

Abstract
Purpose
Painful peripheral neuropathy is a frequent toxicity associated with bortezomib therapy. This study aimed to identify loci that affect susceptibility to this toxicity.
Experimental Design
A genome-wide association study (GWAS) of 370,605 SNPs was performed to identify risk variants for developing severe bortezomib-induced peripheral neuropathy (BiPN) in 469 multiple myeloma (MM) patients who received bortezomib-dexamethasone therapy prior to autologous stem-cell in randomized clinical trials of the Intergroupe Francophone du Myelome (IFM) and findings were replicated in 114 MM patients of the HOVON-65/GMMG-HD4 clinical trial.
Results
A single SNP in the PKNOX1 gene was associated with BiPN in the exploratory cohort (rs2839629; OR, 1.89, 95% CI: [1.45-2.44]; P = 7.6 × 10−6) and in the replication cohort (OR, 2.04; 95% CI = [1.11-3.33]; P= 8.3 × 10−3). In addition, rs2839629 is in strong linkage disequilibrium (r2 = 0.87) with rs915854, located in the intergenic region between PKNOX1 and CBS. Expression quantitative trait loci mapping showed that both rs2839629 and rs915854 genotypes impact PKNOX1 expression in nerve tissue while rs2839629 affects CBS expression in skin and blood.
Conclusions
The use of GWAS in MM pharmacogenomics has identified a novel candidate genetic locus mapping to PKNOX1 and in the immediate vicinity of CBS at 21q22.3 associated with the severe bortezomib-induced toxicity. The proximity of these two genes involved in neurologic pain whose tissue-specific expression is modified by the two variants provides new targets for neuro-protective strategies.
Introduction
Some patients with multiple myeloma (MM) have subclinical or even clinical peripheral neuropathy (PN) at diagnosis. This PN can be related to co-morbidities, such as diabetes mellitus, or associated with the M-protein itself. In the course of the disease, PN is mostly induced by therapies, especially thalidomide (thalidomide-induced peripheral neuropathy, TiPN) and bortezomib (bortezomib-induced peripheral neuropathy, BiPN), which may be considered as distinct clinical entities (1). TiPN may arise after prolonged administration of thalidomide (in 30 to 55% of patients treated for 12 months, including 15 to 25% with grade 2 or higher PN) and appears to be due to a cumulative effect. Initial symptoms include sensory changes, such as paraesthesia and hyperaesthesia, later followed by motor symptoms and autonomic dysfunction. BiPN is characterized by neuropathic pain and a length-dependent distal sensory neuropathy with suppression of reflexes. Motor neuropathy may follow and infrequently results in mild to severe distal weakness in the lower limbs. There may also be a significant autonomic component, which manifests as dizziness, hypotension, diarrhoea or constipation and/or extreme fatigue. BiPN is thought to occur at a certain threshold of treatment (within five cycles but rarely beyond) in 40 to 60% of the patients, including 15 to 40% who will develop severe PN (grade 2 or higher). This drug-induced toxicity is well known by physicians and nurses, and patients are now systematically informed about these potential side-effects. The use of subcutaneous bortezomib reduces the incidence of BiPN but does not abrogate this toxicity (2). Since no effective prophylactic treatment is available, prompt action in case of symptoms, including dose-reduction and weekly administration of bortezomib, is crucial to manage this severe toxicity, which may dramatically affect the quality of life (3-5). Therefore the identification of patients at risk of developing BiPN or TiPN is an important issue. This is especially true since the triplet combination of bortezomib-thalidomide-dexamethasone (VTD) is considered one of the best induction regimens prior to high-dose therapy and autologous stem cell transplantation for the treatment of younger patients with de novo MM (6).
The interindividual differences in the onset of BiPN or TiPN is in agreement with an underlying genetic susceptibility to this toxicity. Rare variants in bortezomib or thalidomide target proteins could affect patient's sensitivity to these drugs. Among the pharmacogenomics methods to discover genetic loci associated with drug-induced toxicities, the candidate gene approach has shown a significant genetic contribution to the risk of developing TiPN or BiPN (7-10). However a genome-wide association study (GWAS) has the capacity to identify new genetic variants that will have a direct or indirect effect on drug sensitivity. Here we report the results of a GWAS of 583 MM patients treated with bortezomib to discover genetic variants associated with severe BiPN. This is the first GWA pharmacogenomics study of bortezomib treatment toxicity and provides novel insights into bortezomib-related pathways.
Patients and methods
Clinical samples
Peripheral-blood DNA samples were collected from 598 patients with newly diagnosed MM who received bortezomib-dexamethasone (VD) induction therapy. Patients were treated in randomized clinical trials of the Intergroupe Francophone du Myelome (IFM) (IFM 2005-01, IFM2007-02) or routine practice in France (n= 482) and in a randomized clinical trial of the Dutch/Belgian Haemato-Oncology Foundation for Adults in The Netherlands (HOVON) and the German-Speaking Myeloma Multicenter Group(GMMG) (HOVON-65/GMMG-HD4) (n= 116) . The IFM VD treatment consisted of four 3-week cycles of bortezomib 1.3 mg/m2 administered intravenously on days 1, 4, 8, and 11 plus dexamethasone 40 mg days 1-4 (all cycles) and days 9-12 (cycles 1 and 2). The HOVON 65/GMMG-HD4 VD treatment consisted of three cycles of bortezomib 1.3 mg/m2 administered intravenously on days 1, 4, 8, and 11 plus dexamethasone 40 mg on days 1-4, 9-12 and 17-20 (patients enrolled in the HOVON-65/GMMG-HD4 trial received doxorubicin 9 mg/m2 per day on days 1 through 4, in addition to VD according to the PAD regimen. Adverse events including PN were graded by National Cancer Institute Common Toxicity Criteria Version 3.0. All patients provided written informed consent for both the treatment and companion protocols.
Genotyping
Data quality assessment and control steps carried out during GWAS are summarized in the Supplementary Fig. S1. A total of 482 MM samples in the exploratory IFM cohort and 116 MM samples in the Dutch and German replication cohort were genotyped using Affymetrix SNP6.0 Human DNA chips. Affymetrix CEL files were analyzed either by using Affymetrix Genotyping Console software v4.0 (GTC 4.0), followed by application of the Affymetrix Birdseed algorithm v2.0 to generate SNP genotype calls for the IFM exploratory cohort (GEO accession GSE65777) or by application CRLMM v2 algorithm to generate SNP genotype calls for the replication cohort (GEO accession GSE66903).
Samples quality control (QC)
Stringent QC thresholds were applied to filter out poorly genotyped subjects: if contrast QC <0.4, call rate <97% and outlying heterozygosity rate (het_rate > mean het_rate +3SD) the individual was removed. Principal component analysis (PCA) was performed to visualize the genetic ancestry of the IFM samples that passed the QC and assess whether population adjustment should be made (Supplementary Fig. S2). Random 60,000 genotypes of IFM subjects (IFM, n= 469) and unrelated individuals from 3 HapMap phase III populations representing Northwest European (CEU, n= 162), African (YRI, n= 163), and Chinese (CHB, n = 82) ancestries were combined to calculate the PCA. This method identified samples not clustered with Northwest European group (IFM outliers, n=34), given that these patients were equally distributed between the cases and controls groups (Fisher's exact test P = 0.36) no adjustment was needed and therefore they were kept for the GWAS. Inspection of the observed and expected distribution of the neuropathy association statistic showed absence of hidden population substructure (Cochran-Armitage test of association; genomic inflation factor λ = 1.05).
Marker QC
SNP QC was conducted in 4 steps to remove sub optimal markers of the GWA data (Supplementary Fig. S1). 1) unannotated SNPs according to hg19 na32 SNP6 Affymetrix annotations (n= 130) along with SNPs from mitochondrial and sex chromosomes (n= 37,326) were not considered in the study, 2) SNPs with missing genotype in more than 5% of the subjects (n= 16,743), 3) SNPs of low minor allele frequency (MAF) less than 5% (n= 483,984), 4) SNPs showing extensive deviation from Hardy-Weinberg equilibrium (HWE) with a HWE P value less 1×10−5 (n= 834).
Statistical analysis
Statistical analyses were performed using SNPTEST v2.5 (11). Firstly, we compared 370,605 genotypes from 155 grade ≥ 2 BiPN IFM patients to 314 control IFM patients defined as grade 1 BiPN or no BiPN patients. Secondly, we performed a validation using the HOVON-65/GMMG-HD4 cohort for the highest associated SNPs (Ptrend< 10−5) as identified in the exploratory cohort. We compared 41 bortezomib-treated grade ≥ 2 BiPN patients with 75 bortezomib-treated control patients. We applied a one-sided logistic regression with 10,000 label swapping permutations to correct for multiple testing in order to confirm BiPN association in this independent cohort. The predictive value of the SNP validated in the external series was assessed on the overall population (n= 583 i.e. 195 cases and 388 controls) with 107 label swapping permutations.
Results
We conducted a pharmacogenomics GWA study to identify genetic variants associated with bortezomib toxicity in newly diagnosed MM who received VD induction therapy. Using SNP 6.0 Affymetrix arrays, we genotyped 909,622 tagging SNPs in 482 MM cases. Of the 482 DNA samples genotyped, 469 cases passed strict QC criteria (Supplementary Fig. S1). We considered only the 370,605 autosomal SNPs with homozygosity in at least 5% of patients, a genotype call in at least 95% of patients and with a Hardy-Weinberg equilibrium P > 10−5. We compared the genetic contribution of patients who developed BiPN of grade ≥ 2 (n = 155) with that of patients who did not develop severe BiPN or without BiPN (n= 314). We separated grade 0, 1 versus 2 or more based on the clinical impact of such a toxicity. Grade 1 neuropathy requires a careful follow-up, but doses of bortezomib are not modified. While, doses of bortezomib in the routine clinical practice must be adapted (from 1.3 mg/m2 to 1 mg/m2, or from the biweekly to the weekly schedule administration) according to the onset of grade 2 peripheral neuropathy, or stopped in case of grade 3 or more, and resumed in case of recovery. The GWA study showed association for six SNPs with odds ratios (OR) > 1.8 and Ptrend< 10−5 (Table 1; Supplementary Figs. S3 and S4; Supplementary Table 1), although none reached the actual significance in a GWA study (0.05/370,065 = 1.35 × 10−7). To replicate these findings, a validation was performed using SNP 6.0 Affymetrix arrays in 114 newly diagnosed MM patients enrolled in the HOVON-65/GMMG-HD4 clinical trial who received VD induction therapy. A significant association was seen for rs2839629 (OR, 2.04; 95% CI = 1.11-3.33; P = 8.3 × 10−3; Table 2) which maps within the 3' UTR of PKNOX1 (transcription factor PBX/knotted 1 homeobox 1). The overall estimate of BiPN associated with rs2839629 was an OR of 1.89 (95% CI = 1.45-2.44; P = 5 × 10−7). Moreover, rs2839629 is in strong linkage disequilibrium (LD) with rs76516641 (r2 = 0.94) and rs915854 (r2 = 0.86) which map within the intergenic region of 19.5 kb between PKNOX1 and CBS (Fig. 1).
Table 1.
SNPTEST results for exploratory population for the six highest associated SNPs.
| BiPN (grade ≥ 2) | No BiPN or grade 1 BiPN | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr. | Allele A | Allele B | #AA | #AB | #BB | #AA | #AB | #BB | ORAB/AA (95% CI) | ORBB/AA (95% CI) | ORglobal (95% CI) | Ptrend |
| rs10862339 | 12 | A | C | 40 | 79 | 36 | 147 | 135 | 32 | 2.15 (1.38-3.36) | 4.13 (2.29-7.46) | 2.05 (1.55-2.71) | 5.47 × 10−7 |
| rs1344016 | 12 | A | G | 41 | 79 | 35 | 145 | 135 | 34 | 2.07 (1.33-3.23) | 3.64 (2.03-6.54) | 1.94 (1.47-2.56) | 3.81 × 10−6 |
| rs2414277 | 15 | T | C | 21 | 70 | 64 | 86 | 155 | 73 | 1.93 (1.25-3.03) | 3.57 (2.00-6.25) | 1.92 (1.45-2.56) | 6.17 × 10−6 |
| rs2839629 | 21 | G | A | 33 | 79 | 43 | 128 | 137 | 49 | 1.52 (0.93-2.50) | 3.45 (1.96-5.88) | 1.89 (1.45-2.50) | 7.64 × 10−6 |
| rs4776196 | 15 | T | C | 21 | 70 | 64 | 85 | 155 | 74 | 1.92 (1.23-2.94) | 3.45 (1.96-6.25) | 1.89 (1.43-2.50) | 9.31 × 10−6 |
| rs11145770 | 9 | G | A | 46 | 72 | 37 | 141 | 144 | 29 | 2.56 (1.45-4.55) | 3.85 (2.17-7.14) | 1.89 (1.43-2.50) | 9.70 × 10−6 |
Table 2.
One-sided logistic regression in the validation cohort to test whether the direction of association found in the exploratory cohort can be confirmed.
| SNP | ORglobal (95% CI) | P | P pointwise | P FWER |
|---|---|---|---|---|
| rs10862339 | 1.02 (0.58-1.79) | 0.53 | 0.54 | 0.96 |
| rs1344016 | 1.05 (0.60-1.85) | 0.43 | 0.44 | 0.91 |
| rs2414277 | 1.20 (0.71-2.08) | 0.24 | 0.22 | 0.66 |
| rs2839629 | 2.04 (1.11-3.33) | 9.6 × 10−3 | 8.3 × 10−3 | 0.036 |
| rs4776196 | 1.19 (0.71-2.08) | 0.24 | 0.27 | 0.70 |
| rs11145770 | 1.43 (0.75-2.33) | 0.14 | 0.13 | 0.46 |
ORglobal = the estimated odds ratio by logistic regression; P = uncorrected parametric P value; Ppointwise = pointwise P value as determined by permutation; PFWER = permuted P value (family wise error rate correction).
Figure 1.

21q22.3 locus showing genome-wide level of evidence of BiPN in MM. Illustration of the locus with the local linkage disequilibrium and recombination rate over 500-kb centred on rs2839629 (blue triangle). Each diamond, triangle, circle or square represents a SNP found in this locus, rs76516641 and rs915854 are indicated (red diamond). The figure was generated using the web-based tool SNiPA (http://www.snipa.org; 22).
Both PKNOX1 and CBS appear to be strong candidates for BiPN susceptibility genes. PKNOX1 is known to modulate transcriptional activity of chemokine monocyte chemoattractant protein-1 (MCP-1) gene (12-15). Through interaction with its cognate receptor CCR2, MCP-1 contributes to paclitaxel CIPN through changes in dorsal root ganglion (DRG) neurons (16). MCP-1 is universally increased in different models of neuropathic pain and may be considered as a biomarker of chronic pain (17). MCP-1 is an important mediator of macrophage-related neural damage in different animal models of inherited neuropathies and acute inflammatory demyelinating neuropathy (18-19). CBS encodes the endogenous H2S-producing enzyme cystathionine-ß-synthetase. CBS-H2S signaling pathway is implicated in the pathogenesis of a variety of neurodegenerative and inflammatory disorders, diabetic gastric hypersensitivity and plays a crucial role in inflammatory pain in temporomandibular joint (20-23).
To explore the possibility that this association might be mediated through differential expression of PKNOX1 or CBS or both, we examined the correlations between rs2839629, rs76516641 and rs915854 genotypes and tissue-specific gene expression levels by using the expression quantitative trait locus (eQTL) analysis available on the SNiPA portal (http://www.snipa.org; 24) that used GTEX Portal v6 (25) and MuTHER consortium (26) as primary sources. PKNOX1 expression was significantly associated with rs2839629 and rs915854 genotypes in nerve-tibial tissue (P = 5.6 × 10−8 and P = 1.9 × 10−7 respectively; Supplementary Table S2-S3) with higher expression associated with rs2839629 risk alleles (Fig. 2) whereas CBS expression was significantly associated only with rs2839629 in skin (P = 2.6 × 10−15) and in blood (P = 3.1 × 10−8) (Supplementary Table S2). rs915854 is annotated with a regulatory feature cluster characterized by histone marks H3K27ac and H3K4me1 enrichment in blood and cervix cells (Supplementary Table S3). In addition, rs915854 minor allele is predicted to disrupt binding site for the general pioneer factor FOXA1, this could render the enhancer less active for target gene expression such as PKNOX1 (Fig. 3). Conversely, rs76516641 genotype has no significant effect on both PKNOX1 and CBS gene expression (data not shown).
Figure 2.
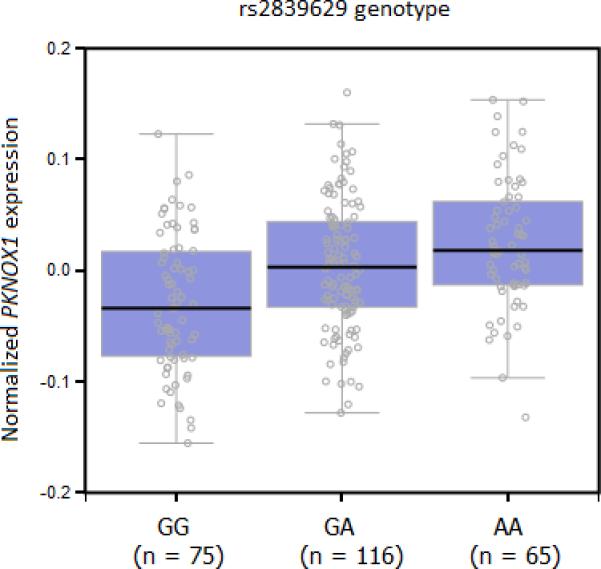
Relationship between nerve-tibial PKNOX1 expression and rs2839629 genotype from the GTEX Portal v6 (http://www.gtexportal.org; 23).
Figure 3.
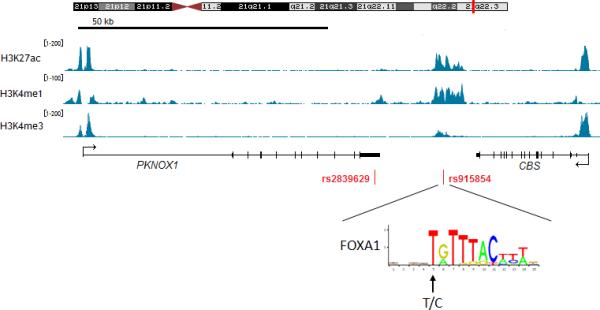
UCS Genome Browser views of histone marks H3K27ac and H3K4me1 enrichment and H3K4me3 depletion within the region covering PKNOX1 and CBS in Hela cells. Arrow indicates the variant sequence and location in position-weight matrix for FOXA1.
Since we show that the risk allele A for rs2839629 is associated with higher levels of PKNOX1expression and previous report have demonstrated that PKNOX1 binds preferentially to the −2578G (rs1024611G) polymorphism leading to increase MCP-1 levels (14), we analysed the relationship between rs2839629A and rs1024611G in the IFM exploratory cohort. We found a significant association between rs2839629 A/ A homozygous genotype and rs1024611G –bearing allele (Fisher's exact test P = 0.01) suggesting a possible epistatic interaction between rs2839629 and rs1024611to regulate MCP-1 expression.
The present pharmacogenomics GWA study also confirmed the modest association of the rs619824 genotype with BiPN (P =0.043) previously identified by Corthals and colleagues (8). Although there has been no overlap with a previous study on late-onset of BiPN associated variants reported by Broyl and colleagues (10) as shown in Supplementary Table S4. This lack of overlap could reflect the potential complexity of predisposition to BiPN. More importantly, the design of the custom SNP chip used previously only contained 3404 SNPs in 983 hypothesis driven genes which were thought to be functionally relevant in abnormal cellular functions, inflammation and immunity, as well as drug responses rather than adverse drug reactions which are less obvious candidates (27).
Discussion
To date there is no established predictors of BiPN, it is impossible to predict which patient will develop neuropathy. Previous studies performed by our group and others using candidate gene approach have revealed significant association between SNP and BiPN, however the clinical relevance of these findings is not clear (8-10). In order to increase our chance to discover variants that might provide new insights in the mechanisms underlying gene-phenotype, we used a hypothesis-free approach. GWAS in cancer pharmacogenomics is challenging and few reports have been published to date. This is mainly due to insufficient statistical power in studies (28). To partially overcome these limitations, we designed our analysis to identify high-effect SNP (OR > 1.8) with MAFs greater than 0.05, in a large cohort of IFM MM patients (n= 469) uniformly treated in order to achieve convincing statistical power (29) and we verified our findings in an independent cohort of Dutch and German MM patients. Furthermore our GWAS approach eliminates selection case-control bias since both case-control studies included patients in cohort studies i.e. IFM or HOVONGMMG clinical trials cohorts. When evaluating toxicity, it is sometimes difficult to distinguish between BiPN and neuropathic pain in general. It is also recognized that the sole use of the NCI CTC for assessment of sensory PN is suboptimal. It is also recognized that detailed patient-reported symptom data and a quality of life assessment more accurately describes this toxicity and that physician-reported NCI-CTC grading underreports PN. These systematic evaluations are difficult to apply in a multicenter study in the context of pharmacogenomics analyses. Of note, our study has enrolled patients without peripheral neuropathy at baseline, and patients were treated with the doublet combination of bortezomib and dexamethasone, and did not receive other neurotoxic agents.
Our analysis revealed a SNP associated with BiPN (rs283962; OR, 1.89; P = 7.6 × 10-6) that was replicated in an independent cohort (OR, 2.04; P= 8.3 × 10−3) in high LD with SNP rs915854. Both variants are in noncoding regions; rs2839629 is located in the 3′UTR of PKNOX1 and rs915854 is in the intergenic region between PKNOX1 and CBS. Expression quantitative trait loci showed that these variants alter PKNOX1 and CBS expression presumably via cis regulatory elements in the case of rs915854 since it falls within a regulatory region (Fig. 3). Finally, we found a significant association between rs2839629A and rs1024611G that could impact MCP-1 expression levels. Given that these genes encode proteins - directly or indirectly - involved in neuropathic (16) and inflammatory pain (23), the functional significance of these predictive SNPs is established. This discovery opens the way to investigate novel pathways linked to PKNOX1 and CBS activities for a better understanding of mechanisms underlying this neurotoxicity.
This work generated new hypothesis regarding neurotoxicity mechanisms and provides new targets for neuro-protective strategies however additional international collaborative efforts including non European countries are warranted to confirm or refute these findings and examine the impact of differential expression of both PKNOX1 and CBS effects on bortezomib exposure in cell model. Our results are preliminary and cannot be proposed yet for a systematic use in the routine clinical practice. Nevertheless, our findings are one of the first steps that may allow for the identification of patients at increased risk of severe BiPN and these patients may benefit from the use of alternative drugs, such as carfilzomib, and/or a more focused clinical management of this toxicity.
Supplementary Material
Translational Relevance.
Bortezomib-based induction regimens are currently the standard of care prior to autologous stem-cell transplantation for the treatment of younger patients with de novo multiple myeloma. One of the most important side-effect of these regimens is peripheral neuropathy that can preclude the administration of therapies and dramatically affects patient's quality of life. The identification of high-quality biomarkers that predict severe bortezomib-induced toxicity is still lacking. Herein, using a genome-wide association study in young myeloma patients treated with bortezomib-dexamethasone, we show that the genetic variant rs2839629 is an important predictor for this neuropathy. In addition, this variant impacts expression of two neighbour genes PKNXO1 and CBS known to be involved directly or indirectly in neuropathic and inflammatory pain. Discovery of this genetic variant reveals novel pathways for this neurotoxicity provides attractive targets for neuroprotective strategies.
Acknowledgements
This study was supported by Intergroupe Francophone du Myélome; the French Institute National du Cancer Grant POBNIMM PROG/08/10 (to H.A.L., S.M.); and National Institutes of Health Grant PO1CA155258-01 (to S.M., H.A.L., N.C.M.). We would like to thank Elise Douillard, Magali Devic, Emilie Maurenton and Nathalie Roi, for excellent technical expertise.
Footnotes
Author contributions
F.M., R.K., H.A.-L., M. V. D., C.D., L.C. and S.M. designed the study and analyzed the data. W.G., C.G.-C., A.A., L.V., H.E., J.S., H.D.S, N.C.M. analyzed the data, M.A., P.S., C.D. and P.M. provided study materials or patients and collected clinical follow-up data. S.M., P.M and M. V.D. wrote the report.
Competing financial interest
PS- honoraria: Janssen-Cilag, Celgene, Onyx; research funding: Millenium, Onyx, Celgene. Other authors declare no conflict of interest.
References
- 1.Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol. 2010;6:657–66. doi: 10.1038/nrneurol.2010.160. [DOI] [PubMed] [Google Scholar]
- 2.Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12:431–40. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- 3.Delforge M, Blade J, Dimopoulos MA, Facon T, Kropff M, Ludwig H, et al. Treatment-related peripheral neuropathy in multiple myeloma: the challenge continues. Lancet Oncol. 2010;11:1086–95. doi: 10.1016/S1470-2045(10)70068-1. [DOI] [PubMed] [Google Scholar]
- 4.Mohty B, El-Cheikh J, Yakoub-Agha I, Moreau P, Harousseau JL, Mohty M. Peripheral neuropathy and new treatments for multiple myeloma: background and practical recommendations. Haematologica. 2010;95:311–9. doi: 10.3324/haematol.2009.012674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richardson PG, Delforge M, Beksac M, Wen P, Jongen JL, Sezer O, et al. Management of treatment-emergent peripheral neuropathy in multiple myeloma. Leukemia. 2012;26:595–608. doi: 10.1038/leu.2011.346. [DOI] [PubMed] [Google Scholar]
- 6.Moreau P, San Miguel J, Ludwig H, Schouten H, Mohty M, Dimopoulos M, et al. Multiple myeloma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24:133–7. doi: 10.1093/annonc/mdt297. [DOI] [PubMed] [Google Scholar]
- 7.Johnson DC, Corthals SL, Walker BA, Ross FM, Gregory WM, Dickens NJ, et al. Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. J Clin Oncol. 2011;29:797–804. doi: 10.1200/JCO.2010.28.0792. [DOI] [PubMed] [Google Scholar]
- 8.Corthals SL, Kuiper R, Johnson DC, Sonneveld P, Hajek R, van der Holt B, et al. Genetic factors underlying the risk of bortezomib-induced peripheral neuropathy in multiple myeloma patients. Haematologica. 2011;96:1728–32. doi: 10.3324/haematol.2011.041434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Favis R, Sun Y, van de Velde H, Broderick E, Levey L, Meyers M, et al. Genetic variation associated with bortezomib-induced peripheral neuropathy. Pharmacogenet Genomics. 2011;21:121–9. doi: 10.1097/FPC.0b013e3283436b45. [DOI] [PubMed] [Google Scholar]
- 10.Broyl A, Corthals SL, Jongen JL, van der Holt B, Kuiper R, de Knegt Y, et al. Mechanisms of peripheral neuropathy associated with bortezomib and vincristine in patients with newly diagnosed multiple myeloma: a prospective analysis of data from the HOVON-65/GMMG HD4 trial. Lancet Oncol. 2010;11:1057–65. doi: 10.1016/S1470-2045(10)70206-0. [DOI] [PubMed] [Google Scholar]
- 11.Marchini J, Howie B, Myers S, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 12.Rovin BH, Lu L, Saxena R. A novel polymorphism in the MCP-1 gene regulatory region that influences MCP-1 expression. Biochem Biophys Res Commun. 1999;259:344–48. doi: 10.1006/bbrc.1999.0796. [DOI] [PubMed] [Google Scholar]
- 13.Wright EK, Jr., Page SH, Barber SA, Clements JE. Prep1/Pbx2 complexes regulate CCL2 expression through the -2578 guanine polymorphism. Genes Immun. 2008;9:419–30. doi: 10.1038/gene.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Page SH, Wright EK, Jr, Gama L, Clements JE. Regulation of CCL2 expression by an upstream TALE homeodomain protein-binding site that synergizes with the site created by the A-2578G SNP. PLoS One. 2011;6:e22052. doi: 10.1371/journal.pone.0022052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pham MH, Bonello GB, Castiblanco J, Le T, Sigala J, He W, Mummidi S. The rs1024611 regulatory region polymorphism is associated with CCL2 allelic expression imbalance. PLoS One. 2012;7(11):e49498. doi: 10.1371/journal.pone.0049498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Boyette-Davis JA, Kosturakis AK, Li Y, Yoon SY, Walters ET, et al. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2013;14:1031–44. doi: 10.1016/j.jpain.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, de Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–83. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- 18.Groh J, Heinl K, Kohl B, Wessig C, Greeske J, Fischer S, et al. Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot-Marie-Tooth 1X. Hum Mol Genet. 2010;19:3530–43. doi: 10.1093/hmg/ddq269. [DOI] [PubMed] [Google Scholar]
- 19.Yuan F, Yosef N, Lakshmana Reddy C, Huang A, Chiang SC, Tithi HR, et al. CCR2 Gene Deletion and Pharmacologic Blockade Ameliorate a Severe Murine Experimental Autoimmune Neuritis Model of Guillain-Barre' Syndrome. PLoS One. 2014;9:e90463. doi: 10.1371/journal.pone.0090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Qu R, Hu S, Xiao Y, Jiang X, Xu GY. Upregulation of Cystathionine β-Synthetase Expression Contributes to Visceral Hyperalgesia Induced by Heterotypic Intermittent Stress in Rats. PLoS One. 2012;7:e53165. doi: 10.1371/journal.pone.0053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schemann M, Grundy D. Role of hydrogen sulfide in visceral nociception. Gut. 2009;58:744. doi: 10.1136/gut.2008.167858. [DOI] [PubMed] [Google Scholar]
- 22.Zhang HH, Hu J, Zhou YL, Hu S, Wang YM, Chen W, et al. Promoted interaction of nuclear factor-κB with demethylated cystathionine-β-synthetase gene contributes to gastric hypersensitivity in diabetic rats. J Neurosci. 2013;33:9028–38. doi: 10.1523/JNEUROSCI.1068-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miao X, Meng X, Wu G, Ju Z, Zhang HH, et al. Upregulation of cystathionine-β-synthetase expression contributes to inflammatory pain in rat temporomandibular joint. Mol Pain. 2014;10:9. doi: 10.1186/1744-8069-10-9. doi: 10.1186/1744-8069-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnold M, Raffler J, Pfeufer A, Suhre K, Kastenmüller G. SNiPA: an interactive, genetic variant-centered annotation browser. Bioinformatics. 2015;31:1334–6. doi: 10.1093/bioinformatics/btu779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grundberg E, Small KS, Hedman ÅK, Nica AC, Buil A, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Ness B, Ramos C, Haznadar M, Hoering A, Haessler J, Crowley J, et al. Genomic variation in myeloma: design, content, and initial application of the Bank On A Cure SNP Panel to detect associations with progression-free survival. BMC Med. 2008;6:26. doi: 10.1186/1741-7015-6-26. doi: 10.1186/1741-7015-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low SK, Takahashi A, Mushiroda T, Kubo M. Genome-wide association study: a useful tool to identify common genetic variants associated with drug toxicity and efficacy in cancer pharmacogenomics. Clin Cancer Res. 2014;20:2541–52. doi: 10.1158/1078-0432.CCR-13-2755. [DOI] [PubMed] [Google Scholar]
- 29.Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet. 2005;6:109–18. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.