

Abstract
The host innate immune system serves as the first line of defense against viral infections. Germline encoded pattern recognition receptors (PRRs) detect molecular patterns associated with pathogens and activate innate immune responses. Of particular relevance to viral infections are those PRRs that activate type I interferon (IFN) responses, which establish an antiviral state. The Order Mononegavirales is comprised of viruses that possess single-stranded, non-segmented, negative-sense (NNS) RNA genomes and are important human pathogens that consistently antagonize signaling related to IFN responses. NNS viruses (NNSVs) have limited encoding capacity compared to many DNA viruses. And as a likely consequence, most open reading frames (ORFs) encode multifunctional viral proteins that interact with host factors in order to evade host cell defenses while promoting viral replication. In this review, we will discuss the molecular mechanisms of innate immune evasion by select NNSVs. A greater understanding of these interactions will be critical in facilitating the development of effective therapeutics and viral countermeasures.
Keywords: Mononegavirales, interferon antagonist, innate immune evasion, viral antagonism
Graphical Abstract
Introduction
The onset of viral infections are first detected by the host innate immune system that stimulates the production of type I interferons (IFNs) and pro-inflammatory cytokines, which can limit multiple stages of the viral life cycle. Viruses can subvert host defenses by encoding for proteins that inhibit innate immune responses. Single-stranded, non-segmented, negative-sense RNA viruses (NNSVs) in the Order Mononegavirales include many efficient pathogens which encode multifunctional proteins. Versatile NNSV proteins have the ability to participate in multiple host-virus interactions in order to evade, antagonize, and disrupt host antiviral protein functions. A detailed understanding of host-virus relationships is crucial to the development of countermeasures to mitigate NNSV infections. Here we focus on the mechanistic basis of molecular interactions occurring between NNSV-encoded proteins and the host proteins of the IFN signaling cascade. This review is organized and presented in two major categories: 1) type I IFN induction due to viral infection and NNSV-mediated inhibition of IFN induction, and 2) type I IFN responses to viral infection and NNSV-mediated inhibition of IFN responses; this is followed by a brief discussion on NNSV-mediated modulation of the host ubiquitination system. While much work has been done in this area, NNSVs use few but multifunctional proteins to disrupt essentially every step in the IFN pathway. The key host-viral interactions described in this review will be important targets to develop novel countermeasures against NNSVs.
Genome organization of NNSVs
NNSVs are divided into five viral families: Bornaviridae, Filoviridae, Paramyxoviridae, Rhabdoviridae, and Nyamiviridae, where the genomes range from 8 to 19 kilobases in size and contain 6 to 11 open reading frames (ORFs) within a single strand of RNA (Fig. 1). The proteins common to all NNSVs include nucleoprotein (N/NP), phosphoprotein (P), matrix protein (M), glycoprotein (G/GP), and the RNA-dependent RNA polymerase (RdRp) also known as large (L) protein. These proteins generally have conserved functions. For example, N/NP coats the viral RNA forming the nucleocapsid whereas G/GP permits attachment and fusion to the host cell. The M protein facilitates viral assembly and budding, and the L polymerase forms part of the viral replication complex along with the P protein [1, 2]. Bornaviruses and Nyamiviruses have an X protein that is involved in regulation of gene synthesis and immune inhibition [3–6]. Filoviruses and paramyxoviruses have more complex genomes that encode for several additional proteins. In filoviruses, VP35 and VP40 are the counterparts to the P and M proteins. In addition, filoviruses contain VP24 and VP30 proteins, which function as the minor matrix protein and to regulate replication/transcription, respectively. Some paramyxoviruses, including henipaviruses, rubulaviruses, pneumoviruses, and metapneumoviruses, also produce a fusion (F) protein and a small hydrophobic (SH) protein that are associated with the viral envelope to facilitate viral entry. The M2 protein found in pneumoviruses and metapneumoviruses are involved in regulation of replication/transcription. Finally, the non-structural proteins NS1 and NS2 are unique only to pneumoviruses that are not packaged as part of the virion, but are critical for inhibition of IFN induction and signaling [7, 8].
Figure 1. Genome organization of NNSVs.

Shown are the different viral genomes included within the Order Mononegavirales. Examples of genus and species (in parentheses) of viruses with the corresponding genome organization are listed on the right. Gene products common to all NNSVs are similarly colored, while genes unique to a specific family are different. Species abbreviations are as follows: Borna disease virus (BDV), Ebolavirus (EBOV), Nipah virus (NiV), Parainfluenza virus (PIV), respiratory syncytial virus (RSV), human metapneumovirus (MPV), rabies virus (RABV), and Midway virus (MIDWV).
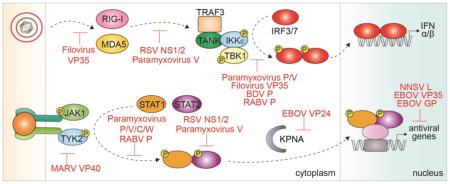
The limited RNA genome of NNSVs in some ways can restrict the genomic diversity and redundancy that is often observed in DNA viruses [9]. As a potential mechanism to overcome limited coding capacity of NNSVs, many NNSV encoded proteins perform multiple functions at distinct stages of the viral replication cycle. Recent studies are beginning to elucidate the molecular basis of the common and unique strategies employed by these viral proteins to evade host innate immune responses while promoting viral replication. In the following sections, we will provide an overview of the host type I interferon responses and discuss the different immune evasion strategies of NNSVs that target various points of the IFN α/β signal transduction pathway (Fig. 2).
Figure 2. Viral inhibitors of the type I IFN signaling pathway.

A simplified representation of the type I IFN signal transduction pathway. A. IFN induction. Viral PAMPS are detected by host PRRs, such as RLRs and TLRs, which lead to the production of type I IFNs and pro-inflammatory cytokines. B. IFN response. IFNα/β binding to IFNAR1/2 activates the Jak/STAT pathway leading to the expression of ISGs. Different steps of the pathway targeted by NNSV proteins are highlighted in red.
Type I IFN induction during viral infection
The innate immune system is an evolutionarily conserved branch of the host response that serves as the first line of defense against invading pathogens. Detection of viral infection is mediated by pattern recognition receptors (PRRs), which recognize pathogen associated molecular patterns (PAMPs) absent in the host. Two major types of PRRs are RIG-I like receptors (RLRs) and Toll-like receptors (TLRs) [1, 10]. RLRs, including retinoic acid inducible gene I (RIG-I) and melanoma associated differentiation factor 5 (MDA5), are cytoplasmic proteins containing N-terminal caspase activation and recruitment domains (CARDs), a central DExD/H box helicase domain, and C-terminal RNA binding domains that recognize PAMPs from viral genomic material, including those generated due to viral replication within the host cells [11, 12]. RIG-I is activated by short double stranded RNA (dsRNA) with 5′-ppp or 5′-OH groups whereas MDA5 preferentially binds to longer lengths of dsRNA [11–17]. Signaling through CARD-CARD interaction between RLRs and the mitochondrial activator of viral signaling (MAVS, also known as IPS-1, VISA, or Cardif) located on the mitochondria [18, 19] (Fig. 2A) results in the activation of TANK binding kinase (TBK1) and inhibitor of nuclear factor kappa-B kinase subunit ε (IKKε) through association with TANK (TRAF family member associated NFκB activator) and TRAF3 (TNF receptor associated factor 3). TBK1/IKKε kinases phosphorylate the transcription factors interferon regulatory factors 3 and 7 (IRF3 and IRF7), which dimerize and translocate into the nucleus to activate the expression of type I IFNs [10].
TLRs are integral membrane glycoproteins that are present in cell, endosomal, and lysosomal membranes. TLR molecules bind to PAMPs through their extracellular domains, which contain leucine rich repeats. TLR2/4 molecules located in cell membranes bind to viral envelope proteins. TLR7/8 and TLR3 detects single stranded RNA (ssRNA) and dsRNA, respectively, in the endosomal compartments [17]. TLR3 activation followed by recruitment of the TIR domain containing adaptor-inducing IFN-β (TRIF) or TLR7/8 activation and the recruitment of adaptor proteins myeloid differentiation factor 88 (MyD88) can also induce type I IFN production by activating IRF3 and nuclear factor kappa-B (NFκB) [17]. Signaling through MyD88 leads to the phosphorylation of inhibitor of kappa light polypeptide gene enhancer in B-cells kinase beta (IKKβ). MyD88-mediated signaling ultimately leads to the activation of NF-κB that stimulate the expression of proinflammatory cytokines, which limit virus replication through a variety of mechanisms (Fig. 2A) [20, 21].
NNSV-mediated inhibition of IFN induction
Type I IFN signaling functions to restrict viral infection and therefore, it is not surprising that viruses have developed strategies to circumvent or inhibit this system. Because PRRs provide immune surveillance to the cell and are the first to detect invasion by foreign particles, these receptors are highly targeted by viral proteins. However, as discussed in the examples below, NNSVs have developed mechanisms that target many critical steps in the IFN signaling cascade.
Sequestration of viral RNA PAMPs
One of best studied examples of PAMP sequestration by NNSVs is in filoviruses. Ebola virus (EBOV) and Marburg virus (MARV) VP35 proteins bind viral dsRNA to prevent their recognition by PRRs RIG-I and MDA5 [22, 23]. Structural studies of the C-terminal IFN inhibitory domain (IID) of EBOV VP35 in complex with an 8 bp in vitro transcribed RNA showed that a series of conserved basic residues facilitate binding of EBOV VP35 to the phosphodiester backbone of dsRNA (Fig. 3A and B) [23]. Mutational analysis of these residues, particularly Arg312, shows that these basic residues are important for dsRNA binding and IFN inhibition [22]. In fact, substitution of Lys319 and Arg322 with alanines renders a guinea pig adapted EBOV avirulent [24]. EBOV VP35 also caps the blunt ends of dsRNA through hydrophobic residues Phe235 and Phe239[22]. This dual interaction provides a mechanism that allows EBOV VP35 to efficiently sequester dsRNA from detection by and activation of RIG-I and MDA5. Similarly, MARV VP35 coats the dsRNA backbone. Although MARV VP35 is structurally homologous to EBOV VP35, with a backbone rmsd < 1.0 Å [23], MARV VP35 is unable to endcap dsRNA and preferentially binds longer dsRNA that are targeted by the RLR MDA5 [19, 23, 25] (Fig. 3C). These differences in MARV VP35 recognition of dsRNA motifs manifests in less efficient inhibition of RIG-I signaling compared to EBOV VP35 [26]. The impact of these interactions in vivo is likely to be more complex than indicated by the in vitro studies. However, the correspondence between in vitro studies that evaluate RNA sequestration with in vivo studies of corresponding mutant viral infections support the relevance of these mechanisms.
Figure 3. PAMP sequestration by filoviral VP35 proteins.

A. Crystal structure showing four molecules of Ebola virus VP35 (EBOV VP35) IID bound to an 8bp dsRNA (magenta) (PDB 3L25) [22]. The area marked with a black square is shown in greater detail in B. Residues in EBOV VP35 IID makes specific contacts to both the RNA phosphodiester backbone and the blunt ends. Residues colored blue make important protein-RNA backbone contacts while residues colored yellow contact the blunt ends. C. Crystal structure of Marburg virus VP35 (MARV VP35) IID bound to 18bp dsRNA (PDB 4GHL) showing that MARV VP35 IID does not end cap but coats the double stranded RNA backbone [23].
Direct binding to RLR
The V proteins of paramyxoviruses can bind to MDA5 and LGP2, but not RIG-I, in order to disrupt downstream signaling responsible for IFN induction [27–31]. The crystal structure of the parainfluenza virus 5 (PIV5) V protein in complex with MDA5 provides the molecular basis of how the V protein disrupts MDA5 function [27] (Fig. 4). The V protein C-terminal domain undergoes a large conformational change upon binding to MDA5 and refolds into a long β-hairpin stabilized by two zinc ions. This long hairpin displaces in MDA5 two β-strands β1 and β6 and the ATPase motif VI and is stabilized by two key interactions, a salt bridge between Glu174 of V and Arg803 of MDA5/LGP2, and a hydrophobic interaction between Trp179 of V and Gly805 of MDA5/LGP2. Consequently, V protein binding disrupts the ATPase activity of MDA5, which is required in the formation of a signaling platform with the CARD domains of MAVS [27, 32]. In RIG-I, Glu803 and Gly805 residues are substituted with a leucine and a glutamate, thus precluding its complex formation with V proteins [27, 31]. These structural observations suggest that specific inhibition of MDA5 and LGP2, but not RIG-I, is likely important for some paramyxoviruses in order to evade host innate immunity. While direct binding provides a highly effective mechanism, modulation of post transcriptional modifications can also facilitate evasion of host immunity (See RLR dephosphorylation section).
Figure 4. Structural rearrangement of MDA5 upon binding to the PIV5 V protein.

A. Crystal structure of the SF2 domain of human MDA5 (PDB 4GL2) [127]. B. Crystal structure of the SF2 domain of porcine MDA5 (yellow) bound to PIV5 V protein residues 168 to 219 (red) (PDB 4I1S) [27]. The key interactions that stabilize the complex are shown as sticks.
The non-structural proteins NS1 and NS2 of respiratory syncytial virus (RSV) also appear to directly target the interaction between RIG-I and MAVS. Live cell imaging of a GFP expressing strain of RSV showed that the virus localized to the mitochondria, which is a signaling hub for IFN production [33]. Furthermore, immunofluorescence and electron microscopy show that transfected RSV NS1 colocalizes with MAVS, consistent with immunoprecipitation studies showing that MAVS binds to NS1 and prevents MAVS binding to RIG-I [33] (Fig. 5A). Similarly, RSV NS2 has been reported to disrupt RIG-I and MAVS binding through interaction with the N-terminal CARD domains of RIG-I (Fig. 5A). Coimmunoprecipitation studies show a loss of interaction between a truncated RIG-I containing only the CARD domains and MAVS in the presence of RSV NS2 proteins [34]. Direct targeting of the CARD domains potentially allows the virus to control the effector function of MAVS, which can limit signaling and antiviral responses.
Figure 5. Mechanisms of NNSV mediated inhibition of IFN induction.

A. The non-structural proteins NS1 and NS2 from respiratory syncytial virus (RSV) interact with MAVS and RIG-I, respectively, to disrupt RLR signaling. B. Measles virus (MeV) V protein binds to protein phosphatase 1 α/γ (PP1α/γ) and inhibits dephosphorylation and activation of RLRs. C. Ebolavirus VP35 (EBOV VP35) protein binds to PACT and inhibits PACT-induced activation of RIG-I. D. Filoviral VP35 proteins bind to kinases TBK1 and IKKe and act as decoy substrates to inhibit the phosphorylation of IRF3/7. Paramyxoviral V protein and Bornavirus and Rhabdovirus P protein also inhibit IRF3/7 phosphorylation in a similar manner.
Suppression of RLR dephosphorylation
Activation of RLRs requires binding of PAMPs and posttranslational modifications, such as dephosphorylation by protein phosphatase 1 (PP1) isoforms [35–37]. The V proteins from paramyxoviruses Nipah virus (NiV) and measles virus (MeV) can bind to PP1α/γ to inhibit the dephosphorylation of MDA5 at Ser88 [38] (Fig. 5B). The C-terminal tail of MeV V protein contains a consensus PP1-binding motif within residues 288 to 291 consisting of amino acids Arg-Ile-Trp-Tyr-Thr. Deletion of this region results in enhanced dephosphorylation of MDA5 at Ser88 in human dendritic cells and negatively impacts viral propagation in human lung epithelial cells [38]. In vitro experiments further show that PP1 is capable of dephosphorylating MeV V proteins as MeV V is phosphorylated at several locations [39–42], suggesting that MeV V acts as a decoy substrate of PP1 to divert the dephosphorylation of MDA5 [38]. However, a consensus PP1 binding site is not present in NiV V protein and inhibition of phosphorylation by NiV and MeV V proteins is limited to MDA5 [38]. Biochemical and cell-biological studies provide important insights into regulatory mechanisms, but structural insights of this process are currently unexplored.
Suppression of PACT induced RIG-I activation
RIG-I can be activated by the cellular protein Protein Kinase R activator (PACT) [43–45], although the exact molecular mechanism is poorly understood. Some viral proteins like EBOV and MARV VP35 proteins target PACT in order to inhibit RIG-I signaling [26, 45] (Fig. 5C). PACT induces potent activation of RIG-I dependent IFNβ promoter activity, which is correlated with an increase in RIG-I ATPase activity. Expression of the EBOV VP35 C-terminal domain suppresses RIG-I ATPase activity as well as IFNβ promoter activity [26, 45]. Coimmunoprecipitation studies show that EBOV VP35 binds PACT and disrupts the interaction between PACT and RIG-I [45]. Moreover, EBOV VP35 residues critical for dsRNA binding, including Arg312, Arg322, and Phe239, are required for PACT binding. Interestingly, dsRNA binding does not appear to mediate the interaction between EBOV VP35 and PACT. Further studies are needed to elucidate upon the molecular mechanism of how PACT binding to VP35 regulates RIG-I activity as well as the role of PACT binding to VP35 on viral polymerase activity as VP35 functions as a cofactor for the filoviral replication complex. [45–47]. The function of PACT in promoting translational inhibition through PKR activation requires additional studies in order to define the cellular role of its impact.
Viral inhibition of transcription factors (IRF3/7)
A large body of evidence suggests that several viral proteins, including Borna disease virus (BDV) and Rabies virus (RABV) P proteins, filoviral VP35 protein, and paramyxoviral V protein, block the phosphorylation of IRF3/7 by IKKε/TBK1 kinases by acting as surrogate substrates, thereby preventing the translocation of IRF3/7 into the nucleus [39, 48–51] (Fig. 2A & Fig. 5D). These studies show that coexpression of the viral P/VP35/V proteins with IKKε/TBK1 prevent phosphorylation of IRF3/7 in a dose-dependent manner. In addition, these viral proteins in turn are phosphorylated by IKKε/TBK1. In the case of EBOV VP35, the binding site involves the kinase domain of IKKε [50]. Incidentally, the kinase domain of TBK1 is highly homologous to that of IKKε, which explains how VP35 can target both IKKε and TBK1. However, it is currently unknown if these viral proteins act alone or in concert with other viral or cellular factors to block IKKε/TBK1 activity.
EBOV VP35 has also been shown to interact with IRF7 and the SUMO E3 enzyme PIAS1 [52] through the N-terminus. VP35 binding potentially enhances the SUMOylation of IRF7 by forming a quaternary complex with IRF7, the E2 enzyme Ubc9, and PIAS1 [52]. This leads to the downregulation of IRF7 and inhibit IFN production.
Type I IFN responses to viral infection
Type I IFNs, α and β, are master regulators of antiviral responses [53, 54]. IFNα is predominantly produced by hematopoietic cells, including plasmacytoid dendritic cells, while IFNβ is more broadly expressed [55, 56]. IFNα/β can act in an autocrine or paracrine fashion and bind to the IFNα/β receptor (IFNAR) to activate the Janus kinase 1 (JAK1) and Tyrosine kinase 2 (Tyk2), which phosphorylate STAT1 and STAT2 (Fig. 2B). Phosphorylated STAT1 is recognized by a subset of the karyopherin α (KPNA) family of nuclear transport proteins, the NPI-1 subfamily, which translocate STAT1-containing complexes to the nucleus [57, 58]. The phosphorylated STAT1/STAT2 heterodimer forms a ternary complex along with IRF9 in the nucleus that stimulates transcription of IFN stimulated genes (ISGs) through the IFN-stimulated gene response elements (ISREs). ISGs can inhibit different stages of viral infection including entry, replication, transcription, translation, assembly, and egress [59, 60]. As a result of the actions of ISG expression, an overall antiviral state is achieved within the infected and neighboring cells. Some specific functions of ISGs include inhibition of viral replication and transcription, degradation of viral nucleic acids, and modulation of lipid metabolism. Some well-studied ISGs include myxovirus resistance 1 (Mx1), IFN-inducible dsRNA-dependent protein kinase R (PKR), 2′-5′-oligoadenylate synthetase (OAS), interferon induced proteins with tetratricopeptide repeats (IFITs), apolipoprotein B mRNA–editing enzyme catalytic polypeptide (APOBEC1), tripartite motif containing proteins (TRIM) molecules, and tetherin [20, 59–67].
NNSV mediated inhibition of interferon response
In addition to the interferon induction signaling pathway, viral pathogens encode for proteins that target the IFNα/β response signaling pathway at various points. In the examples described below, NNSVs have developed ways to inhibit JAK/STAT activity, interfere with the nuclear transport of transcription factor STAT1/STAT2, as well as inhibit the activity of IFN stimulated genes.
Inhibition of phosphorylation of JAK/STAT pathway proteins
IFNα/β binding to IFNAR leads to the phosphorylation of JAK1 and TYK2. Although MARV and EBOV have similar genome organization, only MARV VP40 inhibits JAK1 dependent signaling pathways [68] (Fig. 2B). Expression of MARV VP40 inhibits the tyrosine phosphorylation of JAK1; TYK2, STAT1, and STAT2 in response to IFNγ; and IL6-mediated phosphorylation of STAT1 and STAT3 [68]. This is similar to a JAK1-deficient phenotype, suggesting that MARV VP40 targets JAK1, and not TYK2. Furthermore, MARV VP40 residues Ala57 and Ala165 appear to be important for inhibition of IFN signaling as mutation of these residues results in loss of Jak1 inhibition [68].
Paramyxoviruses, including NiV, Hendra virus (HeV), MeV, and Newcastle disease virus (NDV), also inhibit phosphorylation and activation of STAT1 in response to IFN stimulation (Fig. 2B). These viruses use the P ORF to encode V and W proteins through RNA editing by viral RdRp, and C proteins, which is a result of alternative AUG codon use, in addition to full-length P proteins. P, V, W, and C can target STAT1 to restrict its functions, albeit using independent mechanisms [69, 70]. Structural and biochemical studies of a complex of STAT1 N-terminal domain and the Sendai virus (SeV) C protein has revealed the molecular basis of one such strategy for STAT1 inhibition [70–72]. The C-terminal region (residues 99 to 204) of SeV C protein directly binds to the N-terminal domain of STAT1 (residues 1 to 126). The crystal structure of a STAT1 dimer bound to a dimer of C protein (Fig. 6A) shows how residues Met150 and Arg154 of SeV C protein stabilize this complex [70]. Arg154 makes electrostatic interactions with His58 of STAT1 (Fig. 6B), whereas Met150 inserts into a hydrophobic pocket within STAT1 (Fig. 6C). SeV C proteins carrying mutations at Arg154 and Met150 are unable to bind STAT1 and cannot inhibit IFNα signaling as efficiently as wild type C proteins. Further, structural comparisons of the STAT1-C complex with free STAT1 dimers revealed that C-binding induces conformational changes that allow STAT1 to form a tighter dimer [70]. STAT1 dimers bound to C are also stabilized by additional electrostatic interactions, including an inter-subunit interaction between residues Glu29 and Lys85 and an intra-subunit interaction between Lys85 and Asn82. Molecular models of parallel and antiparallel STAT1 dimers, bound to a monomer or dimer of SeV C protein respectively, were derived from the hetero-tetrameric structure of the STAT1 dimer and SeV C dimer. These models suggest that binding of two C proteins might restrict the STAT1 dimer in a parallel conformation that is not conducive to dephosphorylation at Tyr701. As a result, phosphorylated STAT1 could potentially accumulate in the cytoplasm, forming high molecular weight complexes [70–73].
Figure 6. SeV C protein targets STAT1.

A. Crystal structure of SeV C protein residues 99 to 204 bound to human STAT1 N-terminal domains (residues 1–126) (PDB 3WWT) [70]. B. Expanded view of a key electrostatic interaction between SeV C protein residue Arg154 and STAT1 residue His58, which highlights the role of charge complementarity at the host-viral interface. C. Expanded view of a hydrophobic pocket. SeV C residue Met150 inserts into the hydrophobic pocket within STAT1.
While P/V/C mediated STAT1 binding retains the latter in the cytoplasm, W binds STAT1 and translocates into the nucleus. This is due to the presence of a classical nuclear localization signal “KKAR” within the C-terminus of W, formed by the basic residues Lys439, Lys440, and Arg442, which allows it to bind to KPNA3 and KPNA4 [69]. In both cases, interaction of paramyxoviral P/V/C or W proteins leads to the sequestration of STAT1 away from the receptor kinase complexes and prevents phosphorylation of STAT1. Notably, the ability of P to inhibit STAT1 phosphorylation is much less pronounced compared to V and W, which may be due to the additional role of P as a cofactor for the viral polymerase complex. In addition, residues 114 to 140 in NiV P/V/W proteins are critical for STAT1 binding as mutation of these residues abolishes the ability of these proteins to bind to and inhibit STAT1 phosphorylation, and thereby IFN signaling [69].
Inhibition of nuclear transport of STATs
In contrast to MARV VP40, which inhibits JAK1, EBOV VP24 proteins block the nuclear translocation of phosphorylated STAT1 (pY-STAT1) complexes by targeting the STAT1 transporter KPNA [74–76]. Recent biochemical and structural studies have elucidated how EBOV VP24 affects STAT1 nuclear accumulation while maintaining other KPNA-mediated cargo delivery [76] (Fig. 2B, 7). All KPNA recognize cargo containing a classical nuclear localization signal (cNLS) through a major site on armadillo repeats (ARM) 2–4 and a minor site on ARM 6–8 [77–80]. However, pY-STAT1 is transported by the NPI-subfamily, including KPNA1, KPNA5, and KPNA6, which can recognize a relatively uncharacterized non-classical NLS (ncNLS) [58]. EBOV VP24 binds to KPNA with a significantly higher affinity than pY-STAT1, suggesting that EBOV VP24 competes with pY-STAT1 for binding to KPNA [76]. The crystal structure of EBOV VP24 in complex with the minimal binding region of KPNA5 encompassing ARM 7 to10 shows a large surface area of interaction with a hydrophobic core and high shape complementarity [76]. The binding surface is formed by residues in KPNA that are conserved only among the NPI-subfamily and residues of EBOV VP24 that vary in the closely related MARV VP24 [76]. Use of the ncNLS allows STAT1 transport to occur independently of other nucleocytoplasmic trafficking of cargoes containing cNLSs. This may be important in EBOV pathogenesis by maintaining certain cellular functions that can facilitate viral replication.
Figure 7. Inhibition of the JAK/STAT pathway by EBOV VP24.

A. Crystal structure of EBOV VP24 (red) in complex with ARM 7–10 of KPNA5 (dark blue) (PDB 4U2X) [76]. ARM 7–10 is aligned to the structure of full length KPNA (cyan) (PDB 1BK5) [77] as reference. B. Model showing how EBOV VP24 targets the non-classical nuclear localization signal (ncNLS) recognition of KPNA and blocks nuclear accumulation of pY-STAT1 while leaving classical NLS interactions intact.
The P protein encoded by RABV of the Rhabdoviridae family also targets the JAK/STAT pathway in order to block nuclear trafficking of STATs [49] (Fig. 2B). Like EBOV, RABV infection does not affect the phosphorylation of STAT1 or STAT2 in human epithelial cells, but greatly reduced the levels of activated STATs and IRF9 levels upon stimulation with IFNα and IFNγ while increasing cytoplasmic accumulation of STAT1 and STAT2[49]. Further studies suggest that the C-terminal domain of RABV P protein is critical for STAT1/2 binding, which may be mediated by RABV P residues W265 and M287 [81]. Mutation of Trp265 and Met287 greatly reduces RABV pathogenicity [81].
Inhibition of IFN stimulated genes (ISGs)
Type I IFNα/β signaling through the JAK/STAT pathway stimulates the production of more than 200 interferon stimulated genes (ISGs), which together modulate cellular function to limit viral propagation. One ISG product is the Protein Kinase R (PKR), which is activated upon binding to dsRNA or PKR activator (PACT) [82]. Activated PKR phosphorylates eukaryotic initiation factor 2α (eIF2α) leading to the inhibition of mRNA synthesis that limits viral protein synthesis [82]. However, EBOV infection in human kidney cells coincides with the absence of PKR phosphorylation of eIF2α [83]. Although the precise mechanism of EBOV mediated PKR inhibition is not known, the C-terminal IID domain of VP35 may be involved in the process [84]. Under basal conditions, transfection of plasmid DNA into mammalian cells activates PKR, which suppresses the expression of transfected DNA. In contrast, expression of VP35 is sufficient to relieve this suppression. Furthermore, residues Arg305, Lys309, and Arg312 within VP35 IID are important for PKR inhibition. However, inhibition of PKR may be independent of dsRNA binding since a mutated VP35 IID with a Arg312 to Ala substitution retains the ability to inhibit PKR even though it does not bind dsRNA [84]. The effect of VP35 and PACT interaction on the activity of PKR has not been studied. Nonetheless, the observation that phosphorylation status of eIFα can affect EBOV replication is suggestive that PKR may be involved [83].
The role of tetherin, a type II transmembrane protein with a phosphatidylinositol group at the C-terminus [85], has been well studied in viral egress in retroviruses, herpesviruses, arenaviruses, and filoviruses [86–90]. Activated tetherin forms a parallel homodimer that can insert one end into the cell membrane and the other into the viral membrane, thereby blocking progeny release [91]. Tetherins have been shown to block the release of EBOV VP40 mediated virus like particles (VLPs) [86, 92, 93], but this inhibition can be relieved by EBOV glycoprotein (GP) [92]. In contrast to HIV Vpu, filoviral GP are able to inhibit tetherin from multiple species and in a cell-specific manner without downregulating tetherin expression [67]. In-cell fluorescence resonance energy transfer (FRET) and coimmunoprecipitation assays show that tetherin associated with the GP2 transmembrane region of EBOV GP proteins, suggesting a unique mechanism of inhibition by GP2 [67]. However, it appears that GP does not target tetherin in a sequence-specific manner [94].
More recently, the IFN induced proteins with tetratricopeptide repeats or IFITs have been extensively studied [64, 65, 95]. Four classes of IFIT genes, IFIT1 (or ISG56), IFIT2 (or ISG54), IFIT3 (or ISG60, or IFIT4), and IFIT5 (or ISG58), can be induced by type I and type II IFNs [96]. IFIT1 and IFIT2 can also directly target the elongation factor eIF3E to perturb the formation of the translation preinitiation complex [97, 98]. IFITs recognize specific RNA motifs found in viral RNA but absent in host mRNA species. One example is the 5′-end cap that lacks 2′-O-methylation at the N7 position, a motif that is recognized by IFIT1 [99]. Binding of IFIT1 to the 5′-ends of viral mRNA precludes their interaction with host elongation factors like eIF4E thereby blocking viral translation [100, 101]. In order to avoid detection by IFITs, many viruses have devised a variety of mechanisms, including de novo synthesis of 2′-O methylated caps by West Nile Virus [102, 103], cap snatching from the host by influenza virus [104], hijacking the host capping machinery by HIV [105], or use of internal ribosome entry sites by picornaviruses [106]. NNSVs typically replicate in the cytoplasm where the host capping machinery is unavailable. To circumvent this hurdle, NNSVs possess a unique cap synthesis mechanism where the L polymerase contains guanylyl transferase activity that can transfer a GDP cap to the 5′-end of viral mRNAs [107]. The NNSV mRNA cap is usually methylated at two positions, G-N7 and 2′-O [10, 108]. Studies in the VSV L polymerase show that methylation at both G-N-7 and 2′O positions occur through a single S-adenosyl-L-methionine binding site within a catalytic triad conserved among other 2′O methylases [10, 107]. This region was mapped to the C-terminal region of VSV L within residues Gly1670 to Gly1675 and Asp 1735 [10].
Modulation of the host ubiquitin system
Ubiquitination is required at several steps in IFN production and signaling and results in both recognition of viral pathogens as well as restriction of viral infection. For example, TRIM25 mediated ubiquitination of the CARD domains of RIG-I and MAVS are necessary for their activation and downstream signaling [32, 35, 109–111]. Further, activation of transcription factor NF-κB is dependent on the ubiquitination and proteasomal degradation of the inhibitor kinase IκB [112]. NNSVs encode for proteins that reprogram the ubiquitin system to facilitate degradation of host innate immune molecules that would otherwise restrict viral infection [113]. The V protein of paramyxoviruses, such as simian virus 5 (SV5), mumps virus, human parainfluenza virus 2 (PIV2), and PIV5, targets the damage-specific DNA binding protein 1 (DDB1)-Cullin4-Rbx1 E3 ligase complex to ubiquitinate and mediate proteasomal degradation of STATs [114–116] (Fig. 8A). Recent three-dimensional structures of DDB1 bound to the V protein from the SV5 reveals how the multifunctional V protein interacts with the E3 ligase complex [117–119] (Fig. 8B–D). DDB1 is composed of three β-propeller domains, BPA, BPB, and BPC. BPB binds to Cullin4-Rbx1 complex which links substrates to E2 ubiquitin conjugating enzyme, whereas the BPA and BPC module binds to a variety of substrate recognition adaptors, WD40 domain containing proteins [118]. SV5 V protein displaces the canonical substrate adaptors to bind to DDB1 at BPA-BPC and hijack the DDB1-Cul4-Rbx1 complex to reroute ubiquitination of host antiviral molecules [114–116]. SV5 V protein inserts its N-terminal α-helix α1 into a pocket formed by the loop regions of BPA and BPC [117] (Fig. 8D). This interaction seems to be supported by the C-terminal region or core of V protein, where β4 makes extensive hydrophobic interactions with BPC. The conserved Tyr127 on α2 of V protein also makes multiple van der Waals interactions with α1 and BPC that are important in stabilizing the V-DDB1 complex (Fig. 8D). The solvent exposed outer face of V protein is lined by relatively conserved hydrophobic and nonpolar residues along with Asn100 which are thought to be important in the ubiquitination of STAT proteins [117, 118]. SV5 V forms a complex with both STAT1 and STAT2 with the later providing species-specific contacts [115]. This is the basis for the inability of SV5 to ubiquitinate murine STATs. In fact, mutation of Asn100 can abolish the interaction between V and human STAT2 or even impart SV5 with the ability to antagonize murine STATs [117].
Figure 8. Viral modulation of host ubiquitination.

[70]
A. Cartoon model depicting the mechanism of SV5 V protein mediated ubiquitination of STAT2. B. Crystal structure of the complex of SV5 V protein (red), DDB1 (green), Cullin4 (grey), and Rbx1 (Blue) (PDB 2HYE) [118]. BPB of DDB1 binds to the N-terminal domain of Cullin4, while Rbx1 binds to the C-terminal domain of Cullin4. C. Crystal structure of Simian virus 5 (SV5) V protein (red) and DDB1 (green) showing the three β-propellers BPA, BPB, and BPC (PDB 2B5l) [117]. The SV5 V protein binds at a pocket within the BPA and BPC. D. Close up of the V binding pocket. N-terminal helix α1 of V protein inserts into the hydrophobic pocket of DDB1. Tyr127 of V protein makes important inter-subunit interactions with BPC.
The NS1 and NS2 proteins from RSV are also thought to induce proteasomal degradation of many host immune molecules, including RIG-I, IRF3/7, and STAT2 [8, 120, 121]. Moreover, NS1 and NS2 seem to affect the cellular expression levels of different proteins in the IFN signaling pathway, and when expressed together, the two non-structural proteins produce the strongest effect [8]. It was further observed that the proteasomal inhibitor MG132 could partially relieve the NS1/2 mediated inhibition of host protein expression [8, 121]. However, the exact molecular mechanism underlying these observations have not been fully elucidated.
Conclusions and future directions
While the genomes of NNSVs are limited in their coding capacity, it is becoming increasingly clear that NNSVs encode viral proteins that are capable of antagonizing or rewiring the function of many host genes in order to promote viral pathogenesis. The multifunctional nature of NNSV proteins combined with their high mutation rates allows these pathogens to target multiple cellular factors and adapt to specific hosts. Molecular biology tools, like the reverse genetics system, developed over the last two decades have been instrumental in our understanding of the basic functions of the NNSV genome. However, we are now beginning to appreciate the complex interplay that occurs between viral pathogens and the host innate immune system. A detailed molecular understanding of these host-virus interactions will likely be key to the development of effective antiviral therapies and management of viral outbreaks. Recent advances in functional genomics, along with transcriptomics and proteomics, are proving useful in the investigation of host-virus interactions in the context of specific tissue and cell types, closely related viral species, and a variety of cellular signals [122]. For example, several genome-wide and druggable genome screens using short interfering RNA (siRNA) and CRISPR/Cas are starting to uncover novel genes required for NNSV infections [123–126]. While these advances are identifying novel targets, mechanistic understanding of these key host-viral interactions and use of such information in countermeasure development has significantly lagged. These limitations are highlighted by the lack of well-defined interaction interfaces, which can not only provide insight into regulatory mechanisms, but inform on druggabiltiy as well as the potential to form escape mutants. Thus, studies aimed at providing molecular mechanisms will be critical to form the knowledge base to better understand NNSV immune evasion mechanisms and to develop countermeasures.
Type I interferons are important for host innate immune responses.
Non-segmented negative sense RNA viruses are important human pathogens.
Recent studies are uncovering key host-virus interactions in immune evasion.
These studies provide novel targets for development of countermeasures.
Acknowledgments
Work in our laboratories are supported by in part by NIH grants (R01AI107056 (Leung), R01AI123926 (Amarasinghe), R01AI114654 (Basler), U191099565 (Ting), U19AI109945 (Basler), U19AI109664 (Basler), and by Department of the Defense, Defense Threat Reduction Agency grants HDTRA1-14-0013 (Basler) and HDTRA1-12-1-0051 (Basler). SC is funded by American Heart Association Postdoctoral Fellowship (15POST25140009). The content of the information does not necessarily reflect the position or the policy of the federal government and no official endorsement should be inferred.
Abbreviations
- APOBEC1
Apolipoprotein B mRNA–editing enzyme catalytic polypeptide
- ARM
Armadillo
- ATPase
Adenylpyrophosphatase
- BDV
Borna disease virus
- BPA/B/C
Beta-propeller A/B/C
- CARD
Caspase activation and recruitment domain
- CAS
CRISPR associated protein 9
- CRISPR
Clustered regularly interspaced short palindromic repeats
- ds/ss/mRNA
double-stranded/single-stranded/messenger ribonucleic acid
- DDB1
Damage-specific DNA binding protein 1
- EBOV
Ebola virus
- eIF2α
Eukaryotic initiation factor 2 alpha
- F
Fusion protein
- FRET
Fluorescence resonance energy transfer
- G/GP
Glycoprotein
- HIV
Human immunodeficiency virus
- IFIT
IFN induced proteins with tetratricopeptide repeat
- IFN
Interferon
- IFNAR
IFN alpha/beta receptor
- IID
IFN inhibitory domain
- IKKβ
Inhibitor of kappa light polypeptide gene enhancer in B-cells
- IKKε
Inhibitor of nuclear factor kappa-B kinase subunit epsilon
- IPS
IFN beta promoter stimulator protein 1
- IRF3/7/9
IFN regulatory factor 3/7/9
- ISG
IFN stimulated gene
- ISRE
IFN stimulated gene response element
- JAK1
Janus kinase 1
- KPNA
Karyopherin alpha
- L
Large polymerase
- LGP2
Laboratory of genetics and physiology 2
- M
Matrix protein
- MAPK
Mitogen activated protein kinase
- MARV
Marburg virus
- MDA5
Melanoma differentiation associated protein 5
- MyD88
Myeloid differentiation primary response gene 88
- MeV
Measles virus
- Mx1
Myxovirus resistance 1
- (n)cNLS
(non) classical nuclear localization signal
- NDV
Newcastle disease virus
- NFκB
Nuclear factor kappa B
- NiV
Nipah virus
- N/NP
Nucleoprotein
- NNSV
Non-segmented, negative-sense, single-stranded RNA virus
- NPI-1
Nucleoprotein interacting protein 1
- NS1/2
Non-structural protein 1/2
- OAS
2′-5′-oligoadenylate synthetase
- OH
Hydroxyl
- ORF
Open reading frame
- P
Phosphoprotein
- PACT
PKR activator
- PAMP
Pathogen associated molecular pattern
- PIAS1
Protein inhibitor of activated STAT 1
- PKR
Protein kinase R
- PP1
Protein phosphatase 1
- PRR
Pattern recognition receptor
- RABV
Rabies virus
- Rbx1
Really interesting new gene (RING) box protein 1
- RdRP
RNA dependent RNA polymerase
- RIG-I
Retinoic acid inducible gene I
- RLR
RIG-I-like receptor
- RMSD
Root mean square deviation
- SeV
Sendai virus
- SH
Small hydrophobic
- siRNA
Small interfering RNA
- STAT1/2
Signal transducer and activator of transcription 1
- SUMO
Small ubiquitin-like modifier
- SV5
Simian virus 5
- TANK
TRAF family member associated NFκB activator
- TBK1
TANK binding kinase 1
- TIR
Toll-interleukin 1 receptor
- TLR
Toll-like receptor
- TRAF3
Tumor necrosis factor receptor associated factor 3
- TRIF
TIR domain containing adaptor-inducing interferon-beta
- TRIM
Tripartite motif containing proteins
- TYK2
Tyrosine kinase 2
- VISA
Virus-induced signaling adapter
- VP24/30/35/40
Viral protein 24/30/35/40
- Vpu
Viral protein unique
- VSV
Vesicular stomatitis virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gerlier D, Lyles DS. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol Mol Biol Rev. 2011;75:468–90. doi: 10.1128/MMBR.00007-11. second page of table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortin J, Martin-Benito J. The RNA synthesis machinery of negative-stranded RNA viruses. Virology. 2015;479–480:532–44. doi: 10.1016/j.virol.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Poenisch M, Burger N, Staeheli P, Bauer G, Schneider U. Protein X of Borna disease virus inhibits apoptosis and promotes viral persistence in the central nervous systems of newborn-infected rats. J Virol. 2009;83:4297–307. doi: 10.1128/JVI.02321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herrel M, Hoefs N, Staeheli P, Schneider U. Tick-borne Nyamanini virus replicates in the nucleus and exhibits unusual genome and matrix protein properties. J Virol. 2012;86:10739–47. doi: 10.1128/JVI.00571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poenisch M, Wille S, Ackermann A, Staeheli P, Schneider U. The X protein of borna disease virus serves essential functions in the viral multiplication cycle. J Virol. 2007;81:7297–9. doi: 10.1128/JVI.02468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wensman JJ, Munir M, Thaduri S, Hornaeus K, Rizwan M, Blomstrom AL, et al. The X proteins of bornaviruses interfere with type I interferon signalling. J Gen Virol. 2013;94:263–9. doi: 10.1099/vir.0.047175-0. [DOI] [PubMed] [Google Scholar]
- 7.Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol. 2005;79:9315–9. doi: 10.1128/JVI.79.14.9315-9319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goswami R, Majumdar T, Dhar J, Chattopadhyay S, Bandyopadhyay SK, Verbovetskaya V, et al. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013;23:1025–42. doi: 10.1038/cr.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol. 2003;3:36–50. doi: 10.1038/nri980. [DOI] [PubMed] [Google Scholar]
- 10.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Leung DW, Basler CF, Amarasinghe GK. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends Microbiol. 2012;20:139–46. doi: 10.1016/j.tim.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–8. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A. 2009;106:12067–72. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Lu C, Stewart M, Xu H, Strong RK, Igumenova T, et al. Structural basis of double-stranded RNA recognition by the RIG-I like receptor MDA5. Arch Biochem Biophys. 2009;488:23–33. doi: 10.1016/j.abb.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 18.Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–42. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31:1714–26. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13:214–22. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 22.Leung DW, Shabman RS, Farahbakhsh M, Prins KC, Borek DM, Wang T, et al. Structural and functional characterization of Reston Ebola virus VP35 interferon inhibitory domain. J Mol Biol. 2010;399:347–57. doi: 10.1016/j.jmb.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramanan P, Edwards MR, Shabman RS, Leung DW, Endlich-Frazier AC, Borek DM, et al. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc Natl Acad Sci U S A. 2012;109:20661–6. doi: 10.1073/pnas.1213559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prins KC, Delpeut S, Leung DW, Reynard O, Volchkova VA, Reid SP, et al. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J Virol. 2010;84:3004–15. doi: 10.1128/JVI.02459-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A. 2011;108:21010–5. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards MR, Liu G, Mire CE, Sureshchandra S, Luthra P, Yen B, et al. Differential Regulation of Interferon Responses by Ebola and Marburg Virus VP35 Proteins. Cell Rep. 2016;14:1632–40. doi: 10.1016/j.celrep.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motz C, Schuhmann KM, Kirchhofer A, Moldt M, Witte G, Conzelmann KK, et al. Paramyxovirus V proteins disrupt the fold of the RNA sensor MDA5 to inhibit antiviral signaling. Science. 2013;339:690–3. doi: 10.1126/science.1230949. [DOI] [PubMed] [Google Scholar]
- 28.Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101:17264–9. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Childs K, Randall R, Goodbourn S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J Virol. 2012;86:3411–21. doi: 10.1128/JVI.06405-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parisien JP, Bamming D, Komuro A, Ramachandran A, Rodriguez JJ, Barber G, et al. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J Virol. 2009;83:7252–60. doi: 10.1128/JVI.00153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, et al. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Wu B, Hur S. How RIG-I like receptors activate MAVS. Curr Opin Virol. 2015;12:91–8. doi: 10.1016/j.coviro.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyapalle S, Wong T, Garay J, Teng M, San Juan-Vergara H, Mohapatra S, et al. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS One. 2012;7:e29386. doi: 10.1371/journal.pone.0029386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ling Z, Tran KC, Teng MN. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J Virol. 2009;83:3734–42. doi: 10.1128/JVI.02434-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol. 2010;84:3220–9. doi: 10.1128/JVI.02241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–92. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity. 2013;38:437–49. doi: 10.1016/j.immuni.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis ME, Wang MK, Rennick LJ, Full F, Gableske S, Mesman AW, et al. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe. 2014;16:19–30. doi: 10.1016/j.chom.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu LL, Puri M, Horvath CM, Sen GC. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J Biol Chem. 2008;283:14269–76. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludlow LE, Lo MK, Rodriguez JJ, Rota PA, Horvath CM. Henipavirus V protein association with Polo-like kinase reveals functional overlap with STAT1 binding and interferon evasion. J Virol. 2008;82:6259–71. doi: 10.1128/JVI.00409-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pfaller CK, Conzelmann KK. Measles virus V protein is a decoy substrate for IkappaB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J Virol. 2008;82:12365–73. doi: 10.1128/JVI.01321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shiell BJ, Gardner DR, Crameri G, Eaton BT, Michalski WP. Sites of phosphorylation of P and V proteins from Hendra and Nipah viruses: newly emerged members of Paramyxoviridae. Virus Res. 2003;92:55–65. doi: 10.1016/s0168-1702(02)00313-1. [DOI] [PubMed] [Google Scholar]
- 43.Kok KH, Lui PY, Ng MH, Siu KL, Au SW, Jin DY. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9:299–309. doi: 10.1016/j.chom.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 44.Iwamura T, Yoneyama M, Koizumi N, Okabe Y, Namiki H, Samuel CE, et al. PACT, a double-stranded RNA binding protein acts as a positive regulator for type I interferon gene induced by Newcastle disease virus. Biochem Biophys Res Commun. 2001;282:515–23. doi: 10.1006/bbrc.2001.4606. [DOI] [PubMed] [Google Scholar]
- 45.Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol. 2010;84:10581–91. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Becker S, Rinne C, Hofsass U, Klenk HD, Muhlberger E. Interactions of Marburg virus nucleocapsid proteins. Virology. 1998;249:406–17. doi: 10.1006/viro.1998.9328. [DOI] [PubMed] [Google Scholar]
- 48.Unterstab G, Ludwig S, Anton A, Planz O, Dauber B, Krappmann D, et al. Viral targeting of the interferon-{beta}-inducing Traf family member-associated NF-{kappa}B activator (TANK)-binding kinase-1. Proc Natl Acad Sci U S A. 2005;102:13640–5. doi: 10.1073/pnas.0502883102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brzozka K, Finke S, Conzelmann KK. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol. 2005;79:7673–81. doi: 10.1128/JVI.79.12.7673-7681.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prins KC, Cardenas WB, Basler CF. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol. 2009;83:3069–77. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irie T, Kiyotani K, Igarashi T, Yoshida A, Sakaguchi T. Inhibition of interferon regulatory factor 3 activation by paramyxovirus V protein. J Virol. 2012;86:7136–45. doi: 10.1128/JVI.06705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, et al. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog. 2009;5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–3. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 54.O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–28. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.KTCaMG Snapshot: Interferon signaling. Cell. 2015;163:1808. doi: 10.1016/j.cell.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 57.McBride KM, Banninger G, McDonald C, Reich NC. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. 2002;21:1754–63. doi: 10.1093/emboj/21.7.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997;16:7067–77. doi: 10.1093/emboj/16.23.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–68. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haller O, Staeheli P, Schwemmle M, Kochs G. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015;23:154–63. doi: 10.1016/j.tim.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 62.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, et al. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70:1032–60. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rebouillat D, Hovanessian AG. The human 2′,5′-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J Interferon Cytokine Res. 1999;19:295–308. doi: 10.1089/107999099313992. [DOI] [PubMed] [Google Scholar]
- 64.Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol. 2013;13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vladimer GI, Gorna MW, Superti-Furga G. IFITs: Emerging Roles as Key Anti-Viral Proteins. Front Immunol. 2014;5:94. doi: 10.3389/fimmu.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ozato K, Shin DM, Chang TH, Morse HC., 3rd TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–60. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuhl A, Banning C, Marzi A, Votteler J, Steffen I, Bertram S, et al. The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor tetherin. J Infect Dis. 2011;204(Suppl 3):S850–60. doi: 10.1093/infdis/jir378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valmas C, Grosch MN, Schumann M, Olejnik J, Martinez O, Best SM, et al. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 2010;6:e1000721. doi: 10.1371/journal.ppat.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shaw ML, Garcia-Sastre A, Palese P, Basler CF. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J Virol. 2004;78:5633–41. doi: 10.1128/JVI.78.11.5633-5641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oda K, Matoba Y, Irie T, Kawabata R, Fukushi M, Sugiyama M, et al. Structural Basis of the Inhibition of STAT1 Activity by Sendai Virus C Protein. J Virol. 2015;89:11487–99. doi: 10.1128/JVI.01887-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gotoh B, Komatsu T, Takeuchi K, Yokoo J. The C-terminal half-fragment of the Sendai virus C protein prevents the gamma-activated factor from binding to a gamma-activated sequence site. Virology. 2003;316:29–40. doi: 10.1016/s0042-6822(03)00590-7. [DOI] [PubMed] [Google Scholar]
- 72.Gotoh B, Takeuchi K, Komatsu T, Yokoo J. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J Virol. 2003;77:3360–70. doi: 10.1128/JVI.77.6.3360-3370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takeuchi K, Komatsu T, Yokoo J, Kato A, Shioda T, Nagai Y, et al. Sendai virus C protein physically associates with Stat1. Genes Cells. 2001;6:545–57. doi: 10.1046/j.1365-2443.2001.00442.x. [DOI] [PubMed] [Google Scholar]
- 74.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. 2006;80:5156–67. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol. 2007;81:13469–77. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu W, Edwards MR, Borek DM, Feagins AR, Mittal A, Alinger JB, et al. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe. 2014;16:187–200. doi: 10.1016/j.chom.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conti E, Uy M, Leighton L, Blobel G, Kuriyan J. Crystallographic analysis of the recognition of a nuclear localization signal by the nuclear import factor karyopherin alpha. Cell. 1998;94:193–204. doi: 10.1016/s0092-8674(00)81419-1. [DOI] [PubMed] [Google Scholar]
- 78.Conti E, Kuriyan J. Crystallographic analysis of the specific yet versatile recognition of distinct nuclear localization signals by karyopherin alpha. Structure. 2000;8:329–38. doi: 10.1016/s0969-2126(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 79.Chook YM, Blobel G. Karyopherins and nuclear import. Curr Opin Struct Biol. 2001;11:703–15. doi: 10.1016/s0959-440x(01)00264-0. [DOI] [PubMed] [Google Scholar]
- 80.Conti E, Izaurralde E. Nucleocytoplasmic transport enters the atomic age. Curr Opin Cell Biol. 2001;13:310–9. doi: 10.1016/s0955-0674(00)00213-1. [DOI] [PubMed] [Google Scholar]
- 81.Wiltzer L, Okada K, Yamaoka S, Larrous F, Kuusisto HV, Sugiyama M, et al. Interaction of rabies virus P-protein with STAT proteins is critical to lethal rabies disease. J Infect Dis. 2014;209:1744–53. doi: 10.1093/infdis/jit829. [DOI] [PubMed] [Google Scholar]
- 82.Pindel A, Sadler A. The role of protein kinase R in the interferon response. J Interferon Cytokine Res. 2011;31:59–70. doi: 10.1089/jir.2010.0099. [DOI] [PubMed] [Google Scholar]
- 83.Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol. 2007;81:182–92. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schumann M, Gantke T, Muhlberger E. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol. 2009;83:8993–7. doi: 10.1128/JVI.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, Banting G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4:694–709. doi: 10.1034/j.1600-0854.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 86.Jouvenet N, Neil SJ, Zhadina M, Zang T, Kratovac Z, Lee Y, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83:1837–44. doi: 10.1128/JVI.02211-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gupta RK, Mlcochova P, Pelchen-Matthews A, Petit SJ, Mattiuzzo G, Pillay D, et al. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc Natl Acad Sci U S A. 2009;106:20889–94. doi: 10.1073/pnas.0907075106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jia B, Serra-Moreno R, Neidermyer W, Rahmberg A, Mackey J, Fofana IB, et al. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009;5:e1000429. doi: 10.1371/journal.ppat.1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Le Tortorec A, Neil SJ. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J Virol. 2009;83:11966–78. doi: 10.1128/JVI.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sakuma T, Noda T, Urata S, Kawaoka Y, Yasuda J. Inhibition of Lassa and Marburg virus production by tetherin. J Virol. 2009;83:2382–5. doi: 10.1128/JVI.01607-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139:499–511. doi: 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A. 2009;106:2886–91. doi: 10.1073/pnas.0811014106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Neil SJ, Sandrin V, Sundquist WI, Bieniasz PD. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe. 2007;2:193–203. doi: 10.1016/j.chom.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lopez LA, Yang SJ, Hauser H, Exline CM, Haworth KG, Oldenburg J, et al. Ebola virus glycoprotein counteracts BST-2/Tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J Virol. 2010;84:7243–55. doi: 10.1128/JVI.02636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Diamond MS. IFIT1: A dual sensor and effector molecule that detects non-2′-O methylated viral RNA and inhibits its translation. Cytokine Growth Factor Rev. 2014;25:543–50. doi: 10.1016/j.cytogfr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kusari J, Sen GC. Transcriptional analyses of interferon-inducible mRNAs. Mol Cell Biol. 1987;7:528–31. doi: 10.1128/mcb.7.1.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hui DJ, Bhasker CR, Merrick WC, Sen GC. Viral stress-inducible protein p56 inhibits translation by blocking the interaction of eIF3 with the ternary complex eIF2.GTP.Met-tRNAi. J Biol Chem. 2003;278:39477–82. doi: 10.1074/jbc.M305038200. [DOI] [PubMed] [Google Scholar]
- 98.Terenzi F, Hui DJ, Merrick WC, Sen GC. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J Biol Chem. 2006;281:34064–71. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- 99.Habjan M, Hubel P, Lacerda L, Benda C, Holze C, Eberl CH, et al. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 2013;9:e1003663. doi: 10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kimura T, Katoh H, Kayama H, Saiga H, Okuyama M, Okamoto T, et al. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J Virol. 2013;87:9997–10003. doi: 10.1128/JVI.00883-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumar P, Sweeney TR, Skabkin MA, Skabkina OV, Hellen CU, Pestova TV. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res. 2014;42:3228–45. doi: 10.1093/nar/gkt1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–6. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M, Jr, et al. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8:e1002698. doi: 10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature. 2009;458:914–8. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 105.Wilusz J. Putting an ‘End’ to HIV mRNAs: capping and polyadenylation as potential therapeutic targets. AIDS Res Ther. 2013;10:31. doi: 10.1186/1742-6405-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jang SK, Pestova TV, Hellen CU, Witherell GW, Wimmer E. Cap-independent translation of picornavirus RNAs: structure and function of the internal ribosomal entry site. Enzyme. 1990;44:292–309. doi: 10.1159/000468766. [DOI] [PubMed] [Google Scholar]
- 107.Das K, Arnold E. Negative-Strand RNA Virus L Proteins: One Machine, Many Activities. Cell. 2015;162:239–41. doi: 10.1016/j.cell.2015.06.063. [DOI] [PubMed] [Google Scholar]
- 108.Ogino T. In vitro capping and transcription of rhabdoviruses. Methods. 2013;59:188–98. doi: 10.1016/j.ymeth.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pauli EK, Chan YK, Davis ME, Gableske S, Wang MK, Feister KF, et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci Signal. 2014;7:ra3. doi: 10.1126/scisignal.2004577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Castanier C, Zemirli N, Portier A, Garcin D, Bidere N, Vazquez A, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Inn KS, Gack MU, Tokunaga F, Shi M, Wong LY, Iwai K, et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell. 2011;41:354–65. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kanarek N, London N, Schueler-Furman O, Ben-Neriah Y. Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000166. doi: 10.1101/cshperspect.a000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Randow F, Lehner PJ. Viral avoidance and exploitation of the ubiquitin system. Nat Cell Biol. 2009;11:527–34. doi: 10.1038/ncb0509-527. [DOI] [PubMed] [Google Scholar]
- 114.Didcock L, Young DF, Goodbourn S, Randall RE. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J Virol. 1999;73:9928–33. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Parisien JP, Lau JF, Horvath CM. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol. 2002;76:6435–41. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol. 2002;76:4190–8. doi: 10.1128/JVI.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li T, Chen X, Garbutt KC, Zhou P, Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–17. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 118.Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–3. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 119.Li J, Wang JT, Whelan SP. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2006;103:8493–8. doi: 10.1073/pnas.0509821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bossert B, Marozin S, Conzelmann KK. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol. 2003;77:8661–8. doi: 10.1128/JVI.77.16.8661-8668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol. 2007;81:3428–36. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ramage H, Cherry S. Virus-Host Interactions: From Unbiased Genetic Screens to Function. Annu Rev Virol. 2015;2:497–524. doi: 10.1146/annurev-virology-100114-055238. [DOI] [PubMed] [Google Scholar]
- 123.Panda D, Das A, Dinh PX, Subramaniam S, Nayak D, Barrows NJ, et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc Natl Acad Sci U S A. 2011;108:19036–41. doi: 10.1073/pnas.1113643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee AS, Burdeinick-Kerr R, Whelan SP. A genome-wide small interfering RNA screen identifies host factors required for vesicular stomatitis virus infection. J Virol. 2014;88:8355–60. doi: 10.1128/JVI.00642-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Clemente R, Sisman E, Aza-Blanc P, de la Torre JC. Identification of host factors involved in borna disease virus cell entry through a small interfering RNA functional genetic screen. J Virol. 2010;84:3562–75. doi: 10.1128/JVI.02274-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cote M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–8. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–89. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]