

Abstract
Apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (A3G) is a host restriction factor that impedes HIV-1 replication. Viral integrity is salvaged by HIV-1 virion infectivity factor (Vif), which mediates A3G polyubiquitination and subsequent cellular depletion. Previous studies have implied that A3G polyubiquitination is essential for Vif-induced degradation. However, the contribution of polyubiquitination to the rate of A3G degradation remains unclear. Here we show that A3G polyubiquitination is essential for degradation. Inhibition of ubiquitin-activating enzyme E1 by PYR-41 or blocking the formation of ubiquitin chains by over-expressing the lysine to arginine mutation of ubiquitin K48 (K48R) inhibited A3G degradation. Our A3G mutagenesis study showed that lysine residues 297, 301, 303 and 334 were not sufficient to render lysine free A3G sensitive to Vif mediated degradation. Our data also confirms that Vif could induce ubiquitin chain formation on lysine residues interspersed throughout A3G. Notably, A3G degradation relied on the lysine residues involved in polyubiquitination. Although A3G and the A3G C-terminal mutant interacted with Vif and were modified by ubiquitin chains, the latter remained more resistant to Vif-induced degradation. Furthermore, the A3G C-terminal mutant, but not the N-terminal mutant, maintained potent antiviral activity in the presence of Vif. Taken together, our results suggest that the location of A3G ubiquitin modification is a determinant for Vif-mediated degradation, implying that in addition to polyubiquitination, other factors may play a key role in the rate of A3G degradation.
Keywords: host restriction factor, polyubiquitination, ubiquitin-activating enzyme E1 inhibitor PYR-41, APOBEC3G lysine free mutant, proteasomal degradation
Graphical Abstract
Introduction
Apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (A3G) is a host restriction factor that impedes HIV-1 replication in the absence of the viral encoded protein, Vif [1]. A3G is a member of the APOBEC family of cytidine deaminase proteins, which range from A3A-A3H, with A3G recognized as exerting the most potent antiviral activity against HIV-1 [2]. In the absence of functional Vif, A3G recognizes and binds the nucleocapsid of HIV-1 Gag polyprotein, to successfully encapsidate itself into progeny virus in order to perform its antiviral activity [3–7]. As a cytidine deaminase protein, A3G induces C-to-U mutations in newly synthesized viral DNA [8–10]. These mutations lead to G-to-A hypermutations, which may introduce premature stop codons as a result of altered reading frames, and ultimately lead to the diminished integrity of the virus. In addition, A3G has been shown to inhibit HIV-1 nuclear import and integration into the host genome [2, 11, 12]. When present, functional Vif facilitates the formation of the Cullin 5-Vif-A3G ubiquitin E3 ligase complex, consisting of core binding factor beta (CBFβ), Elongin B (ELOB), Elongin C (ELOC) and ring box subunit (RBX2) [13–15] to mediate A3G proteasomal degradation, prohibit efficient packaging of A3G into budding virions and overcome A3G antiviral function [13, 16–20].
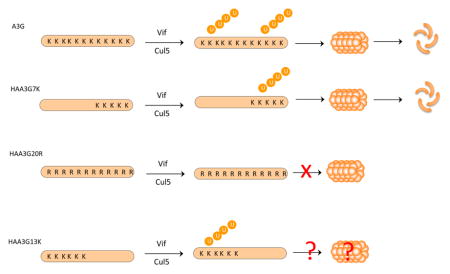
The substrate N-terminus, cysteine, threonine and lysine residues are all potential sites for ubiquitin modification, with lysine recognized as the common target for polyubiquitination [21–29]. A3G has twenty lysine residues which can potentially be targeted by ubiquitin. However, the preferred A3G polyubiquitination modification site(s) for Vif-mediated degradation remains controversial. Findings by Iwatani et al. suggested that spatial constraints between A3G and the Vif-Cullin 5 E3 ligase complex mediate the polyubiquitination of only four lysine residues in the A3G C-terminal domain [30]. Lysine to arginine mutations of these four residues have been shown to block A3G polyubiquitination/proteasomal degradation [30]. Conversely, data by Albin et al. suggested that lysine residues interspersed throughout A3G are modified by ubiquitin [31]. Less is known, however, about the fate of A3G following ubiquitin modification at these disparate sites. In our study, we confirmed that ubiquitin modification occurred on lysine residues dispersed throughout A3G N- and C- terminal regions. Surprisingly, even though both A3G N- and C- terminal regions were polyubiquitinated, polyubiquitination of the A3G N-terminal domain (HAA3G13K) was found to contribute less to Vif-induced proteasomal degradation. Consequently, lysine to arginine substitutions in A3G C-terminal domain (HAA3G13K) increased A3G viral packaging and blocked HIV-1 replication. Our results suggest that ubiquitin modification of A3G is not the sole determinant of A3G degradation. Another factor downstream of A3G polyubiquitination may also play a role in determining the rate of its degradation.
Results
Inhibition of ubiquitin-activating enzyme E1 blocks A3G degradation
Attachment of ubiquitin conjugates to substrate lysine residues influences protein stability, location and function [32, 33]. We previously reported that A3G polyubiquitination is essential for degradation [34]. Here this finding was further confirmed by using an ubiquitin-activating enzyme E1 inhibitor, PYR-41, which irreversibly inhibits E1 activity. A3G-V5 was cotransfected with VR1012 (empty vector) or Vif-cMyc expression plasmid into 293T cells. Two days post-transfection, DMSO or PYR-41 at increasing concentrations was used to treat the cells. MG132 (proteasomal inhibitor) was used as a control. As expected, Vif mediated A3G degradation with DMSO treatment alone (Fig. 1A, lane 2 vs. lane 1); however, increasing concentrations of PRY-41 recovered A3G stability in a dose-dependent manner (Fig. 1A, lanes 3, 4 and 5). In addition, inhibition of the proteasome by MG132 rendered A3G protein levels unchanged compared to the control (Fig 1A, lane 1 vs. lane 6). These results suggest that A3G polyubiquitination is a critical precedent for degradation. Studies from other groups suggested that ubiquitin linkage through ubiquitin K48 is responsible for A3G degradation [35–38]. We also tested this notion by using the dominant negative mutants ubiquitin K48 (UbK48R) and ubiquitin K63 (UbK63R). The 293T cell line was cotransfected with A3G and Vif in the presence of either overexpressed wild-type ubiquitin (Ub-HA), UbK48R or UbK63R. A3G proteolysis was observed when A3G was cotransfected with wild-type Ub-HA or UbK63R; however, UbK48R inhibited Vif-mediated A3G degradation (Fig. 1B, lanes 3 and 4). These results suggest that UbK48, but not UbK63, is the primary ubiquitin conjugate needed for A3G degradation since the dominant negative mutant UbK48R inhibited A3G degradation.
Figure 1. Ubiquitin E1 inhibitor, PYR-41 and K48R block A3G degradation.

(A) 293T cells were cotransfected with A3G and either empty vector, VR1012 or Vif-cMyc. At 24 h post-transfection, the cells were treated with DMSO, MG132 or specific Ubiquitin E1 Inhibitor, PYR-41, overnight. Cells were lysed for Western blotting. (B) 293T cells were cotransfected with A3G and either empty vector, VR1012 or Vif-cMyc in the presence of wild-type ubiquitin (Ub-HA), UbK48R or UbK63R. At 48 h post-transfection, cells were lysed for Western blotting. All data shown throughout this paper are representative of three independent experiments.
A3G lysine residues 297, 301, 303 and 334 are not the sole prerequisites for Vif-mediated degradation
In a previous study, Iwatani et al. showed that A3G C-terminal lysine residues (297, 301, 303 and 334) were critical for A3G sensitivity to Vif, and lysine to arginine mutations of these residues blocked A3G degradation [30]. To determine if the location of modified lysine residues enhances Vif-mediated degradation, we created revertant mutants in the background of our lysine-free A3G mutant, HA-A3G20K/RΔ2K (referred to as HAA3G20R hereafter). Each revertant mutant harbors an HA-tag (YDYVPDYA) fused to the N-terminus (schematic shown in Fig. 2A), as we previously showed that the N-terminus of A3G is an essential ubiquitin acceptor site [27]. When an HA-tag was fused to the N-terminus of lysine-free A3G (HAA3G20R), the A3G mutant was found to be resistant to Vif-induced degradation and its polyubiquitination modification was drastically reduced [27]. Therefore, using HAA3G20R as a template, we generated single arginine to lysine point reversions in A3G positions 297, 301, 303 and 334 (referred to as HAA3G297_K, HAA3G301_K, HAA3G303_K and HAA3G334_K, respectively). In addition, a mutant was created with all four arginine residues reverted back to lysine (referred to as HAA3G4K) as shown in Fig. 2A. To determine if these accessible A3G lysine residues would restore Vif-induced degradation, we cotransfected A3G, HAA3G20R, HAA3G297_K, HAA3G301_K, HAA3G303_K, HAA3G334_K and HAA3G4K into 293T cells with Vif or VR1012. Forty-eight hours post-transfection, the cells were harvested for Western blot analysis. As shown in Fig. 2B, none of the individual point mutations or HAA3G4K was sufficient to restore A3G sensitivity to Vif.
Figure 2. A3G lysine residues 297, 301, 303 and 334 remain Vif insensitive.

(A) Schematic of arginine to lysine reversions generated from A3G lysine-free mutant, HAA3G20R. (B) A3G, HAA3G20R, HAA3G297_K, HAA3G301_K, HAA3G303_K, HAA3G334_K and HAA3G4K were cotransfected with Vif or VR1012 into 293T cells. Forty-eight hours post-transfection, the cells were harvested for Western blot analysis. (C) HAA3G4K, HAA3G20R and A3G were cotransfected with increasing concentrations of Vif (0, 1, 1.5 μg) into 293T cells. At 48 h post-transfection, cells were harvested for Western blotting. (D) 293T cells were cotransfected with A3G, HAA3G20R or HAA3G4K with HA-Ubiquitin-WT and either VR1012 or Vif-c-Myc. At 24 h post-transfection, cells were treated overnight with 10 μM MG132. Ni-NTA beads were used to precipitate A3G under denaturing conditions and analyzed by Western blotting.
To confirm that HAA3G4K is resistant to Vif-induced degradation, 293T cells were cotransfected with A3G, HAA3G20R and HAA3G4K with increasing concentrations of Vif (0, 1, 1.5 μg). Forty-eight hours post-transfection, cells were harvested for Western blot analysis. As expected, HAA3G20R remained stable, while A3G was degraded by Vif in a dose-dependent manner. HAA3G4K remained stable even with the highest Vif concentration (Fig. 2C). These results once again indicate that HAA3G4K is resistant to Vif mediated degradation.
Inhibition of ubiquitin modification of HAA3G4K may block Vif induced degradation. Therefore, to determine if HAA3G4K remains polyubiquitinated, we cotransfected ubiquitin (tagged with HA) with A3G, HAA3G20R and HAA3G4K with either Vif-cMyc or VR1012 into 293T cells. Twenty-four hours post-transfection, 293T cells were treated with the proteasome inhibitor MG132 (10 μM) overnight. Cells were then disrupted in lysis buffer as described in Materials and Methods. Urea was added to the lysates of each sample, and Ni-NTA agarose was used to precipitate A3G under denaturing and native hybrid conditions according to the Invitrogen Ni-NTA purification system manual. Western blot was performed to analyze polyubiquitinated forms of A3G using an anti-HA antibody. As expected, wild-type A3G was heavily modified by ubiquitin chains, while almost no ubiquitin modification was observed in the lysine-free mutant, HAA3G20R (Fig. 2D, lane 2 and 4, respectively). To our surprise, we did not detect the ubiquitin modification of HAA3G4K in our system (Fig. 2D, lane 6).
HAA3G13K increases A3G resistance to Vif
As HAA3G4K was not sufficient to mediate A3G degradation (Fig. 2), we asked the question whether increasing the number of accessible lysine residues would render A3G sensitive to Vif. Therefore, two additional A3G mutants, HAA3G13K and HAA3G7K, were generated using a gene swapping method. HAA3G13K contains 13 lysine residues (N-terminal domain), while HAA3G7K contains 7 lysine residues (C-terminal domain). Each mutant was fused to an HA N-terminal tag to ensure that ubiquitin modification of HAA3G13K and HAA3G7K would occur on internal lysine residues within the N-terminus and C-terminus of A3G, respectively (schematic shown in Fig. 3A). We first tested whether the mutants remained sensitive to Vif-induced degradation. A3G, HAA3G20R, HAA3G7K or HAA3G13K was cotransfected with Vif-cMyc expression plasmid or VR1012 into 293T cells. Forty-eight hours post-transfection, cell lysates were analyzed by Western blot. As expected, A3G was degraded by Vif, while HAA3G20R remained resistant to Vif-mediated degradation (Fig. 3B, lane 1 vs. lane 2; lane 3 vs. lane 4). HAA3G7K was also slightly degraded by Vif; however; HAA3G13K remained insensitive to Vif (Fig. 3B, lane 5 vs. lane 6; lane 7 vs. lane 8). To further confirm these data, HAA3G20R, HAA3G7K or HAA3G13K were cotransfected with increasing concentrations of Vif (0, 1, 1.5 μg), and A3G was cotransfected with 1.5 μg Vif as a control. Forty-eight hours post-transfection, cell lysates were analyzed by Western blot. HAA3G7K degradation was observed in a Vif dose-dependent manner (Fig. 3C, lanes 4, 5 and 6). HAA3G20R and HAA3G13K remained resistant to the highest concentration of Vif (Fig. 3C, lanes 1, 2, 3 for HAA3G13K; lanes 7, 8, 9 for HAA3G20R). These results suggest that the location of accessible lysine residues in A3G influence the rate of its degradation.
Figure 3. HAA3G13K increases A3G resistance to Vif.

(A) Schematic of A3G lysine to arginine substitutions indicated by R. Arginine reversions to lysine are indicated by K. (B) 293T cells were cotransfected with A3G, HAA3G20R, HAA3G7K or HAA3G13K and VR1012 or Vif. At 48 h post-transfection, cells were collected for Western blot analysis. (C) A3G, HAA3G20R, HAA3G7K or HAA3G13K (each 1 μg) were cotransfected with increasing concentrations of Vif (0, 1, 1.5 μg) into 293T cells. At 48 h post-transfection, cells were collected for Western blot analysis.
HAA3G7K and HAA3G13K have a prolonged half-life in the presence of HIV-1 Vif
To observe the half-life of A3G mutants co-expressed with Vif, the steady-state level of A3G mutants was tested using the protein synthesis inhibitor cycloheximide (CHX). First, A3G, HAA3G20R, HAA3G7K or HAA3G13K were cotransfected with Vif into 293T cells. Forty-eight hours post-transfection, cells were treated with CHX (100 μg/ml), and samples were harvested at the indicated time points to detect A3G expression by Western blotting. As expected, wild-type A3G was rapidly degraded by Vif with a half-life of approximately 30 min. Meanwhile, HAA3G7K remained more stable than A3G with a half-life of approximately 110 min. Levels of both HAA3G20R and HAA3G13K remained unchanged by the final time point of 180 min (Fig. 4). These findings once again suggest that the location of accessible A3G lysine residues contributes differently to Vif mediated degradation.
Figure 4. HAA3G7K and HAA3G13K have a prolonged half-life in the presence of HIV- Vif.

293T cells were cotransfected with Vif and either A3G, HAA3G20R, HAA3G7K or HAA3G13K. At 48 h post-transfection, cells were treated with CHX (100 μg/ml), and samples were harvested at the indicated time points.
HAA3G13K efficiently incorporates into wild-type HIV-1 virions and blocks viral replication
If HAA3G13K is more resistant to Vif-induced degradation, it would be expected to package efficiently into wild-type HIV-1 virions and reduce viral infectivity compared to wild-type A3G. Therefore, we next analyzed the packaging efficiency of HAA3G13K into virions. A3G or HAA3G13K were cotransfected with pcDNA3.1, HIV-1 wild-type proviral construct (HXB2) or HIV-1 Vif-deficient proviral construct (HXB2ΔVif) into 293T cells. Forty-eight hours post-transfection, cell samples and culture supernatant samples were harvested. Virus-containing supernatant was used to precipitate viral particles by ultracentrifugation. Virus samples were analyzed by Western blot to measure A3G viral incorporation. As expected, A3G packaging was only detectable with HXB2ΔVif virus (Fig. 5A, lane 3), while HAA3G13K efficiently packaged into both HXB2 and HXB2ΔVif virions (Fig. 5A, lanes 5 and 6). A3G and HAA3G13K did not precipitate in the absence of HIV-1 (Fig. 5A, lanes 1 and 4), indicating the specificity of A3G virion packaging in our system. With efficient viral packaging of HAA3G13K, HAA3G13K was expected to be more effective in hindering wild-type HIV-1 replication compared to A3G. To this end, the effect of A3G mutants on viral infectivity was examined. We cotransfected pcDNA3.1, A3G, HAA3G20R, HAA3G13K or HAA3G7K with either HXB2 or HXB2ΔVif into 293T cells. Forty-eight hours post-transfection, the virus-containing supernatant was harvested and used to infect TZM-bl cells to measure viral infectivity in a multinuclear activation of a galactosidase indicator (MAGI) assay. Viral particles were normalized by HIV-1 p24 enzyme-linked immunosorbent assay (ELISA). A3G and HAA3G7K only efficiently inhibited Vif-deficient virus (Fig. 5C). In agreement with the Vif-resistant phenotype of HAA3G20R and HAA3G13K, shown in the Western blot (Fig. 5B), both mutants drastically inhibited the infectivity of Vif-deficient and wild-type viruses (Fig. 5C).
Figure 5. HAA3G13K packages into HIV-1 virions and blocks HIV-1 replication.

(A) 293T cells were cotransfected with A3G and HAA3G13K in combination with HXB2 or HXB2ΔVif. At 48 h post-transfection, virus-containing supernatant was precipitated by ultracentrifugation, and cells were harvested for Western blot analysis. (B) 293T cells were cotransfected with A3G, HAA3G20R, HAA3G7K and HAA3G13K in combination with HXB2 or HXB2ΔVif. At 48 h post-transfection, cells were harvested for Western blot analysis. (C) Cell culture supernatants were used to infect TZM-bl indicator cells to measure viral infectivity. The amount of virus input was normalized by p24 ELISA.
A3G mutants bind to Vif
The Vif-A3G interaction is essential for Vif to induce A3G degradation [13, 16, 18–20], and changes in the binding strength between these molecules has been suggested to affect Vif-mediated degradation of A3G [38]. To determine if Vif maintains its interaction with HAA3G13K, we performed a co-immunoprecipitation assay. Vif-cMyc was cotransfected with pcDNA3.1, A3G, HAA3G20R, HAA3G7K, HAA3G13K or HAA3G4K into 293T cells. Twenty-four hours post-transfection, 293T cells were treated with the proteasome inhibitor MG132 (10 μM) overnight to evade A3G degradation. Cells were then disrupted in lysis buffer as described in Materials and Methods, and anti-V5 beads were used to immunoprecipitate A3G. Western blot was performed to analyze the A3G and Vif-cMyc interaction. Notably, in the absence of A3G, we did not detect Vif in the immunoprecipitated sample, suggesting that nonspecific binding of Vif did not occur in our system (Fig. 6, lane 1). When wild-type A3G and A3G mutants were present, Vif was efficiently precipitated, suggesting that the mutations in A3G did not interfere with Vif binding.
Figure 6. A3G, HAA3G20R, HAA3G7K, HAA3G13K and HAA3G4K bind to Vif.
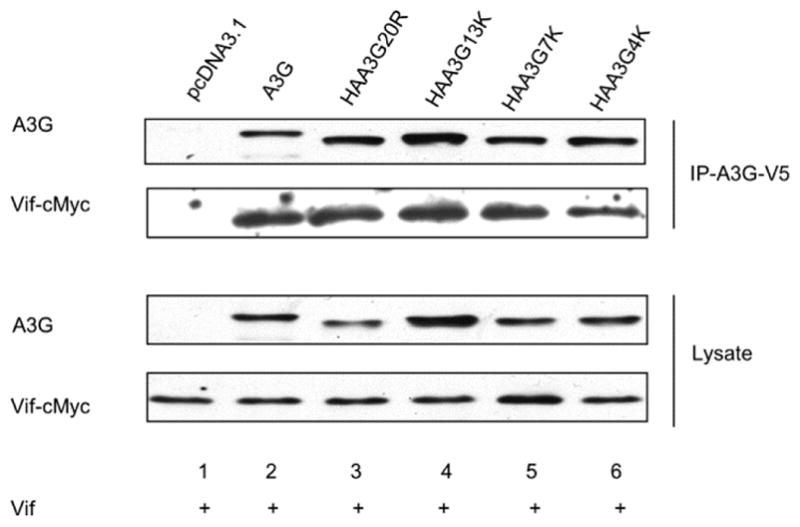
pcDNA3.1, A3G, HAA3G20R, HAA3G7K, HAA3G13K and HAA3G4K were cotransfected into 293T cells with Vif. At 24 h post-transfection, cells were treated with 10 μM MG132 overnight. Samples were subjected to anti-V5 affinity beads to precipitate A3G, and the A3G-Vif interaction was analyzed by Western blotting.
Vif-induced polyubiquitination occurs on lysine residues dispersed throughout A3G
Since our data confirms that polyubiquitination is essential for A3G proteasomal degradation, we set out to determine if the arginine residues in A3G N-terminal domain (HAA3G7K) or C-terminal domain (HAA3G13K) inhibit A3G ubiquitin modification. We cotransfected ubiquitin (tagged with HA) with A3G, HAA3G20R, HAA3G7K and HAA3G13K with either Vif-cMyc or VR1012 into 293T cells. A polyubiquitination assay was performed as described in Materials and Methods. As shown previously, wild-type A3G was heavily modified by ubiquitin chains, while we observed almost no ubiquitin modification of HAA3G20R (Fig. 7, lane 2 and 4, respectively). Surprisingly both HAA3G13K and HAA3G7K were heavily polyubiquitinated (Fig. 7, lanes 6 and 8). This data confirms that lysine residues interspersed throughout A3G are targeted for ubiquitin modification. Once again, we did not detect Vif in the precipitated sample, suggesting that Vif did not contribute to the observed polyubiquitination.
Figure 7. Vif-induced polyubiquitination occurs on lysine residues dispersed throughout A3G.
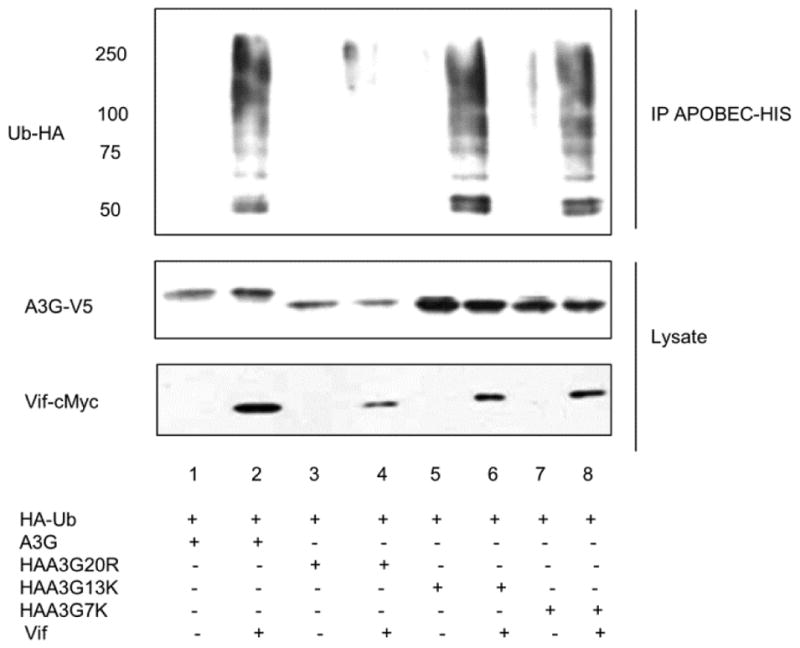
293T cells were transfected with A3G, HAA3G20R, HAA3G7K and HAA3G13K with HA-Ubiquitin-WT and either with empty vector, VR1012, or Vif-c-Myc. At 24 h post-transfection, cells were treated with 10 μM MG132 overnight. Samples were applied to Ni-NTA beads to precipitate A3G under denaturing conditions and analyzed by Western blotting.
Discussion
Polyubiquitination is generally believed to be required for Vif-mediated degradation of A3G, but determining which of the twenty lysine residues in A3G are the essential ubiquitin target sites has been controversial. Data by Iwatani et al. suggest that A3G lysine residues 297, 301, 303 and 334 are the critical determinants for Vif-induced A3G degradation [30]. When Iwatani et al. mutated all four lysine residues to arginine, A3G polyubiquitination was inhibited, suggesting that these four lysine residues were essential ubiquitin acceptor sites [30]. Meanwhile, Albin et al. reported five lysine residues in the N-terminal domain and five residues in the C-terminal domain of A3G that were modified by ubiquitin in the presence of Vif [31]. This observation suggests that lysine residues interspersed throughout A3G contribute to Vif-mediated degradation [31]. We also sought to determine the essential lysine residues for A3G polyubiquitination/degradation by using our lysine-free A3G mutant, HAA3G20R (schematic shown in Fig. 3A), which was previously reported to be resistant to Vif-induced degradation [27]. Based on this lysine-free construct, revertant mutants were created by substituting arginine to lysine at positions 297, 301, 303 and 334 (HAA3G4K) (Fig. 2). HAA3G4K interacted with Vif (Fig. 6) but was not heavily modified by ubiquitin (Fig. 2D lane 6) and remained resistant to Vif-induced degradation (Fig. 2). Using mass spectrometry, Albin et al. reported ubiquitin modification of A3G lysine residues 297, 303 and 334 [31]. Notably, polyubiquitination of residue 301 was not detected and the authors reported slight variation between experiments [31]. The exact reason for the discrepancy between our findings and those reported previously warrants further investigation. To further determine the critical A3G ubiquitin target sites for Vif-mediated degradation, we introduced three additional arginine to lysine mutations at positions 249, 270 and 344 (referred to as HAA3G7K). Additionally, we generated HAA3G13K which contains 13 lysine residues in A3G N-terminal domain. Both HAA3G7K and HAA3G13K increased A3G resistance to Vif. However, while HAA3G7K was partially degraded by Vif (Figs. 3, 4 and 5), HAA3G13K remained resistant to Vif induced degradation, efficiently packaged into HIV-1 virions and reduced viral replication (Figs. 3, 4, 5A and 5C). Surprisingly, our polyubiquitination assay suggests that HAA3G13K and HAA3G7K were modified by ubiquitin to comparable levels as wild-type A3G (Fig. 7). These data suggest that the downstream rate of proteolysis following A3G polyubiquitination depends on the location of the ubiquitin-modified lysine residues.
K-48-linked chain formation is recognized as a signal for substrate translocation to the proteasome for degradation. However, in the case of A3G, we found that HAA3G13K was modified by polyubiquitination but more resistant to Vif induced degradation. Previous work suggests that ubiquitin modification is not a prerequisite for proteasomal degradation [39–41]. For instance, transcription factor Met4 is the critical target of ubiquitin ligase Cdc34/SCFMet30. Although Cdc34/SCFMet30 mediates Met4 polyubiquitination, instead of degradation by the proteasome, ubiquitin modification results in direct inhibition of Met4 as a transcriptional regulator [41]. Yet, how ubiquitin-modified proteins escape proteasomal degradation remains unknown. Work by Zhao et al. suggests that proteasomal degradation requires multifaceted coordination between deubiquitinases, substrate unfolding, ATP hydrolysis and interactions between proteins for substrate recognition [42]. The conformational structure of modified proteins has been proposed to prevent the substrate from tethering to the 19S regulatory subunit of the proteasome or from entering into the 20S catalytic core [43]. Alternatively, the translocation of substrates to the proteasome may be impeded. In addition, whether the structure of ubiquitin modification (branched or linear) alters the rate of the degradation of the substrate remains unknown [44, 45]. Further research is warranted to determine the mechanism for how HAA3G13K renders A3G more stable despite being heavily polyubiquitinated.
Ultimately, our results suggest that the degradation outcome of A3G polyubiquitination depends on the particular lysine residues modified by ubiquitin. This finding reveals novel insight into the different consequences following A3G ubiquitin chain formation.
Materials and Methods
Plasmids, antibodies and reagents
The expression constructs A3G-V5, HAA3G20R (previously named HAA3G20K/RΔ2K), Vif-cMyc, proviral construct HXB2 and Vif-deficient proviral construct HXB2ΔVif were described previously [27, 34]. HAA3G13K was derived using BamH I restriction enzyme digestion and gene swapping between HAA3G20R (vector) and HAA3G (insert). The HAA3G7K mutant was created using BamH I restriction enzyme digestion and gene swapping between A3G (vector) and HAA3G20R (insert). All A3G mutants were expressed with an HA tag in the N-terminus, and the two lysine residues in the C-terminal tag region on the vector were removed using a PCR-based method as previously described [34]. A3G point mutations HAA3G297_K, HAA3G301_K, HAA3G303_K, HAA3G334_K and the combined four lysine (HAA3G4K) mutants were generated using HAA3G20R as a template and the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent Technologies). The following primers were used to generate the A3G point mutations: HAA3G297_K, 5′-CTGTGCCCAGGAAATGGCTAAGTTCATTTCACGCAACCGCC-3′; HAA3G301_K, 5′-AGGAAATGGCTCGCTTCATTTCAAAGAACCGCCACGTGAGC-3′; HAA3G303_K, 5′-CGCTTCATTTCACGCAACAAGCACGTGAGCCTGTGCATC-3′; HAA3G334_K, 5′-CCTGGCCGAGGCTGGGGCCAAGATTTCAATAATGACATACAG-3′. The following primers were used to generate HAA3G4K: 5′-GCTTCAGCTGTGCCCAGGAAATGGCTAAGTTCATTTCAAAGAACAAGCACGTGAGC CTGTGCATCTTCACTGC-3′and CCTGGCCGAGGCTGGGGCCAAGATTTCAATAATGACATACAG. The N-terminal HA-tagged ubiquitin expression construct (pRK5-HA-Ubiquitin-WT), pRK5-HA-ubiquitin K48R and pKR5-HA-ubiquitin K63R were obtained from Ted Dawson through Addgene. Monoclonal anti-c-Myc (4A6), anti-HA (HA11), anti-V5 and anti β-actin antibodies were obtained from Millipore, Covance, Invitrogen and Sigma, respectively. Rabbit anti-Vif antibody, rabbit anti-A3G antibody [46] and HIV-1 p24 monoclonal antibody [47] were obtained from the the NIH AIDS Research and Reference Reagent Program. Proteasome inhibitor (MG132), CHX and E1 activating enzyme inhibitor (PYR-41) were purchased from Sigma.
Cell culture, Western blot analysis and viral infectivity (MAGI) assay
Human embryonic kidney (HEK) 293T cells and TZM-bl cells (NIH AIDS Research and Reference Reagent Program) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) in a 5% CO2 atmosphere. Plasmid DNA transfection was performed with polyethylenimine (PEI) as described previously [27]. At 48 h post-transfection, virus-containing supernatant was harvested for the MAGI assay and virus preparation. HIV-1 virions were prepared by centrifugation at 1,000 × g for 15 min to remove cellular debris and filtered through a 0.2 μm pore size membrane. Next, viral particles were ultracentrifuged at 100,000 × g for 2 h on a 20% sucrose cushion. Viral infection was determined by a MAGI assay as previously described [48–50]. Western blot analyses of cell lysates and virion lysates were performed as previously described [27]. All data shown throughout this paper are representative of three independent experiments.
Affinity precipitation assay
pcDNA3.1, A3G, HAA3G20R, HAA3G13K, HAA3G7K and HAA3G4K were cotransfected with a Vif expression vector into 293T cells. Forty-eight hours post-transfection, cells were disrupted in lysis buffer (50 mM sodium phosphate, pH 8.0, 500 mM NaCl, 1% Triton X-100, and EDTA-free protease inhibitor cocktail from Roche) at 4°C for 30 min. Following centrifugation at 16,000 × g for 30 min at 4°C, the lysates were incubated with anti-V5 beads (Sigma) for 4 h and washed five times with wash buffer (50 mM sodium phosphate, pH 8.0, 500 mM NaCl, 20 mM imidazole). The beads were boiled in SDS-PAGE sample buffer, and samples were subsequently analyzed by Western blotting.
CHX assay
The CHX assay was used to observe the half-life of wild-type A3G and A3G mutants. A3G, HAA3G20R, HAA3G7K and HAA3G13K were cotransfected with a Vif expression plasmid into 293T cells. Forty-eight hours post-transfection, cells were treated with CHX (100 μg/ml), and samples were harvested at the indicated time points for Western blot analysis. The intensity of the protein bands was analyzed by densitometry.
In vivo polyubiquitination assay
The 293T cell line was transfected with the indicated A3G-V5 expression constructs, pRK5-HA-Ubiquitin-WT, and either empty vector, VR1012, or Vif expression vectors. Twenty-four hours post-transfection, MG132 (10 μM) was used to treat the cells overnight. Treated cells were then disrupted in lysis buffer (20 mM sodium phosphate, pH 7.8, 500 mM NaCl, 1% Triton X-100, 10 mM β-mercaptoethanol, 10 μM MG132, 5 mM iodoacetanide, 20 mM imidazole and EDTA-free protease inhibitor cocktail from Roche) and applied to Ni-NTA agarose beads to precipitate APOBEC under denaturing/native hybrid conditions as described in the manual of the Invitrogen Ni-NTA Purification System. A3G was eluted from the beads using 50 mM sodium phosphate, pH 8.0, 500 mM NaCl and 250 mM imidazole. The ubiquitin modification of A3G was analyzed by Western blot.
Highlights.
K48 linked polyubiquitination is essential for Vif induced A3G degradation
A3G lysine residues 297, 301, 303 and 334 were not sufficient to render A3G sensitive to Vif- mediated degradation.
Ubiquitin-modified A3G lysine residues contribute differently to HIV-1 Vif induced A3G degradation.
Ubiquitin modification occurs on lysine residues interspersed throughout A3G
Acknowledgments
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: anti-HIV-1 HXB2 Vif polyclonal antibody; TZM-bl from John C. Kappes, Xiaoyun Wu and Tranzyme Inc.; HIV-1 p24 monoclonal antibody from Bruce Chesebro and Kathy Wehrly; anti-ApoC17 from Klaus Strebel. This work was partially supported by NIH grants SC1GM089269, G12MD007586 and P30AI110527 (TN-CFAR) to BL. TMT is supported by T32AI007281.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–50. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 2.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364:675–87. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Douaisi M, Dussart S, Courcoul M, Bessou G, Vigne R, Decroly E. HIV-1 and MLV Gag proteins are sufficient to recruit APOBEC3G into virus-like particles. Biochem Biophys Res Commun. 2004;321:566–73. doi: 10.1016/j.bbrc.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Alce TM, Popik W. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J Biol Chem. 2004;279:34083–6. doi: 10.1074/jbc.C400235200. [DOI] [PubMed] [Google Scholar]
- 5.Cen S, Guo F, Niu M, Saadatmand J, Deflassieux J, Kleiman L. The interaction between HIV-1 Gag and APOBEC3G. J Biol Chem. 2004;279:33177–84. doi: 10.1074/jbc.M402062200. [DOI] [PubMed] [Google Scholar]
- 6.Luo K, Liu B, Xiao Z, Yu Y, Yu X, Gorelick R, et al. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J Virol. 2004;78:11841–52. doi: 10.1128/JVI.78.21.11841-11852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schafer A, Bogerd HP, Cullen BR. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology. 2004;328:163–8. doi: 10.1016/j.virol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–9. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 9.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–8. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 12.Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81:7099–110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 14.Stanley DJ, Bartholomeeusen K, Crosby DC, Kim DY, Kwon E, Yen L, et al. Inhibition of a NEDD8 Cascade Restores Restriction of HIV by APOBEC3G. PLoS Pathog. 2012;8:e1003085. doi: 10.1371/journal.ppat.1003085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Du J, Evans SL, Yu Y, Yu XF. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature. 2012;481:376–9. doi: 10.1038/nature10718. [DOI] [PubMed] [Google Scholar]
- 16.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–13. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 17.Enfissi A, Codrington J, Roosblad J, Kazanji M, Rousset D. Zika virus genome from the Americas. Lancet (London, England) 2016 doi: 10.1016/S0140-6736(16)00003-9. [DOI] [PubMed] [Google Scholar]
- 18.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 19.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 20.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–7. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Saadon R, Fajerman I, Ziv T, Hellman U, Schwartz AL, Ciechanover A. The tumor suppressor protein p16(INK4a) and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system. Direct evidence for ubiquitination at the N-terminal residue. J Biol Chem. 2004;279:41414–21. doi: 10.1074/jbc.M407201200. [DOI] [PubMed] [Google Scholar]
- 22.Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115:71–82. doi: 10.1016/s0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- 23.Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998;17:5964–73. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadwell K, Coscoy L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science. 2005;309:127–30. doi: 10.1126/science.1110340. [DOI] [PubMed] [Google Scholar]
- 25.Ciechanover A, Ben-Saadon R. N-terminal ubiquitination: more protein substrates join in. Trends Cell Biol. 2004;14:103–6. doi: 10.1016/j.tcb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Kuo ML, den Besten W, Bertwistle D, Roussel MF, Sherr CJ. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 2004;18:1862–74. doi: 10.1101/gad.1213904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Shao Q, Yu X, Kong W, Hildreth JE, Liu B. N-terminal hemagglutinin tag renders lysine-deficient APOBEC3G resistant to HIV-1 Vif-induced degradation by reduced polyubiquitination. J Virol. 2011;85:4510–9. doi: 10.1128/JVI.01925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Herr RA, Chua WJ, Lybarger L, Wiertz EJ, Hansen TH. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J Cell Biol. 2007;177:613–24. doi: 10.1083/jcb.200611063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Hong Y, Wang W, Wu W, Chi Y, Zong H, et al. HSP70 protects BCL2L12 and BCL2L12A from N-terminal ubiquitination-mediated proteasomal degradation. FEBS Lett. 2009;583:1409–14. doi: 10.1016/j.febslet.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Iwatani Y, Chan DS, Liu L, Yoshii H, Shibata J, Yamamoto N, et al. HIV-1 Vif-mediated ubiquitination/degradation of APOBEC3G involves four critical lysine residues in its C-terminal domain. Proc Natl Acad Sci U S A. 2009;106:19539–44. doi: 10.1073/pnas.0906652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Albin JS, Anderson JS, Johnson JR, Harjes E, Matsuo H, Krogan NJ, et al. Dispersed sites of HIV Vif-dependent polyubiquitination in the DNA deaminase APOBEC3F. J Mol Biol. 2013;425:1172–82. doi: 10.1016/j.jmb.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–6. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 33.Fushman D, Wilkinson KD. Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep. 2011;3:26. doi: 10.3410/B3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao Q, Wang Y, Hildreth JE, Liu B. Polyubiquitination of APOBEC3G is essential for its degradation by HIV-1 Vif. J Virol. 2010;84:4840–4. doi: 10.1128/JVI.01911-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–8. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 36.DeHart JL, Bosque A, Harris RS, Planelles V. Human immunodeficiency virus type 1 Vif induces cell cycle delay via recruitment of the same E3 ubiquitin ligase complex that targets APOBEC3 proteins for degradation. J Virol. 2008;82:9265–72. doi: 10.1128/JVI.00377-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature. 2012;481:371–5. doi: 10.1038/nature10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baig TT, Feng Y, Chelico L. Determinants of efficient degradation of APOBEC3 restriction factors by HIV-1 Vif. J Virol. 2014;88:14380–95. doi: 10.1128/JVI.02484-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verdier F, Valovka T, Zhyvoloup A, Drobot LB, Buchman V, Waterfield M, et al. Ruk is ubiquitinated but not degraded by the proteasome. Eur J Biochem. 2002;269:3402–8. doi: 10.1046/j.1432-1033.2002.03031.x. [DOI] [PubMed] [Google Scholar]
- 40.Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol. 2001;2:870–5. doi: 10.1038/ni0901-870. [DOI] [PubMed] [Google Scholar]
- 41.Kaiser P, Flick K, Wittenberg C, Reed SI. Regulation of transcription by ubiquitination without proteolysis: Cdc34/SCF(Met30)-mediated inactivation of the transcription factor Met4. Cell. 2000;102:303–14. doi: 10.1016/s0092-8674(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 42.Zhao M, Zhang NY, Zurawel A, Hansen KC, Liu CW. Degradation of some polyubiquitinated proteins requires an intrinsic proteasomal binding element in the substrates. J Biol Chem. 2010;285:4771–80. doi: 10.1074/jbc.M109.060095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang L, Monteiro MJ. Defective Proteasome Delivery of Polyubiquitinated Proteins by Ubiquilin-2 Proteins Containing ALS Mutations. PLoS One. 2015;10:e0130162. doi: 10.1371/journal.pone.0130162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HT, Kim KP, Uchiki T, Gygi SP, Goldberg AL. S5a promotes protein degradation by blocking synthesis of nondegradable forked ubiquitin chains. EMBO J. 2009;28:1867–77. doi: 10.1038/emboj.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer HJ, Rape M. Enhanced protein degradation by branched ubiquitin chains. Cell. 2014;157:910–21. doi: 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kao S, Miyagi E, Khan MA, Takeuchi H, Opi S, Goila-Gaur R, et al. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology. 2004;1:27. doi: 10.1186/1742-4690-1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chesebro B, Wehrly K, Nishio J, Perryman S. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol. 1992;66:6547–54. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:2855–64. doi: 10.1128/jvi.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, et al. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74:8358–67. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]