

Abstract
Objective(s):
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignant tumor. Administration of chemical compounds influencing apoptosis and T cell development has been discussed as promising novel therapeutic strategies. Valproic acid (VPA) as a recently emerged anti-neoplastic histone deacetylase (HDAC) inhibitor and pioglitazone (PGZ) as a high-affinity peroxisome proliferator-activated receptor-gamma (PPARγ) agonist have been shown to induce apoptosis and cell cycle arrest in different studies. Here, we aimed to investigate the underlying molecular mechanisms involved in anti-proliferative effects of these compounds on human Jurkat cells.
Materials and Methods:
Treated cells were evaluated for cell cycle progression and apoptosis using flowcytometry and MTT viability assay. Real-time RT-PCR was carried out to measure the alterations in key genes associated with cell death and cell cycle arrest.
Results:
Our findings illustrated that both VPA and PGZ can inhibit Jurkat E6.1 cells in vitro after 24 hr; however, PGZ 400 μM presents the most anti-proliferative effect. Interestingly, treated cells have been arrested in G2/M with deregulated cell division cycle 25A (Cdc25A) phosphatase and cyclin-dependent kinase inhibitor 1B (CDKN1B or p27) expression. Expression of cyclin D1 gene was inhibited when DNA synthesis entry was declined. Cell cycle deregulation in PGZ and VPA-exposed cells generated an increase in the proportion of aneuploid cell population, which has not reported before.
Conclusion:
These findings define that anti-proliferative effects of PGZ and VPA on Jurkat cell line are mediated by cell cycle deregulation. Thus, we suggest PGZ and VPA may relieve potential therapeutic application against apoptosis-resistant malignancies.
Keywords: Pioglitazone, Proliferation, T-cell leukemia, Valproic acid
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignant tumor arising from T-cell hematopoietic progenitors with poor prognosis. ALL accounts for almost 15% of recently diagnosed ALL cases in children and 25% in adults (1). The relapse rate in T-ALL patients has been reported as 30% and the outcome has remained extremely poor (2). Conventional chemotherapy as a routinely adopted therapeutic method has shown minimal promising improvement on survival and quality of life for T-ALL patients (3, 4). Therefore, novel and effective therapeutic strategies for T-ALL treatment especially biological compounds targeting cell signaling pathways thereupon influencing pathogenesis and development of T cells should be regarded (5). Aberrant histone acetylation has been reported in the development of several malignancies (6). Conformational state of chromatin and consequently gene expression levels could be altered through induction of histone acetylation. The recruitment of histone deacetylase (HDAC) inhibits the cell cycle distribution and induces apoptosis and cell cycle arrest and increased radiation sensitivity in malignant tumors (7). Therefore, HDAC inhibitors could be introduced as a novel class of therapeutic agents in hematologic and other malig-nancies (8-10). Valproic acid (VPA) is a recently emerged antineoplastic HDAC inhibitor that is clinically used in the treatment of epilepsy (8). Antineoplastic effects of VPA are mediated through inhibition of class I HDAC (11). Induction of apoptosis and cell cycle arrest has been demonstrated in several VPA-treated cancer types and leukemia (8, 12-14). Peroxisome proliferator-activated receptor-gamma (PPARγ) is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors that is expressed in several cell types of the immune system including inflammatory immune cells such as macrophages and T-lymphocytes (15, 16). PPARγ is involved in different physiological and pathological states including glucose homeostasis, cellular differen-tiation, inflammation and cancer (17). Recent studies have documented PPARγ expression in a variety of cancer cells implying its role in the suppression of cell growth and promotion of differentiation and/or apoptosis (18, 19). Pioglitazone (PGZ) is a member of thiazolidinedione (TZD) family, which is a high-affinity PPARγ agonist (20). Although PGZ is able to decrease serum glucose levels in patients with diabetes mellitus type 2 (21), anti-proliferative and anti-inflammatory properties of PGZ have been characterized previously in several cancer types (22-24).
The cell cycle is a mechanism by which eukaryotic cells replicate. Growth-inhibitory signals influ-ence cell cycle progression through their action on a family of cyclin-dependent kinases (CDKs). Cyclins function as regulators of CDK kinases. Different cyclins exhibit distinguished expression and degra-dation patterns that contribute to the temporal coordination of each mitotic event. Cyclin D1 is required for cell cycle G1 to S transition (25). Mutations, amplification, and overexpression of cyclin D1 gene are observed frequently in a variety of tumors and may contribute to tumorigenesis (26). Cdc25A is also a member of the Cdc25 family of phosphatases that is necessary for G1 to S cell cycle progression. It is specifically degraded in response to DNA damage, which hampers cells with chromosomal abnormalities from progressing through cell division (27). CDKN1B (p27) controls the cell cycle progression at G1. The degradation of this protein, which is triggered by its CDK dependent phosphorylation is required for the cellular transition from quiescence to the proliferative state. Mutations of the p27 gene are mostly observed in hematologic malignancies including hairy cell leukemia (HCL) (28). Fas cell surface death receptor (FasR) is a member of the TNF-receptor superfamily. This receptor contains a death domain that has been shown to play a critical role in the physiological regulation of programmed cell death and has been implicated in the pathogenesis of numerous malignancies (29).
The aim of the present study was to evaluate the apoptotic and anti-proliferative effects of VPA and PGZ in Jurkat T-ALL leukemia cell line by quantifying the expression level of cyclin D1, FasR, p27, and Cdc25A genes using real-time RT-PCR method. The results of the present study might introduce VPA and PGZ as probable therapeutic compounds on T-ALL to be further investigated.
Materials and Methods
Materials
PGZ was obtained from Dr Reddy’s Laboratories (India). VPA and RNase A were purchased from Sigma-Aldrich Company (St. Louis, MO, U.S.A). Human Jurkat (clone E6-1) leukemic cells were obtained from NCBI (National Cell Bank of Iran, Pasteur Institute, Tehran, Iran). Fetal Bovine Serum (FBS), High glucose RPMI 1640, penicillin/streptomycin solution, and all other cell culture reagents were purchased from Gibco (Life Technologies, USA). Annexin V-FITC apoptosis detection kit and propidium iodide came from eBioscience Inc. (San Diego, CA, USA). BIOZOL reagent from BioFlux (Bioer, China) was used for total RNA extraction. Real-time PCR (qPCR), SYBR Green Master Mix, and cDNA synthesis kit were purchased from Clontech Laboratories, Inc. (TAKARA, Japan).
Cell culture and treatments
Human Jurkat cells were cultured in RPMI 1640 supplemented with 10% FBS, 100U/ml penicillin, 100 µg/ml streptomycin, and 2 mM Glutamax, then incubated in a fully humidified atmosphere at 37 °C with 5% CO2. They were passed twice a week and prepared for experimental procedures when in log-phase growth. Cells (2-5x105/ml) were washed once with 37 °C PBS and were seeded in 6 well plates followed by resuspension in complete growth media. Cells were then treated with varying concentrations of VPA (2.5 mM and 5 mM) and PGZ (200 µM and 400 µM) obtained from relevant studies (13, 20, 30). Control experiments were carried out by adding PBS to Jurkat cells while non-treated cells were prepared as well. The trypan blue exclusion assay was used to determine cell viability, and the live and dead cells were enumerated using a hemocytometer in each cell culture step. Jurkat cells treated with different concentrations of VPA and PGZ were incubated for 24 hr at 37 °C with 5% CO2. Treated and non-treated cells were then collected by centrifugation at 500 g for 3 min, washed three times and separated into two tubes for cell cycle analysis and RNA extraction. For apoptosis assay, cells were treated the same followed by the MTT methods described below.
Propidium iodide (PI) staining for cell cycle analysis
Cell cycle analysis was performed using propidium iodide staining. Briefly, cells were washed in phosphate-buffered saline (PBS) and fixed dropwise in ice cold 70% ethanol at 20 °C for 2 hr. Then, cells were washed in PBS and treated with 50 μg/ml RNAs A for 30 (30 min, 37 °C) and stained using 20 μg/ml propidium iodide in the presence of 0.1% triton-x 100. Cells were then analyzed by the Partec flowcytometer (Particle Analyzing System, Germany) and the Flowing software v 2.5.1 (Cell Imaging Core, Turku Centre for Biotechnology, Finland).
Flowcytometry analysis for apoptosis quantification
Apoptosis was determined by Annexin V and PI double staining of treated and non-treated Jurkat cells using eBioscience apoptosis kit following manufacturer’s procedures. In brief, harvested cells were washed with cold PBS and then were resuspen-ded in 100 µl of binding buffer followed by incubation with 5 µl of Annexin V-FITC for 15 min at room temperature in the dark. Cells were then washed using a binding buffer, resuspended in 200 µl of binding buffer supplemented with 5 µl of PI and then analyzed using flowcytometer within one hr. In our study, apoptotic cells included early (Annexin V+, PI-) and late (Annexin V+, PI+) apoptosis while viable cells were negative for both Annexin V and PI. As a positive control for apoptosis, Jurkat cells were irradiated with ultraviolet for 10 min and after 3 hr incubation were stained as above.
Real-time RT-PCR
Total RNA was extracted from collected cells using BIOZOL according to the manufacturer’s protocol. One microgram of total RNA was reverse transcribed to cDNA with random hexamer primers. Real-time RT-PCR was performed using Bioer Real-time PCR detection system (Hangzhou high tech, Bioer Technology, China). Glyceraldehyd-3-phospate dehydrogenase (GAPDH) was used as a suitable internal control for gene expression normalization (31). Gene-specific primers for cyclin D1, FasR, p27, Cdc25A (as candidate key regulators of apoptosis and cell cycle transition), and GAPDH are summarized in Table 1. PCR amplifications were performed using TAKARA master mix. For each PCR, 1 μl template cDNA, equivalent to approximately 100 ng total RNA, was mixed with 12.5 μl 2× SYBR Green PCR master mix and 0.4 μM of each forward and reverse primer in a final volume of 20 μl under the following conditions: Initial enzyme activation at 95 °C for 10 min, amplification for 40 cycles (95 °C for 30 sec, 60 °C for 60 sec), followed by a dissociation curve analysis.
Table 1.
Gene-specific primers used for real-time RT-PCR
| Primer (Accession) | Sequence (5’>3’) | Tm | Amplicon size |
|---|---|---|---|
| Cyclin D1 (NM_053056) | F:GAGGCGGAGGAGAAGAAAGAG | 60 | 179 bp |
| R:AGGCGGTAGTAGGACAGGAAG | |||
| FasR (NM_152872) | F:GCTTCCTTCCCATCCTCCTG | 59 | 123 bp |
| R:GTCACTCGTAAACCGCTTCC | |||
| P27 (NM_004064) | F:CAGGAGAGCCAGGATGTCAG | 59 | 178 bp |
| R:GAGTAGAAGAATCGTCGGTT | |||
| Cdc25A (XM_006713435) | F:TTTGGCCAGCAGCATTTCTGTG | 60 | 109 bp |
| R:AGCCACAGTGGGATGAACCAGC | |||
| GAPDH (NM_001289746) | F: GAGCGAGATCCCTCCAAAAT | 60 | 196 bp |
| R: GGCTGTTGTCATACTTCTCATG |
Cell viability (MTT) assay
Cells were seeded at 5x104 cells/well in a 24-well plate with different concentrations of VPA and PGZ and observed after 24 hr of incubation using MTT assay (32-34). Briefly, cells were incubated in triplicate at different concentrations of VPA and PGZ in a final volume of 200 µl of phenol red-free RPMI 1640 for 20 hr at 37 °C with 5% CO2. 20 µl of MTT solution (5 mg/ml) was added to each well and incubated for 4 hr at 37 °C with 5% CO2. Formazan crystals were formed. The 24-well plate was then centrifuged at 400 g for 5 min and media was removed. 300 µl DMSO was then added to each well as a cell lysis solution. Cell viability percentage was assessed using spectrophotometry at 570 nm using the ELx800 Absorbance Reader (Biotek, USA).
Results
PGZ is a more effective inhibitor of proliferation in Jurkat cells
To measure the cytotoxic effects of VPA and PGZ, various increasing concentrations of both were selected and applied to cell culture medium of Jurkat cells for 24 hr. Then, percentages of cell viability were measured by MTT assay (Figure 1). Our results indicated that although PGZ and VPA successfully inhibit cell growth of Jurkat cells at all tested concentrations, they were more effective at higher levels. The most toxicity was observed for 400 μM concentration of PGZ, when about 75% of Jurkat cells were killed followed by VPA 5 mM (55%), PGZ 200 μM (30%), and VPA 2.5 mM (25%).
Figure 1.
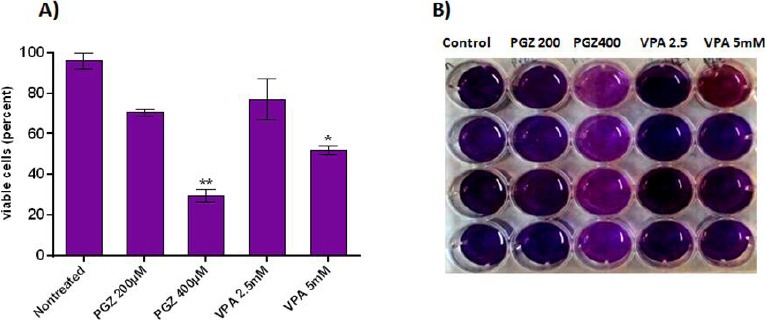
The cytotoxic effects of VPA (2.5 mM and 5 mM) and PGZ (200 µM and 400 µM) measured by MTT Assay. A) The percentage of the viable cell population is visualized in different groups of treated Jurkat cells. B) The MTT assay plate of stained cells. Each column represents different groups. *P<0.05, **P<0.005
Cell growth inhibition of VPA or PGZ treated Jurkat cells is not associated with programmed cell death
In order to address the cytotoxic effects of the selected treatments, PGZ or VPA-treated cells were analyzed for apoptosis phenotype. The surface phos-phatidylserine expression and membrane permeability were measured using the Annexin V/PI staining kit. As a positive control, UV-irradiated apoptotic Jurkat T cells were used to ensure the staining procedure.
Surprisingly, there were no obvious apoptotic Jurkat cells detectable in any of PGZ or VPA-treated samples in comparison with the control cells (Figure 2). These results indicate that although PGZ or VPA exposure for 24 hr can inhibit the growth of Jurkat T cells, this effect is not associated with programmed cell death response. The Jurkat T-cell line with nonfunctional mutant TP53, possesses specific characteristics that determine its apoptotic response.
Figure 2.
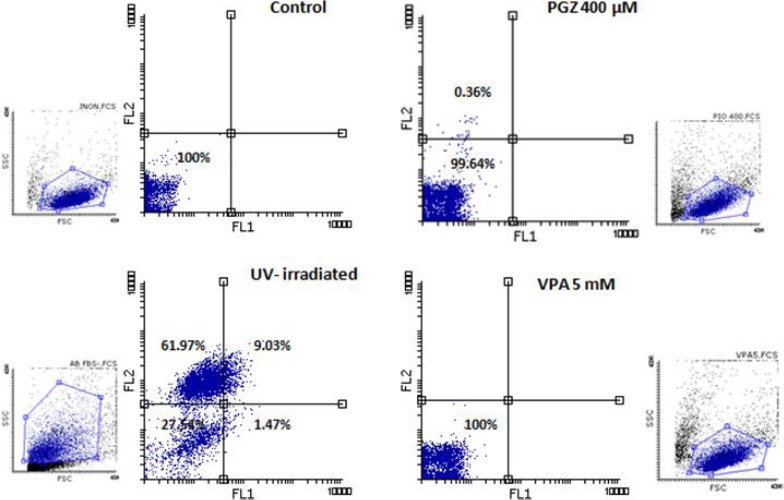
The apoptosis-specific staining of treated cells. FL1 channel indicates Annexin V-FITC and FL2 detects PI signals. Notice the UV-irradiated positive control cells are positive for PI and Annexin V, however, none of treated Jurkat cells express apoptotic signals
A G2/M phase arrest inhibits treated Jurkat cell cycle progression
To explore the underlying mechanism of VPA and PGZ toxic effects more precisely, we studied the cell cycle progression using wider concentrations of drugs for this experiment. Jurkat cells treated with 100, 200, and 400 μM of PGZ or 2.5, 5, and 10 mM of VPA in duplicate were analyzed for DNA content using PI staining followed by flowcytometry. We found an evident general cell cycle progression disruption in treated cells that is presented in Figure 3. As indicated, VPA introduced a sub G1 increase in treated cells which resembles dying cells. All treatments induced a G2/M arrest in the cell cycle of Jurkat cells. Particularly, in VPA 2.5 mM exposure, about 50 percent of cells were stopped in G2/M, which consequently decreases G1 cells. The popu-lation of cells in G1 phase is near control in PGZ however S phase entry is declined in this group drastically. Another alteration is the genotoxic effect of both VPA and PGZ in aneuploidy accumulation. Control Jurkat cells contain about 6% cells naturally, but this increases by treatment up to 14% in PGZ (200 μM) and 12% in VPA (2.5 mM).
Figure 3.
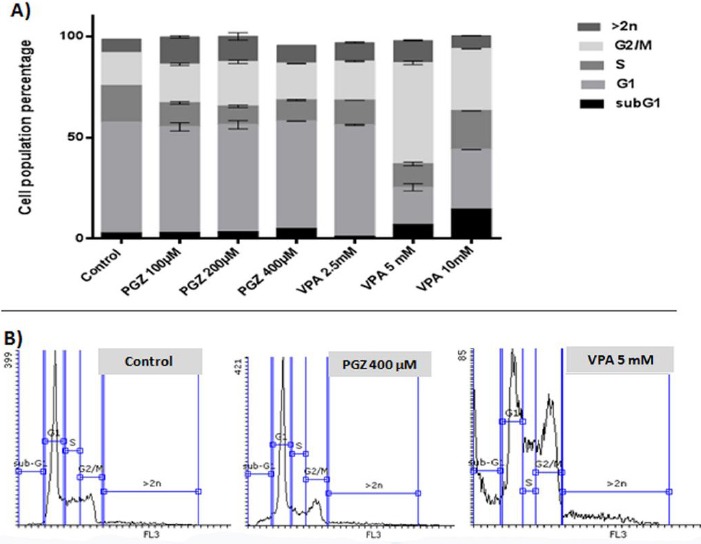
Cell cycle analysis of treated Jurkat cells. A) Each bar shows the percentage of the cell population in each cell cycle phase proportionally for different groups. B) The histogram flowcytometry graphs for PI stained nucleus of control, PGZ 400 μM, and VPA 5 mM groups are presented as sample
Gene expression of cell cycle regulators is deregu-lated in treated Jurkat cells
The general cell cycle disruption led us to measure the related gene expression alterations in treated cells. Different concentrations of PGZ and VPA were applied to Jurkat cells for this experiment and a list of genes (p27, Cdc25A, FasR, and cyclin D1), which are involved in S phase entry and G2/M regulation were candidated for measurement. The results (Figure 3) illustrated that the expression of Cdc25A phosphatase, which is associated with G2/M transition, decreases upon VPA and PGZ treatment, however, a significant increase was observed in p27 expression, one of the cell cycle regulators. This finding is in accordance with G2/M arrest, which was achieved in cell cycle experiments. Indeed the expression of cyclin D1 was declined almost to least in PGZ 400 μM, which was presented as restrained S phase entry. Noticeably, the expression of FasR was up-regulated in higher concentrations of treatments, although no apoptosis was detected.
Discussion
PGZ and VPA have been commonly used as therapeutic chemical compounds in diabetes and epilepsy disorders. Recently, there have been reports of their potential beneficial effects on cancer treatment.
VPA derivatives modulate histone acetylating and have provided promising results in solid tumor clinical trials as epigenetic cancer treatment (12, 35-37). Moreover, in chronic myeloid leukemia (CML), VPA can induce apoptosis and cell arrest (38) and even can restore imatinib sensitivity in resistant cells(39, 40). Here we investigated VPA effect on Jurkat leukemia cells that have a TP53 mutation (41). Our findings illustrated that sodium valproate inhibits Jurkat proliferation in a G2/M arrest depen-dent manner, which is concordant with Cdc25A downregulation. VPA induced cell cycle arrest has been reported for other cell lines previously (30, 42). Indeed, HDAC inhibition can induce a DNA damage response (43), which can amplify the G2/M accumu-lated cells. The observed expressional changes in Cdc25A and p27 can link the cell cycle disruption to damaged DNA in VPA-treated Jurkat cells.
It has been previously reported that PPARγ activation mediated by PGZ, exhibits a differential decrease in viable leukemia cells measured by trypan blue exclusion assay, while normal hematopoietic cells were unaffected (44). It has been suggested that PGZ induces a G1 cell arrest in HL60, another leukemia cell line; however the underlying mechanisms remain to be investigated (45). It has been reported that PGZ can inhibit cancer cell proliferation predominantly by cell cycle arrest with minor apoptotic changes (46). Here, we presented that PGZ can inhibit leukemia Jurkat cells proliferation in an apoptosis-independent manner mainly by G2/M transmission regulation. Similar effects have been reported for troglitazone, another TDZ that induces P27 expression and inhibits cell cycle progression in HCC (47). We found a decline in Cdc25A phosphatase gene expression in response to PGZ treatment that has not been reported before.
The Fas-mediated apoptosis induction has been reported with VPA(11) and PGZ exposure (48). However, we observed an up-regulation of Fas gene expression while no apoptosis was detected. The specific characteristics of Fas-induced extrinsic apoptosis pathway in Jurkat cell line may contribute to this nonfunctional Fas accumulation. Interestingly, the observed S phase inhibition in PGZ 400 μM is concordant with a decrease in cyclin D1 expression, which promotes G1 to S transition.
Proliferation of Jurkat leukemia cells can be stopped by exposure to lower concentrations of ciprofloxacin only by G2/M cell cycle arrest and chromosomal instability or aneuploidy induction, without any apoptosis (49). It has been reported that PGZ can introduce genotoxicity and chromosomal instability in human lymphocytes (50). Similarly, we found such a genotoxic effect for PGZ and VPA on Jurkat leukemia cell line attributed to the increase in >2n nucleus and the cell cycle arrest mediated by p27 and Cdc25A, DNA damage response regulators.
Conclusion
Altogether, our results indicate that PGZ and VPA, two common clinical drugs, can inhibit Jurkat leukemia cells proliferation with a chronic cell cycle deregulation. It seems that the underlying mecha-nism is not affiliated to the apoptosis pathway. The PGZ and VPA may relieve potential therapeutic applications against leukemia and other malignancies considering the suggested apoptosis-independent mechanism.
Acknowledgment
This work was supported by a grant from Golestan University of Medical Sciences, Gorgan, Iran (Grant Number: 930618118). We wish to thank Dr Jahanbakhsh Asadi, Daroo Darman Pars Company, Mrs M Haydari, and Dr S Zhand for their help in accomplishing this study.
References
- 1.Ge J, Liu Y, Li Q, Guo X, Gu L, Ma ZG, et al. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed Environ Sci. 2013;26:902–911. doi: 10.3967/bes2013.019. [DOI] [PubMed] [Google Scholar]
- 2.Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29:551–565. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao M, Gao L, Tao Y, Hou J, Yang G, Wu X, et al. Proteasome inhibitor carfilzomib interacts synergistically with histone deacetylase inhibitor vorinostat in Jurkat T-leukemia cells. Acta Biochim Biophys Sin. 2014;46:484–491. doi: 10.1093/abbs/gmu030. [DOI] [PubMed] [Google Scholar]
- 4.Conter V, Bartram CR, Valsecchi MG, Schrauder A, Panzer-Grümayer R, Möricke A, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115:3206–3214. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 5.Belayachi L, Aceves-Luquero C, Merghoub N, Bakri Y, de Mattos SF, Amzazi S, et al. Retama monosperma n-hexane extract induces cell cycle arrest and extrinsic pathway-dependent apoptosis in Jurkat cells. BMC Complement Altern Med. 2014;14:38. doi: 10.1186/1472-6882-14-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Rassool FV. HDAC inhibitors: roles of DNA damage and repair. Adv Cancer Res. 2012;116:87–129. doi: 10.1016/B978-0-12-394387-3.00003-3. [DOI] [PubMed] [Google Scholar]
- 8.Bollino D, Balan I, Aurelian L. Valproic acid induces neuronal cell death through a novel calpain-dependent necroptosis pathway. J Neurochem. 2015;133:174–186. doi: 10.1111/jnc.13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamparter C, Winn LM. Tissue-specific effects of valproic acid on DNA repair genes and apoptosis in postimplantation mouse embryos. Toxicol Sci. 2014;141:59–67. doi: 10.1093/toxsci/kfu105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mu Q, Ma Q, Lu S, Zhang T, Yu M, Huang X, et al. 10058-F4, a c-Myc inhibitor, markedly increases valproic acid-induced cell death in Jurkat and CCRF-CEM T-lymphoblastic leukemia cells. Oncol Lett. 2014;8:1355–1359. doi: 10.3892/ol.2014.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. Bio Med Res Int. 2010;2010:479364. doi: 10.1155/2010/479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner JM, Hackanson B, Lübbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miao M, Du B, Hu R, Yang Y, Yang W, Liao AJ, et al. [Effect of valproic acid sodium on proliferation and apoptosis of acute T-lymphoblastic leukemia Jurkat cells] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2013;21:343–346. doi: 10.7534/j.issn.1009-2137.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 14.Yoon JY, Ishdorj G, Graham BA, Johnston JB, Gibson SB. Valproic acid enhances fludarabine-induced apoptosis mediated by ROS and involving decreased AKT and ATM activation in B-cell-lymphoid neoplastic cells. Apoptosis. 2014;19:191–200. doi: 10.1007/s10495-013-0906-7. [DOI] [PubMed] [Google Scholar]
- 15.Rosenson RS, Wright RS, Farkouh M, Plutzky J. Modulating peroxisome proliferator–activated receptors for therapeutic benefit? Biology, clinical experience, and future prospects. Am Heart J. 2012;164:672–680. doi: 10.1016/j.ahj.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.da Rocha Junior LF, Dantas AT, Duarte ÂL, de Melo Rego MJ, Pitta Ida R, Pitta MG. PPAR agonists in adaptive immunity: what do immune disorders and their models have to tell us? PPAR res. 2013;2013:519724. doi: 10.1155/2013/519724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavender MA, Lincoff AM. Therapeutic potential of aleglitazar, a new dual PPAR-α/γ agonist. Am J Cardiovasc Drugs. 2010;10:209–216. doi: 10.2165/11539500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Tennis MA, Van Scoyk MM, Freeman SV, Vandervest KM, Nemenoff RA, Winn RA. Sprouty-4 inhibits transformed cell growth, migration and invasion, and epithelial-mesenchymal transition, and is regulated by Wnt7A through PPARγ in non–small cell lung cancer. Mol Cancer Res. 2010;8:833–843. doi: 10.1158/1541-7786.MCR-09-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rashid-Kolvear F, Taboski MA, Nguyen J, Wang DY, Harrington LA, Done SJ. Troglitazone suppresses telomerase activity independently of PPARγ in estrogen-receptor negative breast cancer cells. BMC Cancer. 2010;10:390. doi: 10.1186/1471-2407-10-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barros AL, Aguiar JS, Araújo LC, Peixoto CA, de Medeiros PL, Catanho MTJ, et al. Synergistic anticancer effects of valproic acid, atorvastatin and pioglitazone in human malignant and murine cells. Afr J Pharm Pharmacol. 2014;8:31–39. [Google Scholar]
- 21.Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr J. 2014;13:1. doi: 10.1186/1475-2891-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ninomiya I, Yamazaki K, Oyama K, Hayashi H, Tajima H, Kitagawa H, et al. Pioglitazone inhibits the proliferation and metastasis of human pancreatic cancer cells. Oncol Lett. 2014;8:2709–2714. doi: 10.3892/ol.2014.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Youssef J, Badr M. Peroxisome proliferator-activated receptors and cancer: challenges and opportunities. Br J Pharmacol. 2011;164:68–82. doi: 10.1111/j.1476-5381.2011.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu CH, Cheng SM, Chu KM, Yang SP, Lai JH, Cheng CC. Pioglitazone, a PPAR-γ agonist, downregulates cytokine production and AP-1 DNA-binding protein in human peripheral blood T cells–potential implication in the treatment of atherosclerotic disease. Acta Cardiol Sin. 2009;25:127–133. [Google Scholar]
- 25.Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 26.Borghese L, Dolezalova D, Opitz T, Haupt S, Leinhaas A, Steinfarz B, et al. Inhibition of notch signaling in human embryonic stem cell–derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem cells. 2010;28:955–964. doi: 10.1002/stem.408. [DOI] [PubMed] [Google Scholar]
- 27.Neganova I, Vilella F, Atkinson SP, Lloret M, Passos JF, von Zglinicki T, et al. An important role for CDK2 in G1 to S checkpoint activation and DNA damage response in human embryonic stem cells. Stem Cells. 2011;29:651–659. doi: 10.1002/stem.620. [DOI] [PubMed] [Google Scholar]
- 28.Dietrich S, Hullein J, Lee SC, Hutter B, Gonzalez D, Jayne S, et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood. 2015;126:1005–1008. doi: 10.1182/blood-2015-04-643361. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya K, Samanta SK, Tripathi R, Mallick A, Chandra S, Pal BC, et al. Apoptotic effects of mahanine on human leukemic cells are mediated through crosstalk between Apo-1/Fas signaling and the Bid protein and via mitochondrial pathways. Biochem Pharm. 2010;79:361–372. doi: 10.1016/j.bcp.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Yagi Y, Fushida S, Harada S, Kinoshita J, Makino I, Oyama K, et al. Effects of valproic acid on the cell cycle and apoptosis through acetylation of histone and tubulin in a scirrhous gastric cancer cell line. J Exp Clin Cancer Res. 2010;29:1. doi: 10.1186/1756-9966-29-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki T, Higgins P, Crawford D. Control selection for RNA quantitation. Biotechniques. 2000;29:332–337. doi: 10.2144/00292rv02. [DOI] [PubMed] [Google Scholar]
- 32.van Meerloo J, Kaspers GJ, Cloos J. Cell sensitivity assays: the MTT assay. Methods Mol Biol. 2011;731:237–245. doi: 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- 33.Yazdani Y, Sadeghi H, Alimohammadian M, Andalib A, Moazen F, Rezaei A. Iran J Pharm Res. 2011;10:559–568. [PMC free article] [PubMed] [Google Scholar]
- 34.Yazdani Y, Keyhanvar N, Kalhor HR, Rezaei A. Functional analyses of recombinant mouse hepcidin-1 in cell culture and animal model. Biotechnol Lett. 2013;35:1191–1197. doi: 10.1007/s10529-013-1198-2. [DOI] [PubMed] [Google Scholar]
- 35.Blaheta RA, Nau H, Michaelis M. Valproate and valproate-analogues: potent tools to fight against cancer. Curr Med Chem. 2002;9:1417–1433. doi: 10.2174/0929867023369763. [DOI] [PubMed] [Google Scholar]
- 36.Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol. 2010;2010:479364. doi: 10.1155/2010/479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:212–225. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quintas-Cardama A, Santos F, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25:226–235. doi: 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- 39.Cervera E, Candelaria M, López-Navarro O, Labardini J, Gonzalez-Fierro A, Taja-Chayeb L, et al. Epigenetic therapy with hydralazine and magnesium valproate reverses imatinib resistance in patients with chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2012;12:207–212. doi: 10.1016/j.clml.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Mithraprabhu S, Grigoriadis G, Khong T, Spencer A. Deactylase inhibition in myeloproliferative neoplasms. Investigational new drugs. 2010;28:50–57. doi: 10.1007/s10637-010-9590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013;7:47–60. doi: 10.2147/BTT.S29965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saiki M, Hatta Y, Yamazaki T, Itoh T, Enomoto Y, Takeuchi J, et al. Pioglitazone inhibits the growth of human leukemia cell lines and primary leukemia cells while sparing normal hematopoietic stem cells. Int J Oncol. 2006;29:437–443. [PubMed] [Google Scholar]
- 45.Burton JD, Reichel H, Castillo ME, Blumenthal RD. Growth inhibition of hematologic and epithelial cancer cell lines by both PPARγ antagonist and agonist drugs. Cancer Res. 2005;65:156–156. [Google Scholar]
- 46.Sikka S, Chen L, Sethi G, Kumar AP. Targeting PPARγ signaling cascade for the prevention and treatment of prostate cancer. PPAR Res. 2012;2012:968040. doi: 10.1155/2012/968040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu CW, Farrell GC, Yu J. Functional role of peroxisome-proliferator-activated receptor γ in hepatocellular carcinoma. J Gastroenterol Hepatol. 2012;27:1665–1669. doi: 10.1111/j.1440-1746.2012.07213.x. [DOI] [PubMed] [Google Scholar]
- 48.Koga H, Selvendiran K, Sivakumar R, Yoshida T, Torimura T, Ueno T, et al. PPARγ potentiates anticancer effects of gemcitabine on human pancreatic cancer cells. Int J Oncol. 2012;40:679–685. doi: 10.3892/ijo.2011.1237. [DOI] [PubMed] [Google Scholar]
- 49.Koziel R, Szczepanowska J, Magalska A, Piwocka K, Duszynski J, Zablocki K. Ciprofloxacin inhibits proliferation and promotes generation of aneuploidy in Jurkat cells. J Physiol Pharmacol. 2010;61:233. [PubMed] [Google Scholar]
- 50.Alzoubi K, Khabour O, Hussain N, Al-Azzam S, Mhaidat N. Evaluation of vitamin B12 effects on DNA damage induced by pioglitazone. Mutat Res Genet Toxicol Environ Mutagenesis. 2012;748:48–51. doi: 10.1016/j.mrgentox.2012.06.009. [DOI] [PubMed] [Google Scholar]