

Abstract
Genetic variation can affect drug response in multiple ways, although it remains unclear how rare genetic variants affect drug response. The electronic Medical Records and Genomics (eMERGE) Network, collaborating with the Pharmacogenomics Research Network, began eMERGE‐PGx, a targeted sequencing study to assess genetic variation in 82 pharmacogenes critical for implementation of “precision medicine.” The February 2015 eMERGE‐PGx data release includes sequence‐derived data from ∼5,000 clinical subjects. We present the variant frequency spectrum categorized by variant type, ancestry, and predicted function. We found 95.12% of genes have variants with a scaled Combined Annotation‐Dependent Depletion score above 20, and 96.19% of all samples had one or more Clinical Pharmacogenetics Implementation Consortium Level A actionable variants. These data highlight the distribution and scope of genetic variation in relevant pharmacogenes, identifying challenges associated with implementing clinical sequencing for drug treatment at a broader level, underscoring the importance for multifaceted research in the execution of precision medicine.
It is widely accepted that genetic variation impacts drug metabolism, efficacy, and adverse event risk.1, 2, 3 Several medical centers have begun to routinely offer genetic testing and clinical decision support for common variants in a small number of genes associated with drug dosing or adverse events.4, 5, 6, 7 As whole exome and whole genome sequencing are increasingly used in the clinical setting, the number of variants in these genes (and the number of genes) that can be considered for patient care will undoubtedly increase. However, mechanisms to understand the relationship between these variants and drug response have not yet been put into global clinical practice.
The impact and interpretation of this potential deluge variants is currently unclear. While efforts such as the Pharmacogenomics Research Network (PGRN), the Pharmacogenomics Knowledge Base (PharmGKB), and the Clinical Pharmacogenetics Implementation Consortium (CPIC) have led the discovery and systematic documentation of some findings,8, 9, 10 it is clear that the bulk of variation in pharmacological response and metabolism currently remains unexplained.11, 12, 13 Low‐frequency variants that affect gene function may account for some unexplained differences in pharmacological response and metabolism. As a result, new studies of pharmacogenomic traits and novel initiatives that implement pharmacogenomics in clinical care are transitioning from intensity‐based genotyping arrays14, 15 to next‐generation sequencing technologies.16, 17 While there is much enthusiasm for sequencing‐based studies for precision medicine and pharmacogenomics,18, 19, 20 and for the potential to discover low‐frequency variants that influence drug‐related traits,21 little is known about the location and distribution of genetic variation over genes with established pharmacological impact, much less their relationship to variable drug responses.
The documentation of observed variation within genes known to influence drug response and metabolism is essential to enable new molecular studies of potentially functional variants and to improve the understanding of how key pharmacogenes tolerate genetic changes. To document rare and common variation in key genes of pharmacogenomic relevance, the electronic Medical Records and Genomics (eMERGE) Network22, 23, 24 sequenced 84 genes across ~9000 individuals from nine participating biorepositories linked to electronic health records (EHRs). We describe here the first iteration of the resulting dataset from the project, known as eMERGE‐PGx,25 including processes for variant calling, annotation, and aggregate data access in the Sequence and Phenotype Integration Exchange (SPHINX), a web‐based tool for exploring eMERGE‐PGx data for hypothesis generation with an emphasis on drug response implications of genetic variation (www.emergesphinx.org). We describe sequence variation within the key pharmacogenes captured by PGRNseq,26 explore the potential therapeutic impact of established pharmacogenomic variants, catalog the potential for ongoing pharmacogenomic discovery relative to frequently prescribed drugs,25 and provide example uses for the SPHINX resource. eMERGE‐PGx data indicate that the vast majority of patients sequenced will harbor many genetic variants likely to impact currently prescribed drugs, highlighting the opportunities for improving drug response and the need for downstream functional studies, clinical application guidelines, and continued drug development to ensure a diversity of treatment options given the genetic diversity of the patient population.
RESULTS
Allelic discovery in 82 pharmacogenes
As of February 2015, a total of 5,639 samples have been sequenced from nine eMERGE sites (Table 1) using the PGRNseq targeted exome platform26 (see Methods). The PGRNseq platform was developed by the Pharmacogenomics Research Network (PGRN) to maximize their ability to assay important pharmacogenes across the PGRN. The gene selection was through nomination by PGRN sites and vetted through the network. For the design of each of the 82 genes, PGRNseq included all exons (based on all transcript models) as well as 2 kb upstream and 1 kb downstream of their untranslated regions (UTRs) to allow for discovery and assessment of nearby potential regulatory variation. Details of this assay can be found elsewhere.26 In eMERGE‐PGx, the PGRNseq platform generated a total of 968,004 bp of sequence per individual. Variant sites were well‐sequenced, with an average read depth of 200 reads per site (25th percentile = 152.64, median = 211.09, 75th percentile = 257.31). Sequencing PGx samples revealed 42,010 single nucleotide variants (SNVs), with 149 dropped due to allelic imbalance (ABFilter), 137 dropped due to insufficient quality by depth, 22 dropped due to poor genotype call quality, and 696 failing two or more of these criteria; 41,006 SNVs passed all quality control filters. We further removed 447 variants having a genotype call rate less than 95%, and 10 variants were removed due to mismatches with the reference sequence. After all filtering, 40,549 SNVs remained, and of these, 78 showed the reference allele at low frequency (<0.5%).
Table 1.
Demographics of the eMERGE‐PGx project
| Female (N=2958) | Male (N=2674) | Combined (N=5632)b | |
|---|---|---|---|
| AGE a | 57/61/71 | 57/64/71 | 57/63/71 |
| RACE | |||
| American Indian or Alaska Native | 1% (15) | 0% (7) | 0% (22) |
| Asian | 2% (72) | 2% (41) | 2% (113) |
| Black or African American | 14% (414) | 9% (246) | 12% (660) |
| Native Hawaiian or other Pacific Islander | 0% (4) | 0% (1) | 0% (5) |
| Other | 0% (2) | 0% (3) | 0% (5) |
| Unknown | 8% (227) | 6% (152) | 7% (379) |
| White | 75% (2224) | 83% (2224) | 79% (4448) |
| ETHNICITY | |||
| Hispanic or Latino | 7% (195) | 4% (113) | 5% (308) |
| Not Hispanic or Latino | 89% (2639) | 91% (2433) | 90% (5072) |
| Unknown | 4% (124) | 5% (128) | 5% (252) |
| CLINICAL ATTRIBUTES | |||
| Avg Record Length in years (s.d.) | 17.1 (9.14) | 16.21 (9.36) | 16.66 (9.25) |
| Avg Distinct ICD9 Codes (s.d.) | 106.7 (69.99) | 83.93 (58.54) | 95.5 (65.60) |
| Avg Medication Count (s.d.)c | 9.0 (8.09) | 9.20 (8.49) | 9.09 (8.27) |
birth year was collected, so age is an approximation. Ages are given as lower quartile range, median, and upper quartile range.
demographic information missing on some samples
Medications were restricted to a list of most prescribed medications (see methods).
Comparison of annotation methods (VEP vs. SNPeff)
Of the 40,549 high‐quality SNVs, 27,965 were annotated by VEP to the canonical transcript for one of the PGRNseq targeted genes (Table 2). Of these annotated variants, 8,126 were coding (4,858 missense, 3,169 synonymous, 99 stop gained) and 19,923 were noncoding (5,231 intronic, 5,981 upstream variants, 3,444 downstream variants, 4,165 3′UTR variants, 903 5′UTR variants, and 199 other).
Table 2.
Counts of Ensembl consequence type for variants mapped to canonical transcripts of PGRNseq captured genes
| ENSEMBL consequence type | IN PGx | IN 1KG | IN EXAC | NOVEL |
|---|---|---|---|---|
| Upstream Gene Variant | 6,094 | 2,122 | 23 | 3,924 |
| Intron Variant | 5,542 | 2,016 | 460 | 3,038 |
| Missense Variant | 4,806 | 1,485 | 1,792 | 2,212 |
| 3 Prime UTR Variant | 4,245 | 1,539 | 65 | 2,629 |
| Downstream Gene Variant | 3,574 | 1,239 | 44 | 2,219 |
| Synonymous Variant | 3,147 | 1,335 | 1,255 | 1,163 |
| 5 Prime UTR Variant | 931 | 287 | 59 | 597 |
| Missense Variant, Splice Region Variant | 147 | 48 | 62 | 60 |
| Splice Region Variant, Intron Variant | 142 | 60 | 49 | 54 |
| Stop Gained | 97 | 20 | 31 | 54 |
| Splice Region Variant, Synonymous Variant | 90 | — | 36 | 40 |
| Splice Acceptor Variant | 18 | 5 | 3 | 1 2 |
| Splice Donor Variant | 15 | 3 | 6 | 8 |
| Splice Region Variant, 5 Prime UTR Variant | 14 | 3 | 3 | 10 |
| Initiator Codon Variant | 11 | 2 | 2 | 7 |
| Stop Gained, Splice Region Variant | 3 | 1 | 1 | 2 |
| Stop Lost | 2 | — | — | 2 |
| Stop Retained Variant | 1 | 1 | — | — |
| Splice Region Variant, 3 Prime UTR Variant | 1 | 1 | — | — |
| Total | 28,880 | 10,167 | 3,891 | 16,019 |
Counts of variants previously discovered in the 1000 Genomes Project (1KG), the Exome Aggregation Consortium (EXAC), and novel variants in the eMERGE PGx project (PGx) are also shown.
Compared to dbSNP (build 141), 415 variants were previously observed (52 missense, 26 synonymous, 58 intronic). We also performed comparisons with other large‐scale sequencing projects; 15,163 variants were previously observed by the 1000 Genomes Phase 3 project (1,446 missense, 1,315 synonymous, and 2,075 intronic), and 10,998 variants were reported in the ExAC dataset (3,009 missense, 2,137 synonymous, 773 intronic). Across all three reference sets, 20,886 (51.5% of the total 40,549 SNVs identified) variants from the eMERGE‐PGx dataset were observed previously, and 19,663 (48.5%) are novel, including 1,445 missense, 769 synonymous, and 2,848 intronic variants.
Relative to the Ensembl canonical transcript, VEP annotates 27,434 with SNPEff annotating 19,895 variants, a complete subset of the VEP annotation calls. Comparing these 19,895 variant annotations, the results are highly concordant, with 99.25% of variant consequence calls concordant between SNPEff and VEP. Of the 150 discordant annotations, 105 were considered “stop gained” by SNPEff, but “5′UTR variant” by VEP. There were only 44 other discordant annotations, 35 downstream—3′UTR, 9 intron—splice region between SNPEff and VEP, respectively. More critically, 7,901 annotations spanning 21 genes were made by VEP but not by SNPEff. These included 2,050 intron variants, 1,288 upstream variants, 1,212 downstream variants, 1,138 3′UTR variants, 1,097 missense variants, 864 synonymous variants, 253 5′UTR variants, and 134 others. These discordant annotations are likely due to subtle differences in the definitions of the canonical transcript used by the two software programs. Discordant annotations of predicted variant function is a known issue in the field.27 Because of this issue, we have chosen to provide a single annotation, specifically SNPEff annotations, in SPHINX.
Molecular characteristics of variants in pharmacogenes
As expected, the majority of these variants were diallelic (39,778, 98.1%), although 759 (1.9%) were triallelic, and 12 sites showed all four alleles. Of the diallelic SNVs, there were 2,102 common, 1,230 low‐frequency, 9,465 rare, 4,606 doubleton, and 22,124 singleton variants identified; the full spectrum of allele frequencies for diallelic SNVs annotated to PGRNseq genes is shown in Figure 1.
Figure 1.
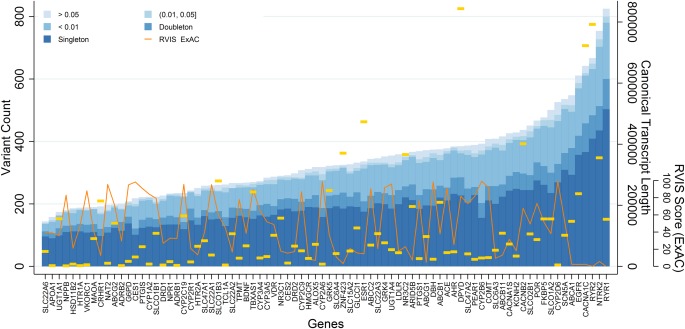
Allelic spectrum of eMERGE‐PGx variants. Counts of genomic variants mapping to the canonical transcript of PGRNseq captured genes are plotted by frequency class (over all samples) by gene (x‐axis) in ascending order. Gold horizontal lines indicate the size of the canonical transcript in basepairs. The inset line plot is a percentile rank of genic intolerance (RVIS) scores computed using the ExAC dataset.
There was a significant linear relationship between gene length and the number of discovered low‐frequency variants (minor allele frequency (MAF) <5%) (P < 0.0001), with an average increase of 0.35 variants per kilobase of gene length (Supplementary Table S1). Nevertheless, there was variability in this relationship: RYR1, the gene with the second largest canonical transcript coding region (15,011 bp), has the largest number of variants, 667, with 409 of them (61%) singletons. SLC22A6 contains the fewest variants, 144, despite having a transcript length of 2,141 bp, three times larger than the smallest captured. We also see a significant and somewhat stronger association between the genic intolerance scores for these genes (based on the ExAC data) and the number of low‐frequency variants, with an estimated decrease of 46.3 variants per intolerance score unit (P < 0.0001).
Variants in multiple ethnic groups
We recalculated this frequency spectrum within administratively reported African‐American (n = 650), European (n = 4373), Asian (n = 112), and Hispanic/Latino (n = 310) groups (Figure S1). Black or African‐American samples show the largest number of variants per person. European‐American samples (the largest sample set) show a much lower median number of variants per person, although this sample set has great variability in both high and low variant counts. Cumulative minor allele frequencies over all variants are shown in Figure S2. In European‐descent samples, cumulative MAF (CMAF) range from 2.88% for SLC22A6 to 26.11% for NTRK2. African‐American samples had a much lower and narrower CMAF range from 1.55% for CYP2R1 to 4.95% for ABCA1.
Potential therapeutic impact
Nearly every captured gene (95.12%) has one or more variants with a scaled Combined Annotation‐Dependent Depletion (CADD) score above 20 (Figure 2). The RYR2 gene had the highest CADD scoring variant (56), while BDNF variants had the highest median scaled CADD Score (∼10), with the calcium channels RYR1 and CACNA1S also harboring variants with high scaled CADD scores, with 24 variants in these genes scoring above 30. Importantly, 96.19% (5,424) of all samples had one or more CPIC Level A actionable variants, with the median being two actionable variants per individual over the entire sample (2,318 individuals), and 1,273 individuals having variants with only one. Notably, 1,517 individuals had actionable variants within three genes, and 316 had actionable variants within four or more genes (293 with variants within four genes, 22 with five genes, and one with six genes). We also note other low‐frequency variants (<5%) within the CPIC actionable genes; 1,932 individuals (34.2%) have one or more missense variants in at least one of the seven CPIC genes examined, with the majority (1,616 individuals) having only one gene with missense variation. No individual had missense variants in more than four CPIC genes (six individuals had four missense variable CPIC genes, 52 had three, and 258 had two).
Figure 2.
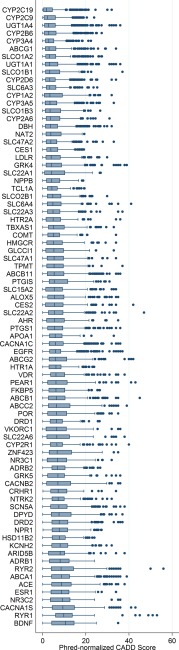
Boxplot of scaled (Phred) CADD score annotations for alleles by gene. Genes are ranked from top to bottom by ascending median CADD score.
Using two sources of drug prescription activity in the US in 2013, 38 genes were found to have some level of evidence from PharmGKB implicating them in the metabolism of one of 31 drugs. Within these 38 genes, 12,637 variants were identified, including 2,208 missense variants, of which 458 were potentially damaging by CADD score. Selecting only these 458 missense variants, we then calculated the CMAF (the frequency of having one or more nonsynonymous variants) by potentially impacted drug. Using this frequency as an estimate of the general US population CMAF, and assuming that the reported prescription counts are distinct individuals, we then estimated the proportion of prescriptions potentially affected for each drug (Figure 3). For example, roughly 4 million of the 27 million prescriptions for rosuvastatin may be affected by one of 407 missense variants within eight genes (ABCB11, ABCG2, CYP2C9, CYP3A5, HMGCR, SLCO1B1, SLCO1B3, SLCO2B1), which occurred in 17.8% of the eMERGE‐PGx sample. When restricted to predicted damaging missense variants, there were 64 variants within genes for rosuvastatin with a CMAF of 9.84%, potentially influencing nearly 600,000 prescriptions in 2013, although their clinical impact is unknown and could range from no effect to severe myopathy. When we examine genes for drugs with a low therapeutic index like digoxin and warfarin, we observe very different results. CYP2C9 (a drug‐metabolizing enzyme) has 54 CADD‐damaging missense variants with a CMAF of 0.84%, VKORC1 (a drug target) has 11 variants with a CMAF of 0.03%, or ABCB1 (a transporter in the case of digoxin), has 85 variants with a CMAF of 0.35%.
Figure 3.
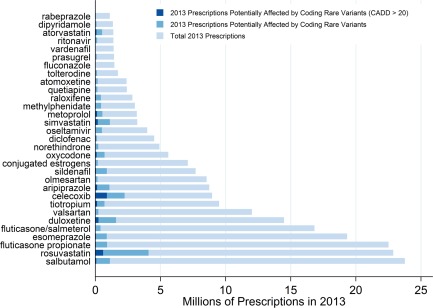
Estimates of prescriptions impacted by rare missense variants within pharmacogenes impacting the metabolism of frequently prescribed drugs.
Similarly, the 25 most dispensed medications encompass over 1.5 billion prescriptions in the US over 2013, of which seven drugs (fluticasone, albuterol, omeprazole, metoprolol, atorvastatin, and simvastatin) account for roughly 410 million prescriptions. These drugs are influenced by genes captured by PGRNseq according to PharmGKB. When computing CMAF of low‐frequency missense variants by drug, an estimated 4% (fluticasone) to 34.6% (simvastatin) of individuals taking these prescriptions harbor one or more variants within genes that potentially influence their action, with an estimated impact on nearly 75 million prescriptions in 2013.
Accessing eMERGE‐PGx data
As described, all of the summary data in eMERGE‐PGx are being made publicly available in SPHINX (www.emergesphinx.org). This web‐based portal to query information by gene, by pathway, or by drug can be used to generate descriptive data and/or hypotheses for future research based on these 82 pharmacogenes. Figure 4 shows an annotated home page for SPHINX. Queries can be made by entering a gene name/symbol, pathway name, or a drug name (full list of available genes, pathways, and drugs are available using the links on the top left corner of the home page). The resulting information is displayed on subsequent webpages organized based on the nature of the search. Searching SPHINX by gene will result in a table of all available variants identified in the eMERGE‐PGx dataset, including chromosome and basepair location, rsID if available, type of variant according to SNPEff, global allele frequency in the complete eMERGE‐PGx dataset, and allele frequency stratified by self‐reported ancestry. This type of query would be useful for individuals who have interest in particular genes or specific variants from these genes to obtain estimates of allele frequency in a large clinical population: for example, the situation where someone had identified a rare variant in their study in the gene ABCA1 and wondered if this rare variant was observed in other datasets. In considering all of the variants in ABCA1 shown in Figure S3, only four of these variants are cataloged in PharmGKB (as denoted by the rsIDs) and all of these have very low frequency in eMERGE‐PGx. These types of queries become most important for variants that are not yet cataloged by other resources like dbSNP. The result enables a researcher to know if the variant has been observed and at what frequency in eMERGE‐PGx. Because of the rich, longitudinal phenotypic data in eMERGE, another possibility for this query might include searching through the eMERGE‐PGx dataset for all patients who have a particular variant in ABCA1 and then perform EHR chart review for that small set of patients to determine if there is any likely clinical significance to that variant.
Figure 4.
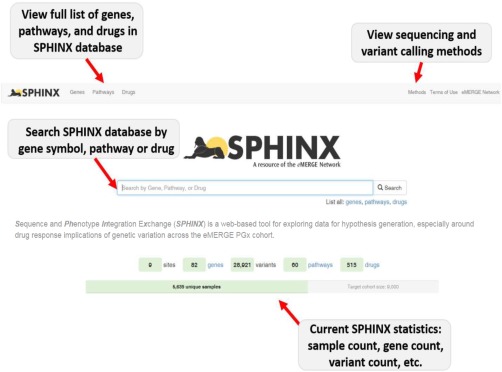
Screenshot of SPHINX website (http://emergesphinx.org).
Consider another use case in which a researcher is interested in all variations in genes from a particular pathway of interest, such as ABC transporters (shown in Figure S4). If the research question involves how much genetic variation exists in these genes and which genes would be appropriate targets for subsequent genotyping or sequencing, the pathway query capability may be of great utility. From this view, an investigator can view information about the specific gene and variant as shown in Figure S4. Finally, searching by drug will provide a list of all genes from PharmGKB linked to that particular drug. Figure S5 shows an example from 1,25 dihydroxyvitamin d3. An investigator who works on a particular drug/compound can search for variant information for all genes linked to their drug of interest. These types of queries will enable researchers in the scientific community to search a public database resource of summary data cataloging all variations identified in the eMERGE‐PGx project. Individual‐level DNA sequence data from this project with key pharmacologic response phenotypes available from electronic medical records will also be made available via dbGaP for the research community.
DISCUSSION
In this study we examined sequence variation within the key pharmacogenes in an eMERGE‐PGx dataset, potential therapeutic impact of established pharmacogenomic variants, potential for ongoing pharmacogenomic discovery, and example uses for the SPHINX resource. By examining a diverse clinical population of over 5,000 people, we report the largest targeted sequencing study of established pharmacogenes to date, with data queryable from the SPHINX database. Variation is frequent within these clinically relevant genes, with most individuals having multiple clinically actionable variants. Hundreds of additional variants with potential pharmacogenomic function were identified and made available online to the research community, setting the stage for future association studies within the eMERGE network.
Compared to other sequencing studies and variant repositories, nearly half of all variants identified were novel, illustrating that existing exome‐based resources, even those from large studies, may not characterize genetic variation as well as the targeted methods used for PGRNseq genes with a large sample size and very high depth of coverage (∼200 reads on average). The majority of identified variants are singletons and doubletons, extremely low‐frequency variants that will require new analytic or high‐throughput molecular strategies to fully elucidate their function. Future studies of these variants within eMERGE using EMR‐based phenotypes may improve our understanding of their function on a phenotypic level. Computational predictions of variant pathogenicity (such as the CADD algorithm) may also prove useful for variant prioritization, or for the exploration of specific phenotypes. For example, the RYR2 gene has been implicated with level 3 evidence from PharmGKB in rhabdomyolysis following cerivastatin treatment.28 This gene showed the highest score for any gene‐annotated variant. The BDNF gene, inconsistently implicated in impacting drug efficacy for a variety of psychiatric disorder treatments, shows the highest median CADD score.29, 30, 31, 32 In addition, a more thorough examination of the distribution of types of variation in different drug classes would be extremely valuable. Perhaps we would observe different patterns in transporters, phase I enzymes, phase II enzymes, channels, pharmacologic targets, and/or drugs with low therapeutic index that would highlight relevant biological or evolutionary hypotheses about these genes.
Considerable care must be taken, however, when interpreting such scores for clinical implementation. A recent eMERGE study of SCN5A and KCNH2 found that pathogenic classification of splice and missense variants within these genes can vary broadly, even from commercial laboratories that provide clinical testing for these specific genes.33 Clearly, certain findings may warrant the recontact of study participants to avoid potentially life‐threatening conditions, and the complex ethical issues surrounding return of research results have been previously noted34 and are a continual focus with the eMERGE network.
The eMERGE‐PGx dataset is enriched for established pharmacogenomics variants; prior work by Van Driest et al.35 has shown that nearly all individuals (98%) have at least one known, actionable variant by current CPIC guidelines, which would either alter the dose of a prescribed drug or would suggest an alternative therapy. We recapitulate this result, showing a median of two actionable variants per person, with over 1,800 individuals having three or more actionable variants. As a result, there is a strong possibility that this information could influence the clinical care of a patient over his or her lifetime. This key finding highlights the importance and potential clinical impact of the cataloged genetic variation. Importantly, we also observed that genes with established CPIC guidelines harbor many more potentially deleterious missense variants that have not been previously characterized or reported.
To further explore the potential for pharmacogenomic discovery, we used resources from the PharmGKB database to build connections between PGRNseq captured genes and frequently prescribed drugs. While these drug–gene relationships are based on much weaker levels of evidence than CPIC recommendations, we estimate that missense variants within these genes have the potential to affect metabolism and efficacy of millions of US prescriptions annually. Based on using gene sets with annotations by drug in PharmGKB, we explored the relationships between types of variants in the genes indicated as relevant for each drug. Even when restricting this analysis to only predicted damaging missense variants, 2.6% of individuals have a variant within the genes that affect rosuvastatin according to PharmGKB (22 million prescriptions annually), and 9.8% of individuals have variants within genes that affect celecoxib (9 million prescriptions annually). While additional research will be required to establish clinical effects and guidelines, with 34% of individuals harboring multiple variants within CPIC‐associated genes, there is great potential for pharmacogenomic discovery within eMERGE‐PGx. To encourage the similar use of eMERGE‐PGx data in the broader pharmacogenomics community, variant‐level data are viewable on SPHINX with each data release through the online SPHINX portal (http://www.emergesphinx.org). Through linkages with the PharmGKB database, variant data can be queried by gene, variant, pathway, and drug. SPHINX does not yet have any phenotypic data deposited, but this is an active area of development for eMERGE.
There are several limitations to this study. Participants were recruited from clinical settings and as a result may be enriched for alleles that influence disease or treatment. As described in Rasumussen‐Torvik et al.,25 each eMERGE site used a unique recruitment strategy for eMERGE‐PGx. Some sites specifically ascertained participants who were prescribed medications with pharmacogenes of interest on the gene panel. Others recruited based on disease. As a consequence of this ascertainment strategy, the study sample (while multiethnic) has limited population diversity, which limits our ability to detect rare alleles isolated to non‐European descent populations. With respect to variant annotation, for simplicity our strategy examined variant consequences in the context of the Ensembl canonical transcript only; many variants will have different consequences relative to different transcripts, so assessments of variant consequences are likely underestimates of their most severe impact.
While it is unclear specifically how many of the identified variants influence clinical outcomes, it is clear that surveys of these critically important genes using sequencing technologies will reveal large numbers of rare variants, each with the potential to impact pharmacogenomic traits. Future studies within the eMERGE‐PGx project will explore these relationships with the ultimate goal of informing clinical care with genetic variation.
METHODS
Sequencing and quality control
As of February 2015, a total of 5,639 samples have been sequenced from nine eMERGE sites (Table 1) (more details in Supplemental Material). Samples were sequenced by the Center for Inherited Disease Research (CIDR), University of Washington, Mayo Clinic,36 Icahn School of Medicine at Mount Sinai, or Children's Hospital of Philadelphia (CHOP). Sequencing was performed using the PGRNseq targeted exome platform, using 100 bp paired end runs on a HiSeq2500, and aligned to the GRCh37 reference with decoy sequences with Burrows‐Wheeler Aligner (BWA).37 Reads were further processed using GATK HaplotypeCaller v. 3.3‐0 according to the GATK best practices38 with multisample calling. Reads for the two targeted HLA genes (HLA‐B and HLA‐DQB3) were excluded due to general poor alignment, thus all further results refer to 82 pharmacogenes. Although both insertion/deletions (INDELs) and SNVs were called, only SNV calls were used for subsequent analyses and are currently provided in SPHINX. Raw variant calls failing any of the following filters were dropped: QUAL <50; ABHet >0.75; QD <5.0. Raw genotype calls failing any of the following filters were also dropped: GQ <50; Heterozygous call with AB >0.75.
Variant frequencies
Variants were partitioned into five mutually exclusive frequency classes: common (MAF >0.05), low frequency (>0.01, 0.05), rare (<0.01), doubleton (observed only twice), and singleton (observed only once). For all variants, we required at least 10,714 chromosomal observations (nonmissing genotype calls), equivalent to 95% genotyping efficiency. Consistent with the use of rare‐variant burden tests, we computed a CMAF, indicating the frequency at which individuals have one or more nonreference alleles at low frequency (<0.05) within a gene. We considered loci showing nonreference alleles at high‐frequency (>0.95) as likely errors in the reference sequence and included the reference allele as the minor allele for CMAF calculations. Residual Variation Intolerance Scores (RVISs) for captured genes relative to the ExAC release 0.3 were accessed online (http://chgv.org/GenicIntolerance/). Linear regression examining the relationship between gene length and the number of identified variations was performed using STATA 12.0 (STATA, College Station, TX).
Variant annotation
We performed variant annotation using the Ensembl Variant Effect Predictor (VEP) v. 74, build39, 40 and SNPEff41 v. 3.5c (build 2014‐02‐21), annotated against the GRCh37.71 database, and restricted annotations to the Ensembl canonical transcript of PGRNseq captured genes only. CADD42 PhRED‐normalized scores were retrieved online and mapped to variants by chromosome, position, and alternate allele. On the PhRED scale, substitutions are assigned scores according to percentile, where the highest 10% of all scores are assigned values ≥C10, the highest 1% are assigned values ≥C20, etc.42 We also compared identified variants to other established catalogs of genetic variation, including dbSNP build 141 (accessed online in VCF format 3/4/2015), 1000 Genomes Project phase 3 data (accessed online in VCF format 2/19/2015), and the Exome Aggregation Consortium (ExAC) dataset release 0.3 (accessed online in VCF format 1/13/2015).
To annotate variants by pharmacogenomics impact, recommendations were accessed for nine genes with CPIC “Level A” evidence, which provide specific clinical actionability (Table 3). CPIC Level A indicates that “Genetic information should be used to change prescribing of affected drug”8 and can be found at https://www.pharmgkb.org/page/cpic. Variants were mapped to CPIC alleles by chromosome, basepair position, and alternate allele. Defining the star (*) alleles10 for all of the relevant genes is currently ongoing.
Table 3.
Clinical Pharmacogenetics Implementation Consortium (CPIC) actionable variants for selected genes
| GENE | CPIC PUBMED IDS | RS number | Number of eMERGE PGx samples with at least one nonreference allele |
|---|---|---|---|
| CYP2C19 | 23486447;21716271; | 4244285 | 1,578 |
| CYP2C19 | 23698643 | 4986893 | 20 |
| CYP2C19 | 12248560 | 2,087 | |
| CYP2C19 | 28399504 | 37 | |
| CYP2C19 | 41291556 | 19 | |
| CYP2C19 | 72552267 | 3 | |
| CYP2C9 | 25099164; 21900891 | 1057910 | 635 |
| CYP2C9 | 1799853 | 1,186 | |
| CYP2D6 | 16947 | 4,767 | |
| CYP2D6 | 1065852 | 2,061 | |
| CYP2D6 | 1135840 | 3,686 | |
| CYP2D6 | 3892097 | 1,783 | |
| CYP2D6 | 28371706 | 238 | |
| CYP2D6 | 28371725 | 926 | |
| DYPD | 23988873 | 3918290 | 54 |
| DYPD | 55886062 | 8 | |
| DYPD | 67376798 | 53 | |
| G6PD | 24787449 (Table S4) | 1050828 | 144 |
| G6PD | 1050829 | 349 | |
| G6PD | 5030868 | 2 | |
| G6PD | 137852339 | 2 | |
| SLCO1B1 | 22617227;24918167 | 2306283 | 3,940 |
| SLCO1B1 | 4149015 | 599 | |
| SLCO1B1 | 4149056 | 1,486 | |
| TPMT | 21270794;23422873 | 1142345 | 481 |
| TPMT | 1800460 | 383 | |
| TPMT | 1800462 | 22 | |
| TPMT | 1800584 | 1 | |
| VKORC1 | 21900891 | 9923231 | 3,280 |
| CYP2C19 | 23486447;21716271; | 4244285 | 1,578 |
| CYP2C19 | 23698643 | 4986893 | 20 |
| CYP2C19 | 12248560 | 2,087 |
To further examine the implications for pharmacogenomics discovery, we accessed two sources of prescription activity in the US from the IMS Institute for Healthcare Informatics, a National Prescription Audit listing the 100 most frequently prescribed brand name drugs with nationwide prescription numbers from April 2013 to March 2014,43 and a subsequent review of medication use in 2013 which lists the 25 most dispensed medications.44 Brand names and/or active ingredients of these drugs were matched to brand names and/or active ingredients listed in PharmGKB.45 PharmGKB reports gene–drug interactions with multiple levels of supporting evidence, including clinical annotation, variant annotation, “very important pharmacogenes,” and pathways. Using PharmGKB, we extracted reported interactions between these drugs and genes captured by the PGRNseq platform with any level of evidence as a potential pharmacogene for a given drug.
Data availability
Summary level data from the most current version of the eMERGE‐PGx project data are viewable in SPHINX. First released in December 2013, SPHINX provides allelic variation identified by the sequencing and variant calling pipelines reported here. Users can search identified variants by a variety of criteria, including basic attributes such as gene symbol. More advanced searches use data from PharmGKB and other public data sources to enable queries by drug and metabolic pathway, allowing higher‐level hypotheses to be investigated. Variant information includes chromosome, position, SNP ID (if known), SNPEff41 annotated consequence (e.g., downstream, 3′UTR, nonsynonymous, etc.), and allele frequencies calculated globally across the entire cohort and by population for European and African descent groups.
WEB RESOURCES
SPHINX: http://www.emergesphinx.org; SNPeff: http://snpeff.sourceforge.net/download.html#databases; ExAC: ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3/; PharmGKB: https://www.pharmgkb.org/page/cpicStatusLegend; ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/VCF/; ftp://ftp‐trace.ncbi.nih.gov/1000genomes/ftp/release/20130502/. The two sources of top prescribed drugs are available here: http://www.medscape.com/viewarticle/825053#vp_2; http://www.imshealth.com/en/thought‐leadership/ims‐institute/reports/use‐of‐medicines‐in‐the‐us‐2013.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
M.D.R., W.S.B., D.R.C., S.J.B., D.C.C., S.S., T.M., and D.R. wrote the manuscript; M.D.R., W.S.B., D.R.C., K.W., R.L., T.M., E.B., R.L.C., J.L.H., G.P.J., J.C.D., L.R‐T., and D.R. designed the research; M.D.R., W.S.B., D.R.C., A.O‐O., J.R.W., B.A., M.K.B., S.J.B., D.S.C., J.C., D.C.C., K.F.D., C.J.G., A.S.G., B.K., J.K., T.E.K., S.F.M., A.R.M., V.P., C.L.P., J.F.P., C.A.P., J.R., S.A.S., A.S., M.S., S.S., T.V., W.W., S.V., K.W., R.L., T.M., E.B., M.H.B., D.C., R.L.C., C.C., J.L.H., H.H., J.B.H., I.A.H., I.J.K., G.P.J., E.B.L., C.A.M., M.S.W., J.C.D., L.R‐T., and D.R. performed the research; W.S.B., D.R.C., A.O‐O., J.R.W., A.S.G., S.A.S., S.S., J.L.H., G.P.J., J.C.D., and L.R‐T. analyzed the data; J.R.W., K.F.D., A.S.G., and G.P.J. contributed new reagents/analytical tools.
Supporting information
Supporting Information Figure 2.
Supporting Information
ACKNOWLEDGMENTS
The eMERGE Network was initiated and funded by NHGRI through the following grants: U01HG006828 (Cincinnati Children's Hospital Medical Center/Boston Children's Hospital); U01HG006830 (Children's Hospital of Philadelphia); U01HG006389 (Essential Institute of Rural Health, Marshfield Clinic Research Foundation and Pennsylvania State University); U01HG006382 (Geisinger Clinic); U01HG006375 (Group Health Cooperative/University of Washington); U01HG006379 (Mayo Clinic); U01HG006380 (Icahn School of Medicine at Mount Sinai); U01HG006388 (Northwestern University); U01HG006378 (Vanderbilt University Medical Center); and U01HG006385 (Vanderbilt University Medical Center serving as the Coordinating Center). The PGRNSeq dataset (eMERGE PGx), please also add U01HG004438 (CIDR) serving as a Sequencing Center. This work was also supported in part by the Mayo Clinic Center for Individualized Medicine, National Institutes of Health grants U19 GM61388 (the Pharmacogenomics Research Network), R01 GM28157, U01 HG005137, R01 CA138461, and R01 AG034676 (the Rochester Epidemiology Project).
The first two authors contributed equally to this work.
References
- 1. Wang L., McLeod, H.L. & Weinshilboum, R.M. Genomics and drug response. N. Engl. J. Med. 364(12), 1144–1153 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Meyer, U.A. , Zanger, U.M. & Schwab, M. Omics and drug response. Annu. Rev. Pharmacol. Toxicol. 53, 475–502 (2013). [DOI] [PubMed] [Google Scholar]
- 3. Meyer, U.A. Pharmacogenetics — five decades of therapeutic lessons from genetic diversity. Nat. Rev. Genet. 5(9), 669–676 (2004). [DOI] [PubMed] [Google Scholar]
- 4. Pulley J.M. et al Operational implementation of prospective genotyping for personalized medicine: the design of the Vanderbilt PREDICT project. Clin. Phamacol. Ther. 92(1), 87–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson, J.A. et al Institutional profile: University of Florida and Shands Hospital Personalized Medicine Program: clinical implementation of pharmacogenetics. Pharmacogenomics. 14(7), 723–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shuldiner A.R. et al Implementation of pharmacogenetics: the University of Maryland Personalized Anti‐platelet Pharmacogenetics Program. Am. J. Med. Genet. C Semin. Med. Genet. 166C(1), 76–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. He Y.J. & McLeod, H.L. Ready when you are: easing into preemptive pharmacogenetics. Clin. Phamacol. Ther. 92(4), 412–414 (2012). [DOI] [PubMed] [Google Scholar]
- 8. Caudle, K.E. et al Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 15(2), 209–217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whirl‐Carrillo, M. et al Pharmacogenomics knowledge for personalized medicine. Clin. Phamacol. Ther. 92(4), 414–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Relling, M.V. & Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Phamacol. Ther. 89(3), 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roden, D.M. , Wilke, R.A. , Kroemer, H.K. & Stein, C.M. Pharmacogenomics: the genetics of variable drug responses. Circulation. 123(15), 1661–1670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chhibber, A. et al Genomic architecture of pharmacological efficacy and adverse events. Pharmacogenomics. 15(16), 2025–2048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klein, K. & Zanger, U.M. Pharmacogenomics of cytochrome P450 3A4: recent progress toward the “missing heritability” problem. Front. Genet. 4, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oetjens, M.T. et al Assessment of a pharmacogenomic marker panel in a polypharmacy population identified from electronic medical records. Pharmacogenomics. 14(7), 735–744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deeken, J. The Affymetrix DMET platform and pharmacogenetics in drug development. Curr. Opin. Mol. Ther. 11(3), 260–268 (2009). [PubMed] [Google Scholar]
- 16. Mizzi, C. et al Personalized pharmacogenomics profiling using whole‐genome sequencing. Pharmacogenomics. 15(9), 1223–1234 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Esplin, E.D. , Oei, L. & Snyder, M.P. Personalized sequencing and the future of medicine: discovery, diagnosis and defeat of disease. Pharmacogenomics. 15(14), 1771–1790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Musunuru, K. Personalized genomes and cardiovascular disease. Cold Spring Harb Perspect Med. 5(1), a014068–a014068 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mooney, S.D. Progress towards the integration of pharmacogenomics in practice. Hum Genet. 134, 459–465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ong, F.S. , Lin, J.C. , Das, K. , Grosu, D.S. & Fan, J.‐B. Translational utility of next‐generation sequencing. Genomics. 102(3), 137–139 (2013). [DOI] [PubMed] [Google Scholar]
- 21. Wagner, M.J. Rare‐variant genome‐wide association studies: a new frontier in genetic analysis of complex traits. Pharmacogenomics. 14(4), 413–424 (2013). [DOI] [PubMed] [Google Scholar]
- 22. McCarty, C.A. et al The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genomics. 4, 13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crawford, D.C. et al eMERGEing progress in genomics‐the first seven years. Front. Genet. 5, 184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gottesman, O. et al The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet. Med. Off. J. Am. Coll. Med. Genet. 15(10), 761–771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rasmussen‐Torvik, L.J. et al Design and anticipated outcomes of the eMERGE‐PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin. Phamacol. Ther. 96(4), 482–489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gordon, A.S. et al PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet. Genomics. (2016); e‐-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCarthy, D.J. et al Choice of transcripts and software has a large effect on variant annotation. Genome Med. 6(3), 26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marciante, K.D. et al Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet. Genomics. 21(5), 280–288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCarthy, M.J. , Leckband, S.G. & Kelsoe, J.R. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics. 11(10), 1439–1465 (2010). [DOI] [PubMed] [Google Scholar]
- 30. Zou, Y.‐F. et al Association of brain‐derived neurotrophic factor genetic Val66Met polymorphism with severity of depression, efficacy of fluoxetine and its side effects in Chinese major depressive patients. Neuropsychobiology. 61(2), 71–78 (2010). [DOI] [PubMed] [Google Scholar]
- 31. Murphy, G.M. et al BDNF and CREB1 genetic variants interact to affect antidepressant treatment outcomes in geriatric depression. Pharmacogenet. Genomics. 23(6), 301–313 (2013). [DOI] [PubMed] [Google Scholar]
- 32. Niitsu, T. , Fabbri, C. , Bentini, F. & Serretti, A. Pharmacogenetics in major depression: a comprehensive meta‐analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry. 45, 183–194 (2013). [DOI] [PubMed] [Google Scholar]
- 33. Van Driest, S.L. et al Association of Arrhythmia‐Related Genetic Variants With Phenotypes Documented in Electronic Medical Records. JAMA. 315(1), 47–57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fullerton, S.M. et al Return of individual research results from genome‐wide association studies: experience of the Electronic Medical Records and Genomics (eMERGE) Network. Genet. Med. Off. J. Am. Coll. Med. Genet. 14(4), 424–431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Driest, S.L. et al Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther. 95(4), 423–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bielinski, S.J. et al Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time‐using genomic data to individualize treatment protocol. Mayo Clin. Proc. 89(1), 25–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinforma Oxf. Engl. 25(14), 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van der Auwera, G.A. et al Current Protocols in Bioinformatics 11.10.1–11.10.33 (eds. Bateman A. et al) Current protocols in bioinformatics / editoral board, Baxevanis, A.D., et al. (John Wiley & Sons, Hoboken, NJ; 2002). [Google Scholar]
- 39. Zheng, X. et al A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinforma Oxf. Engl. 28(24), 3326–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McLaren, W. et al Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinforma Oxf. Engl. 26(16), 2069–2070 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cingolani, P. et al A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin). 6(2), 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kircher, M. et al A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46(3), 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Top 100 Most Prescribed, Top Selling Drugs [Internet]. <http://www.medscape.com/viewarticle/825053>. Accessed May 5, 2015.
- 44. Medicine use and shifting costs of healthcare. 2014.
- 45. Hewett, M. et al PharmGKB: the Pharmacogenetics Knowledge Base. Nucleic Acids Res. 30(1), 163–165 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 2.
Supporting Information
Data Availability Statement
Summary level data from the most current version of the eMERGE‐PGx project data are viewable in SPHINX. First released in December 2013, SPHINX provides allelic variation identified by the sequencing and variant calling pipelines reported here. Users can search identified variants by a variety of criteria, including basic attributes such as gene symbol. More advanced searches use data from PharmGKB and other public data sources to enable queries by drug and metabolic pathway, allowing higher‐level hypotheses to be investigated. Variant information includes chromosome, position, SNP ID (if known), SNPEff41 annotated consequence (e.g., downstream, 3′UTR, nonsynonymous, etc.), and allele frequencies calculated globally across the entire cohort and by population for European and African descent groups.