

Abstract
Rationale
Activation of NLRP3 inflammasome mediating IL-1β secretion has emerged as an important component of inflammatory processes in atherosclerosis. Mitochondrial DNA (mtDNA) damage is detrimental in atherosclerosis and mitochondria are central regulators of the NLRP3 inflammasome. Human atherosclerotic plaques express increased mtDNA damage. The major DNA glycosylase OGG1, is responsible for removing the most abundant form of oxidative DNA damage.
Objective
To test the role of OGG1 in development of atherosclerosis in mouse.
Methods and Results
We observed that Ogg1 expression decreases over time in atherosclerotic lesion macrophages of Ldlr KO mice fed a western diet. Ogg1−/−Ldlr−/− mice fed a western diet resulted in an increase in plaque size and lipid content. We found increased oxidized mtDNA, inflammasome activation, and apoptosis in atherosclerotic lesions and also higher serum IL-1β and IL-18 in Ogg1−/−Ldlr−/− mice compared with Ldlr−/−. Transplantation with Ogg1−/− bone marrow (BM) into Ldlr−/− mice led to larger atherosclerotic lesions and increased IL-1β production. However, transplantation of Ogg1−/−Nlrp3−/− BM reversed the Ogg1−/− phenotype of increased plaque size. Ogg1−/− macrophages showed increased oxidized mtDNA and had greater amounts of cytosolic mtDNA and cytochrome c, increased apoptosis, and more IL-1β secretion. Finally, we found that proatherogenic miR-33 can directly inhibit human OGG1 expression and indirectly suppress both mouse and human OGG1 via AMPK.
Conclusions
OGG1 plays a protective role in atherogenesis by preventing excessive inflammasome activation. Our study provides insight into a new target for therapeutic intervention based on a link between oxidative mtDNA damage, OGG1, and atherosclerosis via NLRP3 inflammasome.
Keywords: Ogg1, atherosclerosis, DNA repair, mitochondria, inflammasome, macrophage
Subject Terms: Vascular Disease, Atherosclerosis, Animal Models of Human Disease, Inflammation
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease that arises from an imbalance in lipid metabolism and a maladaptive immune response driven by the accumulation of cholesterol-laden macrophages in the artery wall. Rupture of atheromatous lesions results in thrombotic occlusion of coronary and cerebral vessels, producing the clinical complications of atherosclerosis1, 2. There is a growing understanding that blood monocytes are recruited to the inflamed vascular wall develop into inflammatory macrophages (M1-like phenotype) and foam cells, which contribute to pathogenesis at many stages of this disease and, therefore, represent a target for therapeutic interventions3, 4. In addition to hyperlipidemia and other genetic factors, inflammation is now known as a critical contributor to atherogenesis. The activation of NLRP3 inflammasome mediating IL-1β secretion has recently emerged as an important component of inflammatory processes underlying atherosclerosis5–7. The NLRP3 inflammasome can be activated by many diverse danger signals and mitochondria (mt) function as the central hub for integrating these various signals5, 8, 9. We recently showed that during apoptosis/pyroptosis, mitochondrial DNA (mtDNA) damaged by oxidative stress activates the NLRP3 inflammasome, releasing active IL-1β10. Mt are a major site of ROS production and mtDNA is a vulnerable target for ROS-mediated oxidative damage due to its close proximity and the lack of protective histones. Human atherosclerotic plaques showed increased mtDNA damage compared with normal vessels and leukocyte mtDNA damage was associated with higher-risk plaques11, 12.
Oxidative damage in DNA is repaired primarily via the enzymes of the base excision repair (BER) pathway. Among these, OGG1, the major DNA glycosylase, is responsible for removal of 7,8-dihydro-8-oxo-2′-deoxyguanosine (8-OH-dG), one of the most frequent endogenous base lesions formed in the DNA of aerobic organisms13. OGG1 deficiency leads to elevated levels 8-OH-dG in mtDNA14, 15. In contrast, mitochondrial overexpression of Ogg1 improves mitochondrial function, cell survival and fewer mtDNA deletions through increased repair of 8-OH-dG under oxidative stress conditions in vitro16. A recent study reported in an abstract format showed that loss of OGG1 led to increased apoptosis and larger fatty streaks in mice on western diet and overexpression of Ogg1 reversed this phenotype, suggesting that Ogg1 may play a protective role in atherosclerosis17.
MicroRNAs (miRNAs) are critical post-transcriptional regulators of their target genes. miR-33 targets a number of genes involved in regulating cellular cholesterol export and fatty acid oxidation, including Prkaa1 (protein kinase, AMP-activated, alpha 1 catalytic subunit) whose gene product is commonly referred to as AMPKα1 (5′ AMP-activated protein kinase alpha subunit 1), which is a metabolic master switch and a target of several drugs used for metabolic diseases in patients18–20. Interestingly, AMPK also regulates Ogg1 expression21. Antagonizing miR-33a/b may be an effective strategy for raising plasma HDL levels and protecting against atherosclerosis in mice and non-human primates22, 23.
In this work, we show the atheroprotective role of OGG1 in a western diet-induced model of atherosclerosis using Ldlr−/− mice. The loss of OGG1 resulted in increased mtDNA damage, increased NLRP3 inflammasome activation in macrophages, increased IL-1β production, and more apoptosis in atherosclerotic plaques, which led to accelerated atherogenesis. NLRP3 deficiency counteracted the loss of OGG1, evidenced by smaller atheromatous plaque lesions.
METHODS
Animals and diets
Ogg1−/− mice were kindly provided from Dr. Christi A. Walter (Univ. Texas Health Science Center at San Antonio). Nlrp3−/− mice were provided by Dr. K. A. Fitzgerald (University of Massachusetts Medical School, Worcester, MA). We have crossed for at least 8 generations and established Ogg1−/−Ldlr−/− and Ogg1−/−Nlrp3−/− DKO mice. Starting at 8 weeks of age, mice were fed a western diet (WD) (TD88137, Harlan Teklad) for 16 weeks. For bone marrow (BM) transplantation, BM from Ogg−/−, WT, and Ogg1−/−Nlrp3−/− mice was transplanted into irradiated Ldlr−/− mice. After recovery (8 wks), chimeric mice were placed on a WD for 12 wks. Whole blood was collected to confirm the efficiency of the BM transplant. All animal experimental procedures were conducted in strict compliance with the policies on animal welfare of the National Institute of Health. The protocol was approved by the Animal Care and Use Committee at Cedars-Sinai Medical Center.
Assessment of atherosclerotic lesions in the aorta and aortic sinus
The aortas were dissected and the adherent (adventitial) fat was gently removed. Whole aortas were opened longitudinally from the aortic arch to the iliac bifurcation, mounted en face, and stained for lipids with Oil Red O. Hearts were embedded in OCT (Tissue-Tek; Sakura, Torrance, CA) and serial 7 μm–thick cryosections from the aortic sinus were mounted and stained with Oil Red O and hematoxylin. Image analysis was performed by a trained observer blinded to the genotype of the mice. Representative images were obtained and lesion areas were quantified with Image analysis software using a BZ-9000 microscope (Keyence, Itasca, IL). The lesion area in the aorta en face preparations was expressed as a percent of the aortic surface area, as previously reported. The lesion area and lipid-stained areas in the aortic sinus were measured24.
Immunofluorescent analysis of cryosections of the aortic sinus
Apoptotic cells in lesions were detected by TUNEL after proteinase K and EDTA treatment, using the In Situ Cell Death Detection Kit (Millipore). Caspase-1 activity was detected by FLICA staining. For Immunohistochemical staining for frozen sections, fixing and antigen blocking was performed using immunoglobulin from the species of the secondary antibodies. Next, the sections were incubated with primary antibodies overnight at 4′C, followed by incubation with an appropriate secondary antibodies conjugated with fluorescent dyes. For assessment of macrophage co-localization, macrophages were detected using anti-F4/80 antibody (eBioscience), and anti-CD45 (eBioscience); nuclei were counterstained with DAPI. For some analysis anti-8OH-dG (Bioss) was used, along with anti-TOM20 (Santa Cruz, CA). Images were captured using BZ-9000 microscope (Keyence, Itasca, IL) and analyzed by BZ analyzer software.
Laser-capture microdissection (LCM)
Laser-capture microdissection was performed using a LMD instrument (Leica LMD 700, Leica Biosystem) as previously described25. To visualize F4/80+ cells, a guide slide was prepared by staining for F4/80 as described above. Cells corresponding to F4/80+ area in serial sections were collected, and RNA was extracted using the RNA Isolation Kit (Clontech). cDNA was synthesized and SYBR Green 2xPCR mixture (Clontech) was used in qPCR according to the manufacturer’s instructions. Ogg1 primers: Fw: 5′-tgtgtaccgaggagacgaca-3′, Rv: 5′- ctgtgccaggctgacatcta -3′.
Statistics
All data are expressed as means ± SD. Statistical differences were measured using either an unpaired Student t test or 1-way analysis of variance (ANOVA) when appropriate with Tukeys post-hoc test. In some cases, the area under the curve was determined, followed by one-way ANOVA with Tukeys post-hoc test. A P value of ≤0.05 was considered statistically significant. Data analysis was performed using Prism software version 5.0a (GraphPad, San Diego, CA). Asterisks in the figures represent the following: *: P ≤ 0.05; **: P ≤ 0.01; and ***: P ≤ 0.001.
See online Supplement for remaining Methods.
RESULTS
OGG1 deficiency promotes apoptotic and inflammatory responses in macrophages
Somatic mtDNA damage has been reported to accumulate with ageing associated coronary atherosclerotic heart disease26. OGG1 removes the most common oxidative DNA lesion, 8-OH-dG, which we have previously linked to NLRP3 inflammasome activation10. We wanted to determine the effect of OGG1 deficiency on oxidized mtDNA damage in macrophages. Ogg1−/− bone marrow derived macrophages (BMDM) treated with menadione, a ROS inducer, had greater amounts of oxidative damage in their mtDNA as measured by 8-OH-dG dot blot analysis compared with WT BMDMs (Figure 1A). Failure to repair oxidative DNA damage in OGG1 deficient mice can lead to further mt dysfunction and apoptosis27. We therefore examined the effect of the cytotoxic oxysterol, 7-ketocholesterol (7-KC), which is found in oxidized LDL and atherosclerotic lesions, on apoptosis28. 7-KC treatment resulted in more cytochrome c (cyt c) and mtDNA in the cytosol of OGG1 deficient macrophages compared to WT cells (Figure 1B and C). 7-KC can activate the NLRP3 inflammasome29, 30. To assess whether mtDNA defects could promote an inflammatory phenotype caused by 7-KC, we examined caspase1 activation by fluorochrome-labeled inhibitors of caspases assay (FLICA). 7-KC treatment elicited more caspase-1 activation in Ogg1−/− macrophages than WT controls (data not shown). We assessed cell viability by MTT assay after incubating with 7-KC for 24 hours. Exposure to 7-KC resulted in a concentration-dependent increase in cell death that was significantly greater in Ogg1−/− cells (Figure 1D). The NLRP3 inflammasome is activated by many diverse danger signals and mt function is the central hub for integrating these signals, resulting in inflammation. We previously showed that oxidized mtDNA binds to and activates the NLRP3 inflammasome10. Thus, in Ogg1−/− BMDM, where there is more oxidized mtDNA damage, there should be greater NLRP3 inflammasome activation. BMDM were pretreated with LPS and then stimulated with various NLRP3 activators such as ATP, 7-KC, and cholesterol crystals. Ogg1−/− macrophages secreted significantly more IL-1β, but not TNF-α, compared with WT cells (Figure 1E). We next investigated whether we could rescue this phenotype by adding mitochondrial specific Ogg1 back into Ogg1−/− BMDM. Retrovirus expressing mtOgg1 introduced into the Ogg1−/− BMDMs produced less IL-1β in response to LPS +ATP stimulation compared with control virus transduced cells (Figure 1F). The efficiency of retroviral infection was checked by eGFP expression under fluorescent microscope (Supplemental Figure IA). Taken together, these results indicate that macrophages deficient in OGG1 exhibit greater mitochondrial dysfunction, increased inflammasome activity, and more apoptosis when exposed to 7-ketocholesterol than WT cells.
Figure 1. OGG1-deficient macrophages are more apoptotic and inflammatory.
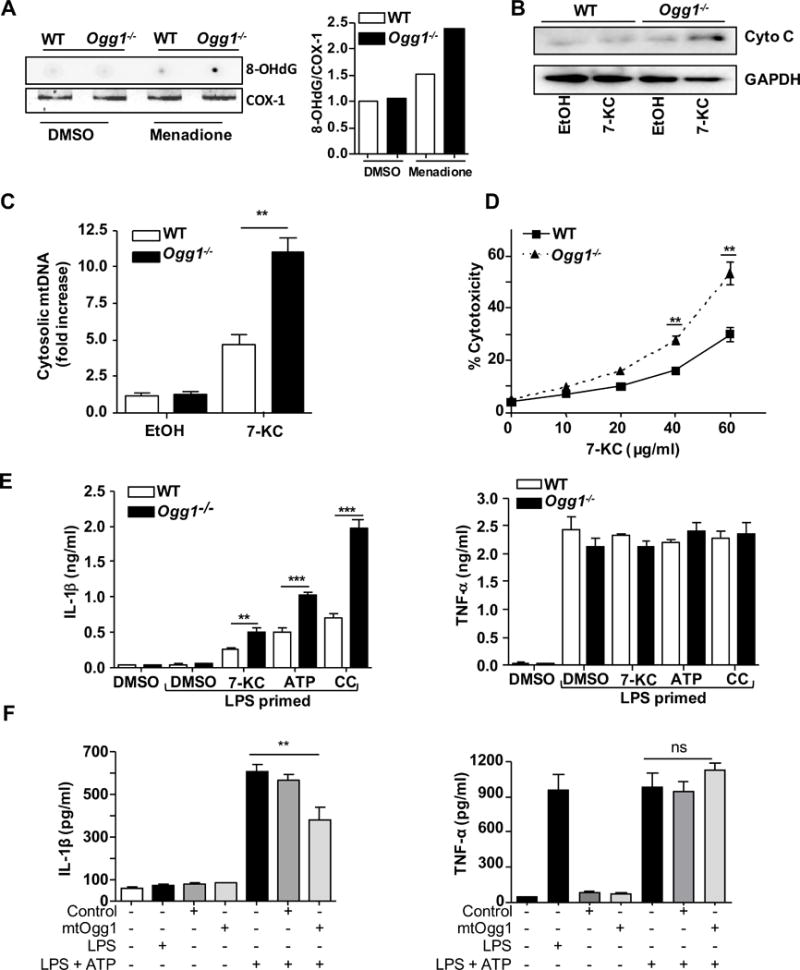
(A) The level of 8-OHdG in mtDNA. BMDM derived from WT or Ogg1−/− mice were stimulated with ROS inducer (menadione 25 μM for 30 min), followed by 2h incubation to allow for DNA repair. mtDNA was extracted and 8-OH-dG was measured by dot blot analysis and COX-1 PCR served as a loading control. Densitometry is shown in the right panel. Similar results were observed in 3 independent experiments. (B–C) BMDM derived from WT or Ogg1−/− mice were stimulated with 40 μg/ml 7-KC or control for (4 h). (B) Cytochrome c release into the cytosol was assessed by Western blot analysis. Similar results were observed in 3 independent experiments. (C) mtDNA content in the cytosol was assessed by RT-PCR. (D) Cell viability was determined by MTT assay after indicated doses of 7-KC treatment for 24 h. (E) 4 h LPS-primed BMDM from WT or Ogg1−/− mice were stimulated with 5 mM ATP (1 h), 20 μg/ml 7-KC (6 h), or 2 mg/ml cholesterol crystals (16 h). IL-1β and TNF-α concentrations in the culture supernatant were determined by ELISA. (F). Ogg1 KO BMDM cells were transduced with control and mtOgg1 expressing retrovirus for 48 h before the treatment of 400 ng/ml of LPS for 4 h plus 5 mM of ATP in the last hour. IL-1β and TNF-α concentrations in the culture supernatant were determined by ELISA. (C–F) All data are mean±SD and representative of 3 independent experiments in triplicate. Significance was determined using Student’s t-test *p<0.05, **p<0.01, ***p<0.001.
The expression of OGG1 diminishes as atherosclerotic lesions progress
mtDNA damage has been correlated with the extent of atherosclerosis and usually precedes atherogenesis31. Oxidative damage to mtDNA can be detected by strong immunoreactivity for 8-OH-dG, is found in all cell types of the plaque and correlates with atherosclerosis progression32, 33. However, the expression of Ogg1 in the lesion during diet-induced atherogenesis is not known. At 8 weeks of age, Ldlr−/− mice were fed western diet (WD), blood and tissue were harvested at the indicated time points. As expected, serum total cholesterol and aortic root plaque size increased over time (Figure 2A and B). OGG1 was detected in the lesions by immunofluorescence and was readily visible in the lesion after 4 weeks on WD (Figure 2C and D). However, OGG1 protein decreased as the lesion progressed, with staining in few cells by 16 weeks WD. The reduction in OGG1 staining was also specifically observed in F4/80 positive macrophages (Figure 4C and D). Recent studies have found that some smooth muscle cells (SMCs) may upregulate F4/80 on their surface as they become more macrophage like34. However, in our system, the majority of F4/80 positive cells co-expressed CD45, indicative of their hematopoietic origin (Supplemental Figure IIA). We confirmed our immunofluorescence results using laser-capture microdissection (LCM). F4/80+ macrophages were isolated from atherosclerotic plaques of Ldlr−/− mice fed WD for 4 and 16 weeks. Total RNA was extracted and mRNA expression analysis performed by RT-PCR. Notably, Ogg1 mRNA expression levels decreased in lesional macrophages at 16 weeks compared with 4 weeks (Figure 2E). Taken together, these data suggest that Ogg1 expression diminishes in the plaque lesions generally, and in macrophages specifically, as the atheroma progresses.
Figure 2. The expression of OGG1 diminishes in macrophages in atherosclerotic lesions overtime.
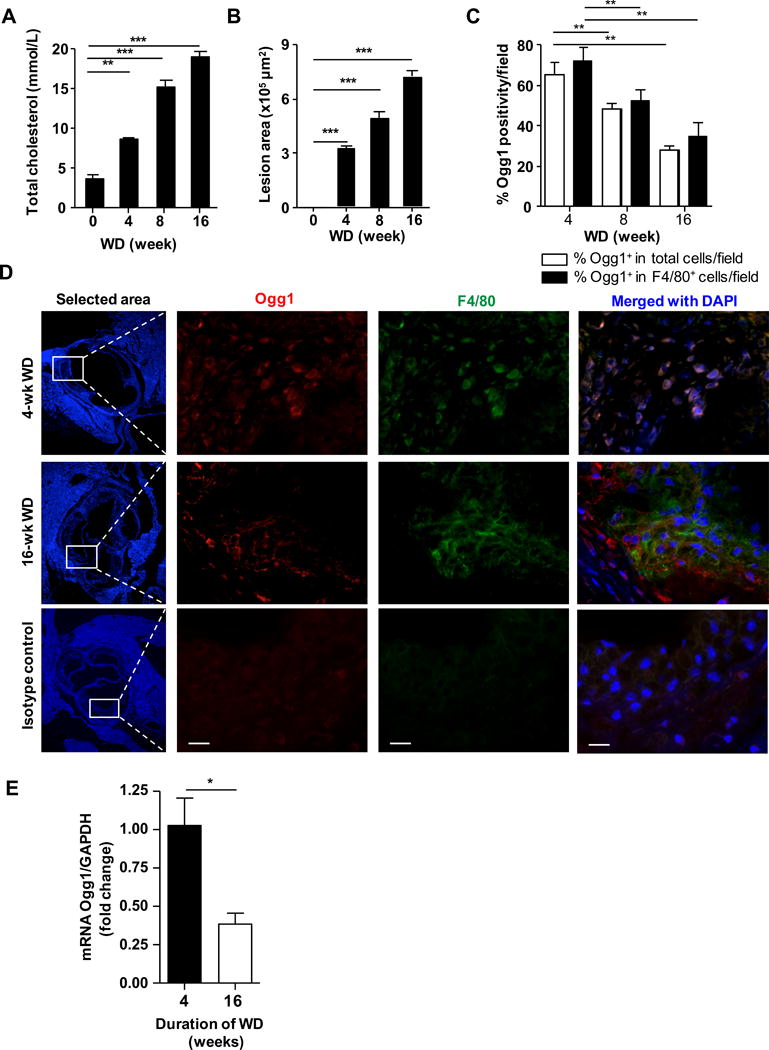
Ldlr−/− mice fed western diet for 0, 4, 8 or 16 weeks (n=6 per group). (A) Total plasma cholesterol concentrations. All data are mean±SD and each sample in duplicate. (B) Quantification of aortic root plaque area. (C) Quantification of OGG1+ cells within aortic root plaque as measured by immunofluorescence. (D) Representative images of immunofluorescent staining for OGG1 in aortic root plaques. Macrophage marker F4/80 (green), Nuclei (blue) and OGG1 (red). Magnification 40×. Scale bar = 20 μm. (E) Expression of Ogg1 mRNA in plaque F4/80+ macrophages isolated by laser-capture microdissection and analyzed by qRT-PCR. All data are mean±SD and representative of 2 independent experiments in triplicate. Significance was determined using One-Way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001.
OGG1 deficiency leads to accelerated atherosclerosis
To understand the significance of OGG1 in development and progression of atheromas, we generated Ogg1−/−Ldlr−/− double knockout (DKO) mice and fed them a WD diet for 16 weeks starting at 8 weeks of age. Representative images of the aortic root lesions are shown in Figure 3A. We found that the atherosclerotic lesion area and its macrophage content in cross-sections of the aortic root were increased significantly in Ogg1−/−Ldlr−/− mice compared with Ldlr−/− controls (Figure 3B and C). The critical features of advanced atheromatous lesions are increases in TUNEL positive apoptotic cells and necrotic area35, 36, and a corresponding decrease in collagen content. OGG1 deficiency resulted in a significant increase in both TUNEL positive cells and necrotic area, as well as a decrease in collagen content in their lesions (Figure 3D–F, Supplemental Figure IIB). En face analyses of the aorta showed an increase in the total surface area occupied by lesions in DKO mice compared with controls (Figure 3G and H), suggesting the loss of OGG1 accelerates diet-induced atheroma development in Ldlr−/− mice. We did observe a gene dose effect as Ogg1+/−Ldlr−/− mice displayed an intermediate phenotype. Thus OGG1 deficiency leads to larger more advanced atheromatous lesions on the Ldlr−/− background.
Figure 3. OGG1 deficiency leads to accelerated atherosclerosis in LDLR-deficient mice.
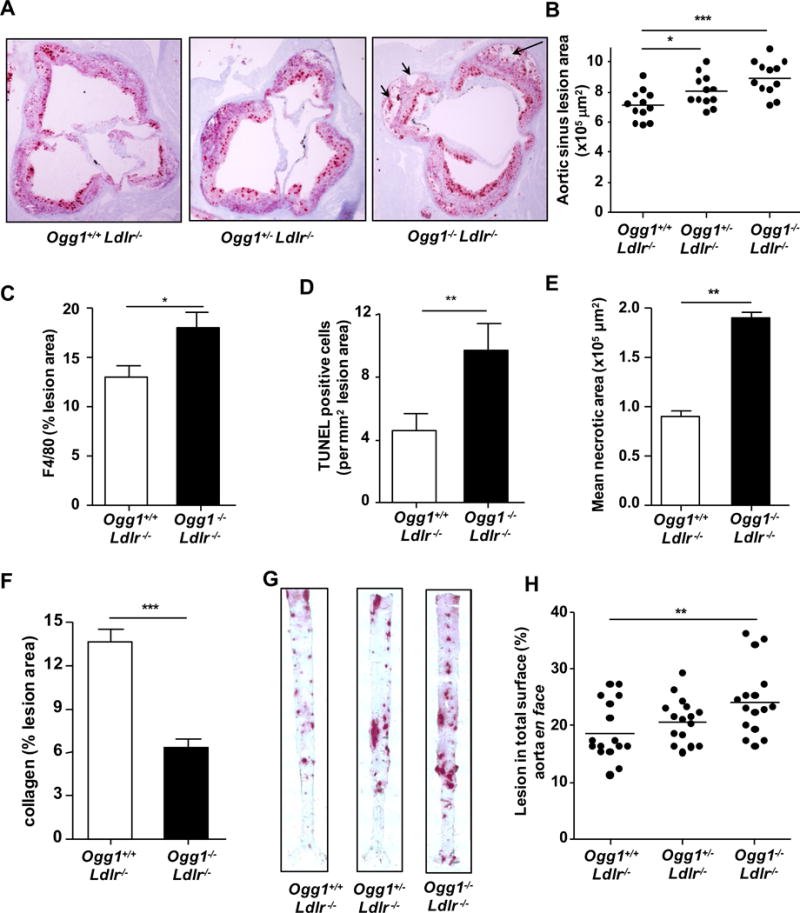
Ogg1−/−Ldlr−/−, Ogg1+/−Ldlr−/− and control Ogg1+/+Ldlr−/− mice were fed WD for 16 weeks (n=12). (A) Representative pictures of aortic root plaques stained with Oil Red O (40×). Black arrows show necrotic cellular areas. (B) Aortic root plaques area quantification. (C) Macrophage content. (D) Quantification of TUNEL positivity area (E) Necrotic core area in aortic root lesions (F) Percent collagen staining of aortic root using Mason’s Trichrome stain. (G) Representative images of aorta en face stained with Oil Red O. (H) Quantification of aortic en face lesion area (n=16). All data are mean±SD. Significance was determined using Student’s t-test and One-Way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001.
OGG1 deficiency leads to increased damaged mtDNA (8-OH-dG) and inflammasome activity in atherosclerotic lesions
Since OGG1 removes 8-OH-dG from DNA, we expected that we would find increased accumulation of 8-OH-dG modifications of DNA in the plaques in our Ogg1−/−Ldlr−/− mice. Immunofluorescence revealed a ~4-fold greater accumulation of 8-OH-dG in the plaques of DKO mice, compared with controls (Figure 4A and B). Using Tom20 as a mitochondrial marker, colocalization analysis indicated that >80% of the 8-OH-dG staining was mitochondrial, implicating mtDNA as the likely target (Figure 4B, Supplemental Figure IIC). Additionally, F4/80 positive macrophages were observed to have increased 8-OH-dG staining in the lesions as well (Figure 4C).
Figure 4. OGG1 deficiency leads to increased oxidative DNA damage (8-OH-dG) dominantly in mitochondria and caspase-1 activity in macrophages atherosclerotic lesions.
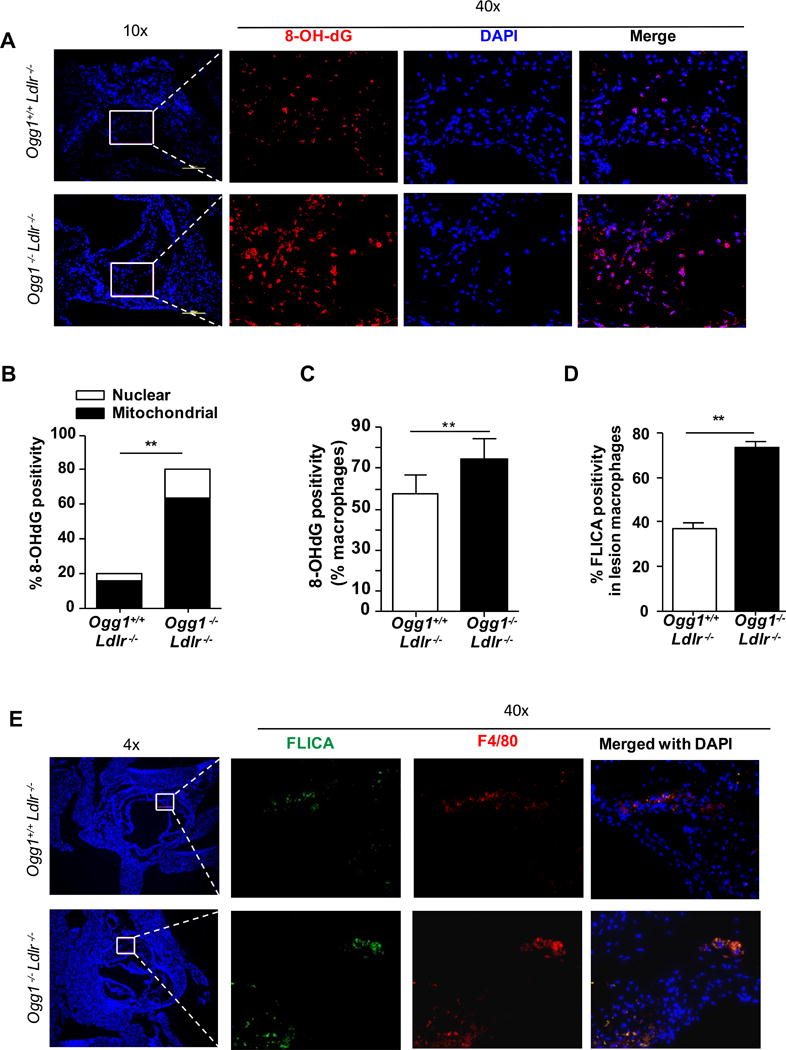
Ogg1−/−Ldlr−/− and control Ogg1+/+Ldlr−/− mice were fed WD for 16 weeks. Aortic root plaques were analyzed by immunofluorescence. (A) DNA damage was measured by immunostaining for 8-OH-dG (red) and nuclei (blue). (B) Quantitative analysis of 8-OH-dG+ cells in selected area. Increased 8-OH-dG found mainly in mitochondria (TOM20) (C) 8-OH-dG positive macrophages in lesion. (D) Quantification of active caspase-1+ cells in lesional macrophages. (E) Caspase-1 activity was assessed by FLICA (green) and by labelling macrophages with anti-F4/80 (red) and nuclei (blue). All data (in B, C, and D) are mean±SD and representative of 2 independent experiments (n=10). Significance was determined using Student’s t-test. **p<0.01.
Since we found an increase in 8-OH-dG in the lesions, and concomitantly an increase in apoptotic cells, we reasoned that we would also find an increase in IL-1β producing cells as apoptotic signals along with oxidized mtDNA can activate the NLRP3 inflammasome10. To elucidate the contribution of Nlrp3 inflammasome in the accelerated atherogenesis that we observed in OGG1 deficient Ldlr−/− mice (DKO mice), caspase-1 activation was investigated by FLICA in lesions of the aortic roots of both groups of mice. Nearly 2-fold more FLICA-positive F4/80+ macrophages were detected in the plaques of DKO mice compared with controls (Figure 4D and E). Taken together, these results indicate that the loss of OGG1 results in advanced plaque formation characterized by increased oxidized DNA damage (8-OH-dG), more apoptotic cells, and more cells with active caspase-1, which can produce IL-1 and IL-18, cytokines that are both strongly pro-atherogenic.
Lipid accumulation and cell death within the lesions contribute to activation of inflammatory cells that release proinflammatory and proatherogenic mediators into the serum37, 38. Additionally, our data suggests increased inflammasome activity in OGG1 deficient mice. Analysis of key cytokines and chemokines in the plasma revealed significantly higher concentrations of IL-1β, IL-18, and MCP-1 in DKO mice, whereas the concentrations of IL-12p40, IL-6, and TNF-α were indistinguishable between the two groups (Figure 5). Both IL-1β and IL-18 are dependent on the inflammasome for their secretion.
Figure 5. Plasma levels of cytokines and chemokines.
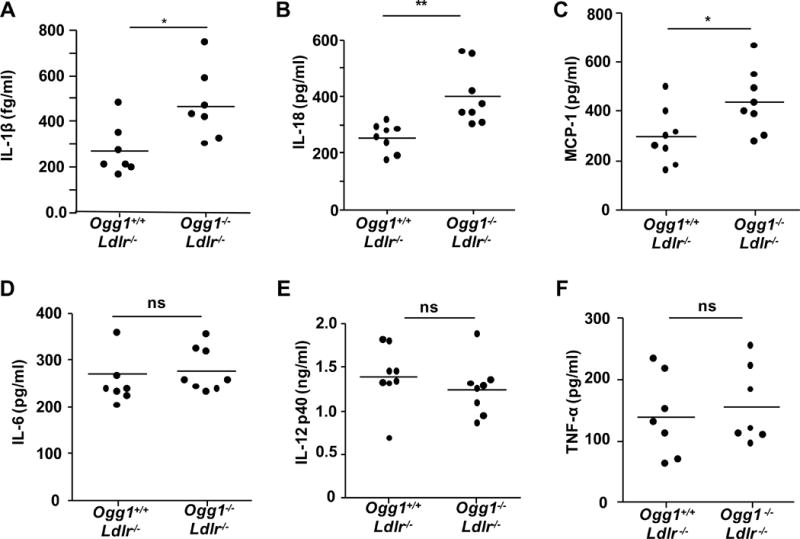
(A–D) Plasma concentrations of IL-1β measured by MSD and IL-18, MCP-1, IL-6, IL-12p40 and TNF-α were detected by ELISA (n=7–8). All data are expressed as mean±SD and performed in triplicate. Student’s t-test was used *p<0.05. ns= not significant.
A prior study reported that Ogg1−/− mice may develop metabolic syndrome39. In order to determine if the differences we observed were due to possible metabolic changes, we tested the mice after 16 weeks of WD for glucose tolerance and insulin resistance and found no difference between the genotypes (Supplemental Figure IIIA). The body weight gains and plasma cholesterol levels (triglycerides and lipoprotein profiles) were also similar (Supplemental Figure IIIB and C). Thus, the accelerated atheroma development in the OGG1-deficient mice in this study cannot be attributed to metabolic changes such as more severe dyslipidemia or altered glucose homeostasis.
NLRP3 significantly contributes to OGG1 deficiency-induced acceleration of atherosclerosis in hematopoietic cells
We hypothesized that OGG1 deficiency and the consequent accumulation of 8-OH-dG in mtDNA would result in more NLRP3 inflammasome activation as a critical driver of the accelerated atherosclerosis that we observed. Therefore, we next investigated whether NLRP3 is required for the acceleration of atherosclerosis Ogg1−/− mice. We generated Ogg1−/−Nlrp3−/− mice, and then created bone marrow (BM) chimeric mice using BM cells from WT, Ogg1−/−, Ogg1−/−Nlrp3−/− or Nlrp3−/− mice into Ldlr−/− recipient mice and 8 weeks after BM transplant, placed them on WD for 12 weeks. Mice that received Ogg1−/− BM developed significantly larger, more complex lesions in aortic root and abdominal aorta than mice receiving WT BM (Figure 6A–E). In agreement with our hypothesis, OGG1 deficiency-accelerated lesion size was reduced in Ldlr−/− mice that received OGG1 and NLRP3 doubly deficient BM cells (Figure 6A–E). Indeed, while the lesion size was significantly reduced in the Ogg1−/−Nlrp3−/− chimeras compared with Ogg1−/− alone, there was no significant difference between Ogg1−/−Nlrp3−/− and Nlrp3−/− highlighting the importance of the NLRP3 pathway in the Ogg1−/− mice for atherosclerotic lesion size (Figure 6B, E). Finally, Nlrp3−/− chimeras alone did not have significantly smaller lesions than WT mice, although they did trend towards a reduction (Figure 6B,C,E), consistent with a recent study5.
Figure 6. NLRP3 is required for OGG1 deficiency-induced acceleration of atherosclerosis in hematopoietic cells.
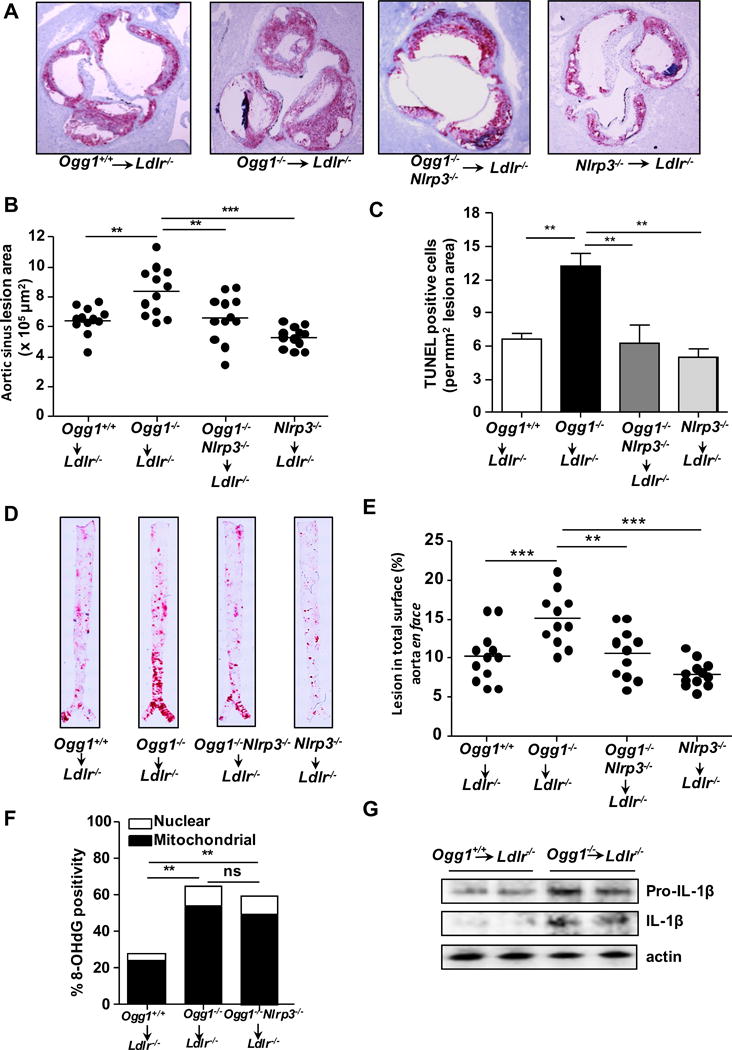
BM from WT, Ogg1−/−, Ogg1−/−Nlrp3−/−, or Nlrp3−/− mice were transferred into irradiated Ldlr−/− mice and after 8 week reconstitution, mice were fed a WD for 12 weeks (n=11–12). (A) Representative Oil Red O staining of aortic root plaques (10×). (B) Quantification of aortic root lesion area. (C) Quantification of TUNEL positivity (D) Representative images of Oil Red O staining on Aorta en face. (E) Quantification of aortic en face lesion area. (F) Quantification of 8-OH-dG positivity in aortic root plaque by immunofluorescent staining. (G) Mature IL-1β in the aortic arch. Each lane represents the pooled aortic arch from 2 mice lysates and analyzed by western blot for IL-1β. All data are mean±SD. Significance was determined using One-Way ANOVA with Tukey’s post-hoc test (B, C, E, and F). *p<0.05, **p<0.01.
Consistent with the role of OGG1 to remove oxidized guanine residues, we noted a greater accumulation of 8-OH-dG in the plaques of Ogg1−/− and Ogg1−/−Nlrp3−/− BM chimeras (Figure 6F). The accumulation of 8-OH-dG was not affected by the presence or absence of NLRP3. We then investigated whether we could detect increased amounts of mature IL-1β in the lesions. Ldlr−/− mice that received Ogg1−/− BM cells indeed expressed more mature cleaved IL-1β as compared with WT BM (Figure 6G). Importantly, atheroma development was substantially reduced in the chimeric mice that received Ogg1−/−Nlrp3−/− BM despite no difference in glucose tolerance, insulin resistance and lipid metabolism compared with the WT chimeras (Supplemental Figure IIID and E). Collectively, these data suggest that OGG1 deficiency in hematopoietic cells accelerates plaque progression at least partially through the Nlrp3 inflammasome.
miR-33 downregulates OGG1 in human and mouse cells by different mechanisms, leading to increased 8-OH-dG accumulation and IL-1β production
OGG1 content in the aortic root diminished significantly as atherosclerosis progressed, suggesting that atherosclerosis inversely affected Ogg1 expression or stability. Therefore, we examined the role of AMPK, an upstream regulator of Ogg121. We confirmed that AMPK activation induced Ogg1 transcription by treating mouse BMDM cells with AICAR and Metformin, both potent AMPK activators. Indeed, both treatments led to increased Ogg1 mRNA (Figure 7A). We next assessed whether activated AMPK levels changed during atherogenesis. Consistent with the decreased expression of Ogg1 during atherogenesis, the amount of phospho- and AMPK in aortic arch lysate of Ldlr−/− mice a fed a 16 week-WD was also diminished (Figure 7B). Thus it is likely that the decrease in AMPK leads to reduced Ogg1 expression during atherosclerosis.
Figure 7. The regulation of Ogg1 expression.
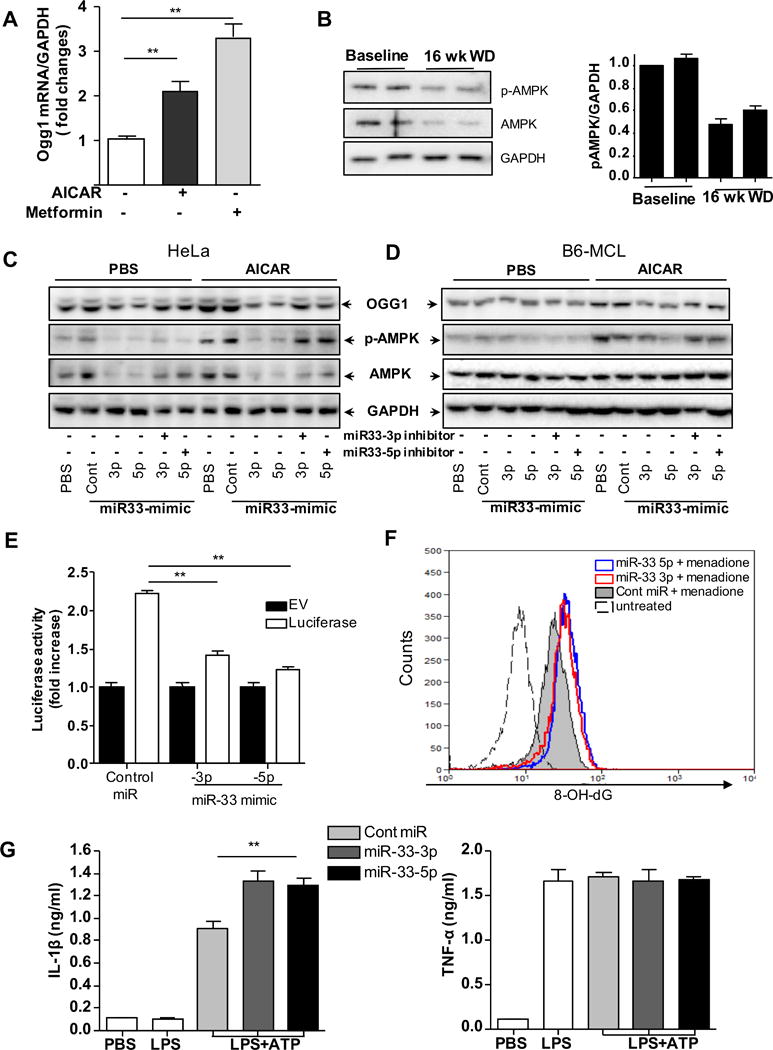
(A) mRNA Ogg1 expression was examined by RT-PCR. The cells were treated with AMPK activators (1 mM AICAR for 4 h and 7.5 μM Metformin for 8h). (B) p-AMPK and AMPK protein in the aortic arch. Each lane represents the pooled aortic arch lysates from two Ldlr−/− mice and analyzed by western blot. GAPDH used as a loading control. Band densitometry is shown in the right panel. (C and D) Western blot analysis of Ogg1, p-AMPK, AMPK and GAPDH protein expression transfected with the control mimic (control), miR-33-3p and -5p mimics, and/or mimic inhibitors for 48 h treated with or without 1 mM AICAR for 2 h. (E) Luciferase reporter activity in HEK 293 co transfected with the miR control, miR-33a -3p and -5p with Luciferase reporter (contains 3′ UTR of OGG1 (human)) plasmid for 48 h. (F) HEK 293 cells were transfected with miR-33-3p and -5p mimic and control miRNA before 25 μM menadione (ROS inducer) treatment for 30 min, followed by 4hr incubation to allow for DNA repair. DNA damage was measured by intracellular immunostaining for 8-OH-dG and measured by flow cytometry. (B–D and F) Similar results were observed in 3 independent experiments. (G) MCL cells were transfected with control miR, miR-33-3p and miR-33-5p mimic for 48 h before the treatment of 400 ng/ml of LPS for 4 h plus 5 mM of ATP in the last hour. (A, E, and G) All data are means ± SD and representative of 3 experiments in duplicate. Significance was determined using One-Way ANOVA with Tukey’s post-hoc test. **p<0.01.
Analysis of the 3′ UTR regions of human OGG1 identified putative binding sites for miR-33 at nucleotides 312–318 and 367–373, but not in mouse Ogg1 (Supplemental Figure IVA). miR-33 is known to play an important pro-atherogenic role, negatively affecting lipid metabolism and cholesterol export, as well as the master regulatory switch AMPK40–42. We therefore asked if these miR-33 binding sites were functional and regulated OGG1 levels in the cell either directly, or through AMPK. miR-33 mimics as well as their specific inhibitors were transfected into both human and mouse cells to test for their effects on OGG1 expression. As predicted, densitometric quantification of OGG1 protein amounts normalized by GAPDH amounts showed that the miR-33 mimics were able to inhibit OGG1 expression in HeLa cells, but not in B6-MCL cells (Figure 7C, Supplemental Figure IVB), suggesting that miR-33a could directly regulate human OGG1, but not mouse Ogg1. This was especially the case for miR-33-5p, while miR-33-3p was less effective in its inhibition. Inclusion of the mimic inhibitors abrogated the effects of the miR-33 mimics (Figure 7C and D). Next we assessed protein expression by treating the cells with 1 mM AICAR (AMPK activator) for 2h, and observed that this led to increased phosphorylation of AMPKα as well as increased expression of Ogg1, but the addition of miR-33-3p and -5p mimics significantly inhibited them in both human and mouse cells (Figure 7C and D). As before, co-transfection of miR-33-3p or -5p specific inhibitors reversed the effect of miR-33-3p and -5p mimics in these cells (Figure 7C and D). To assess the functionality of these sequences, we transfected HEK293 cells with a reporter construct, which expressed a luciferase coding sequence fused to the 3′UTR of OGG1 (human). Cotransfection of miR-33-3p and -5p mimics markedly suppressed the activity of the OGG1 3′UTR luciferase construct, compared with empty vector control (Figure 7E). Next, we examined the effect of miR-33 inhibition on OGG1 activity to repair oxidized DNA damage. HeLa cells were transfected with miR-33-3p and -5p mimic and 48h later, they were treated with 25μM of menadione to induce mtROS16. The level of 8-OH-dG was determined by flow cytometry. We found that menadione treatment led to more 8-OH-dG in miR-33 mimic transfected cells, compared with control cells (Figure 7F). Finally, similar to Ogg1−/− macrophages, MCL transfected with miR-33 mimics produced more IL-1β in response to LPS + ATP stimulation (Figure 7G), but produced similar amounts of TNF-α. Taken together, these data suggest that miR-33a downregulates Ogg1 expression through AMPK in both mice and humans, and also directly targets human but not mouse Ogg1. Thus reduction in OGG1 by miR-33 leads to increased 8-OH-dG accumulation and greater IL-1β production.
It was recently reported that miR-33 is upregulated in human atherosclerotic plaques43. As our data showed that OGG1 diminishes over time during atherogenesis in mice, and it’s expression can be regulated miR-33 and AMPK, we next investigated whether OGG1 was also reduced in human atherosclerotic plaques. Indeed, in agreement with our mouse data, we found that OGG1 transcript was indeed reduced in human coronary and carotid plaques compared with normal vessels (Supplemental Figure V).
DISCUSSION
Atherosclerosis is a disease characterized by lipid accumulation in the vessel wall, cell death and chronic inflammation. In our study we found that WD-fed Ogg1−/−Ldlr−/− mice and Ogg1−/− BM chimeric mice developed bigger, more complex lesions with larger necrotic cores. However, this increase was abrogated in Ogg1−/−Nlrp3−/− BM chimeric mice, suggesting that the mechanism by which Ogg1 deficiency leads to accelerated atherogenesis is at least in part through the NLRP3 inflammasome. While some human Ogg1 polymorphisms have been linked with various pathologies such as large artery atherosclerotic stroke in smokers44 and SLE severity45, 46, no studies have yet demonstrated its role in atherosclerosis.
Apoptosis in both vascular SMCs and macrophages may promote inflammation and alter plaque composition, although the significance of apoptosis in atherosclerosis depends on the stage of the plaque, localization, and the cell types involved47. Previously, Ogg1 KO mice showed decreased intracellular ATP content48 and were more susceptible to apoptosis in response to oxidative stress49, 50. Here, we show that loss of OGG1 in macrophages results in increased mtDNA damage, greater cytochrome c release, increased cytosolic mtDNA, and caspase1 activation, leading to increased apoptosis, increased IL-1β secretion and inflammation. Since intrinsic apoptosis is regulated by mitochondria, mt dysfunction due to mtDNA damage can lead to more cell death51, 52. In the case of atheromas, macrophage apoptosis contributes to necrotic core formation, which can trigger acute thrombotic events53, 54. Apoptotic cells display a variety of recognition signals that lead to their swift removal by professional phagocytes. However, improper removal of these cells can lead to secondary necrosis and subsequent inflammation55. As Ogg1 expression diminishes over time as atherosclerosis progresses, more cell death may occur, resulting in progressively larger necrotic cores.
OGG1 is responsible for repairing 8-OH-dG lesions in DNA, and is particularly important for clearing these lesions from mtDNA. While there is more nuclear DNA in the cell compared with mtDNA, mtDNA is much more susceptible to oxidative damage due to its lack of protective histones and its close proximity to mtROS. Additionally, mitochondria only have base excision repair, of which OGG1 is a member, while the nucleus possesses both base excision repair as well as nucleotide excision repair56,57. Thus OGG1 deficiency would likely have a much greater effect on mtDNA than nuclear DNA, with which our data was in agreement. Ogg1 deficiency resulted in a substantial increase in oxidized mtDNA, which resulted in NLRP3 activation, IL-1β secretion, and accelerated atherosclerosis. Several reports have investigated the causes and pathological consequences of excessive mtROS in atherosclerosis in both humans and mouse models36, 58. It is reported that 8-OH-dG accumulation in mtDNA is increased 20-fold in livers of Ogg1−/− mice compared to WT mice59. The human Ser326Cys (C8055G) OGG1 polymorphism is associated with decreased repair activity60 and is linked to large artery atherosclerotic stroke in smokers44; the C1245G polymorphism correlates with increased 8-OH-dG in leukocyte DNA and more severe manifestations of systemic lupus erythematosus (SLE)45, 46. Atherosclerosis is a well-recognized complication of SLE, but underlying mechanisms are complex and remain areas of active investigation. Thus growing evidence supports the potential role of Ogg1 in modifying atherosclerosis. Oxidation of mtDNA is directly attributed to reactive oxygen species derived from mt respiration, making mitochondria both the source and the target of oxidative stress. Several reports have linked mitochondrial damage to atherosclerosis in both humans and mouse models36, 58. While mtDNA damage might be expected to result in mitochondrial dysfunction including decreased ATP production and increased generation of reactive oxygen species, OGG1-null mice up to the age of 14 months had normal mitochondrial function61. We suggest that the proatherogenic effect of OGG1 deficiency is more likely to be a consequence of activation of NLRP3 by overabundant 8-OH-dG. Our finding that loss of NLRP3 mitigated the proatherogenic effect of OGG1 deficiency on plaque size supports this notion. The NLRP3 inflammasome is activated by many diverse stimuli making NLRP3 the most versatile and importantly, also the most clinically implicated inflammasome62, 63. Furthermore, IL-1β is known as a critical component of inflammation that accelerates atherosclerosis64–66, and our data is in agreement with this concept.
It was previously reported that Ogg1−/− mice on a WD develop obesity and metabolic syndrome39. However, in our study, the genetic loss of Ogg1 did not significantly exacerbate metabolic alterations after 16 weeks on WD, compared with Ldlr−/− control mice. Importantly, adoptive transfer of Ogg1−/− BM cells into irradiated Ldlr−/− mice also resulted in increased atherogenesis, increased apoptosis, and increased inflammasome activity, with no changes in metabolic readouts. While our data suggests a strong link between OGG1 and the NLRP3 inflammasome during atherosclerosis, it is also possible that Ogg1 acts through other pathways. Indeed, while lesion size (aortic sinus and aorta en face) in Ogg1−/− mice seemed to mainly be driven by the NLRP3 pathway, the lipid content of the lesions was increased in Ogg1−/− mice, but was not affected by NLRP3 deletion at twelve weeks WD (data not shown) suggesting that OGG1 may affect lipid accumulation in the plaque through a non-NLRP3 inflammasome related pathways. Any new mechanisms by which OGG1 may affect the progress of atherosclerosis that is NLRP3 independent will require further studies.
It is intriguing that another study using an acute injury model, LPS-induced organ failure model, suggested a pro-inflammatory role for OGG1 as Ogg1−/− mice had reduced inflammation. The authors, proposed that single strand breaks made by OGG1 in the DNA may serve as a danger signal that induces inflammation in their model of acute inflammation67. However, other studies have shown that OGG1 has an anti-inflammatory effect in different disease models68, in agreement with our findings. It could be that the pro- or anti-inflammatory effects of OGG1 are disease dependent with OGG1 being anti-inflammatory in more chronic disorders, such as atherosclerosis, while OGG1 may be pro-inflammatory in certain acute diseases. Further research is required to distinguish between these two possibilities.
The role of miR-33 in atherogenesis has increasingly been recognized69–71. Treatment with miR-33 antagonist oligonucleotide in mouse and nonhuman primates, as well studies in miR-33 KO mice, showed protection against atherosclerosis by increasing reverse cholesterol transport, leading to higher plasma HDL levels72. Additionally miR-33α/β was found to be markedly increased in human carotid atherosclerotic plaques compared with normal arteries73. Furthermore, miR-33 controls energy metabolism via repression of key mitochondrial genes to limit ATP production and dampen cholesterol efflux74, 75. Also, miR-33 was shown to skew towards M1 polarization of macrophages, which is proinflammatory and proatherogenic, while antagonism of miR-33 leads to M2 polarization, which is atheroprotective75. Mitochondrial activity and macrophage polarization are linked, with miR-33 influencing both processes43, 75. OGG1, which is required for mtDNA repair, may therefore play a regulating role in both these pathways. Finally, miR-33 can also regulate Il1β expression levels in atherosclerotic lesions76. However, Goedeke L et al77 reported that long-term anti-miR-33 therapy in mice might be associated with side effects such as hypertriglyceridemia and moderate hepatic steatosis, but overall, miR-33 is regarded as a potential therapeutic target for atherosclerosis40, 70, 78, Therefore, it is of great potential interest that in this study we discovered that miR-33 also targets OGG1 and downregulates its expression directly in human cells, and indirectly in both human and mice.
However, while the complete repertoire of miR-33 target genes is unknown, miR-33 targets a number of metabolic pathways, including AMPKα. The expression of AMPKα is diminished during atherogenesis, which leads to decreased autophagy and paradoxical upregulation of SREBP-1c and SREBP-2, as well as further progression of atherosclerosis79, 80. Restoring AMPKα expression protects from atherogenesis81, 82. Importantly, AMPKα acts upstream of Ogg1, thus miR-33 can also act on Ogg1 indirectly and this pathway is conserved in both humans and mice. Indeed, we also found that hOGG1 expression was also reduced in human atherosclerotic plaques in diseased arteries compared with normal controls.
Several publications have reported that stress responsive transcription factors, Nrf-2 (nuclear factor erythroid 2 [NF-E2]-related factor 2) and NF-YA (nuclear transcription Y subunit alpha), also regulate Ogg1 expression83, 84. Coincidentally, they are also regulated by AMPK85. Furthermore, NF-Ys function as a heterotrimeric transcriptional complex composed of three subunits, NF-YA, NF-YB, and NF-YC. A recent study showed NF-YC is also a miR-33 target gene as it was markedly increased in mice administered with miR-33 anti-sense oligonucleotide compared to control miRNA-treated mice77. Thus it is likely that miR-33 plays an important role in vivo controlling Ogg1 during atherogenesis and further studies are required to understand the complex interaction with miR-33 and OGG1 during plaque progression.
There is a growing understanding on the key roles of macrophages in the initiation, progression and resolution of atherosclerotic inflammation43, 86. Our study further highlights the importance of macrophages as OGG1 deficiency caused a significant increase in oxidative damage in mtDNA leading to increased caspase 1 activation, most notably in macrophages, leading to greater IL-1β production and accelerated atherogenesis. Two isoforms of OGG1 protein exist (mitochondrial and nuclear), but the importance of mitochondrial OGG1 in the protection of oxidative stress is clearly demonstrated in this study. Furthermore, mtDNA damage is significantly more abundant and persists longer than nuclear DNA (nDNA) damage following exposure to oxidative stress87. Human atheromatous plaques show increased mtDNA damage, particularly associated with higher-risk plaques43, 88. In conclusion, this work provides direct evidence for a causal link between oxidative DNA damage repair enzyme, OGG1, and atherosclerotic plaque development in part via the NLRP3 inflammasome and IL-1β. Our study reveals a novel therapeutic target to limit oxidative mtDNA damage, which then down-regulates the NLRP3/IL-1β axis and prevent or treat atherosclerosis.
Supplementary Material
Novelty and Significance.
What Is Known?
Mitochondrial DNA damage has been implicated in NLRP3 inflammasome activation and in atherogenesis, and is increased in human atherosclerotic plaques.
OGG1 (oxy-guanine glycosylase 1) is the main enzyme responsible for repairing mitochondrial oxidative DNA damage.
miR-33 negatively regulates several anti-atherosclerotic pathways and is considered to be a potential anti-atherosclerosis therapeutic target.
What New Information Does This Article Contribute?
Loss of OGG1 leads to increased mitochondrial DNA damage and NLRP3 inflammasome activation in plaque macrophages.
This study shows that OGG1 plays a protective role in atherogenesis by preventing excessive inflammasome activation.
OGG1 is reduced in plaque macrophages as the lesion progresses and is negatively regulated by the proatherogenic miR-33.
Recent evidence suggests that oxidative stress and mitochondrial DNA (mtDNA) damage is present in atherosclerotic plaques, directly promoting NLRP3 inflammasome activation and multiple pro-atherogenic processes through mitochondrial dysfunction. Using mice deficient in both OGG1 and the LDL receptor (LDLR) we found that; 1) genetic deletion of OGG1 leads to larger and more advanced plaques; 2) increased accumulation of the oxidative DNA damage product 8-OH-dG localized to the mitochondria of plaque macrophages; and 3) increased caspase-1 activation. By using OGG1/NLRP3 double knockout mice as donors for bone marrow chimera experiments we found that the OGG1 deficiency-mediated increase in atherosclerotic plaque progression was predominately NLRP3-dependent. We observed that OGG1 expression diminishes in atherosclerotic plaque macrophages in mice as the lesion progresses and in human atherosclerotic plaques. Finally, we discovered that miR-33, a proathogenic microRNA, which is known to be increased in atherosclerotic lesions, downregulates OGG1 expression. These findings demonstrate that OGG1 is atheroprotective and that targeting mtDNA repair may provide a new strategy for reducing plaque development by harnessing pre-existing protective processes.
Acknowledgments
We would like to thank G. Huang and P. Sun for excellent technical assistance.
SOURCES OF FUNDING
This work was supported by the National Institutes of Health Grants AI105845 (to M.A.) and HL111483 (to S.C.).
Nonstandard Abbreviations and Acronyms
- AMPK
AMP-activated protein kinase
- ATP
Adenosine triphosphate
- HDL
High-density lipoprotein
- LDL
Low-density lipoprotein
- Ldlr
Low density lipoprotein receptor
- miRNA
microRNA
- mtDNA
Mitochondrial DNA
- Nlrp3
nucleotide binding domain and leucine-rich repeat (NLR) pyrin domain containing 3
- Ogg1
8-Oxoguanine glycosylase
- PCR
Polymerase Chain Reaction
- TG
Triglyceride
- WD
Western diet
- 8-OH-dG
7,8-dihydro-8-oxo-2′-deoxyguanosine
Footnotes
DISCLOSURES
None.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nature reviews Immunology. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: Subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL. Activation of nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: Implication of a novel role of inflammasome in atherogenesis. PloS one. 2014;9:e87552. doi: 10.1371/journal.pone.0087552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gage J, Hasu M, Thabet M, Whitman SC. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein e-null mice. Can J Cardiol. 2012;28:222–229. doi: 10.1016/j.cjca.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly N, Mielke LA, Harris J, Coll RC, Mills KH, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LA. Activation of the nlrp3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced il-1beta in type 2 diabetes. Nature immunology. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurung P, Lukens JR, Kanneganti TD. Mitochondria: Diversity in the regulation of the nlrp3 inflammasome. Trends in molecular medicine. 2015;21:193–201. doi: 10.1016/j.molmed.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the nlrp3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu E, Calvert PA, Mercer JR, Harrison J, Baker L, Figg NL, Kumar S, Wang JC, Hurst LA, Obaid DR, Logan A, West NE, Clarke MC, Vidal-Puig A, Murphy MP, Bennett MR. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128:702–712. doi: 10.1161/CIRCULATIONAHA.113.002271. [DOI] [PubMed] [Google Scholar]
- 12.Gray K, Kumar S, Figg N, Harrison J, Baker L, Mercer J, Littlewood T, Bennett M. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circulation research. 2015;116:816–826. doi: 10.1161/CIRCRESAHA.116.304921. [DOI] [PubMed] [Google Scholar]
- 13.Dianov G, Bischoff C, Piotrowski J, Bohr VA. Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J Biol Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- 14.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (ogg1) gene and 8-oxoguanine accumulates in the mitochondrial dna of ogg1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 15.Russo MT, De Luca G, Degan P, Parlanti E, Dogliotti E, Barnes DE, Lindahl T, Yang H, Miller JH, Bignami M. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the myh and ogg1 DNA glycosylases. Cancer Res. 2004;64:4411–4414. doi: 10.1158/0008-5472.CAN-04-0355. [DOI] [PubMed] [Google Scholar]
- 16.Torres-Gonzalez M, Gawlowski T, Kocalis H, Scott BT, Dillmann WH. Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in h9c2 cells under oxidative stress conditions. Am J Physiol Cell Physiol. 2014;306:C221–229. doi: 10.1152/ajpcell.00140.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruchko M, Sukhanov S, Gorodnya O, Pastukh V, Danchuk S, Rachek L, Gillespie M. Involvement of mitochondrial (mt) DNA oxidative damage and circulating fragments in early stages of atherogenesis. Faseb Journal. 2015;29 [Google Scholar]
- 18.Shirwany NA, Zou MH. Ampk in cardiovascular health and disease. Acta Pharmacol Sin. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong Y, Zhang M, Liang B, Xie Z, Zhao Z, Asfa S, Choi HC, Zou MH. Reduction of amp-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Xu S, Jiang B, Cohen RA, Zang M. Activation of sterol regulatory element binding protein and nlrp3 inflammasome in atherosclerotic lesion development in diabetic pigs. PloS one. 2013;8:e67532. doi: 10.1371/journal.pone.0067532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Habib SL, Kasinath BS, Arya RR, Vexler S, Velagapudi C. Novel mechanism of reducing tumourigenesis: Upregulation of the DNA repair enzyme ogg1 by rapamycin-mediated ampk activation and mtor inhibition. Eur J Cancer. 2010;46:2806–2820. doi: 10.1016/j.ejca.2010.06.117. [DOI] [PubMed] [Google Scholar]
- 22.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. The Journal of clinical investigation. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, Horiguchi M, Nakamura T, Chonabayashi K, Hishizawa M, Hasegawa K, Kume N, Yokode M, Kita T, Kimura T, Ono K. Microrna-33 deficiency reduces the progression of atherosclerotic plaque in apoe−/− mice. J Am Heart Assoc. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Shimada K, Zhang W, Huang G, Crother TR, Arditi M. Il-17a is proatherogenic in high-fat diet-induced and chlamydia pneumoniae infection-accelerated atherosclerosis in mice. Journal of immunology (Baltimore, Md: 1950) 2010;185:5619–5627. doi: 10.4049/jimmunol.1001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trogan E, Choudhury RP, Dansky HM, Rong JX, Breslow JL, Fisher EA. Laser capture microdissection analysis of gene expression in macrophages from atherosclerotic lesions of apolipoprotein e-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2234–2239. doi: 10.1073/pnas.042683999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat Res. 1992;275:169–180. doi: 10.1016/0921-8734(92)90021-g. [DOI] [PubMed] [Google Scholar]
- 27.Ricci C, Pastukh V, Leonard J, Turrens J, Wilson G, Schaffer D, Schaffer SW. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol. 2008;294:C413–422. doi: 10.1152/ajpcell.00362.2007. [DOI] [PubMed] [Google Scholar]
- 28.Terasaka N, Wang N, Yvan-Charvet L, Tall AR. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via abcg1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15093–15098. doi: 10.1073/pnas.0704602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lizard G, Lemaire S, Monier S, Gueldry S, Neel D, Gambert P. Induction of apoptosis and of interleukin-1beta secretion by 7beta-hydroxycholesterol and 7-ketocholesterol: Partial inhibition by bcl-2 overexpression. FEBS Lett. 1997;419:276–280. doi: 10.1016/s0014-5793(97)01473-7. [DOI] [PubMed] [Google Scholar]
- 30.Indaram M, Ma W, Zhao L, Fariss RN, Rodriguez IR, Wong WT. 7-ketocholesterol increases retinal microglial migration, activation, and angiogenicity: A potential pathogenic mechanism underlying age-related macular degeneration. Sci Rep. 2015;5:9144. doi: 10.1038/srep09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 32.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappab-mediated inflammation in macrophages. Circulation research. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenfeld ME. Converting smooth muscle cells to macrophage-like cells with klf4 in atherosclerotic plaques. Nat Med. 2015;21:549–551. doi: 10.1038/nm.3875. [DOI] [PubMed] [Google Scholar]
- 35.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Cell metab. United States: 2012 Elsevier Inc; 2012. Macrophage autophagy plays a protective role in advanced atherosclerosis; pp. 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Tabas I. Emerging roles of mitochondria ros in atherosclerotic lesions: Causation or association? J Atheroscler Thromb. 2014;21:381–390. doi: 10.5551/jat.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tedgui A, Mallat Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 38.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sampath H, Vartanian V, Rollins MR, Sakumi K, Nakabeppu Y, Lloyd RS. 8-oxoguanine DNA glycosylase (ogg1) deficiency increases susceptibility to obesity and metabolic dysfunction. PloS one. 2012;7:e51697. doi: 10.1371/journal.pone.0051697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez-Hernando C, Suarez Y, Rayner KJ, Moore KJ. Micrornas in lipid metabolism. Curr Opin Lipidol. 2011;22:86–92. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moore KJ, Rayner KJ, Suarez Y, Fernandez-Hernando C. Micrornas and cholesterol metabolism. Trends Endocrinol Metab. 2010;21:699–706. doi: 10.1016/j.tem.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of mir-33a/b in non-human primates raises plasma hdl and lowers vldl triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karunakaran D, Thrush AB, Nguyen MA, Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P, Ouimet M, Pezacki JP, Moore KJ, Perisic L, Maegdefessel L, Hedin U, Harper ME, Rayner KJ. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-mir33 in atherosclerosis. Circulation research. 2015;117:266–278. doi: 10.1161/CIRCRESAHA.117.305624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shyu HY, Shieh JC, Ji-Ho L, Wang HW, Cheng CW. Polymorphisms of DNA repair pathway genes and cigarette smoking in relation to susceptibility to large artery atherosclerotic stroke among ethnic chinese in taiwan. J Atheroscler Thromb. 2012;19:316–325. doi: 10.5551/jat.10967. [DOI] [PubMed] [Google Scholar]
- 45.Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. The role of hogg1 c1245g polymorphism in the susceptibility to lupus nephritis and modulation of the plasma 8-ohdg in patients with systemic lupus erythematosus. International journal of molecular sciences. 2015;16:3757–3768. doi: 10.3390/ijms16023757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HT, Lin CS, Lee CS, Tsai CY, Wei YH. Increased 8-hydroxy-2′-deoxyguanosine in plasma and decreased mrna expression of human 8-oxoguanine DNA glycosylase 1, anti-oxidant enzymes, mitochondrial biogenesis-related proteins and glycolytic enzymes in leucocytes in patients with systemic lupus erythematosus. Clin Exp Immunol. 2014;176:66–77. doi: 10.1111/cei.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kockx MM, Herman AG. Apoptosis in atherosclerosis: Beneficial or detrimental? Cardiovasc Res. 2000;45:736–746. doi: 10.1016/s0008-6363(99)00235-7. [DOI] [PubMed] [Google Scholar]
- 48.Bacsi A, Chodaczek G, Hazra TK, Konkel D, Boldogh I. Increased ros generation in subsets of ogg1 knockout fibroblast cells. Mechanisms of ageing and development. 2007;128:637–649. doi: 10.1016/j.mad.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruchko MV, Gorodnya OM, Zuleta A, Pastukh VM, Gillespie MN. The DNA glycosylase ogg1 defends against oxidant-induced mtdna damage and apoptosis in pulmonary artery endothelial cells. Free radical biology & medicine. 2011;50:1107–1113. doi: 10.1016/j.freeradbiomed.2010.10.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruchko M, Gorodnya O, LeDoux SP, Alexeyev MF, Al-Mehdi AB, Gillespie MN. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288:L530–535. doi: 10.1152/ajplung.00255.2004. [DOI] [PubMed] [Google Scholar]
- 51.Cha MY, Kim DK, Mook-Jung I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med. 2015;47:e150. doi: 10.1038/emm.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 2010;1797:113–128. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 53.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature reviews Immunology. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: The importance of lesion stage and phagocytic efficiency. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 56.Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. Journal of Biological Chemistry. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- 57.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free radical biology & medicine. 2002;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 58.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circulation research. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 59.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (ogg1) gene and 8-oxoguanine accumulates in the mitochondrial dna of ogg1-defective mice. Cancer research. 2001;61:5378–5381. [PubMed] [Google Scholar]
- 60.Janik J, Swoboda M, Janowska B, Ciesla JM, Gackowski D, Kowalewski J, Olinski R, Tudek B, Speina E. 8-oxoguanine incision activity is impaired in lung tissues of nsclc patients with the polymorphism of ogg1 and xrcc1 genes. Mutat Res. 2011:709–710. 21–31. doi: 10.1016/j.mrfmmm.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 61.Stuart JA, Bourque BM, de Souza-Pinto NC, Bohr VA. No evidence of mitochondrial respiratory dysfunction in ogg1-null mice deficient in removal of 8-oxodeoxyguanine from mitochondrial DNA. Free Radic Biol Med. 2005;38:737–745. doi: 10.1016/j.freeradbiomed.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 62.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced nlrp3-asc inflammasome activation interferes with insulin signaling. Nature immunology. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin-1beta decreases the severity of atherosclerosis in apoe-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:656–660. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 65.Ozaki E, Campbell M, Doyle SL. Targeting the nlrp3 inflammasome in chronic inflammatory diseases: Current perspectives. J Inflamm Res. 2015;8:15–27. doi: 10.2147/JIR.S51250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu X, Kakkar V. Inflammasome and atherogenesis. Curr Pharm Des. 2014;20:108–124. doi: 10.2174/13816128113199990586. [DOI] [PubMed] [Google Scholar]
- 67.Boldogh I, Hajas G, Aguilera-Aguirre L, Hegde ML, Radak Z, Bacsi A, Sur S, Hazra TK, Mitra S. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J Biol Chem. 2012;287:20769–20773. doi: 10.1074/jbc.C112.364620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hashizume M, Mouner M, Chouteau JM, Gorodnya OM, Ruchko MV, Potter BJ, Wilson GL, Gillespie MN, Parker JC. Mitochondrial-targeted DNA repair enzyme 8-oxoguanine DNA glycosylase 1 protects against ventilator-induced lung injury in intact mice. Am J Physiol Lung Cell Mol Physiol. 2013;304:L287–297. doi: 10.1152/ajplung.00071.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramirez CM, Goedeke L, Rotllan N, Yoon JH, Cirera-Salinas D, Mattison JA, Suarez Y, de Cabo R, Gorospe M, Fernandez-Hernando C. Microrna 33 regulates glucose metabolism. Mol Cell Biol. 2013;33:2891–2902. doi: 10.1128/MCB.00016-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rayner KJ, Fernandez-Hernando C, Moore KJ. Micrornas regulating lipid metabolism in atherogenesis. Thromb Haemost. 2012;107:642–647. doi: 10.1160/TH11-10-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Madrigal-Matute J, Rotllan N, Aranda JF, Fernandez-Hernando C. Micrornas and atherosclerosis. Curr Atheroscler Rep. 2013;15:322. doi: 10.1007/s11883-013-0322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moore KJ, Rayner KJ, Suarez Y, Fernandez-Hernando C. Micrornas and cholesterol metabolism. Trends Endocrinol Metab. 2010;21:699–706. doi: 10.1016/j.tem.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karunakaran D, Thrush AB, Nguyen M-A, Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P, Ouimet M, Pezacki JP, Moore KJ, Perisic L, Maegdefessel L, Hedin U, Harper M-E, Rayner KJ. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-mir33 in atherosclerosis. Circulation Research. 2015;117:266–278. doi: 10.1161/CIRCRESAHA.117.305624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Price NL, Fernandez-Hernando C. Novel role of mir-33 in regulating of mitochondrial function. Circulation research. 2015;117:225–228. doi: 10.1161/CIRCRESAHA.117.306949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K, Rayner KJ, Steinberg GR, Zamore PD, Fisher EA, Loke P, Moore KJ. Microrna-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. The Journal of clinical investigation. 2015;125:4334–4348. doi: 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA. Mir33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circulation research. 2014;115:759–769. doi: 10.1161/CIRCRESAHA.115.304164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goedeke L, Salerno A, Ramirez CM, Guo L, Allen RM, Yin X, Langley SR, Esau C, Wanschel A, Fisher EA, Suarez Y, Baldan A, Mayr M, Fernandez-Hernando C. Long-term therapeutic silencing of mir-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol Med. 2014;6:1133–1141. doi: 10.15252/emmm.201404046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rotllan N, Ramirez CM, Aryal B, Esau CC, Fernandez-Hernando C. Therapeutic silencing of microrna-33 inhibits the progression of atherosclerosis in ldlr−/− mice–brief report. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1973–1977. doi: 10.1161/ATVBAHA.113.301732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheng J, Qiao L, Xu X, Zhai C, Zhao K, Ji X, Chen W. Lower amp-activated protein kinase level is associated with the vulnerability of coronary atherosclerotic plaques by attenuating the expression of monocyte autophagy. Coron Artery Dis. 2015;26:322–327. doi: 10.1097/MCA.0000000000000243. [DOI] [PubMed] [Google Scholar]
- 80.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. Ampk phosphorylates and inhibits srebp activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate amp-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic ldl receptor-deficient mice. Diabetes. 2006;55:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- 82.Forouzandeh F, Salazar G, Patrushev N, Xiong S, Hilenski L, Fei B, Alexander RW. Metformin beyond diabetes: Pleiotropic benefits of metformin in attenuation of atherosclerosis. J Am Heart Assoc. 2014;3:e001202. doi: 10.1161/JAHA.114.001202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee MR, Kim SH, Cho HJ, Lee KY, Moon AR, Jeong HG, Lee JS, Hyun JW, Chung MH, You HJ. Transcription factors nf-ya regulate the induction of human ogg1 following DNA-alkylating agent methylmethane sulfonate (mms) treatment. J Biol Chem. 2004;279:9857–9866. doi: 10.1074/jbc.M311132200. [DOI] [PubMed] [Google Scholar]
- 84.Bartz RR, Suliman HB, Fu P, Welty-Wolf K, Carraway MS, MacGarvey NC, Withers CM, Sweeney TE, Piantadosi CA. Staphylococcus aureus sepsis and mitochondrial accrual of the 8-oxoguanine DNA glycosylase DNA repair enzyme in mice. American journal of respiratory and critical care medicine. 2011;183:226–233. doi: 10.1164/rccm.200911-1709OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salminen A, Kaarniranta K. Amp-activated protein kinase (ampk) controls the aging process via an integrated signaling network. Ageing research reviews. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 86.Randolph GJ. Mechanisms that regulate macrophage burden in atherosclerosis. Circulation research. 2014;114:1757–1771. doi: 10.1161/CIRCRESAHA.114.301174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yakes FM, VanHouten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Botto N, Rizza A, Colombo MG, Mazzone AM, Manfredi S, Masetti S, Clerico A, Biagini A, Andreassi MG. Evidence for DNA damage in patients with coronary artery disease. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2001;493:23–30. doi: 10.1016/s1383-5718(01)00162-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.