

Abstract
Syndecan-1 (Sdc1/CD138) expression is linked to disease severity in multiple myeloma (MM), although the causal basis for this link remains unclear. Here we report that capture of the IGF1 receptor (IGF1R) by Sdc1 suppresses ASK1-dependent apoptosis in MM cells. Sdc1 binds two different fractions of IGF1R, one that is constitutively active and a second that is activated by IGF1 ligand. Notably, IGF1R kinase activity in both fractions is blocked by synstatinIGF1R (SSTNIGF1R), a peptide that inhibits IGF1R capture by Sdc1, as well as by a truncated peptide (SSTNIGF1R-T) that appears to be specific for MM cells. Mechanistically, we show that ASK1 is bound to active IGF1R and inhibited by Tyr and Ser83/Ser966 phosphorylation. When IGF1R engagement with Sdc1 is blocked by SSTNIGF1R, ASK1 becomes activated, and initiates JNK- and caspase-3-mediated apoptosis. In pharmacological tests, we find SSTNIGF1R is highly stable in human plasma and displays a half-life of 27 hr in mice, wherein it significantly reduces both the size and neovascularization of CAG myeloma tumor xenografts. Taken together, our results offer a preclinical proof of concept and mechanistic rationale for the exploration of SSTNIGF1R as an experimental therapeutic to dually attack MM tumor cell survival and tumor angiogenesis.
Keywords: Syndecan, myeloma, synstatin, ASK1, JNK, insulin-like growth factor-1 receptor, apoptosis
INTRODUCTION
Multiple myeloma (MM), a disease in which malignant plasma cells thrive in the bone marrow, is the second most prevalent hematologic malignancy in the United States (1,2). The emergence of new therapies (e.g., bortezomib, thalidomide and its derivatives) has greatly improved survival rates in patients with myeloma (2). However, these therapies only slow tumor growth rather than cure the disease. Thus, the need for novel therapies that prevent the progression of the disease and maintain patient quality of life remains a high priority.
Sdc1 (CD138) is highly expressed on malignant plasma cells and has a causal role in disease progression (3–8). Suppressed expression of Sdc1 causes myeloma cells to grow poorly in vitro and in vivo and undergo apoptosis (3,9), but how Sdc1 promotes survival is unclear. Plasma cell and myeloma cell survival is regulated by apoptosis signal-regulating kinase-1 (ASK1), a kinase activated in response to metabolic, genotoxic and endoplasmic reticulum (ER) stress (10). Plasma cells enduring ER stress due to copious immunoglobulin synthesis trigger the unfolded protein response (UPR), which, if prolonged, stimulates ASK1 leading to caspase activation and cell death (10,11). To protect themselves, both normal and malignant plasma cells rely on mechanisms that suppress ASK1 activation. One mechanism is increased expression of Blimp1, which negatively regulates the expression of ASK1 (10). Of note, Blimp1 promotes the expression of Sdc1, which becomes highly expressed in plasma and myeloma cells (12,13), possibly suggesting competing roles for this receptor and ASK1. Other means of suppressing ASK1 activation include inhibitory phosphorylation of the kinase on Ser83 and Ser966 (14–17), removal of an activating phosphorylation of Thr838 (Thr845 in mouse)(18), and tyrosine phosphorylation of its inhibitory domain by the insulin-like-growth factor-1 receptor (IGF1R), a pro-survival-signaling receptor tyrosine kinase (19).
Our prior work has shown that Sdc1 captures and activates IGF1R, suggesting a potential mechanism for suppressing ASK1 activation in myeloma. In breast cancer cells and activated vascular endothelial cells, Sdc1 captures IGF1R together with the αvβ3 or αvβ5 integrin; their docking with Sdc1 activates the IGF1R, which in turn activates the integrins via an inside-out signaling mechanism (20–22). This promotes adhesion and migration of the cells on ligands for these integrins, and is required for the response of endothelial cells to vascular endothelial cell growth factor (VEGF) during the early stages of angiogenesis (20,23,24). Importantly, the formation of the receptor complexes can be blocked by an inhibitory peptide called synstatin (SSTNIGF1R), which mimics the capture motif (amino acids 92–119 in mouse, 93–120 in human) in the extracellular domain of Sdc1 (20,23). Because of the easy availability of its cell surface target, SSTNIGF1R readily disrupts angiogenesis and the growth of carcinoma xenografts when delivered systemically to mice (20,23,24).
IGF1R has an important survival role in many cells, and its high expression correlates with poor prognosis in myeloma (25–27). Although myeloma cells are known to express high levels of Sdc1, the possibility that its high expression is a means to suppress ASK1 and prevent apoptosis has not been explored. In the present work, we show that IGF1R activity is blocked by SSTNIGF1R in myeloma cells, which activates ASK1 and stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK), which acts downstream of ASK1 to cause caspase-3-mediated apoptosis. We find that apoptosis is induced in activated vascular endothelial cells as well. We also show that SSTNIGF1R is highly stable in vitro and in vivo, and blocks the growth of myeloma xenografts in vivo by targeting both the myeloma cells and the endothelial cells that comprise the tumors.
MATERIALS AND METHODS
Reagents
SSTN peptides obtained from LifeTEIN LLC (Hillsborough, NJ) were reconstituted in DMEM (Life Technologies, Grand Island, NY) containing 200 mM HEPES (Sigma-Aldrich, St. Louis, MO) for in vitro studies, or HEPES-buffered 0.9% saline for use in vivo. IGF1R inhibitor Tyrphostin I-OMe-AG538 (AG538) was from Sigma-Aldrich. Monoclonal antibody (mAb) against human Sdc1 (B-A38) was from Acris Antibodies (San Diego, CA), to αvβ3 (LM609), αvβ5 (P1F6), and α4 integrin (P1H4) were from EMD Millipore (Billerica, MA). Other α4 mAbs were 44H6 (LifeSpan Biosciences, Seattle, WA) and 2C11 (Novus Biologicals, Littleton, CO). Monoclonal Ab D24A5 (Cell Signaling Technology, Danvers, MA) to the αvβ5 integrin was used for Western blotting. IGF1R-specific antibodies were the function-blocking mAb 24–57 (ThermoFisher Scientific, Waltham, MA), mouse mAbs JBW902 (EMD Millipore) used for immunoprecipitations/FACS, JY202 (EMD Millipore) to detect pY1131-IGF1R by Western blotting), mAb 33255 (R&D Systems, Minneapolis, MN) for Western blotting, and Alexa647-conjugated mouse mAb K74-218 (BD Biosciences, San Jose, CA) to detect pY1131-IGF1R by FACS. Isotype controls, anti-phosphotyrosine mAb PY20 and MECA32 (used for IHC staining of blood vessels) were from BD Biosciences. Secondary antibodies and Click-iT TUNEL Alexa488 were from Life Technologies. CellTiter-GLO, ApoTox-GLO and Caspase3/7-GLO were from Promega (Madison, WI). The PathScan Stress and Apoptosis Signaling Antibody Array Kit was obtained from Cell Signaling Technology. JNK inhibitors SP600125 and JNK-IN-8 were both obtained from EMD Millipore. Mouse α-tubulin mAb TU-02 was from Santa Cruz Biotechnology (Dallas, TX) and rabbit anti-α/β-tubulin (2148) from Cell Signaling Technology. ASK1 inhibitors, NQDI-1 and TC-ASK-10, were acquired from R&D Systems. Total ASK1 antibodies include mouse mAb 2E4 from Life Technologies (used for Western blotting) and rabbit mAb D11C9 from Cell Signaling Technology (used for immunoprecipitation and Western blotting); phospho-specific polyclonal Western blotting antibodies include 3765S (pThr838/Thr845), 3761S (pSer83) and 3764S (pSer966/Ser967) from Cell Signaling Technology. SAPK/JNK antibodies include mouse mAbs E.665.10 (pThr183/pTyr185) from Life Technologies and 252355 (total JNK) from R&D Systems. Fibronectin (FN) was kindly provided by Dr. Donna Peters, University of Wisconsin-Madison). Vitronectin (VN) was purified from human plasma as described in (28). Human recombinant IGF1 was obtained from Peprotech (Rocky Hill, NJ).
Cell culture
All cells were cultured at 37°C and 92.5% air/7.5% CO2. Myeloma cells were grown in RPMI 1640 (Life Technologies) with 4 mM L-glutamine (ThermoFisher Scientific), 10% FBS (Atlanta Biologicals, Lawrenceville, GA) and 0.05 mM beta-mercaptoethanol (Sigma). RPMI-8226, U266, MM.1R and P3X63Ag8 myeloma cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA). CAG cells were supplemented with 40 μg/mL Geneticin (Corning Life Sciences, Tewksbury, MA). CAG cells were obtained from the Myeloma Institute for Research and Therapy (Little Rock, AR). KMS-12-BM cells (authenticated by Leibniz Institute DSMZ) were kindly provided by Dr. Fotis Asimakopoulos, University of Wisconsin-Madison. MDA-MB-231 breast carcinoma, human umbilical vein (HUVEC), SV40-T immortalized human dermal microvascular endothelial cells (HMEC-1) (29), HaCaT human immortalized keratinocytes and MCF10A human immortalized mammary epithelial cells were grown as described previously (20,21,30). UM-SCC-47 cells were authenticated (STR profile) and kindly provided by Dr. Paul Harari, University of Wisconsin- Madison. SCC-25, and MDA-MB-468, MCF-7 and SKBr3 breast carcinoma cells, were all obtained from ATCC. Carcinoma cells were grown in DMEM (ThermoFisher Scientific) supplemented with 4 mM L-glutamine, 10% FBS (Atlanta Biologicals) and 100 units/mL penicillin and 100 μg/mL streptomycin (Life Technologies). Hydrocortisone (1 μg/mL; BDBiosciences) was added to SCC cultures. All cells were routinely screened for mycoplasma.
Flow cytometry
CAG cells cultured in the presence or absence of serum for 6 hr were treated with 30 μM SSTNIGF1R, 10 μM tyrphostin AG538, or 1.5 μg/mL IGF1R blocking antibody (mAb 24–57) versus an isotype control (mouse IgG1) for 1 hr in the presence or absence of 100 ng/mL IGF1 (15 min stimulation). Cells were then fixed, blocked with CMF-PBS containing 1% heat-denatured BSA plus 20 μg/mL goat anti-mouse Fab fragment, permeabilized in CMF-PBS containing 1% heat-denatured BSA and 0.1% w/v saponin (Sigma) and stained with Alexa-Fluor-647-conjugated mAb K74-218 specific for pY1131 in the activation loop of IGF1R. Cells were then subjected to flow cytometry to assess relative levels of activated IGF1R.
Immunoprecipitations
Immunoprecipitations of Sdc1, IGF1R, the αvβ5 integrin and ASK1 with or without competing SSTN peptides, were carried out as previously described (20,21) using 0.5–1.0 mg of input/sample and lysis buffer supplemented with HALT Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific).
siRNA design and transfection
Lipofectamine RNAiMAX and Silencer Select control (AM4635), human ASK1 (siRNA ID# S8676; Target Sequence: 1262(CA)GCGAGTAGATAATATCGAA1282, GenBank Accession number NM_005923.3) and human IGF1R-specific (siRNA ID# 110754; Target Sequence: 3692GGAATACAGGAAGTATGGA(tt)3710 GenBank Accession Number NM_000875.4) siRNA oligos were acquired from Life Technologies. 106 CAG or 2 × 106 MM.1R cells per 35 mm well were transfected with 100 nM siRNA using Lipofectamine RNAiMAX and Opti-MEM I transfection medium (Life Technologies) in accordance with the manufacturer’s instructions. 6 h post-transfection, wells were supplemented with 10% FBS and 3 mL of complete growth medium. Cells were harvested 48 h post-transfection by western blot or Caspase3/7-GLO.
Cell growth and survival assays
2.5 × 104 myeloma cells; 1–2 × 103 adherent cells/well were plated in 96-well plates in complete medium in the presence or absence of SSTN peptides for 24, 48 or 72 hr. In some experiments, cells were pre-treated with JNK inhibitors for 1 hr prior to the addition of SSTN peptide to the wells. Cell number and caspase activation/apoptosis were measured using CellTiter-GLO, ApoTox-GLO, Caspase3/7-GLO and Click-iT TUNEL Alexa488 in accordance with the manufacturer’s instructions.
Cell stress and apoptosis marker array
Cells were plated in 6-well plates in complete medium and treated with or without SSTNIGF1R peptide for 1, 3, 9, or 24 hr. All cells were harvested and processed in accordance with the PathScan Stress and Apoptosis Signaling Antibody Array Kit instructions manual using whole cell lysates from equal cell equivalents. Relative fluorescent intensity (700 nm) for each target was measured using a LI-COR Biosciences Odyssey flat-top digital imaging scanner and LI-COR Image Studio v3.1 analysis software in two separate experiments.
SSTN Stability and PK/PD studies
For stability assays, SSTNIGF1R and GRGDSP peptide (EMD Millipore) were incubated in either human plasma (obtained from the UW-Madison Hospital Blood Bank) or sterile CMF-PBS (control) for 0, 8, 24 or 72 hr and their activity (ability to block endothelial cell adhesion) measured against a standard curve using HUVECs plated on wells coated with either VN (SSTNIGF1R) or FN (GRGDSP). For in vivo stability (steady-state accumulation) assays, female BALB/c mice (3 mice/treatment condition) were serially injected every 24 hr subcutaneously with biotinylated SSTNIGF1R (0.365 mg/kg) for up to 6 total injections. Mice were then terminally bled under anesthesia at 0, 24, 48, 72, 96, 120 and 144 hr post-injection. Alternatively, mice were given a single bolus of SSTNIGF1R (0.365 mg/kg) and were terminally bled at 0, 0.1, 0.3, 1, 3, 6, 12 and 24 hr post-injection. Levels of SSTNIGF1R in serum samples spotted on nitrocellulose membranes were quantified by IR scanning using anti-biotin antibody on a LI-COR Biosciences Odyssey digital imaging system and LI-COR Image Studio v4.0 analysis software. Pharmacokinetic and pharmacodynamic variables were calculated using the WinnonLin, Phoenix 64 software package.
Tumor formation in animals
All animal studies were performed in accordance with UW and NIH guidelines after institutional review and approval. Under aseptic conditions, 106 CAG myeloma cells were subcutaneously injected into the right and left flanks of 6–8-wk-old female, athymic Foxn1nu outbred nude mice (Harlan Laboratories). Tumors were allowed to form for 1 week, then animals were randomized and surgically implanted with Alzet (Durect Corp., Cupertino, CA) osmotic pumps (Model 2004, 0.25 μl/h) containing either PBS, PBS plus 4 mM SSTNIGF1R or PBS plus 4 mM SSTNIGF1R-T. After four weeks treatment, the mice were euthanized and tumors harvested and tumor volume (V = 0.524 × length × width2) was determined (31). Serial frozen sections (8 μm) were cut, fixed in periodate-lysine-paraformaldehyde, quenched in 0.05% NaBH4 followed by 0.1M glycine and then permeabilized/blocked in IHC buffer (0.02% NaN3, 0.1% BSA, 0.2% Triton X-100, 0.05% Tween-20 in PBS) containing 10% goat serum. Sections were stained with rat mAb MECA32 (5 μg/mL) to quantify blood vessel density followed by an Alexa488-conjugated goat anti-rat secondary (2 μg/mL). Images were acquired using a PlanFluor 40X objective (1.30 NA; Nikon, Melville, NY) and a Roper Scientific Photometrics CoolSnap ES camera on a Nikon Eclipse TE2000U microscopy system. Mean MECA-32 staining intensity was calculated across 6 representative fields for each cohort (i.e., control vs. SSTN-treated mice) using Metamorph software.
RESULTS
Myeloma cells express the Sdc1-coupled IGF1R receptor complex
RPMI-8226, U266, CAG and MM.1R myeloma cells were screened for cell surface expression of IGF1R, as well as the αvβ3 or αvβ5 integrin that is required as an obligate partner for IGF1R capture by Sdc1. High cell surface expression of Sdc1 is observed, as expected, along with the IGF1R and αvβ5 integrin (Fig. 1A). However, the cells express little or no αvβ3 integrin. Screening of primary Sdc1-positive myeloma cells isolated from twelve patient bone marrow aspirates confirms the expression of IGF1R, the αvβ3 and the αvβ5 integrin (Fig. S1) in diseased cells in vivo. IGF1R and αvβ5 integrin are present as a complex with Sdc1, as they co-precipitate with the syndecan from CAG, RPMI-8226 and MM-1R cells (Fig. 1B, D). Furthermore, the receptor complex in each of the three cell lines is disrupted by 1 μM SSTNIGF1R (20,21), comprised of amino acids 92–119 in mouse Sdc1 (Fig. 1B, D). Surprisingly, a truncated peptide (SSTNIGF1R-T), comprising amino acids 94–119, shown previously to be ineffective as an inhibitor in carcinoma and endothelial cells (20), also disrupts capture of IGF1R and αvβ5 integrin by Sdc1, although it is 3–10-fold less effective than SSTNIGF1R. Shown as a control, VLA-4 (α4β1 integrin) does not immunoprecipitate with Sdc1 from myeloma cells growing in suspension (Fig. 1C).
FIGURE 1. Myeloma cells express IGF1R coupled to Sdc1 and αvβ5 integrin.

A. RPMI-8226, U266, CAG and MM.1R human myeloma cells are analyzed by flow cytometry for relative cell surface expression of Sdc1 (B-A38), αvβ5 integrin (P1F6), αvβ3 integrin (LM609) and IGF1R (JBW902). B. Lysates of CAG cells grown in suspension culture are subjected to immunoprecipitation using isotype-matched control mIgG1, B-A38 (Sdc1), P1F6 (αvβ5 integrin) and/or JBW902 (IGF1R) in the presence of SSTNIGF1R (1 μM) or SSTNIGF1R-T (1, 3 or 10 μM). Immunoprecipitates are probed for presence of Sdc1 (B-A38), IGF1R (33255) and αvβ5 integrin (D24A5). C. CAG cell lysates are subjected to immunoprecipitation as in (B) using B-A38 or 44H6 specific for VLA-4 (α4 integrin), and analyzed for the presence of VLA-4. D. Lysates of RPMI-8226 or MM.1R cells are subjected to immunoprecipitation with B-A38 containing no competitor, or 1 μM SSTNIGF1R or 10 μM SSTNIGF1R-T and analyzed as in (B).
Sdc1-coupled IGF1R comprises both constitutively active and IGF1-stimulated fractions
IGF1R coupled to Sdc1 is active, shown by phosphorylation of Y1131 in its kinase activation loop, which is abolished by tyrphostin AG538, an IGF1R-specific kinase inhibitor (Fig. 2A). Not unexpectedly, IGF1R is activated by IGF1 present in the serum, as it is blocked by IGF1R blocking antibody or serum starvation, and is activated in the absence of serum by exogenous IGF1 (Fig. 2A–C). However, a constitutively active receptor fraction is also observed that does not depend on IGF1 and is insensitive to IGF1R blocking antibody (Fig. 2A–C). Importantly, activation of IGF1R in both fractions, namely the constitutively active and IGF1-stimulated fraction, depends on coupling of IGF1R to Sdc1, as both are inhibited by SSTNIGF1R.
FIGURE 2. IGF1R activation in myeloma is inhibited by SSTNIGF1R.
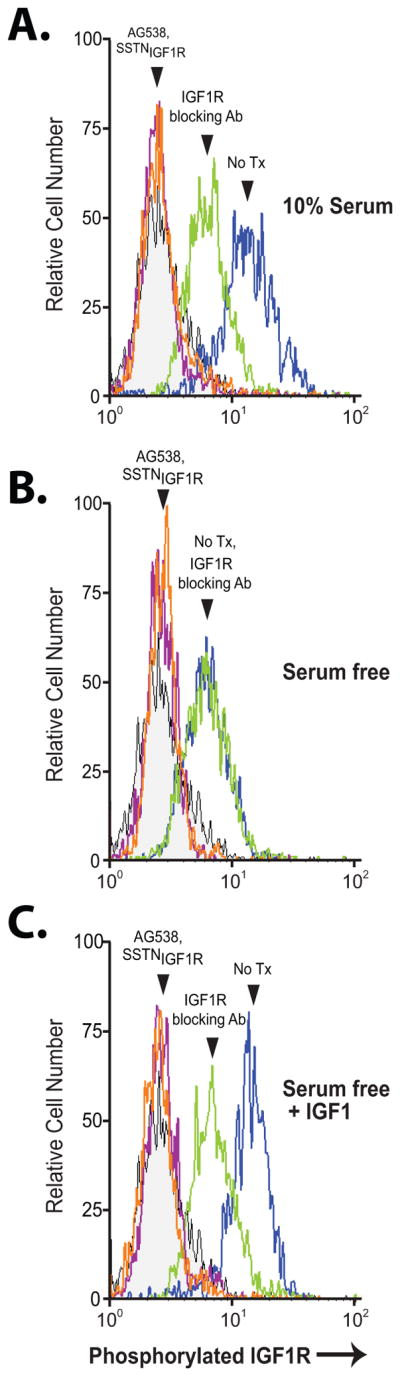
CAG cells cultured in serum-containing medium (A) or serum-free medium for 6 hr (B, C) were treated with 30 μM SSTNIGF1R, 10 μM tyrphostin AG538, or 1.5 μg/mL IGF1R blocking antibody (24–57) versus isotype-matched control mIgG1 for 1 hr in the presence (C) or absence (A, B) of supplementary 100 ng/mL IGF1. Cells were then fixed, blocked, permeabilized, stained with mAb K74-218 specific for pY1131 in the activation loop of IGF1R and analyzed by flow cytometry for levels of activated IGF1R.
Sdc1-coupled IGF1R is critical for the growth and survival of tumor and vascular endothelial cells
Because IGF1R performs critical survival functions in tumor cells, we examined the growth of myeloma cells in the presence of SSTNIGF1R. The growth of human CAG, RPMI-8226, KMS-12, U266, MM.1R and the mouse P3X63Ag8 myeloma cells was significantly reduced by a three-day treatment with 30 μM SSTNIGF1R (Fig. 3A). SSTNIGF1R-T also caused cell loss (Fig. 3A), but was 3–10-fold less effective, correlating with its reduced ability to disrupt IGF1R docking with Sdc1 (cf. Fig. 1). One function of IGF1R in the ternary receptor complex is to activate the αvβ3 or αvβ5 integrin, with which it is partnered (20,21). In the myeloma cells, however, the αvβ5 integrin appears to be inactive, as it fails to engage its matrix ligands fibronectin or vitronectin (Fig. S2A). In addition, αvβ5-blocking antibody is without effect on myeloma cell growth (Fig. S2B), suggesting that growth inhibition observed with SSTNIGF1R is likely to trace to its inhibition of the IGF1R and not the integrin.
FIGURE 3. SSTN inhibits growth of myeloma, carcinoma and activated endothelial cells.

A. Myeloma cell growth in complete culture medium containing 30 μM SSTNIGF1R or SSTNIGF1R-T is quantified over 3 days and graphed as % increase in cell number relative to untreated cells (set to 100%, top panel); B. Endothelial cells positive (HUVEC (Sdc1+), HMEC-1) and negative (HUVEC(Sdc1−)) for Sdc1 expression were grown in the presence of 30 μM SSTNIGF1R or SSTNIGF1R-T for 3 days and graphed as % increase in cell number compared to untreated cells as in (A); C. Non-transformed HaCaT keratinocytes and MCF10A breast epithelial cells, UM-SCC47 and SCC25 head and neck squamous cell carcinoma, and MDA-MB-468, MDA-MB-231, MCF-7 and SKBr3 breast carcinoma cells grown in 3, 10 or 30 μM SSTNIGF1R for 3 days are graphed as % increase in cell number compared to untreated cells as in (A).
Testing whether or not the inhibition of angiogenesis by SSTNIGF1R (20,21,24) may be explained in part by inhibition of cell survival as well, we find that prolonged SSTNIGF1R inhibits the growth of HUVEC and HMEC-1 endothelial cells (Fig. 3B). The growth of a HUVEC cell isolate shown previously to lack Sdc1 and not rely on Sdc1 for IGF1R activation (20) is insensitive to the peptide (Fig. 3B). In contrast to the myeloma cells, however, and in agreement with prior published reports (20), SSTNIGF1R-T is without effect on the endothelial cells (Fig. 3B). This extends as well to a panel of head and neck and breast carcinoma cells (Fig. 3C), in which SSTNIGF1R causes reduced growth. However, non-transformed HaCaT keratinocytes and MCF10A breast epithelial cells are unaffected by the peptide (Fig. 3C), suggesting that carcinoma cells, but not normal epithelial cells, rely on this receptor complex.
SSTN activates JNK-mediated apoptosis
The marked cell loss observed in the presence of SSTNIGF1R is likely due to apoptosis. Indeed, 24 hr treatment of CAG cells with 1–30 μM SSTNIGF1R causes significant loss of viability, mirrored by executioner caspase activation (Fig. 4A) and DNA fragmentation by TUNEL assay (Fig. 4B). Analysis of apoptosis markers in CAG and MM.1R myeloma cells shows increased phosphorylation of p53 (Ser15), activation of JNK and activation of caspase-3 over 9 hr of treatment (Fig. 4C). Total expression of IkBα is decreased, although Ser32/36 phosphorylation targeting it for destruction is not observed, in fact it is reduced, suggesting that its loss traces to reduced expression, consistent with the ability of either p53 or JNK to increase expression of pro-apoptotic and reduce expression of anti-apoptotic genes (32,33). No major changes in Erk1/2, Akt, p38MAPK or Bad phosphorylation is observed in the myeloma cells.
FIGURE 4. SSTN treatment stimulates JNK-mediated cell death.

A. CAG cells grown for 24 hr in complete culture medium containing 1–30 μM SSTNIGF1R are analyzed using ApoTox-GLO to quantify % cell viability or apoptosis compared to untreated cells; B. Percent of apoptotic cells (untreated vs. 30 μM SSTNIGF1R from part (A)) quantitated by TUNEL assay; C. CAG and MM.1R myeloma cells, MDA-MB-231 breast carcinoma and HUVEC vascular endothelial cells were grown for up to 24 hr in the presence or absence of 30 μM SSTNIGF1R. Cell lysates at various time points are assessed using an antibody array to analyze the relative expression and/or phosphorylation levels of stress and apoptotic markers. D. MM.1R and CAG myeloma cells are grown for 9 hr in complete culture medium and treated with vehicle (DMSO) alone or JNK inhibitor (JNK-IN-8) in the presence or absence of 30 μM SSTNIGF1R. Induction of apoptosis is quantified as fold increase in Caspase 3/7 activity (Caspase3/7-GLO) relative to vehicle alone (set to 1.0).
These findings also extend, with some exceptions, to the HUVECs. Activation of the JNK pathway and caspase-3, and a reduction in levels of IkBα occurs in these cells as well, although they take longer (24 hr) to achieve the same signal level (Fig. 4C). In addition, there is loss of Erk1/2 and Akt activation, perhaps indicative of SSTN inhibiting cell-matrix adhesion signals that are not observed in the suspended myeloma cells. The loss of Akt activity is mirrored by a reduction in phosphorylation of Bad on Ser136, a pro-apoptotic Bcl-2 family member that is a known target of Akt. SSTNIGF1R also induces activation of p38MAPK in the endothelial cells. However, unlike in the myeloma cells, there is no apparent change in p53 phosphorylation. An almost identical apoptotic response to SSTNIGF1R is seen in MDA-MB-231 breast carcinoma cells, which overexpress mutant p53 (34) (Fig. 4C). This, together with the fact that the RPMI-8226, KMS-12 and U266 myeloma cells express mutant p53 yet show diminished survival when treated with SSTNIGF1R, suggests that JNK, and not p53, is the primary inducer of apoptosis in response to SSTNIGF1R.
To test whether or not JNK activation is key to the onset of apoptosis, CAG and MM1.R cells were treated with SSTNIGF1R in the presence or absence of the JNK inhibitor, JNK-IN-8. Treatment with 30 μM SSTNIGF1R alone causes a nearly 5-fold increase in caspase 3/7 activity in both myeloma cell lines (Fig. 4D). However, co-treatment with increasing concentrations of JNK-IN-8 reduces this activation, with complete block of caspase activation seen at 1 μM, despite the continued presence of the SSTNIGF1R peptide (Fig. 4D). Near identical results were obtained using another JNK inhibitor, SP600125 (10 μM; data not shown).
Sdc1-coupled IGF1R regulates ASK1 activation
JNK is commonly activated by apoptosis-signal regulatory kinase (ASK1) in tumor cells responding to cellular stress (32). Testing the hypothesis that docking of IGF1R to Sdc1 suppresses ASK1 activation, we find that ASK1 co-immunoprecipitates with Sdc1 from CAG and MM.1R myeloma cells, which is prevented by SSTNIGF1R (Fig. 5B). ASK1 is inactive when in a complex with the Sdc1-coupled ternary receptor complex, as shown by its inhibitory phosphorylation on Ser83 and Ser966 and on tyrosines (Fig. 5A, B). Displacement of IGF1R from Sdc1 by SSTNIGF1R, or its inhibition by tyrphostin AG538, blocks the inhibitory phosphorylation of ASK1 and allows its activation by phosphorylation of Thr838 (Fig. 5A, B). Furthermore, silencing IGF1R expression (Fig. 5C) leads to JNK phosphorylation (Fig. 5D) and apoptosis (Fig. 5E) – directly mirroring the effects of SSTNIGF1R treatment (Fig. 4, 5D, 5E) and confirming that IGF1R is critical to Sdc1-coupled survival signaling. Silencing ASK1 expression prevents JNK phosphorylation (Fig. 5D) and induction of apoptosis in the presence of SSTNIGF1R (Fig. 5E) suggesting that the Sdc1-coupled IGF1R complex suppresses JNK-mediated apoptosis by inhibiting ASK1.
FIGURE 5. Sdc1-coupled IGF1R sequesters and inactivates ASK1.

A. ASK1 immunoprecipitates from CAG or MM.1R cells treated for 6 hr with 10 μM SSTNIGF1R or 10 μM IGF1R inhibitor AG538 are probed for inactive (pY, pSer83 and pSer966), active (pThr838) or total ASK1; B. Sdc1 immunoprecipitates from CAG or MM.1R cells treated with 10 μM SSTNIGF1R or 10 μM AG538 are probed for the presence of total ASK1, inactive (pY) or active (pThr838) ASK1, active IGF1R (pY1131) or total IGF1R, and total Sdc1 (mAb B-A38); C. RNAi knock-down of ASK1 and IGF1R protein levels in CAG and MM.1R cells 48 hr after control (Ctrl) or target-specific siRNA transfection; D. Equal cell equivalents from control, ASK1 or IGF1R siRNA-transfected CAG and MM.1R cells (48 hr post-transfection) treated with or without 30 μM SSTNIGF1R for 8 hr are analyzed for levels of active JNK (pThr183/pTyr185) and total JNK; E. Active Caspase 3/7 is quantified in control, IGF1R or ASK1 siRNA-transfected CAG and MM.1R cells treated as in (D).
SSTNIGF1R is highly stable in vitro and in vivo
Unlike many peptides that are rapidly destroyed and/or cleared in vivo, SSTNIGF1R is relatively stable, supporting its potential as a therapeutic for myeloma and other cancers. It retains full activity even after incubation in human plasma for 72 hr at 37°C (Fig. 6A), as well as in mouse or bovine serum (data not shown). Note that any degradation would be observable as the peptide is already the minimal length necessary to retain full activity (20). In contrast, a GRGDSP integrin inhibitory peptide used for comparison is almost completely destroyed within 8 hr (35) (Fig. 6A). The peptide is also very stable and displays excellent pharmacokinetics and pharmacodynamics in vivo. The half-life of a single intravenous injection (0.365 mg/kg) of biotinylated SSTNIGF1R into recipient mice is 27 hr (Fig. 6B), with a calculated Cmax of 0.016 μg/ml, AUC of 0.467 μg/ml*hr, Tmax of 0.08 hr, clearance of 39 ml/hr, and Vd of 1442 ml. Daily subcutaneous injection of 0.365 mg/kg/day reaches a steady-state plateau of 3.5 μM in the plasma (Fig. 6C).
FIGURE 6. Stability of SSTNIGF1R in vitro and in vivo.
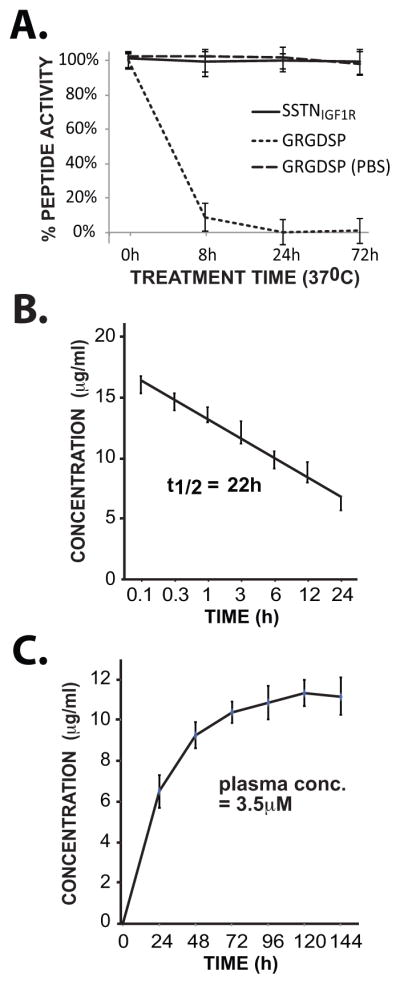
A. Activity of SSTNIGF1R in an endothelial cell adhesion assay following incubation in human plasma at 37°C for up to 72 hr. Activity is compared to an integrin-inhibitory GRGDSP peptide incubated in either human plasma or PBS. B. Stability of biotinylated SSTNIGF1R in the serum of mice following intravenous injection of a single bolus of peptide (0.365 mg/kg); C. Accumulation of biotinylated SSTNIGF1R in the serum of mice injected every 24 hr with 0.365 mg/kg of peptide. Serum samples are collected every 24 hr prior to injection of a new bolus.
SSTN blocks the growth of CAG myeloma xenografts
SSTNIGF1R has shown efficacy as an inhibitor of angiogenesis and breast carcinoma growth in vivo (20). But whether or not it acts on tumor growth by targeting the tumor cell as well as angiogenesis is not clear. Our finding that SSTNIGF1R-T acts on the myeloma cell and not endothelial cells provides the opportunity to make this determination, as well as to test the efficacy of SSTNIGF1R on myeloma growth in vivo. CAG myeloma xenografts were treated using Alzet pumps to deliver 3.65 mg/kg/day of SSTNIGF1R or SSTNIGF1R-T, a concentration 10-fold higher than used for the PK/PD study (cf. Fig. 6) and therefore likely to approach 30 μM in the blood. During a 4-week treatment of established tumors, SSTNIGF1R caused over 90% reduction in tumor growth, eradicated 3 of the 8 tumors, and reduced blood vessel density in the residual tumors by over 90% (Fig. 7). SSTNIGF1R-T was also highly effective, causing nearly 60% loss in tumor growth (Fig. 7A, B), noting that it is 3–10-fold less effective at displacing IGF1R from Sdc1 in myeloma cells (cf. Fig. 1, 3). In contrast, as expected due to its lack of effect on endothelial cells, SSTNIGF1R-T had no significant effect on tumor angiogenesis (Fig. 7C, D).
FIGURE 7. SSTN blocks the growth of myeloma xenografts by targeting the tumor cell and angiogenesis.

CAG myeloma cells implanted subcutaneously in immunodeficient nude (nu/nu) mice and are allowed to form palpable tumors for 7–10 days. Mice are then implanted with Alzet pumps delivering 0.25 μL/hr of 4 mM SSTNIGF1R, SSTNIGF1R-T or saline alone for an additional 4 weeks. A. Tumors harvested at the end of the 4-week treatment period (8 per cohort shown; ‘X’ denotes no tumor found). B. Quantification of tumor volume. Treated and untreated cohorts were compared using unpaired one-tail t-test. P-values of less than 0.05 were considered significant (**). Data represent mean plus or minus SEM. C. Representative tumors sectioned and stained with MECA-32 antibody specific for the pan-endothelial cell marker PV1/PLVAP. D. Quantification of overall MECA-32 staining intensity. E. Model depicts capture of inactive αvβ5 integrin and IGF1R via a docking site in the extracellular domain of Sdc1 (21,23), which causes autophosphorylation of IGF1R and binding of inactive ASK1 (with inactivating phosphorylation on Ser83, Ser966 (S) and tyrosines (Y)). Displacement of the integrin and IGF1R from Sdc1 inactivates IGF1R, allow ASK1 activation via autophosphorylation of Thr838 (T), and downstream activation of JNK via MKK4 or MKK7, leading to caspase activation and apoptosis.
DISCUSSION
Myeloma is characterized by high expression of Sdc1 (CD138) and many studies have linked this receptor to poor prognosis in this disease (5). Our current findings suggest that a major role of the syndecan is suppression of apoptosis via its capture and activation of IGF1R, which inhibits ASK1 upstream of JNK (see model in Fig. 7E). JNK is a pro-apoptotic member of the MAPK superfamily, consisting of JNK1 and JNK2, which are ubiquitously expressed, and JNK3, which is expressed in neuronal and cardiac tissues (32,36,37). It is activated by phosphorylation on Thr183 and Tyr185 by the dual specificity kinases JNKK1/MKK4/SEK and JNKK2/MKK7, which in turn are activated by upstream MAPKKKs, including ASK1 (also known as M3K5) in response to stress inducers such as lipopolysaccharide, ultraviolet radiation, endoplasmic reticulum and oxidative stress, DNA damage and disrupted calcium homeostasis, leading to programmed cell death (32,37). JNK exerts its pro-apoptotic function through its phosphorylation of transcription factors, including c-Jun, that upregulate the expression of pro-apoptotic genes or downregulate pro-survival genes, as well as by directly phosphorylating Bcl-2 and its family members Bcl-xL, BIM and BAD in the intrinsic apoptosis pathway (32).
Our findings suggest that the Sdc1-coupled IGF1R complex suppresses JNK-mediated apoptosis by inhibiting the stress-sensing ASK1 (16,38,39). ASK1 is a homo-oligomeric serine/threonine protein kinase that is activated by auto-transphosphorylation on Thr838 (Thr845 in mouse) (40). Other cytosolic proteins, such as thioredoxin, glutaredoxin and 14-3-3 protein, prevent activation by binding the enzyme, and serine-threonine kinases, including Raf-1, Akt/protein kinase B, 3-phosphoinositide-dependent protein kinase-1 (PDK1) and IκB kinase, inactivate it by phosphorylating Ser83 and Ser966 (14–17,40). This inhibitory phosphorylation is reversed in response to stress, allowing auto-activation facilitated by apoptosis-promoting tumor necrosis factor receptor-associated factor-2 (TRAF2) and TRAF6 (16,40). This oligomeric complex of proteins, or “signalosome” is localized to key receptors that signal through this stress-sensing mechanism to either induce or suppress apoptosis, including the TNFR (41), the unfolded protein ER sensor IRE1p (42), and, as we show here, the pro-survival growth factor receptor IGF1R (19) when coupled to Sdc1.
Prior work has shown that ASK1 is associated with IGF1R and that IGF1R inhibits the enzyme by phosphorylation of as-yet-unidentified tyrosines within its N-terminal inhibitory domain, causing suppression of TNFα-mediated JNK activation and preventing apoptosis caused by depriving the cells of IGF1 (19). The association of ASK1 with IGF1R appears to be constitutive, as it is not abolished by inactivating mutation of the IGF1R kinase or IGF1 withdrawal (19). Our findings extend the ways in which ASK1 is regulated, as we find that the IGF1R activated by its docking to Sdc1 causes the inhibitory tyrosine phosphorylation of ASK1, likely via IGF1R kinase directly, as well as the inhibitory phosphorylation of Ser83 and Ser966. This is reversed either by chemical inhibition of the IGF1R kinase, or by inhibition of the kinase due to its displacement from Sdc1 by SSTNIGF1R, allowing ASK1 auto-transphosphorylation on Thr838. The serine-threonine kinases that carry out the inhibitory phosphorylation in myeloma remain unidentified. Although Akt and its activating kinase PDK1 are known to be activated downstream of IGF1R and phosphorylate Ser83 and Ser966 in ASK1, respectively (15,43), we find no change in Akt activity levels in myeloma cells treated with SSTNIGF1R. Akt-induced phosphorylation could play a role in the HMEC-1 vascular endothelial cells or MDA-MB-231 breast carcinoma cells, however, in which the induction of JNK activity and apoptosis is matched by a reduction in Akt activation, potentially tracing to the Sdc1-coupled IGF1R/integrin complex acting as a cell adhesion signaling complex in these cells (20,21).
The Sdc1-coupled ternary receptor complex suppresses apoptosis in activated endothelial cells and carcinoma cells, as well as myeloma, whereas normal epithelial cells do not rely on this mechanism. This is likely due to differences in expression of the αvβ3 or αvβ5 integrin necessary for Sdc1-mediated capture of IGF1R (21). The αvβ3 integrin in particular is often upregulated in response to stress, especially in carcinoma or in endothelial cells undergoing angiogenesis (44), whereas epithelial cells that are not under stress express IGF1R and Sdc1, but this integrin. Although it has no apparent adhesion role in the myeloma cells, expression of the αvβ5 integrin in myeloma cells fulfills a similar role, namely, acting as an obligate partner for successful IGF1R capture.
Inhibition of ASK1 is known to play a critical role in the longevity of plasma cells and their malignant myeloma counterparts, tracing to ER stress arising from their expression of copious amounts of immunoglobulin. ASK1 expression is negatively regulated by the transcription factor Blimp-1 in these cells, thereby governing the lifetime of short-lived and long-lived plasma cells responding to immune stimulus, as well as survival of myeloma cells (10). Interestingly, Blimp-1 is a positive regulator of Sdc1 expression (45), which becomes highly expressed in plasma and myeloma cells, potentially, as we demonstrate here, as the organizer of a signaling complex that suppresses ASK1 activity and the onset of JNK-induced apoptosis (summarized in model, Fig. 7E). The ability of SSTNIGF1R to target this major pro-survival mechanism in both tumor cells and activated endothelial cells, without affecting the survival of non-transformed cells, makes it a promising cancer therapeutic, especially due to its remarkable stability in vivo.
Although recent therapeutic advances have prolonged MM patient survival, the disease is ultimately incurable due to resistance that invariably arises. Therefore, there is an urgent need for novel therapies to treat this disease. IGF1R protects tumor cells from diverse stress-inducing stimuli including cytotoxic cytokines, hypoxia-ischemia, oxidative stress and DNA damage (46). IGF1R expression in a number of tumor types correlates with poor prognosis, in part because of its ability to cross-talk with other receptor tyrosine kinases (RTK) and mammalian target of rapamycin (mTOR) rendering tumors refractory to RTK-targeted therapies and other anti-neoplastic drugs. However, downregulation or functional inactivation of IGF1R sensitizes tumor cells to apoptosis (47). As such, apart from its potential as a single-agent therapeutic to target IGF1R survival signals (in both tumor cells and tumor-associated angiogenic endothelial cells), SSTNIGF1R may also be used clinically as an adjunct or adjuvant to increase the efficacy of other apoptosis-inducing therapies, especially in tumors prone to relapse due to innate or acquired resistance to conventional clinical therapies currently in use.
Intriguingly, IGF1R’s strong cytoprotective effects may trace to the anti-apoptotic effects of heat shock proteins (HSPs), in particular HSP70 and HSP90 (48). Upregulated expression of HSP90 has been observed in MM cells interacting with bone marrow stromal cells (BMSC) leading to enhanced cell proliferation and resistance suggesting that HSP90 mediates the protective effects of BMSC (49). Further, both HSPs are associated with the UPR response, are known to interact with and inactivate ASK1 and JNK (43,50) and HSP70/90 expression is decreased after inhibition or knock-down of either IGF1R or Sdc1 (51–53). HSP90 is known to associate with mitochondria in tumor cells, but not with mitochondria in normal cells (54). Indeed, decreased IGF1R signaling and HSP70/90 expression is associated with mitochondrial dysfunction (48,53), leading to an increase in free radical production, oxidative damage and apoptosis through activation of the mitochondrial cell death machinery (i.e., Apaf-1/cytochrome c and Caspase9/3 activation) – the primary means of ASK1-induced cell death. As such, SSTNIGF1R’s disruption of Sdc1-coupled IGF1R survival signals may decrease expression of HSP70/90 and/or relieve HSP70/90-dependent inactivation of ASK1 to induce apoptosis in tumor cells while sparing normal cells.
Another possible consequence of SSTNIGF1R’s disruption of Sdc1-coupled IGF1R survival signals may be inhibition of mTOR signaling. Rapamycin-induced apoptosis has been shown to inhibit protein phosphatase-5 (PP5; negatively regulates ASK1 by dephosphorylating Thr838) and activation of ASK1 leading to JNK activation (55,56). Rescue from rapamycin-induced apoptosis was found to be independent of AKT and Ras/ERKs, but was dependent on PI3K. More recently, IGF1R has been shown to activate PKC isoforms downstream of PI3K leading to phosphorylation of the pro-apoptotic protein BAD at Ser112 and subsequent sequestration and inactivation of BAD (and likely ASK1) by protein 14-3-3 (57,58). Investigation into whether SSTNIGF1R treatment induces apoptosis by relieving HSP70/90 or PI3K/mTOR/PKC-dependent inactivation of ASK1 will be future areas of focus.
Supplementary Material
Acknowledgments
This work was supported by funds from the National Institutes of Health to A.C.R. (R01-CA139872, R01-CA118839, R01-CA163662) and to R.D.S. (CA135075 and CA138340). We thank the University of Wisconsin Carbone Cancer Center’s Trillium Fund for support, and the use of its shared services, supported by NIH/NCI P30 CA014520.
Footnotes
The authors disclose no potential conflicts of interest.
Authorship Contributions: D.M.B. participated in concept design, generation of data and writing the manuscript. O.J. and Y.Y. performed experiments and provided critique of the manuscript, R.D. S. participated in concept design and critique of the manuscript, and A.C.R. oversaw concept design and participated in writing the manuscript.
Conflict of Interest: The authors declare no competing financial interest.
References
- 1.Bergsagel D. The incidence and epidemiology of plasma cell neoplasms. Stem Cells. 1995;13(Suppl 2):1–9. [PubMed] [Google Scholar]
- 2.Laubach JP, Richardson PG, Anderson KC. The evolution and impact of therapy in multiple myeloma. Med Oncol. 2010;27(Suppl 1):S1–6. doi: 10.1007/s12032-010-9442-2. [DOI] [PubMed] [Google Scholar]
- 3.Khotskaya YB, Dai Y, Ritchie JP, MacLeod V, Yang Y, Zinn K, et al. Syndecan-1 is required for robust growth, vascularization, and metastasis of myeloma tumors in vivo. J Biol Chem. 2009;284:26085–26095. doi: 10.1074/jbc.M109.018473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Connell FP, Pinkus JL, Pinkus GS. CD138 (syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am J Clin Pathol. 2004;121:254–263. doi: 10.1309/617D-WB5G-NFWX-HW4L. [DOI] [PubMed] [Google Scholar]
- 5.Sanderson RD, Yang Y. Syndecan-1: a dynamic regulator of the myeloma microenvironment. Clin Exp Metastasis. 2008;25:149–159. doi: 10.1007/s10585-007-9125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Yaccoby S, Liu W, Langford JK, Pumphrey CY, Theus A, et al. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood. 2002;100:610–617. doi: 10.1182/blood.v100.2.610. [DOI] [PubMed] [Google Scholar]
- 7.Jung O, Trapp-Stamborski V, Purushothaman A, Jin H, Wang H, Sanderson RD, et al. Heparanase-induced shedding of syndecan-1 promotes VEGFR2 activation and an invasive phenotype in multiple myeloma. Oncogenesis. 2016 doi: 10.1038/oncsis.2016.5. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, MacLeod V, Dai Y, Khotskaya-Sample Y, Shriver Z, Venkataraman G, et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood. 2007;110:2041–2048. doi: 10.1182/blood-2007-04-082495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu YH, Yang CY, Chien WL, Lin KI, Lai MZ. Removal of Syndecan-1 Promotes TRAIL-Induced Apoptosis in Myeloma Cells. J Immunol. 2012;188:2914–2921. doi: 10.4049/jimmunol.1102065. [DOI] [PubMed] [Google Scholar]
- 10.Lin FR, Huang SY, Hung KH, Su ST, Chung CH, Matsuzawa A, et al. ASK1 promotes apoptosis of normal and malignant plasma cells. Blood. 2012;120:1039–1047. doi: 10.1182/blood-2011-12-399808. [DOI] [PubMed] [Google Scholar]
- 11.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes & development. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Sanderson RD, Lalor P, Bernfield M. B lymphocytes express and lose syndecan at specific stages of differentiation. Cell regulation. 1989;1:27–35. doi: 10.1091/mbc.1.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puckett MC, Goldman EH, Cockrell LM, Huang B, Kasinski AL, Du Y, et al. Integration of apoptosis signal-regulating kinase 1-mediated stress signaling with the Akt/protein kinase B-IkappaB kinase cascade. Molecular and cellular biology. 2013;33:2252–2259. doi: 10.1128/MCB.00047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seong HA, Jung H, Ichijo H, Ha H. Reciprocal negative regulation of PDK1 and ASK1 signaling by direct interaction and phosphorylation. J Biol Chem. 2010;285:2397–2414. doi: 10.1074/jbc.M109.064295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayakawa T, Matsuzawa A, Noguchi T, Takeda K, Ichijo H. The ASK1-MAP kinase pathways in immune and stress responses. Microbes and infection / Institut Pasteur. 2006;8:1098–1107. doi: 10.1016/j.micinf.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, et al. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. The EMBO journal. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galvan V, Logvinova A, Sperandio S, Ichijo H, Bredesen DE. Type 1 insulin-like growth factor receptor (IGF-IR) signaling inhibits apoptosis signal-regulating kinase 1 (ASK1) J Biol Chem. 2003;278:13325–13332. doi: 10.1074/jbc.M211398200. [DOI] [PubMed] [Google Scholar]
- 20.Beauvais DM, Ell BJ, McWhorter AR, Rapraeger AC. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med. 2009;206:691–705. doi: 10.1084/jem.20081278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beauvais DM, Rapraeger AC. Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. J Cell Sci. 2010;123:3796–3807. doi: 10.1242/jcs.067645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McQuade KJ, Beauvais DM, Burbach BJ, Rapraeger AC. Syndecan-1 regulates alphavbeta5 integrin activity in B82L fibroblasts. J Cell Sci. 2006;119:2445–2456. doi: 10.1242/jcs.02970. [DOI] [PubMed] [Google Scholar]
- 23.Rapraeger AC. Synstatin: a selective inhibitor of the syndecan-1-coupled IGF1R-alphavbeta3 integrin complex in tumorigenesis and angiogenesis. The FEBS journal. 2013;280:2207–2215. doi: 10.1111/febs.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rapraeger AC, Ell BJ, Roy M, Li X, Morrison OR, Thomas GM, et al. Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between alphaVbeta3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. The FEBS journal. 2013;280:2194–2206. doi: 10.1111/febs.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bataille R, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P. CD221 (IGF-1R) is aberrantly expressed in multiple myeloma, in relation to disease severity. Haematologica. 2005;90:706–707. [PubMed] [Google Scholar]
- 26.Chng WJ, Gualberto A, Fonseca R. IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia. 2006;20:174–176. doi: 10.1038/sj.leu.2403997. [DOI] [PubMed] [Google Scholar]
- 27.Georgii-Hemming P, Wiklund HJ, Ljunggren O, Nilsson K. Insulin-like growth factor I is a growth and survival factor in human multiple myeloma cell lines. Blood. 1996;88:2250–2258. [PubMed] [Google Scholar]
- 28.Yatohgo T, Izumi M, Kashiwagi H, Hayashi M. Novel purification of vitronectin from human plasma by heparin affinity chromatography. Cell Struct Funct. 1988;13:281–292. doi: 10.1247/csf.13.281. [DOI] [PubMed] [Google Scholar]
- 29.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, et al. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Jin H, Rapraeger AC. Syndecan-1 and Syndecan-4 Capture Epidermal Growth Factor Receptor Family Members and the alpha3beta1 Integrin Via Binding Sites in Their Ectodomains: novel synstatins prevent kinase capture and inhibit alpha6beta4-integrin-dependent epithelial cell motility. J Biol Chem. 2015;290:26103–26113. doi: 10.1074/jbc.M115.679084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 32.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 34.Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocrine-related cancer. 2006;13:293–325. doi: 10.1677/erc.1.01172. [DOI] [PubMed] [Google Scholar]
- 35.Aguzzi MS, D’Arcangelo D, Giampietri C, Capogrossi MC, Facchiano A. RAM, an RGDS analog, exerts potent anti-melanoma effects in vitro and in vivo. PloS one. 2011;6:e25352. doi: 10.1371/journal.pone.0025352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Lin A. Role of JNK activation in apoptosis: a double-edged sword. Cell research. 2005;15:36–42. doi: 10.1038/sj.cr.7290262. [DOI] [PubMed] [Google Scholar]
- 38.Fujino G, Noguchi T, Matsuzawa A, Yamauchi S, Saitoh M, Takeda K, et al. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Molecular and cellular biology. 2007;27:8152–8163. doi: 10.1128/MCB.00227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. Journal of cellular physiology. 2002;191:95–104. doi: 10.1002/jcp.10080. [DOI] [PubMed] [Google Scholar]
- 40.Hattori K, Naguro I, Runchel C, Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell communication and signaling : CCS. 2009;7:9. doi: 10.1186/1478-811X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoeflich KP, Yeh WC, Yao Z, Mak TW, Woodgett JR. Mediation of TNF receptor-associated factor effector functions by apoptosis signal-regulating kinase-1 (ASK1) Oncogene. 1999;18:5814–5820. doi: 10.1038/sj.onc.1202975. [DOI] [PubMed] [Google Scholar]
- 42.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 43.Zhang R, Luo D, Miao R, Bai L, Ge Q, Sessa WC, et al. Hsp90-Akt phosphorylates ASK1 and inhibits ASK1-mediated apoptosis. Oncogene. 2005;24:3954–3963. doi: 10.1038/sj.onc.1208548. [DOI] [PubMed] [Google Scholar]
- 44.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature reviews Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner CA, Jr, Mack DH, Davis MM. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell. 1994;77:297–306. doi: 10.1016/0092-8674(94)90321-2. [DOI] [PubMed] [Google Scholar]
- 46.Kurmasheva RT, Houghton PJ. IGF-I mediated survival pathways in normal and malignant cells. Biochimica et biophysica acta. 2006;1766:1–22. doi: 10.1016/j.bbcan.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 47.Tao Y, Pinzi V, Bourhis J, Deutsch E. Mechanisms of disease: signaling of the insulin-like growth factor 1 receptor pathway--therapeutic perspectives in cancer. Nat Clin Pract Oncol. 2007;4:591–602. doi: 10.1038/ncponc0934. [DOI] [PubMed] [Google Scholar]
- 48.Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. 2008;12:743–761. doi: 10.1111/j.1582-4934.2008.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Kung AL, Davies FE, et al. Antimyeloma activity of heat shock protein-90 inhibition. Blood. 2006;107:1092–1100. doi: 10.1182/blood-2005-03-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hwang JR, Zhang C, Patterson C. C-terminus of heat shock protein 70-interacting protein facilitates degradation of apoptosis signal-regulating kinase 1 and inhibits apoptosis signal-regulating kinase 1-dependent apoptosis. Cell Stress Chaperones. 2005;10:147–156. doi: 10.1379/CSC-90R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boeddeker SJ, Baston-Buest DM, Altergot-Ahmad O, Kruessel JS, Hess AP. Syndecan-1 knockdown in endometrial epithelial cells alters their apoptotic protein profile and enhances the inducibility of apoptosis. Mol Hum Reprod. 2014;20:567–578. doi: 10.1093/molehr/gau009. [DOI] [PubMed] [Google Scholar]
- 52.Hao CN, Geng YJ, Li F, Yang T, Su DF, Duan JL, et al. Insulin-like growth factor-1 receptor activation prevents hydrogen peroxide-induced oxidative stress, mitochondrial dysfunction and apoptosis. Apoptosis. 2011;16:1118–1127. doi: 10.1007/s10495-011-0634-9. [DOI] [PubMed] [Google Scholar]
- 53.Sadaba MC, Martin-Estal I, Puche JE, Castilla-Cortazar I. Insulin-like growth factor 1 (IGF-1) therapy: Mitochondrial dysfunction and diseases. Biochimica et biophysica acta. 2016;1862:1267–1278. doi: 10.1016/j.bbadis.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 54.Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 55.Hosoi H, Dilling MB, Shikata T, Liu LN, Shu L, Ashmun RA, et al. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999;59:886–894. [PubMed] [Google Scholar]
- 56.Huang S, Shu L, Easton J, Harwood FC, Germain GS, Ichijo H, et al. Inhibition of mammalian target of rapamycin activates apoptosis signal-regulating kinase 1 signaling by suppressing protein phosphatase 5 activity. J Biol Chem. 2004;279:36490–36496. doi: 10.1074/jbc.M401208200. [DOI] [PubMed] [Google Scholar]
- 57.Thimmaiah KN, Easton JB, Houghton PJ. Protection from rapamycin-induced apoptosis by insulin-like growth factor-I is partially dependent on protein kinase C signaling. Cancer Res. 2010;70:2000–2009. doi: 10.1158/0008-5472.CAN-09-3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masters SC, Subramanian RR, Truong A, Yang H, Fujii K, Zhang H, et al. Survival-promoting functions of 14-3-3 proteins. Biochem Soc Trans. 2002;30:360–365. doi: 10.1042/bst0300360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.