow A., Ueno T., Umekawa M., Umemiya-Shirafuji R., Underwood B. R., Ungermann C., Ureshino R. P., Ushioda R., Uversky V. N., Uzcátegui N. L., Vaccari T., Vaccaro M. I., Váchová L., Vakifahmetoglu-Norberg H., Valdor R., Valente E. M., Vallette F., Valverde A. M., Van den Berghe G., Van Den Bosch L., van den Brink G. R., van der Goot F. G., van der Klei I. J., van der Laan L. J. W., van Doorn W. G., van Egmond M., van Golen K. L., Van Kaer L., van Lookeren Campagne M., Vandenabeele P., Vandenberghe W., Vanhorebeek I., Varela-Nieto I., Vasconcelos M. H., Vasko R., Vavvas D. G., Vega-Naredo I., Velasco G., Velentzas A. D., Velentzas P. D., Vellai T., Vellenga E., Vendelbo M. H., Venkatachalam K., Ventura N., Ventura S., Veras P. S. T., Verdier M., Vertessy B. G., Viale A., Vidal M., Vieira H. L. A., Vierstra R. D., Vigneswaran N., Vij N., Vila M., Villar M., Villar V. H., Villarroya J., Vindis C., Viola G., Teresa Viscomi M., Vitale G., Vogl D. T., Voitsekhovskaja O. V., von Haefen C., von Schwarzenberg K., Voth D. E., Vouret-Craviari V., Vuori K., Vyas J. M., Waeber C., Lyn Walker C., Walker M. J., Walter J., Wan L., Wan X., Wang B., Wang C., Wang C.-Y., Wang C., Wang C., Wang C., Wang D., Wang F., Wang F., Wang G., Wang H.-j., Wang H., Wang H.-G., Wang H., Wang H.-D., Wang J., Wang J., Wang M., Wang M.-Q., Wang P.-Y., Wang P., Wang R. C., Wang S., Wang T.-F., Wang X., Wang X.-j., Wang X.-W., Wang X., Wang X., Wang Y., Wang Y., Wang Y., Wang Y.-J., Wang Y., Wang Y., Wang Y. T., Wang Y., Wang Z.-N., Wappner P., Ward C., Ward D. M. V., Warnes G., Watada H., Watanabe Y., Watase K., Weaver T. E., Weekes C. D., Wei J., Weide T., Weihl C. C., Weindl G., Weis S. N., Wen L., Wen X., Wen Y., Westermann B., Weyand C. M., White A. R., White E., Whitton J. L., Whitworth A. J., Wiels J., Wild F., Wildenberg M. E., Wileman T., Wilkinson D. S., Wilkinson S., Willbold D., Williams C., Williams K., Williamson P. R., Winklhofer K. F., Witkin S. S., Wohlgemuth S. E., Wollert T., Wolvetang E. J., Wong E., Wong G. W., Wong R. W., Wong V. K. W., Woodcock E. A., Wright K. L., Wu C., Wu D., Wu G. S., Wu J., Wu J., Wu M., Wu M., Wu S., Wu W. K. K., Wu Y., Wu Z., Xavier C. P. R., Xavier R. J., Xia G.-X., Xia T., Xia W., Xia Y., Xiao H., Xiao J., Xiao S., Xiao W., Xie C.-M., Xie Z., Xie Z., Xilouri M., Xiong Y., Xu C., Xu C., Xu F., Xu H., Xu H., Xu J., Xu J., Xu J., Xu L., Xu X., Xu Y., Xu Y., Xu Z.-X., Xu Z., Xue Y., Yamada T., Yamamoto A., Yamanaka K., Yamashina S., Yamashiro S., Yan B., Yan B., Yan X., Yan Z., Yanagi Y., Yang D.-S., Yang J.-M., Yang L., Yang M., Yang P.-M., Yang P., Yang Q., Yang W., Yang W. Y., Yang X., Yang Y., Yang Y., Yang Z., Yang Z., Yao M.-C., Yao P. J., Yao X., Yao Z., Yao Z., Yasui L. S., Ye M., Yedvobnick B., Yeganeh B., Yeh E. S., Yeyati P. L., Yi F., Yi L., Yin X.-M., Yip C. K., Yoo Y.-M., Yoo Y. H., Yoon S.-Y., Yoshida K.-I., Yoshimori T., Young K. H., Yu H., Yu J. J., Yu J.-T., Yu J., Yu L., Yu W. H., Yu X.-F., Yu Z., Yuan J., Yuan Z.-M., Yue B. Y. J. T., Yue J., Yue Z., Zacks D. N., Zacksenhaus E., Zaffaroni N., Zaglia T., Zakeri Z., Zecchini V., Zeng J., Zeng M., Zeng Q., Zervos A. S., Zhang D. D., Zhang F., Zhang G., Zhang G.-C., Zhang H., Zhang H., Zhang H., Zhang H., Zhang J., Zhang J., Zhang J., Zhang J., Zhang J.-p., Zhang L., Zhang L., Zhang L., Zhang L., Zhang M.-Y., Zhang X., Zhang X. D., Zhang Y., Zhang Y., Zhang Y., Zhang Y., Zhang Y., Zhao M., Zhao W.-L., Zhao X., Zhao Y. G., Zhao Y., Zhao Y., Zhao Y.-x., Zhao Z., Zhao Z. J., Zheng D., Zheng X.-L., Zheng X., Zhivotovsky B., Zhong Q., Zhou G.-Z., Zhou G., Zhou H., Zhou S.-F., Zhou X.-j., Zhu H., Zhu H., Zhu W.-G., Zhu W., Zhu X.-F., Zhu Y., Zhuang S.-M., Zhuang X., Ziparo E., Zois C. E., Zoladek T., Zong W.-X., Zorzano A., Zughaier S. M., Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laplante M., Sabatini D. M., mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang S. A., Pacold M. E., Cervantes C. L., Lim D., Lou H. J., Ottina K., Gray N. S., Turk B. E., Yaffe M. B., Sabatini D. M., mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 1236566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korolchuk V. I., Mansilla A., Menzies F. M., Rubinsztein D. C., Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Progida C., Cogli L., Piro F., De Luca A., Bakke O., Bucci C., Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci. 123, 1480–1491 (2010). [DOI] [PubMed] [Google Scholar]
- 50.McIntyre G. F., Erickson A. H., Procathepsins L and D are membrane-bound in acidic microsomal vesicles. J. Biol. Chem. 266, 15438–15445 (1991). [PubMed] [Google Scholar]
- 51.Khundadze M., Kollmann K., Koch N., Biskup C., Nietzsche S., Zimmer G., Hennings J. C., Huebner A. K., Symmank J., Jahic A., Ilina E. I., Karle K., Schöls L., Kessels M., Braulke T., Qualmann B., Kurth I., Beetz C., Hübner C. A., A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLOS Genet. 9, e1003988 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Platt F. M., Boland B., van der Spoel A. C., The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 199, 723–734 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riederer M. A., Soldati T., Shapiro A. D., Lin J., Pfeffer S. R., Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 125, 573–582 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sullivan P. M., Zhou X., Robins A. M., Paushter D. H., Kim D., Smolka M. B., Hu F., The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4, 51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suzuki H., Kaizuka T., Mizushima N., Noda N. N., Structure of the Atg101–Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat. Struct. Mol. Biol. 22, 572–580 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N., Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J.-i., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K., Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Ejlerskov P., Hultberg J. G., Wang J., Carlsson R., Ambjørn M., Kuss M., Liu Y., Porcu G., Kolkova K., Friis Rundsten C., Ruscher K., Pakkenberg B., Goldmann T., Loreth D., Prinz M., Rubinsztein D. C., Issazadeh-Navikas S., Lack of neuronal IFN-β-IFNAR causes Lewy body- and Parkinson’s disease-like dementia. Cell 163, 324–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rocha N. P., de Miranda A. S., Teixeira A. L., Insights into neuroinflammation in Parkinson’s disease: From biomarkers to anti-inflammatory based therapies. Biomed. Res. Int. 2015, 628192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Webster C. P., Smith E. F., Bauer C. S., Moller A., Hautbergue G. M., Ferraiuolo L., Myszczynska M. A., Higginbottom A., Walsh M. J., Whitworth A. J., Kaspar B. K., Meyer K., Shaw P. J., Grierson A. J., De Vos K. J., The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao S., MacNair L., McLean J., McGoldrick P., McKeever P., Soleimani S., Keith J., Zinman L., Rogaeva E., Robertson J., C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res. 10.1016/j.brainres.2016.04.062 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Xiao S., MacNair L., McGoldrick P., McKeever P. M., McLean J. R., Zhang M., Keith J., Zinman L., Rogaeva E., Robertson J., Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann. Neurol. 78, 568–583 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Chen J.-F., Zhang Y., Wilde J., Hansen K. C., Lai F., Niswander L., Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat. Commun. 5, 3885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/9/e1601167/DC1
fig. S1. Isolation of SMCR8-associated proteins.
fig. S2. Identifying C9orf72- or SMCR8-associated Rab GTPases.
fig. S3. Characterization of SMCR8 and C9orf72 shRNA constructs.
table S1. Polypeptide composition through Flag-C9ORF72 immunopurification by mass spectrometry.