

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) identifies a specific lung disorder characterized by chronic, progressive fibrosing interstitial pneumonia of unknown etiology, which lacks effective treatment. According to the current pathogenic perspective, the aberrant proliferative events in IPF resemble those occurring during malignant transformation.
Main body
Receptor tyrosine kinases (RTK) are known to be key players in cancer onset and progression. It has been demonstrated that RTK expression is sometimes also altered and even druggable in IPF. One example of an RTK—the MET proto-oncogene—is a key regulator of invasive growth. This physiological genetic program supports embryonic development and post-natal organ regeneration, as well as cooperating in the evolution of cancer metastasis when aberrantly activated. Growing evidence sustains that MET activation may collaborate in maintaining tissue plasticity and the regenerative potential that characterizes IPF.
Conclusion
The present work aims to elucidate—by applying the logic of simplicity—the bio-molecular mechanisms involved in MET activation in IPF. This clarification is crucial to accurately design MET blockade strategies within a fully personalized approach to IPF.
Keywords: Cancer, Idiopathic pulmonary fibrosis, Invasive growth, Epithelial-to-mesenchymal transition, Precision medicine
Background
Idiopathic pulmonary fibrosis (IPF) is characterized by progressive scarring of the lungs ultimately leading to severe respiratory failure and death [1]. The median survival of patients is only 3 years following diagnosis [2], similar or worse than that of several oncologic diseases. Despite the fact that recently significant progress has been made in identifying of the bio-molecular mechanisms related to the development of IPF, a better understanding of disease pathogenesis is needed to identify more effective therapies and to improve patients’ outcome. According to the current pathogenic perspective, the aberrant proliferative events in IPF resemble that occuring during malignant transformation. Growing evidence supports the cancer-like molecular nature of IPF and this intriguing hypothesis is now also being exploited for therapeutic purposes [3]. The discovery of pathogenic links between the two diseases may have practical consequences in encouraging the use of cancer drugs for treating IPF. Receptor tyrosine kinases (RTK) are known to be key players in cancer onset and progression; it has also been demonstrated that the expression of some RTK family members is also altered and even druggable in IPF [4, 5]. The multi-kinase inhibitor—nintedanib—was initially developed for cancer, and has now been approved for the treatment of IPF thanks to the observation that targeted receptors are also abnormally activated in IPF [6]. The MET proto-oncogene is a RTK that is a key regulator of invasive growth [7], which is the biological program that orchestrates dynamic changes in tissues leading to cell proliferation, survival and migration across the extracellular matrix (ECM) and which can be inappropriately overexpressed in cancer spreading and metastatization. On the other hand, the MET-induced invasive growth is now emerging as potential target in IPF, although some issues require better understanding and clarification. Thus, this review aims to analyze the multiple facets of MET activation in cancer and IPF, under a context-specific perspective to the fibrotic disease.
The empirical evidence: MET structure and signaling
The MET proto-oncogene is a key regulator of the genetic program known as invasive growth. The MET gene, located on chromosome 7q31, encodes for the TK receptor for ‘Scatter Factor’ or Hepatocyte Growth Factor (HGF), which detects adverse micro-environmental conditions and drives cell invasion and metastasis through the transcriptional activation of the invasive growth signature, a genetic program also defined as the epithelial-mesenchymal transition (EMT) [8]. The latter includes cell–cell dissociation and scattering, migration, cellular proliferation, resistance to anoikis and angiogenesis. MET is now a prominent target in cancer therapy, with several compounds in active clinical development (Table 1). Different strategies have been pursued to inhibit MET, each focusing on one of the sequential steps that regulate MET activation. Scatter factors (HGF and Macrophage-stimulating Protein-MSP) belong to the plasminogen family of proteins, which is defined by the presence of at least one characteristic domain known as the kringle domain (an 80 amino-acid double-looped structure formed by three internal disulphide bridges); a serine-protease domain and an activation segment that is located between the kringle and the protease domains. HGF and MSP are unique scatter factors because they lack proteolytic activity; they are secreted as single-chain biologically inert glycoprotein precursors and are converted into their bioactive form in the extracellular environment by specific proteases, which break the bond between two positively charged aminoacids (Arg494–Val495). The mature factors are heterodimers consisting of an α-chain and a β-chain held together by a disulphide bond. The MET receptor for HGF and the RON receptor for MSP are single-pass, disulphide-linked α/β heterodimers that are formed by proteolytic processing of a common precursor in the post-Golgi compartment. In both receptors (which share 63 % overall homology), the α-chains are completely extracellular, whereas the β-chains are transmembrane subunits that contain tyrosine kinase activity. The extracellular region of these receptors displays structural analogies with the extracellular domains of semaphorins (a large family of secreted and membrane-bound proteins) and their receptors plexins. The extracellular region contains the sema domain: a conserved sequence of about 500 amino acids comprising an eight-cysteine peptide module that is conventionally termed MRS (MET-related sequence), together with three glycine-proline rich (G-P) repeats. The intracellular domains include tyrosine kinase catalytic sites that are flanked by distinctive juxtamembrane and carboxy-terminal sequences. Phosphorylation of MET on residues Tyr 1234 and Tyr 1235 within the catalytic sites results in positive modulation of enzyme activity, whereas phosphorylation of a serine residue in the juxtamembrane domain downregulates the kinase. After activation, MET elicits intramolecular phosphorylation of the other two critical tyrosine residues at the carboxy-terminal domains (Tyr 1349 and Tyr 1356), at the C-terminal of the α-chain: these two sites, together with the surrounding aminoacids, constitute the so-called “multifunctional docking site”, a motif which, when activated after phosphorylation, induces a series of biological processes that ultimately lead to invasive growth. The specificity of this unique response is determined by qualitative activation of specific pathways that are responsible for the oncogenic and migratory effects of MET [for a full review see 9–11], (Fig. 1). Moreover activation of the MET receptor is known to promote a cancer-associated thrombo-hemorrhagic syndrome that is mediated by transcriptional up-regulation of the pro-coagulation factors plasminogen activator inhibitor type-1 and cyclooxygenase-2 [12]. In human tumors, MET activation can be induced through different mechanisms, namely: (i) MET over-expression, related to: MET gene amplification; enhanced MET transcription; induction by other oncogenes such as RAS, RET; hypoxia-activated transcription; (ii) structural alteration, such as: point mutations which cause increased kinase activity; oncogene rearrangement (such as chromosomal translocation responsible for the Trp-MET fusion protein); abnormal post-translational processing resulting in a constitutively active molecule exposed on the cell membrane; impaired down-regulation generally due to mutations that prevent binding of the Cbl ubiquitin ligase which is responsible for MET ubiquitination and endocytosis thus leading to increased receptor expression at cell surface and enhanced signal transduction. In addition, naturally truncated and active MET receptors have been detected in malignant human musculoskeletal tumors; (iii) HGF-independent autocrine-paracrine activation. In these contexts, paracrine activation—typical of physiological conditions—can become pathological in the presence of abnormal HGF production by mesenchymal cells. Autocrine activation occurs when tumor cells aberrantly express both HGF and its receptor, as observed in rhabdomyosarcomas, gliomas, carcinomas of thyroid, breast and lung cancers, (iv) HGF-independent mechanisms. Moreover MET phosphorylation can also occur through transactivation by other membrane receptors, including adhesive receptors such as CD44, integrins, signal transducing receptors (RON, EGFR family members, FAS, plexin B9) [for a detailed review see 11, 13]. In addition, it has been demonstrated that MET activation can be mediated by an interaction between MET and microbes, including H. Pylori, associated with gastro-esophageal reflux disease which, in turn, is known to be implicated in the development of IPF, giving rise to recurrent lung insult [14, 15]. Notably, MET is expressed in stem and committed progenitor cells and the MET-driven invasive growth is usurped by cancer stem cells (CSC) [7]. As for normal tissues, tumors are structured according to a hierarchy, which includes two main components; thus tumors are composed of tumor-initiating cells (TICs), known as CSCs, which are the small fraction of cells within a tumor mass featuring self-renewal potential, capability of continuous proliferation and the ability to initiate tumor formation when transplanted. TICs also sustain tumor regeneration, growth and dissemination and can be considered the key target of cancer inhibition. Conversely, the vast majority of the tumor is constituted of cells with limited proliferative properties, which tend to aberrantly differentiate and ultimately die [16, 17]. Rapid progress has been made regarding CSC expression of metabolic regulation markers, growth factors, and transcription factors [18, 19]. A greater knowledge of the biological mechanisms responsible for maintaining the stem phenotype is required to understand more fully how the stem compartment sustains tumor persistence, and leads to recurrence after tumor dormancy and failed therapies. In this way, the clinical management and therapeutic options for cancer can be improved. The MET oncogene is crucial to sustain CSC and self-maintenance of tumors. A number of studies have documented the involvement of MET in TIC plasticity [20–23] in several cancer types and in inducing CSC chemo- and radio-resistance [24–26]. In conclusion the MET-driven invasive growth is necessary for efficient cancer spreading as well as stemness properties. Due to its overlapping biological functions MET activation influences IPF development and progression. We and others have already reported that both myofibroblasts and epithelial cells of fibroblast foci in IPF harbor MET in its activated form [27, 28]. Although the anti-fibrotic effect of the MET-ligand HGF, is well known [11, 29], deregulation of the MET signaling cascade is clearly implicated in the development of IPF but its exact role remains to be clarified.
Table 1.
Details on anti-MET agents already available in the clinical scenario for anticancer therapy
| Drug | Target | Cancer type | References |
|---|---|---|---|
| Anti-MET monoclonal antibodies | |||
| SAIT301 | MET | Advanced MET positive solid tumors | [30]a |
| ARGX-111 | MET | MET protein overexpressing advanced cancer | [31]a |
| MetMab (ornatuzumab) | MET | Advanced or metastatic solid tumors | [32–37]a |
| JNJ-61186372 | MET-EGFR (bispecific ab) | Advanced NSCLCs | [38–40]a |
| ABT-700 | MET | Advanced solid tumors | |
| MET tyrosine kinase inhibitors | |||
| PF-02341066 (crizotinib) | MEK, ALK, ROS 1 (triple inhibitor) | Advanced NSCLCs, gastric cancers, metastatic urothelial cancers,anaplastic large cell lymphoma, colorectal cancers, advanced relapsed/refractory solid tumors, primary CNS tumors | [41–43]a |
| XL-184 (cabozantinib) | MET, VEGFR2 (dual inhibitor) | NSCLCs with brain metastasis, advanced cholangiocarcinoma, metastatic triple negative breast cancers, colorectal cancers, metastatic Merkel cell carcinoma, recurrent endometrial cancers, breast cancers with brain metastasis, metastatic renal cell carcinoma | [44–54]a |
| AZD6094 (Volitinib) | MET | Gastric adenocarcinoma, papillary renal cell carcinoma | [55–58]a |
| GSK1363089 (Foretinib) | MET, VEGFR2 (dual inhibitor) | Papillary renal cell carcinoma, medulloblastoma, metastatic gastric cancers, hepatocellular carcinoma | [59–67]a |
| AMG337 | MET | Advanced gastric and esophageal adenocarcinoma, advanced solid tumors | [68–72]a |
| ARQ-197 (tivantinib) | MET (non ATP-competitive) | Relapsed/refractory multiple myeloma; locally advancer or metastatic colorectal cancers; metastatic triple negative breast cancers; childhood relapsed/refractory solid tumors; recurrent/metastatic head and neck cancers; gastric cancers; metastatic solid tumors; metastatic prostate cancers; metastatic or locally advancer kidney cancers; mesothelioma; small cell lung cancers; hepatocellular carcinoma; | [73–90]a |
| INC280 (capmatinib) | MET | NSCLCs; CRCs; HNSCC; advanced solid tumors, hepatocellular carcinoma; metastatic CRCs; metastatic renal cell carcinoma; recurrent glioblastoma; advanced or metastatic melanoma | [91, 92]a |
| EMD 1204831 | MET | Advanced solid tumors; advanced hepatocellular carcinoma | [93]a |
| MGCD265 | MEK, ALK (dual inhibitor) | Advanced cancers | [94, 95]a |
| MK8033 | MET, RON (ATP-competitive dual inhibitor) | Advanced solid tumors | [96]a |
| PF-04217903 | MET (ATP-competitive) | Advanced cancers | [97–99]a |
aFor more details see: www.clinicaltrials.gow
Fig. 1.
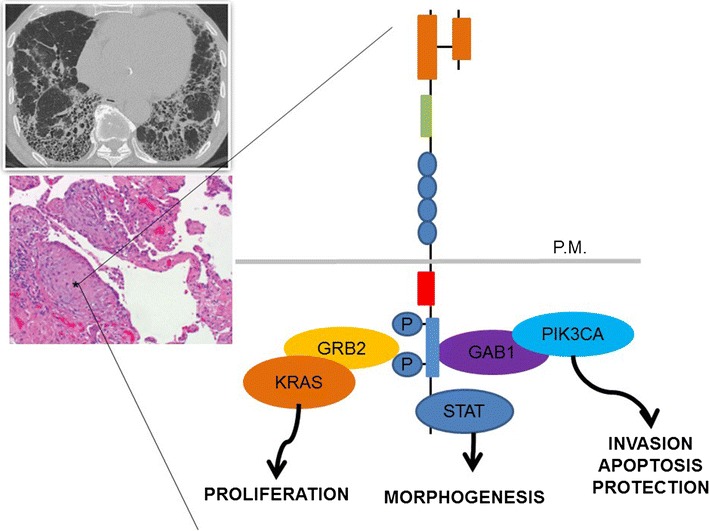
MET signaling pathway in IPF. Enhanced MET activation controls genetic programs leading to cell growth, invasiveness and protection from apoptosis. For both the biological and therapeutic implications of MET activation in myofibroblasts in FF, its KRAS-driven pro-proliferating activity can be separated from the PI3CA-related pro-invasive role. The activity of branching morphogenesis depends on STAT family members, mainly STAT3 (Giordano et al. [100]). STAT3 is known to contribute to lung damage in IPF onset and progression (Pedroza et al. FASEB J 2016; 30(1):129–4)
Competing hypotheses: the role of MET in IPF
IPF is a proliferative disorder affecting the lungs, characterized by aberrant deposition of ECM and consequent remodeling associated with the activation of fibroblasts as a response to still unknown injuries. IPF diagnosis is confirmed by histological identification of the usual interstitial pneumonia (UIP) pattern on surgical (and rarer transbronchial) biopsies, together with detection at high resolution (HR) computerized tomography (CT) scan of bibasilar reticular abnormalities (honeycombing pattern) with minimal or absent ground-glass opacities [1, 101, 102]. The key histological feature of IPF is represented by the so-called fibroblast foci (FF) defined as aggregates of actively proliferating fibroblasts and myofibroblasts. Activated fibroblasts express α-SMA (smooth muscle actin), accounting for to term “myofibroblasts”. In addition they secrete increased levels of ECM-degrading proteases (metalloproteinases MMP2, MMP3, MMP9), facilitating increased ECM turnover and altered ECM deposition; they also secrete growth factors (such as HGF, IGF, NGF, WNT1 and EGF) which can induce proliferative signals within adjacent epithelial cells. Moreover activated fibroblasts behave as modulators of the immune response following tissue injury by secreting cytokines (e.g. IL-1) and chemokines (e.g. MCP1) [103–107]. Activated fibroblasts/myofibroblasts can be found in wound healing processes and sclerosing tissue and as well as in cancers [108]. Through embryogenesis, cells start to move out from developing tissues in order to organize the structure of fetal organs. In a similar fashion, in adult life, during wound healing and tissue repair processes, health cells migrate into the wound to recreate pre-existing tissue patterns [109]. The acquisition of cell motility is required but is not enough to sustain the whole process. Indeed cells need to trigger a number of biological programs, as well as to activate mitotic divisions to repair injured tissues [110]. Thus embryogenesis, tissue repair after wound healing and cancer share similar mechanistic basis, since the same biological activities—cell proliferation, survival and migration, namely the Invasive Growth—are activated in both normal and malignant contexts. During wound healing repair activities as well as in cancer metastatic spreading, several cytokines are secreted in the reactive interstitial compartment. For instance interleukin-1 (IL-1) and 6 (IL-6), tumor necrosis factor alpha (TNF-α) and transforming growth factor beta (TGF-β) are known to induce the transcriptional up-regulation of HGF (in fibroblasts and macrophages) and MET (in epithelial cells) [111, 112]. HGF is also biologically activated, as demonstrated by the overexpression of proteases involved in pro-HGF activation [113, 114]. Moreover, HGF might be activated through an autocrine loop in stromal myofibroblasts. This mechanisms has been well demonstrated during tumor cell invasion [115] but can reasonably be significant in wounds repair as well. Overall, this highly performant HGF assures a proper activation of MET, which is, thus, involved in tissue protective physiological systems. These morphogenetic pathways trigger the EMT by activating biological processes such as cell motility and invasion [116], known as invasive growth program. The aberrant activation of the above described wound healing machinery ultimately characterizes IPF onset and progression. Thus, HGF/MET-driven aberrant morphogenesis plays a crucial role not only in cancer but in IPF, as well. However it should be underlined that its activation and progression in IPF certainly differs from that in cancer, regarding both spatial and temporal characteristics. A proliferating tumor becomes malignant when neoplastic cells move to adjacent environments and settle in tissues and organs that are distant from the original site of growth. In IPF the actively proliferating FFs contrast with neighboring areas of relatively normal parenchyma and move from subpleural regions towards central areas. IPF is overall a lung-specific disease, defined by a centripetal track of disease progression in absence of distant cell scattering. The latter is a key difference with respect to scattering of malignant cells, which essentially means distant and peripheral dissemination. Furthermore IPF is a heterogeneous disease also in the age of lesions, meaning the stage of pathology in different lung parenchymal regions. Thus normal lung tissue is interspersed with interstitial fibrosis, honeycomb cysts and fibroblast foci [1]. On the other hand, it is well known that most tumors tend to become more aggressive in clinical behavior over time, although this time course may be variable. During cancer progression, MET activation generally occurs as a late event, as a consequence to transcriptional up-regulation driven by unfavorable microenvironmental conditions, such as hypoxia or ionizing radiation [7, 117]. Sometimes, rapidly invasive cancers are diagnosed because of appearance of metastatic lesions in absence of a clearly detectable primary mass. Among these highly invasive and malignant tumors, an extremely high mutational frequency of MET coding sequence has been reported; MET mutations have been biologically associated to the observed transformed phenotype [118]. The above described differences between IPF and cancer strictly reflect the differences of cell lineages. Indeed cancer is, by definition, a disease of genes, which evolves through a dynamic process of clonal expansion and selection in of advantageous somatic driver lesions [119, 120]. Each individual tumour is defined by a unique clonal evolution resulting from an intricate connection between genetic and non-genetic/epigenetic factors, leading to phenotypic and genotypic heterogeneity. Among the diversity in tumor-cell population, the CSC compartment brings about tumor maintenance and progression [121]. MET-driven invasive growth is aberrantly activated in cancer, mainly as a late event, leading to distant dissemination and malignant progression. More recent studies have reported that MET amplified cancer clones are selected under therapeutic pressure in a context of molecularly heterogeneous lesions exposed to targeted therapies or radiotherapy [8, 122–125]. In CSC, both the occurrence of genetic lesions (as amplification) and physiological expression of MET can contribute to tumorigenesis and therapeutic resistance, by sustaining the invasive growth phenotype. On the other hand, myofibroblasts within FF in IPF are characterized by cellular and genetic heterogeneity. Notably—very recently, Jones and colleagues elegantly demonstrated that FFs in IPF identify—quite unexpectedly—morphologically complex 3D-structures, each independent from the others [126]. These findings strongly suggest that IPF onset relies on the aberrant local responses that are activated and lead to multifocal injuries. As a consequence diffuse cellular fate conversion and tissue plasticity are associated to IPF. During organ regeneration, MET physiological activation displays protective functions: epithelial cells located at the wound edges exploit invasive growth to enhance cellular division and repopulation of the injured areas [127–129]. When the damage inappropriately persists, as in IPF, the HGF/MET pair actively contrasts myofibroblasts activation and the consequent associated abnormal deposition of extracellular matrix [130]. Moreover it is well known that semaphorins might activate MET in and HGF-independent manner. As already presented, MET and plexins share high homology at the extracellular sema domain. When MET oligomerizes with plexins, it can be activated by semaphorins, even in the absence of its ligand HGF [131, 132]. Growing evidence sustains that semaphorins—and their ligands plexins—have a role in enhancing immune function and angiogenesis as well as in controlling lung fibrogenic diseases [133–136]. As a consequence, fibrosing settings, as IPF, which co-express both HGF and plexins might feature even hyperactive invasive capacities.
Applying the razor: MET as an actionable target for IPF
In a complex and heterogeneous setting, which applies to IPF, the principle of pluralitas non est ponenda sine necessitate (Ockham’s razor, principle attributed to the 14th century logician William of Ockham) can be applied to correctly understand the role of MET-driven invasive growth at disease onset. IPF resembles cancer in many MET-associated behaviors, such as invasive phenotype and pro-coagulant status. However dynamics of malignant divergent clonal selective pressure and heterogeneity clearly differ from those occurring in IPF and impact on the biological significance of MET activation. The RTK MET is phosphorylated in myofibroblasts in FF: in a context-specific regulation of its expression, MET might become a functional marker of IPF and an actionable target. The cytogenetic heterogeneity, that is a hallmark of FF, can be exploited for therapeutic purposes and already commercially available MET-inhibitors can be tested to interfere with IPF progression. MET-mediated events in IPF rely on qualitative differences among physiological signals, whereas no driver genetic lesions, causally implicated in the disease can be clearly demonstrated. Thus MET blockage falls among those therapeutic strategies aimed to impair the “aberrant recapitulation of developmental programs” as Selma and coll. already defined IPF [137]. A dynamic crosstalk between MET and developmental signaling pathways which are known to be activated in IPF is well documented. Among them the most relevant are those driven by Wnt/β-catenin and TGFβ cascades [116, 136]. The evolutionary conserved Wnt signaling canonical pathway is known to be a key player in maintaining tissue homeostasis, cell proliferation and differentiation, and in regulating cell renewal and differentiation. Wnt signaling is also implicated in a variety of cancers [138, 139]. Wnt/β-catenin pathway is expressed in the adult lung epithelium and overexpressed during inappropriate EMT in IPF [116, 140]. Although intensive efforts have been made, the Wnt signaling pathway remains difficult to target. Tight cross-talk among MET and Wnt signaling is known in tissue morphogenesis as well as in several cancer types [141–144]. Conversely, an intricate interaction between TGFβ and MET signaling is well discovered. The TGFβ superfamily is known to play a critical role in the regulation of cell differentiation and proliferation. The TGF cascade mainly involves the activation of the cytoplasmic signaling molecules Smad2 and Smad3 for the TGF/activin pathway and Smad1/5/5/9 for the TGF/bone morphogenetic protein (BMP) pathway [145–149]. In particular the cross talk between TGFβ/BMP pathways is implicated in several biological programs and involved in a number of progressive disease, among which cancer [150–153]. The role of the TGFβ pathway has been extensively studied in IPF. Overexpression of its effects is induced by persistent injury and is, in turn, associated to the aberrant lung remodeling and fibrogenesis by activating myofibroblasts to produce extracellular matrix [130, 154]. Many drugs have been developed to target the TGFβ family signaling cascade [155]. In particular the FDA approved in 2014 the TGFβ1 inhibitor Pirfenidone for IPF therapy [156–159]. HGF can antagonize the TGFβ profibrotic phenotypes by several mechanisms, mainly by transcriptional TGFβ down-modulation [160] and ERK-mediated inhibition of Smad proteins [161, 162]. Nevertheless, more recently it has been shown that overexpression of the HGF receptor MET together with CD44 isoform 6 (CD44v6) sustains the TGFβ signaling in IPF through an autocrine loop [27]. Another relevant issue is that there is considerable experimental evidence that tissue hypoxia is associated with the onset of fibrosis. However, although chronic hypoxemia can clinically characterize IPF, the role of local hypoxia as a driver of the progressive fibrotic nature of the disease has not been fully clarified. Low oxygen tension has variable effects on cellular proliferation depending on the cell type. While arresting alveolar epithelial cell proliferation, low oxygen tension has been shown to promote normal fibroblast proliferation, leading to the possibility that hypoxia may promote IPF fibroblasts proliferation [163–166]. It has been recently reported that a pathological feed-forward loop may exist in the IPF lung, in which hypoxia promotes IPF fibroblasts proliferation via stimulation of miR-210 expression, which in turn worsens hypoxia [167]. More importantly, molecular links are beginning to emerge between hypoxia, EMT and stemness. During embryogenesis hypoxia contributes to the induction of niches that maintain pluripotent cells. During carcinogenesis, hypoxia has the potential to exert significant effect on the maintenance and evolution of cancer stem cells. Moreover solid tumor hypoxia is a well-known factor in tumor aggressiveness and invasive potential [168]. It is plausible that hypoxia-induced MET up-regulation may occur in IPF as well, and can cooperate in triggering the regenerative/reparative processes that define the disease onset and progression. This hypothesis questions the use of anti-angiogenic agents in IPF, as in cancer therapy; deprivation of a blood supply, and thus of oxygen could in fact induce, besides the desirable tissue necrosis, a dangerous “invasive switch”. It would therefore be advisable to combine anti-angiogenic treatments (e.g. nintedanib) with an anti-MET agent to prevent these potential drawbacks. Regarding the rationale of MET therapeutic blockade in IPF, another key point must be underlined. Since IPF is, by definition, polyclonal, the reported MET activation may be independent of on the phenomena of oncogenic addiction associated with structural alterations related to cancer clonal evolution or—considering the role played by HGF—to ligand–receptor autocrine circuits, which frees cells from the need for a paracrine supply of growth factor. In this perspective, cells undergoing EMT often take advantage of the physiological function of MET as an “expedience” [169] to gain a selective advantage in IPF progression.
Conclusion
The molecular pathways involved in the metastatic process in cancer are shared with IPF. Among them the MET-driven invasive growth program plays a crucial role. As discussed above, MET activation governs a number of physiological and pathological processes that modulate dynamic changes and plasticity of tissues. If in cancer MET activation enables cells to overcome damage induced by targeted agents and ionizing radiation, there is enough evidence to sustain that in IPF the versatility of the MET-mediated biological responses may promote tissue remodeling by integrating growth, survival and migration cues in response to abnormal environmental stimuli or cell-autonomous perturbations in absence of addiction phenomena. More likely, MET expression in myofibroblasts behaves as in cancer stem cells, where it sustains the inherent self-renewing, self-preserving and invasive growth phenotype [8, 19]. These notions indicate three clinical implications: (1) MET is a versatile candidate for targeted therapeutic intervention in IPF. (2) Targeted therapies against MET could be effective as a combinatorial approach to restrict disease progression, rather than being used as single front-line approaches. (3) Validation of MET expression as a biomarker is mandatory to developing therapies for IPF based on MET inhibition.
Authors’ contributions
GMS, AG, AB, MM and SB designed the paper structure and wrote the manuscript; FM helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Prof Paolo M Comoglio and Prof Ernesto Pozzi for helpful discussions; Dr. Radhika Srinivasan for the revision of the manuscript. To the memory of Prof Maurizio Luisetti.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- IPF
idiopathic pulmonary fibrosis
- FF
fibroblast foci
- RTK
receptor tyrosine kinases
- EMT
epithelial-mesenchymal transition
- HGF
hepatocyte growth factor
- ECM
extracellular matrix
- IGF
insulin-like growth factor
- NGF
nerve growth factor
- EGF
epidermal growth factor
- MMP
metalloproteinases
Contributor Information
Giulia M. Stella, Phone: +39 0382503369, Email: g.stella@smatteo.pv.it
Alessandra Gentile, Email: alessandra.gentile@ircc.it.
Alice Baderacchi, Email: alice.balderacchi@libero.it.
Federica Meloni, Email: f.meloni@smatteo.pv.it.
Melissa Milan, Email: melissa.milan@ircc.it.
Silvia Benvenuti, Email: silvia.benvenuti@ircc.it.
References
- 1.Raghu G, Rochewerg B, Zhang Y, Garcia CA, Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, et al. ATS/ERS/JRS/ALAT Statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;1838(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3(1):8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laureson S, Sidhu R, Goodall M, Adler AI. NICE guidance for treating idiopathic pulmonary fibrosis. Lancet Resp Med. 2016;4(3):176–177. doi: 10.1016/S2213-2600(16)00022-9. [DOI] [PubMed] [Google Scholar]
- 4.Vancheri C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur Resp Rev. 2013;22(129):265–272. doi: 10.1183/09059180.00003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimminger F, Günther A, Vancheri C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur Resp J. 2015;45(5):1434–1445. doi: 10.1183/09031936.00149614. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, et al. Efficacy and safety of nintedaninb inidiopathic pulmonary fibrosis. N Eng J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 7.Boccaccio C, Comoglio P. Invasive growth: a MET-drive genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6(8):637–645. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 8.Boccaccio C, Comoglio PM. MET, a driver of invasive growth and cancer clonal evolution under therapeutic pressure. Curr Opin Cell Biol. 2014;31:98–105. doi: 10.1016/j.ceb.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Stella GM, Benvenuti S, Comoglio PM. Targeting the MET oncogene in cancer and metastases. Expert Opin Investig Drugs. 2010;19(11):1381–1389. doi: 10.1517/13543784.2010.522988. [DOI] [PubMed] [Google Scholar]
- 10.Boccaccio C, Comoglio PM. A functional role for hemostasis in early cancer development Cancer Res. 2005;65(19):8579–8582. doi: 10.1158/0008-5472.CAN-05-2277. [DOI] [PubMed] [Google Scholar]
- 11.Trusolino L, Bertotti A, Comoglio PM. MET signaling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- 12.Boccaccio C, Sabatino G, Medico E, Girolami F, Follenzi A, Reato G, Sottile A, Naldini L, Comoglio PM. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature. 2005;434(7031):396–400. doi: 10.1038/nature03357. [DOI] [PubMed] [Google Scholar]
- 13.Corso S, Comoglio PM, Giordano S. Cancer therapy: can the challenge be MET? Trends Mol Med. 2005;11(6):284–292. doi: 10.1016/j.molmed.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Schirrmeister W, Gnad T, Wex T, Higashiyama S, Wolke C, Naumann M, Lendeckel U. Ectodomain shedding of E-cadherin and c-Met is induced by Helicobacter pylori infection. Exp Cell Res. 2009;315(20):3500–3508. doi: 10.1016/j.yexcr.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 15.Bennett D, Bargagli E, Refini RM, Campagna MS, Gennari L, Nuti R, Figura N, Rottoli P. Helicobacter pylori seroprevalence in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2007;43(2):635–638. doi: 10.1183/09031936.00104813. [DOI] [PubMed] [Google Scholar]
- 16.Wasik AM, Grabarek J, Pantovic A. Cieslar-Pobuda Asgari HR, Bundgaard-Nielsen C, Rafat M, Dixon IM, Ghavami S, Los MJ. Reprogramming and carcinogenesis-parallels and distinctions. Int Rev Cell Mol Biol. 2014;308:167–203. doi: 10.1016/B978-0-12-800097-7.00005-1. [DOI] [PubMed] [Google Scholar]
- 17.Akbari-Birgani S, Paranjothy T, Zuse A, Janikowsky T, Cieslar-Pobuda A, Likus W, Urasinska E, Schweizer F, Ghavami S, Klonisch T, Los MJ. Cancer stem cells, cancer-initiating cells and methods fo their detection. Drug Discov Today. 2016;21(5):836–842. doi: 10.1016/j.drudis.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Cieslar-Pobuda A, Jain MW, Kratz G, Rzezowska-Wolny J, Ghavami S, Wiechec E. The expression pattern of PFKB3 enzyme distinguishes between induced-pluipotent stem cells and cancer stem cells. Oncotarget. 2015;6(30):29753–29770. doi: 10.18632/oncotarget.4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farahani E, Patra HK, Jangamreddy JR, Rashedi I, Kawalec M. Rao Pariti RK, Batakis P, Wiechec E. cell adhesion molecules and their relation to (cancer) cell stemness. Carcinogenesis. 2014;35(4):747–759. doi: 10.1093/carcin/bgu045. [DOI] [PubMed] [Google Scholar]
- 20.Lau E, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, Chain S, Lin CH, Tsang SY, Ma S, Ng IO, Lee TK. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in hepatocellular carcinoma through c-MET/FRA1/HEY1 signaling. Cell rep. 2016;15(6):1175–1189. doi: 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 21.D’Amico L, Belisario D, Migliardi G, Grange C, Bussolati B, D’Amelio P, Perera T, Dalmasso E, Carbonare LD, Godio L, Comoglio PM, et al. C-met inihibition blocks bone metastais development induced by renal cancer stem cells. Oncotarget. 2016. In Press. [DOI] [PMC free article] [PubMed]
- 22.Hou C, Sun B, Jiang Y, Zheng J, Yang N, Ji C, Liang Z, Shi J, Zhang R, Liu Y, Ye C, Zuo P. Micro-RNA -31 inhibits lung adenocarcinoma stem-like cells via down-regulation of MET-PIK3 K-Akt signaling pathways. Anticancer Agents Med Chem. 2016;16(4):501–518. doi: 10.2174/1871520615666150824152353. [DOI] [PubMed] [Google Scholar]
- 23.Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, 3rd Rideout WM, et al. Met signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014;74(6):1857–1869. doi: 10.1158/0008-5472.CAN-13-2340-T. [DOI] [PubMed] [Google Scholar]
- 24.Delitto D, Vertes-George E, Hughes SJ, Behrns KE, Trevino JG. C-MET signalling in the development of tumorigenesis and chemoresistance: potential application in pancreatic cancer. World J Gastroenterol. 2014;20(26):8458–8470. doi: 10.3748/wjg.v20.i26.8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yashiro M, Nishii T, Hasegawa T, Matsuzaki T, Morisaki T, Fukoka T, Hirakawa K. A c-MET inhibitor increases the chemosensitivity of cancer stem cell to the irinotecan in gastric carcinoma. Br J Cancer. 2013;109(10):2619–2628. doi: 10.1038/bjc.2013.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Bacco F, d’Ambrosio A, Casanova E, Orzan F, Neggia R, Albano R, Verginelli F, Cominelli M, Poliani PL, Luraghi P, et al. Met inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol Med. 2016;8(5):550–568. doi: 10.15252/emmm.201505890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stella GM, Inghilleri S, Pignochino Y, Zorzetto M, Oggionni T, Morbini P, Luisetti M. Activation of oncogenic pathways in idiopathic pulmonary fibrosis. Transl Oncol. 2014;7(5):650–655. doi: 10.1016/j.tranon.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghatak S, Bogatkevic GS, Atnelishvili I, Akter T, Feghali-Bostowick C, Hoffman S, Fresco VM, Fuchs JC, Visconti RP, Markwald RR, et al. Overexpression of c-MET and CD44v6 receptors contributes to autocrineTGF-β1 signaling in interstitial lung disease. J Biol Chem. 2014;289(11):7856–7872. doi: 10.1074/jbc.M113.505065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crestani B, Marchand-Adam S, Quesnel C, Plantiere L, Borensztajn K, Marchal J, Mailleux A, Soler P, Dehoux M. Hepatocyte growth factor and lung fibrosis. Proc Am Thorac Soc. 2012;9(3):158–163. doi: 10.1513/pats.201202-018AW. [DOI] [PubMed] [Google Scholar]
- 30.Lee BS, Kang S, Kim KA, Song YJ, Cheong KH, Cha HY, Kim CH. Met degradation by SAIT301, a Met monoclonal antibody, reduces the invasion and migration of nasopharyngeal cancer cells via inhibition of EGR-1 expression. Cell Death Dis. 2014;5:e115. doi: 10.1038/cddis.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hultberg A, Morello V, Huyghe L, De Jonge N, Blanchetot C, Hanssens V, De Boeck G, Silence K, Festjens E, Heukers R, Roux B, Lamballe F, Ginestier C, Charafe-Jauffret E, Maina F, Brouckaert P, Saunders M, Thibault A, Dreier T, de Haard H, Michieli P. Depleting MET-expressing tumor cells by ADCC provides a therapeutic advantage over inhibiting HGF/MET signaling. Cancer Res. 2015;75(16):3373–3383. doi: 10.1158/0008-5472.CAN-15-0356. [DOI] [PubMed] [Google Scholar]
- 32.Rolfo C, Van Der Steen N, Pauwels P, Cappuzzo F. Onartuzumab in lung cancer: the fall of Icarus? Expert Rev Anticancer Ther. 2015;15(5):487–489. doi: 10.1586/14737140.2015.1031219. [DOI] [PubMed] [Google Scholar]
- 33.Xin Y, Jin D, Eppler S, Damico-Beyer LA, Joshi A, Davis JD, Kaur S, Nijem I, Bothos J, Peterson A, Patel P, Bai S. Population pharmacokinetic analysis from phase I and phase II studies of the humanized monovalent antibody, onartuzumab (MetMAb), in patients with advanced solid tumors. J Cli Pharmacol. 2013;53(11):1103–1111. doi: 10.1002/jcph.183. [DOI] [PubMed] [Google Scholar]
- 34.Xiang H, Bender BC, Reyes AE, 2nd, Merchant M, Jumbe NL, Romero M, Davancaze T, Nijem I, Mai E, Young J, Peterson A, Damico-Beyer LA. Onartuzumab (MetMAb): using nonclinical pharmacokinetic and concentration-effect data to support clinical development. Clin Cancer Res. 2013;19(18):5068–5078. doi: 10.1158/1078-0432.CCR-13-0260. [DOI] [PubMed] [Google Scholar]
- 35.Bendell JC, Ervin TJ, Gallinson D, Singh J, Wallace JA, Saleh MN, Vallone M, Phan SC, Hack SP. Treatment rationale and study design for a randomized, double-blind, placebo-controlled phase II study evaluating onartuzumab (MetMAb) in combination with bevacizumab plus mFOLFOX-6 in patients with previously untreated metastatic colorectal cancer. Clin Colorectal Cancer. 2013;12(3):218–222. doi: 10.1016/j.clcc.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Spigel DR, Edelman MJ, Mok T, O'Byrne K, Paz-Ares L, Yu W, Rittweger K, Thurm H, MetLung Phase III Study Group Treatment rationale study design for the MetLung trial: a randomized, double-blind phase III study of onartuzumab (MetMAb) in combination with erlotinib versus erlotinib alone in patients who have received standard chemotherapy for stage IIIB or IV met-positive non-small-cell lung cancer. Clin Lung Cancer. 2012;13(6):500–504. doi: 10.1016/j.cllc.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 37.Feng Y. Ma PC Anti-MET targeted therapy has come of age: the first durable complete response with MetMAb in metastatic gastric cancer. Cancer Discov. 2011;1(7):550–554. doi: 10.1158/2159-8290.CD-11-0289. [DOI] [PubMed] [Google Scholar]
- 38.Moores SL, Chiu ML, Bushey BS, Chevalier K, Luistro L, Dorn K, Brezski RJ, Haytko P, Kelly T, Wu SJ, Martin PL, Neijssen J, Parren PW, Schuurman J, Attar RM, Laquerre S, Lorenzi MV, Anderson GM. A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 2016;76(13):3942–3953. doi: 10.1158/0008-5472.CAN-15-2833. [DOI] [PubMed] [Google Scholar]
- 39.Zheng S, Moores S, Jarantow S, Pardinas J, Chiu M, Zhou H, Wang W. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. MAbs. 2016;8(3):551–556. doi: 10.1080/19420862.2015.1136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jarantow SW, Bushey BS, Pardinas JR, Boakye K, Lacy ER, Sanders R, Sepulveda MA, Moores SL, Chiu ML. Impact of cell-surface antigen expression on target engagement and function of an epidermal growth factor receptor × c-MET bispecific antibody. J Biol Chem. 2015;290(41):24689–24704. doi: 10.1074/jbc.M115.651653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castellanos EH, Horn L. Re-evaluating progression in an era of progress: a review of first- and second-line treatment options in anaplastic lymphoma kinase-positive non-small cell lung cancer. Oncologist. 2016;21(6):755–761. doi: 10.1634/theoncologist.2015-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ou SH, Agarwal N. Ali SM High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. 2016;98:59–61. doi: 10.1016/j.lungcan.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 43.Ye M, Zhang X, Li N, Zhang Y, Jing P, Chang N, Wu J, Ren X, Zhang J. ALK and ROS1 as targeted therapy paradigms and clinical implications to overcome crizotinibresistance. Oncotarget. 2016;7(11):12289–12304. doi: 10.18632/oncotarget.6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulkarni A, Vijaykumar VE, Natarajan SK, Sengupta S, Sabbisetti S. Sustained inhibition of cMET-VEGFR2 signaling using liposome-mediated delivery increases efficacy and reduces toxicity in kidney cancer. Nanomed. 2016;12(7):1853–1861. doi: 10.1016/j.nano.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Sameni M, Tovar EA, Essenburg CJ, Chalasani A, Linklater ES, Borgman A, Cherba DM, Anbalagan A, Winn ME, Graveel CR, Sloane BF. Cabozantinib (XL184) inhibits growth and invasion of preclinical TNBC models. Clin Cancer Res. 2016;22(4):923–934. doi: 10.1158/1078-0432.CCR-15-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, Krainer M, Bergman A, Hoelzer W, De Wit R, Bögemann M, Saad F, Cruciani G, Vuillemin AT, Feyerabend S, Miller K, Houédé N, Hussain S, Lam E, Polikoff J, Stenzl A, Mainwaring P, Ramies D, Hessel C, Weitzman A, Fizazi K. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016. pii: JCO655597. [DOI] [PubMed]
- 47.Roy S, Narang BK, Rastogi SK, Rawal RK1. A novel multiple tyrosine-kinase targeted agent to explore the future perspectives of anti-angiogenic therapy for the treatment of multiple solid tumors: cabozantinib. Anticancer Agents Med Chem. 2015;15(1):37–47. doi: 10.2174/1871520614666140902153840. [DOI] [PubMed] [Google Scholar]
- 48.Cabanillas ME, Brose MS, Holland J, Ferguson KC, Sherman SI. A phase I study of cabozantinib (XL184) in patients with differentiated thyroid cancer. Thyroid. 2014;24(10):1508–1514. doi: 10.1089/thy.2014.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, Hammers H, Hutson TE, Lee JL, Peltola K, Roth BJ, Bjarnason GA, Géczi L, Keam B, Maroto P, Heng DY, Schmidinger M, Kantoff PW, Borgman-Hagey A, Hessel C, Scheffold C, Schwab GM, Tannir NM, Motzer RJ, METEOR Investigators Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1814–1823. doi: 10.1056/NEJMoa1510016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, Hammers HJ, Donskov F, Roth BJ, Peltola K, Lee JL, Heng DY, Schmidinger M, Agarwal N, Sternberg CN, McDermott DF, Aftab DT, Hessel C, Scheffold C, Schwab G, Hutson TE, Pal S. Motzer RJ; METEOR investigars. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(7):917–927. doi: 10.1016/S1470-2045(16)30107-3. [DOI] [PubMed] [Google Scholar]
- 51.Navis AC, Bourgonje A, Wesseling P, Wright A, Hendriks W, Verrijp K, van der Laak JA, Heerschap A, Leenders WP. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS One. 2013;8(3):e5826. doi: 10.1371/journal.pone.0058262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herreros-Villanueva M, Zubia-Olascoaga A, Bujanda L. c-Met in pancreatic cancer stem cells: therapeutic implications. World J Gastroenterol. 2012;18(38):5321–5323. doi: 10.3748/wjg.v18.i38.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, Engst S, Lee L, Lesch J, Chou YC, Joly AH. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Guessous F, Kofman A, Schiff D, Abounader R. XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs. 2010;13(2):112–121. [PMC free article] [PubMed] [Google Scholar]
- 55.Schuller AG, Barry ER, Jones RD, Henry RE, Frigault MM, Beran G, Linsenmayer D, Hattersley M, Smith A, Wilson J, Cairo S, Déas O, Nicolle D, Adam A, Zinda M, Reimer C, Fawell SE, Clark EA, D'Cruz CM. The MET inhibitor AZD6094 (Savolitinib, HMPL-504) induces regression in papillary renal cell Ccrcinoma patient-derived xenograft models. Clin Cancer Res. 2015;21(12):2811–2818. doi: 10.1158/1078-0432.CCR-14-2685. [DOI] [PubMed] [Google Scholar]
- 56.Gavine PR, Ren Y, Han L, Lv J, Fan S, Zhang W, Xu W, Liu YJ, Zhang T, Fu H, Yu Y, Wang H, Xu S, Zhou F, Su X, Yin X, Xie L, Wang L, Qing W, Jiao L, Su W, Wang QM. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol. 2015;9(1):323–333. doi: 10.1016/j.molonc.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia H, Dai G, Weng J, Zhang Z, Wang Q, Zhou F, Jiao L, Cui Y, Ren Y, Fan S, Zhou J, Qing W, Gu Y, Wang J, Sai Y, Su W. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2,3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J Med Chem. 2014;57(18):7577–7589. doi: 10.1021/jm500510f. [DOI] [PubMed] [Google Scholar]
- 58.Chen HM, Tsai CH, Hung WC. Foretinib inhibits angiogenesis, lymphangiogenesis and tumor growth of pancreatic cancer in vivo by decreasing VEGFR-2/3 and TIE-2 signaling. Oncotarget. 2015;6(17):14940–14952. doi: 10.18632/oncotarget.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faria CC, Golbourn BJ, Dubuc AM, Remke M, Diaz RJ, Agnihotri S, Luck A, Sabha N, Olsen S, Wu X, Garzia L, Ramaswamy V, Mack SC, Wang X, Leadley M, Reynaud D, Ermini L, Post M, Northcott PA, Pfister SM, Croul SE, Kool M, Korshunov A, Smith CA, Taylor MD, Rutka JT. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer Res. 2015;75(1):134–146. doi: 10.1158/0008-5472.CAN-13-3629. [DOI] [PubMed] [Google Scholar]
- 60.Shah MA, Wainberg ZA, Catenacci DV, Hochster HS, Ford J, Kunz P, Lee FC, Kallender H, Cecchi F, Rabe DC, Keer H, Martin AM, Liu Y, Gagnon R, Bonate P, Liu L, Gilmer T, Bottaro DP. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PLoS One. 2013;8(3):e54014. doi: 10.1371/journal.pone.0054014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Logan TF. Foretinib (XL880): c-MET inhibitor with activity in papillary renal cell cancer. Curr Oncol Rep. 2013;15(2):83–90. doi: 10.1007/s11912-013-0299-3. [DOI] [PubMed] [Google Scholar]
- 62.Choueiri TK1, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM, Ottesen LH, Laubscher KH, Sherman L, McDermott DF, Haas NB, Flaherty KT, Ross R, Eisenberg P, Meltzer PS, Merino MJ, Bottaro DP, Linehan WM, Srinivasan R. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181–6. [DOI] [PMC free article] [PubMed]
- 63.Shapiro GI, McCallum S, Adams LM, Sherman L, Weller S, Swann S, Keer H, Miles D, Müller T, Lorusso P. A Phase 1 dose-escalation study of the safety and pharmacokinetics of once-daily oral foretinib, a multi-kinase inhibitor, in patients with solid tumors. Invest New Drugs. 2013;31(3):742–750. doi: 10.1007/s10637-012-9881-z. [DOI] [PubMed] [Google Scholar]
- 64.Seiwert T1, Sarantopoulos J, Kallender H, McCallum S, Keer HN, Blumenschein G Jr. Phase II trial of singleagent foretinib (GSK1363089) in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Invest New Drugs. 2013;31(2):417–24. [DOI] [PMC free article] [PubMed]
- 65.Huynh H, Ong R, Soo KC. Foretinib demonstrates anti-tumor activity and improves overall survival in preclinical models of hepatocellular carcinoma. Angiogenesis. 2012;15(1):59–70. doi: 10.1007/s10456-011-9243-z. [DOI] [PubMed] [Google Scholar]
- 66.Zillhardt M, Park SM, Romero IL, Sawada K, Montag A, Krausz T, Yamada SD, Peter ME, Lengyel E. Foretinib (GSK1363089), an orally available multikinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. Clin Cancer Res. 2011;17(12):4042–4051. doi: 10.1158/1078-0432.CCR-10-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Naing A, Kurzrock R, Adams LM, Kleha JF, Laubscher KH, Bonate PL, Weller S, Fitzgerald C, Xu Y, LoRusso PM. A comparison of the pharmacokinetics of the anticancer MET inhibitor foretinib free base tablet formulation to bisphosphate salt capsule formulation in patients with solid tumors. Invest New Drugs. 2012;30(1):327–334. doi: 10.1007/s10637-010-9536-x. [DOI] [PubMed] [Google Scholar]
- 68.Hughes PE, Rex K, Caenepeel S, Yang Y, Zhang Y, Broome MA, Kha HT, Burgess TL, Amore B, Kaplan-Lefko PJ, Moriguchi J, Werner J, Damore MA, Baker D, Choquette DM, Harmange JC, Radinsky R, Kendall R, Dussault I, Coxon A. In vitro and in vivo activity of AMG 337, a potent and selective MET kinase inhibitor, in MET-dependent cancer models. Mol Cancer Ther. 2016;15(7):1568–79. [DOI] [PubMed]
- 69.Du Z, Caenepeel S, Shen Y, Rex K, Zhang Y, He Y, Tang ET, Wang O, Zhong W, Zhou H, Huang J, Huang E, Hu L, Coxon A, Zhang M. Preclinical evaluation of AMG 337, a highly selective small molecule MET inhibitor, in hepatocellular carcinoma. Mol Cancer Ther. 2016;15(6):1227–1237. doi: 10.1158/1535-7163.MCT-15-0745. [DOI] [PubMed] [Google Scholar]
- 70.Lee J, Tran P, Klempner SJ. Targeting the MET pathway in gastric and oesophageal Cancers: refining tu optimal approach. Clin Oncol. 2016;28(8):e35–e44. doi: 10.1016/j.clon.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 71.Boezio AA, Copeland KW, Rex K, K Albrecht B, Bauer D, Bellon SF, Boezio C, Broome MA, Choquette D, Coxon A, Dussault I, Hirai S, Lewis R, Lin MH, Lohman J, Liu J, Peterson EA, Potashman M, Shimanovich R, Teffera Y, Whittington DA, Vaida KR, Harmange JC. Discovery of (R)-6-(1-(8-Fluoro-6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-a]pyridin-3-yl)ethyl)-3-(2-methoxyethoxy)-1,6-naphthyridin-5(6H)-one (AMG 337), a potent and selective inhibitor of MET with high unbound target coverage and robust in vivo antitumor activity. J Med Chem. 2016;59(6):2328–42. [DOI] [PubMed]
- 72.Aprile G, Leone F, Giampieri R, Casagrande M, Marino D, Faloppi L, Cascinu S, Fasola G, Scartozzi M. Tracking the 2015 gastrointestinal cancers symposium: bridging cancer biology to clinical gastrointestinal oncology. Onco Targets Ther. 2015;8:1149–1156. doi: 10.2147/OTT.S82624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eng C, Bessudo A, Hart LL, Severtsev A, Gladkov O, Müller L, Kopp MV, Vladimirov V, Langdon R, Kotiv B, Barni S, Hsu C, Bolotin E, von Roemeling R, Schwartz B, Bendell JC. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int J Cancer. 2016;139(1):177–186. doi: 10.1002/ijc.30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scagliotti G, von Pawel J, Novello S, Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A, Spigel D, Hirsh V, Shepherd FA, Sequist LV, Sandler A, Ross JS, Wang Q, von Roemeling R, Shuster D, Schwartz B. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2015;33(24):2667–2674. doi: 10.1200/JCO.2014.60.7317. [DOI] [PubMed] [Google Scholar]
- 75.Yoshioka H, Azuma K, Yamamoto N, Takahashi T, Nishio M, Katakami N, Ahn MJ, Hirashima T, Maemondo M, Kim SW, Kurosaki M, Akinaga S, Park K, Tsai CM, Tamura T, Mitsudomi T, Nakagawa K. A randomized, double-blind, placebo-controlled, phase III trial of erlotinib with or without a c-Met inhibitortivantinib (ARQ 197) in Asian patients with previously treated stage IIIB/IV nonsquamous nonsmall-cell lung cancer harboring wild-type epidermal growth factor receptor (ATTENTION study). Ann Oncol. 2015;26(10):2066–72. [DOI] [PubMed]
- 76.Tolaney SM, Tan S, Guo H, Barry W, Van Allen E, Wagle N, Brock J, Larrabee K, Paweletz C, Ivanova E, Janne P, Overmoyer B, Wright JJ, Shapiro GI, Winer EP, Krop IE. Phase II study of tivantinib (ARQ 197) in patients with metastatic triple-negative breast cancer. Invest New Drugs. 2015;33(5):1108–1114. doi: 10.1007/s10637-015-0269-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Porta C, Giglione P, Ferrari A, Reversi F, Liguigli W, Imarisio I, Ganini C. Tivantinib (ARQ197) in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2015;15(6):615–622. doi: 10.1586/14737140.2015.1050383. [DOI] [PubMed] [Google Scholar]
- 78.Zaman S, Shentu S, Yang J, He J, Orlowski RZ, Stellrecht CM, Gandhi V. Targeting the pro-survival protein MET with tivantinib (ARQ 197) inhibits growth of multiple myeloma cells. Neoplasia. 2015;17(3):289–300. doi: 10.1016/j.neo.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okusaka T, Aramaki T, Inaba Y, Nakamura S, Morimoto M, Moriguchi M, Sato T, Ikawa Y, Ikeda M, Furuse J. Phase I study of tivantinib in Japanese patients with advanced hepatocellular carcinoma: Distinctive pharmacokinetic profiles from other solid tumors. Cancer Sci. 2015;106(5):611–617. doi: 10.1111/cas.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xi WH, Yang LY, Cao ZY, Qian Y. Tivantinib (ARQ-197) exhibits anti-tumor activity with down-regulation of FAK in oral squamous cell carcinoma. Biochem Biophys Res Commun. 2015;457(4):723–729. doi: 10.1016/j.bbrc.2015.01.062. [DOI] [PubMed] [Google Scholar]
- 81.Leon LG, Gemelli M, Sciarrillo R, Avan A, Funel N, Giovannetti E. Synergistic activity of the c-Met and tubulin inhibitor tivantinib (ARQ197) with pemetrexed in mesothelioma cell. Curr Drug Targets. 2014;15(14):1331–1340. doi: 10.2174/1389450116666141205160924. [DOI] [PubMed] [Google Scholar]
- 82.Puzanov I, Sosman J, Santoro A, Saif MW, Goff L, Dy GK, Zucali P, Means-Powell JA, Ma WW, Simonelli M, Martell R, Chai F, Lamar M, Savage RE, Schwartz B, Adjei AA. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Invest New Drugs. 2015;33(1):159–168. doi: 10.1007/s10637-014-0167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pant S, Saleh M, Bendell J, Infante JR, Jones S, Kurkjian CD, Moore KM, Kazakin J, Abbadessa G, Wang Y, Chen Y, Schwartz B, Camacho LH. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann Oncol. 2014;25(7):1416–1421. doi: 10.1093/annonc/mdu157. [DOI] [PubMed] [Google Scholar]
- 84.Kang YK, Muro K, Ryu MH, Yasui H, Nishina T, Ryoo BY, Kamiya Y, Akinaga S, Boku N. A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer. Invest New Drugs. 2014;32(2):355–356. doi: 10.1007/s10637-013-0057-2. [DOI] [PubMed] [Google Scholar]
- 85.Feldman DR, Einhorn LH, Quinn DI, Loriot Y, Joffe JK, Vaughn DJ, Fléchon A, Hajdenberg J, Halim AB, Zahir H, Motzer RJ. A phase 2 multicenter study of tivantinib (ARQ 197) monotherapy in patients with relapsed or refractory germ cell tumors. Invest New Drugs. 2013;31(4):1016–1022. doi: 10.1007/s10637-013-9934-y. [DOI] [PubMed] [Google Scholar]
- 86.Santoro A, Rimassa L, Borbath I, Daniele B, Salvagni S, Van Laethem JL, Van Vlierberghe H, Trojan J, Kolligs FT, Weiss A, Miles S, Gasbarrini A, Lencioni M, Cicalese L, Sherman M, Gridelli C, Buggisch P, Gerken G, Schmid RM, Boni C, Personeni N, Hassoun Z, Abbadessa G, Schwartz B, Von Roemeling R, Lamar ME, Chen Y, Porta C. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
- 87.Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, von Pawel J, Schwartz B, Von Roemeling R, Sandler AB. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13(5):391–395. doi: 10.1016/j.cllc.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 88.Previdi S, Abbadessa G, Dalò F, France DS. Broggini M Breast cancer-derived bone metastasis can be effectively reduced through specific c-MET inhibitor tivantinib (ARQ 197) and shRNA c-MET knockdown. Mol Cancer Ther. 2012;11(1):214–223. doi: 10.1158/1535-7163.MCT-11-0277. [DOI] [PubMed] [Google Scholar]
- 89.Rosen LS, Senzer N, Mekhail T, Ganapathi R, Chai F, Savage RE, Waghorne C, Abbadessa G, Schwartz B, Dreicer R. A phase I dose-escalation study of Tivantinib (ARQ 197) in adult patients with metastatic solid tumors. Clin Cancer Res. 2011;17(24):7754–7756. doi: 10.1158/1078-0432.CCR-11-1002. [DOI] [PubMed] [Google Scholar]
- 90.Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S, Ramlau R, Arthur S, Gorbachevsky I, Schwartz B, Schiller JH. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–3315. doi: 10.1200/JCO.2010.34.0570. [DOI] [PubMed] [Google Scholar]
- 91.Moran-Jones K, Brown LM, Samimi G. INC280, an orally available small molecule inhibitor of c-MET, reduces migration and adhesion in ovarian cancer cell models. Sci Rep. 2015;5:1174. doi: 10.1038/srep11749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brandes F, Schmidt K, Wagner C, Redekopf J, Schlitt HJ, Geissler EK, Lang SA. Targeting cMET with INC280 impairs tumour growth and improves efficacy of gemcitabine in a pancreatic cancer model. BMC Cancer. 2015;15:71. doi: 10.1186/s12885-015-1064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bladt F, Faden B, Friese-Hamim M, Knuehl C, Wilm C, Fittschen C, Grädler U, Meyring M, Dorsch D, Jaehrling F, Pehl U, Stieber F, Schadt O, Blaukat A. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res. 2013;19(11):2941–2945. doi: 10.1158/1078-0432.CCR-12-3247. [DOI] [PubMed] [Google Scholar]
- 94.Padda S, Neal JW, Wakelee HA. MET inhibitors in combination with other therapies in non-small cell lung cancer. Transl Lung Cancer Res. 2012;1(4):238–253. doi: 10.3978/j.issn.2218-6751.2012.10.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Belalcazar A, Azaña D, Perez CA, Raez LE, Santos ES. Targeting the Met pathway in lung cancer. Expert Rev Anticancer Ther. 2012;12(4):519–528. doi: 10.1586/era.12.16. [DOI] [PubMed] [Google Scholar]
- 96.Marchion DC, Bicaku E, Xiong Y. Bou Zgheib N, A Sawah E, Stickles XB, Judson PL, Lopez AS, Cubitt CL, Gonzalez-Bosquet J, Wenham RM, Apte SM, Berglund A, Lancaster JM. A novel c-Met inhibitor, MK8033, synergizes with carboplatin plus paclitaxel to inhibit ovarian cancer cell growth. Oncol Rep. 2013;29(5):2011–2018. doi: 10.3892/or.2013.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yeh I, Botton T, Talevich E, Shain AH, Sparatta AJ, de la Fouchardiere A, Mully TW, North JP, Garrido MC, Gagnon A, Vemula SS, McCalmont TH, LeBoit PE, Bastian BC. Activating MET kinase rearrangements in melanoma and Spitz tumours. Nat Commun. 2015;6:7174. doi: 10.1038/ncomms8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cui JJ, McTigue M, Nambu M, Tran-Dubé M, Pairish M, Shen H, Jia L, Cheng H, Hoffman J, Le P, Jalaie M, Goetz GH, Ryan K, Grodsky N, Deng YL, Parker M, Timofeevski S, Murray BW, Yamazaki S, Aguirre S, Li Q, Zou H, Christensen J. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1-yl)ethanol (PF-04217903) for the treatment of cancer. J Med Chem. 2012;55(18):8091–8109. doi: 10.1021/jm300967g. [DOI] [PubMed] [Google Scholar]
- 99.Zou HY, Li Q, Lee JH, Arango ME, Burgess K, Qiu M, Engstrom LD, Yamazaki S, Parker M, Timofeevski S, Cui JJ, McTigue M, Los G, Bender SL, Smeal T, Christensen JG. Sensitivity of selected human tumor models to PF-04217903, a novel selective c-Met kinase inhibitor. Mol Cancer Ther. 2012;11(4):1036–1047. doi: 10.1158/1535-7163.MCT-11-0839. [DOI] [PubMed] [Google Scholar]
- 100.Giordano S, et al. Different point mutations in the met oncogene elicit distinct biological properties. FASEB J. 2000;14:401–408. doi: 10.1096/fasebj.14.2.399. [DOI] [PubMed] [Google Scholar]
- 101.Chung JH, Lynch DA. The value of multidisciplinary approach to the diagnosis of usual interstitail pneumonitis and idiopatic pulmonary fibrosis: radiology, pathology and clinical correlation. AJR Am J Roentgenol. 2016;206(3):463–471. doi: 10.2214/AJR.15.15627. [DOI] [PubMed] [Google Scholar]
- 102.Spagnolo P, Sverzellati N, Rossi G, Cavazza A, Tzouvelekis A, Crestani B, Vancheri C. Idiopathic pulmonary fibrosis: an update. Ann Med. 2015;47(1):15–27. doi: 10.3109/07853890.2014.982165. [DOI] [PubMed] [Google Scholar]
- 103.Huang SK, Horowitz JC. Outstanding their welcome: the persistent myofibroblasts in IPF. Austin J Pulm Resp Med. 2014;1(1):3. [PMC free article] [PubMed] [Google Scholar]
- 104.Moore MW, Herzog EL. Regulation and relevance of myofibroblast responses in idiopatic pulmonary fibrosis. Curr Pathobiol Rep. 2013;1(3):199–208. doi: 10.1007/s40139-013-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- 106.Noble PW, Barkarauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sivakumar P, Ntolios P, Jenkins G, Laurent G. Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr Opin Pulm Med. 2012;18:462–469. doi: 10.1097/MCP.0b013e328356800f. [DOI] [PubMed] [Google Scholar]
- 108.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 109.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 110.Lu P, Werb Z. Patterning mechanisms of branched organs. Science. 2008;322(1506–56):2. doi: 10.1126/science.1162783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Michalopoulos GK, Defances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 113.Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 114.Owen KA, Qiu D, Alves J, Schumacher AM, Kilpatrick LM, Li J, Harris JL, Ellis V. Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease uPA. Biochem J. 2010;426(2):219–228. doi: 10.1042/BJ20091448. [DOI] [PubMed] [Google Scholar]
- 115.Tokonou M, Niki T, Eguchi K, Iba S, Tsuda H, Yamada T, Matsuno Y, Kondo H, Saitoh Y, Imamura H, Hirohashi S. C-MET expression in myofibroblsts: ole in autiocrine activation and prognostic significance in lung adenocarcinoma. Am J Pathol. 2001;158(4):1451–1463. doi: 10.1016/S0002-9440(10)64096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chilosi M, Poletti V, Zamò A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, et al. Aberrant wnt/beta catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162:1497–1502. doi: 10.1016/S0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.De Bacco F, Luraghi P, Medico E, Reato G, Girolami F, Perera T, Gabriele P, Comoglio PM, Boccaccio C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J Natl Cancer Inst. 2011;103(8):645–646. doi: 10.1093/jnci/djr093. [DOI] [PubMed] [Google Scholar]
- 118.Stella GM, Benvenuti S, Gramaglia D, Scarpa A, Tomezzoli A, Cassoni P, Senetta R, Venesio T, Pozzi E, Bardelli A, Comoglio PM. MET mutations in cancers of unknown primary origin (CUPs) Hum Mutat. 2011;32(1):44–50. doi: 10.1002/humu.21374. [DOI] [PubMed] [Google Scholar]
- 119.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 120.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stella GM, Luisetti M, Pozzi E, Comoglio PM. Oncogenes in non-small-cell lung cancer: emerging connections and novel therapeutic dynamics. Lancet Respir Med. 2013;1(3):251–261. doi: 10.1016/S2213-2600(13)70009-2. [DOI] [PubMed] [Google Scholar]
- 122.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hylnd C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;136(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 123.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistance and clonal selction of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Piao Y, Park SY, Henry V, Smith BD, Tiao N, Flynn DL, de GrootJF. Novel MET/TIE2/VEGFR2 inhibits tumor growth and invasiveness in bevcizumab-resistant glioblastoma mouse models. Neuro Oncol. 2016. In Press. [DOI] [PMC free article] [PubMed]
- 125.Stella GM, Senetta R, Inghilleri S, di Cantogno LV, Mantovani C, Piloni D, Scudeller L, Meloni F, Papotti M, Ricardi U, Cassoni P. MET mutation are associated with aggressive and radioresistant brain metastatic non-small-cell lung cancer. Neuro Oncol. 2016;18(4):598–599. doi: 10.1093/neuonc/nov325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jones MG, Fabre A, Schneider P, Cinetto F, Sgalla G, Mavrogordato M, Jogai S, Alzetani A, Marshall BG, O’Really KW, et al. Three-dimensional characterization of fibroblast fociin idiopathic pulmonary fibrosis. JCI Insight. 2016; 1(5). [DOI] [PMC free article] [PubMed]
- 127.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad USA. 2004;101(13):4477–4482. doi: 10.1073/pnas.0306068101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Borowiak M, Garrat AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad USA. 2004;101(29):10608–10613. doi: 10.1073/pnas.0403412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177(1):151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Giordano S, Corso S, Conrotto P, Artigini S, Gilestro G, Barberis D, Tamagnone L, Comoglio PM. The semaphorin 4D receptor controls invasive growth by coupling with MET. Nat Cell Biol. 2002;4(9):720–724. doi: 10.1038/ncb843. [DOI] [PubMed] [Google Scholar]
- 132.Conrotto P, Corso S, Gamberini S, Comoglio PM, Giordano S. Interplay between scatter factor receptors and B plexins controls invasive growth. Oncogene. 2004;23(30):5131–5137. doi: 10.1038/sj.onc.1207650. [DOI] [PubMed] [Google Scholar]
- 133.Peng HY, Gao W, Chong FR, Liu HY, Zhang JI. Semaphorin A4 enhances lung fibrosis through activation of akt via plexinD1 receptor. J Biosci. 2015;40(5):855–862. doi: 10.1007/s12038-015-9566-9. [DOI] [PubMed] [Google Scholar]
- 134.Reilkoff RA, Peng H, Muray LA, Peng X, Russel T, Montgomery R, Feghali-Bostwick C, Shaw A, Horrner RJ, Gulati M, Mathur A, Elias JA, Herzog EL. Semaphorin 7a-regulatoyT cells are associate with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor beta1-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187(2):180–188. doi: 10.1164/rccm.201206-1109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gan Y, Feghali-Bostwick C, Peng X, Russel T, Chen Q, Mathai SK, Homer R, Gulati M, Siner J, Elias J, Bucala R, Herzog E. Role of semaphoring 7a signaling in transforming growth factor beta-induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63(8):2484–2494. doi: 10.1002/art.30386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204(5):1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Selman M, Pardo A, Kaminski N. Idiopatic pulmonary fibrosis: aberrant recapitulation of developmental pograms? PLoS Med. 2008;5(3):e62. doi: 10.1371/journal.pmed.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Holland JD, Klaus A, Garrat AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25(2):254–264. doi: 10.1016/j.ceb.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 139.Anatas JN, Moon RT. WNT signaling patways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 140.Konighshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, Eickelberg O. Functional Wnt signaling is increased in idiopatic pulmonary fibrosis. PLoS ONE. 2008;3(5):e2142. doi: 10.1371/journal.pone.0002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu Y, Chattopadhyay N, Qin S, Szekeres C, Vasylyev T, Mahoney ZX, Taglienti M, Bates CM, Chapman HA, Miner JH, Kreidberg JA. Coordinate integrin and c-Met signaling regulate Wnt gene expression during epithelial morphogenesis. Development. 2009;136:843–853. doi: 10.1242/dev.027805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boon EM, van der Neut R, van de Wetering M, Clevers H, Pals ST. Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer. Cancer Res. 2002;62:5126–5128. [PubMed] [Google Scholar]
- 143.Fodde R, Brabletz T. Wnt/beta catenin signaling inn cancer stemness and malignant behaviour. Curr Opin Cell Biol. 2007;19(2):150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 144.Kim KH, Seol HJ, Kim EH, Rhee J, Jin HJ, Lee Y, Joo KM, Lee J, Nam DH. Wnt/β-catenin signaling is a key downstream mediator of MET signaling glioblastoma stem cells. Neuro Oncol. 2013;1582:161–171. doi: 10.1093/neuonc/nos299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Attisano L, Wrana JL. Signal transduction by the TGFβ superfamily. Science. 2002;296(5573):1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 146.Massague J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGFβ family signaling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 148.Weiss A, Attisano L. The TGFβ superfamily signaling pathway. WIRE Dev Biol. 2013;2:47–63. doi: 10.1002/wdev.86. [DOI] [PubMed] [Google Scholar]
- 149.Shi Y, Massague J. Mechanisms of TGFβ signalling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 150.Guo X, Wang XF. Signaling cross talk betwee TGF & #x03B2;/BMP and other pathways. Cell Res. 2009;18(1):71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Neuzillet C, Tjeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A. Targeting TGFβ pathway for cancer therapy. Pharmacol Therap. 2015;147:22–31. doi: 10.1016/j.pharmthera.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 152.Ye L, Jiang WG. Bone morphogenetic proteins in tumor associated angiogenesis and implication in cancer therapies. Cancer Lett. 2015; pii: S0304–3835(15):0074–1. [DOI] [PubMed]
- 153.De Caestecker MP, Piek E, Roberts AB. Role of transforming growth factor -β signaling in cancer. J Natl Cancer Inst. 2000;92(17):1388–1402. doi: 10.1093/jnci/92.17.1388. [DOI] [PubMed] [Google Scholar]
- 154.Fernandez IE, Eickelberg O. The impact of TGFβ on lung fibrosis. Proc Am Thorc Soc. 2012;9(3):11–116. doi: 10.1513/pats.201203-023AW. [DOI] [PubMed] [Google Scholar]
- 155.Akhurst RJ, Hata A. targeting the TGFβsignalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Raghu G, Johnson C, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone. Am J Resp Crit Care Med. 1999;159:1061–1069. doi: 10.1164/ajrccm.159.4.9805017. [DOI] [PubMed] [Google Scholar]
- 157.Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakat K, Taguchi Y, Nagai S, Itoh H, Ohi M, et al. Double-blind, placebo-controlled trial of pirfenidone in patients wuth idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–1047. doi: 10.1164/rccm.200404-571OC. [DOI] [PubMed] [Google Scholar]
- 158.Kim SE, Keating GM. Pirfenidone: a review of its use in idiopatic pulmonary fibrosis. Adis Drug Eval. 2015;75(2):219–230. doi: 10.1007/s40265-015-0350-9. [DOI] [PubMed] [Google Scholar]
- 159.Selvaggio AS, Noble PW. Pirfenidone iniates a new era in the treatment of idiopathic pulmonary fibrosis. Ann Rev Med. 2016;67:487–495. doi: 10.1146/annurev-med-120214-013614. [DOI] [PubMed] [Google Scholar]
- 160.Ueki T, Kaneda Y, Tsutsui H, Nakasaki K, Sawa Y, Morishita R, Matsumoto K, Nakamura T, Takahashi H, Okamoto E, Fujimoto J. hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat Med. 1999;5(2):226–230. doi: 10.1038/5593. [DOI] [PubMed] [Google Scholar]
- 161.Yang J, Dai C, Liu Y. Hepatocyte growth factor suppresses renal interstitial myofibroblstas activation and intercepts smad signal transduction. M J Pathol. 2003;163(2):621–632. doi: 10.1016/S0002-9440(10)63689-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dai C, Liu Y. Hepatocyte growth factor antagonizes the profibrotic action of TGFβ1 in mesangial cells by stabilizing the Smad transcriptional corepressor TGIF. J Am Soc Nephrol. 2004;15(6):1402–1412. doi: 10.1097/01.ASN.0000130568.53923.FD. [DOI] [PubMed] [Google Scholar]
- 163.Rabbani ZN, Mi J, Zhang Y, Delong M, Jackson IL. Hypoxia inducible factor 1α signaling in fractionated radiation induced lung injury: role of oxidative stress and tissue hypoxia. Radiat Res. 2010;173:165–174. doi: 10.1667/RR1816.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Sayto Y, Johnson RS, Kretzler M, Cohen CD, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Krick S, Eul BG, Hanze J, Savai R, Grimminger F, Seeger W, Rose F. Role of hypoxia-inducible factor-1 alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am J Respir Cell Mol Biol. 2005;32:395–403. doi: 10.1165/rcmb.2004-0314OC. [DOI] [PubMed] [Google Scholar]
- 166.Jain M, Sznajder JI. Effects of hypoxia on the alveolar epithelium. Proc Am Thorac Soc. 2005;2:202–205. doi: 10.1513/pats.200501-006AC. [DOI] [PubMed] [Google Scholar]
- 167.Bodempudi V, Hergert P, Smith K, Xia H, Herrera J, Perteson M, Khalil W, Kahm J, Bitterman PB. Hencke CA.miR-210 promotes IPF fibroblast proliferation in response to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2014;307(4):L283–L294. doi: 10.1152/ajplung.00069.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Alderton GK. Tumorigenesis-pathway to stemness. Nat Rev Cancer. 2016;16:70. [Google Scholar]
- 169.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]