

Abstract
Cephalochordates, the sister group of vertebrates + tunicates, are evolving particularly slowly. Therefore, genome comparisons between two congeners of Branchiostoma revealed so many conserved noncoding elements (CNEs), that it was not clear how many are functional regulatory elements. To more effectively identify CNEs with potential regulatory functions, we compared noncoding sequences of genomes of the most phylogenetically distant cephalochordate genera, Asymmetron and Branchiostoma, which diverged approximately 120–160 million years ago. We found 113,070 noncoding elements conserved between the two species, amounting to 3.3% of the genome. The genomic distribution, target gene ontology, and enriched motifs of these CNEs all suggest that many of them are probably cis-regulatory elements. More than 90% of previously verified amphioxus regulatory elements were re-captured in this study. A search of the cephalochordate CNEs around 50 developmental genes in several vertebrate genomes revealed eight CNEs conserved between cephalochordates and vertebrates, indicating sequence conservation over >500 million years of divergence. The function of five CNEs was tested in reporter assays in zebrafish, and one was also tested in amphioxus. All five CNEs proved to be tissue-specific enhancers. Taken together, these findings indicate that even though Branchiostoma and Asymmetron are distantly related, as they are evolving slowly, comparisons between them are likely optimal for identifying most of their tissue-specific cis-regulatory elements laying the foundation for functional characterizations and a better understanding of the evolution of developmental regulation in cephalochordates.
Keywords: CNE, regulatory element, cephalochordate, asymmetron, amphioxus
Introduction
Noncoding DNA includes several classes of functional elements that are phylogenetically conserved. Some of the best-known are microRNAs (miRNAs) (Hausser and Zavolan 2014) and their target sequences, long noncoding RNAs of several types (Fatica and Bozzoni 2014), and gene regulatory sequences (Guil and Esteller 2012; Nelson and Wardle 2013; Fatica and Bozzoni 2014). Many of these noncoding elements have characteristic sequences that can be used to identify them, but cis-regulatory regions of genes, which consist of constellations of transcription factor binding sites, are less stereotyped. As they typically reside either in regions flanking coding sequences or within introns, the initial strategy to identify them was to clone introns or a few thousand base pairs of flanking sequences into a reporter plasmid and determine if the potential regulatory DNA directs tissue-specific expression in embryos. Once this relatively large piece is determined to contain a transcriptional enhancer, it can be pared down to determine the precise sequences that constitute the enhancer. This traditional strategy works well but is cumbersome.
Recently, high-throughput approaches based on assessing chromatin state (methylation and acetylation) (e.g. ChIP-seq) and accessibility (e.g. DNaseI-seq, ATAC-seq) were applied to identify potential regulatory elements in the whole genome of a single species (Valouev et al. 2008; Buenrostro et al. 2013; Stergachis et al. 2014; Martinez-Morales 2015). However, such high-throughput approaches without concomitant analysis of transcription factor binding can lead to high false positives (Dogan et al. 2015). Neither of these methods directly addresses the question of how enhancers have changed during evolution of new traits and new species.
For understanding the evolution of regulatory DNA, comparative genomics has been useful. As more and more genomes became available, computational comparisons of genomes that are neither too closely nor too distantly related have identified large numbers of potential regulatory sequences, typically a few hundred base pairs long. These conserved noncoding elements (CNEs) can be experimentally tested for regulatory activity by linking them to a reporter gene such as green fluorescent protein (GFP) or LacZ and introducing them into eggs or embryos. This combined approach of in silico sequence conservation profiling, followed by elimination of sequences matching miRNAs and other known elements, followed by in vivo testing of the remaining sequences for gene regulatory activity has been successful in identifying many functional cis-regulatory elements (Woolfe et al. 2005; Pennacchio et al. 2006; McEwen et al. 2009; Hemberg et al. 2012). Moreover, comparisons of these elements across larger phylogenetic distances can reveal their origin and evolutionary fates along different lineages (Lang et al. 2010; Braasch et al. 2016). In general, many CNEs seem to be lineage-specific with high turnover rate even between closely-related species (Meader et al. 2010; Hiller et al. 2012), but some CNEs have been found to be extremely evolutionarily conserved even across different phyla (Royo et al. 2011; Clarke et al. 2012). The evolution of cis-regulatory elements has been considered to be the all-important factor in creating phenotypic diversity (Davidson 2011), and while it has more recently been shown that evolution of proteins is also highly important (Ono et al. 2014), the evolution of cis-regulation remains key to understanding phenotypic diversity.
One important consideration of studying CNEs via comparative genomics is that if the genome sequences of the organisms being compared are too much alike, the background noise will be too high for the CNEs to stand out. Conversely, when the genome sequences are too different, there may be no conservation outside of coding regions. This is the Goldilocks principle. The genome sequences of the organisms being compared must differ just the right amount from one another—not too little or too much. Thus, for vertebrates, whose genomes are evolving moderately slowly, comparisons between human and a teleost fish revealed 1,400 CNEs, 90% of which were functional (Woolfe et al. 2005), while comparisons among somewhat more closely related vertebrates yielded up to 3,000 CNEs, many of which were verified as functional regulatory elements (Ishibashi et al. 2012; Parker et al. 2014; Martinez-Morales 2015; Yousaf et al. 2015). In contrast, relatively few CNEs were shown by comparisons of the genomes of fast-evolving tunicates and vertebrates and many of those were not in syntenic loci (Maeso et al. 2013; Sanges et al. 2013). It took comparisons between two species within the same genus that separated only 3 million years ago (mya) to reveal additional CNEs in tunicates (Doglio et al. 2013). For cephalochordates, which are evolving exceptionally slowly (Yue et al. 2014), comparisons with fish and mouse identified about 670 such CNEs but only half of 42 tested for regulatory activity drove expression in zebrafish embryos (Hufton et al. 2009). However, with >20,000 genes in a cephalochordate genome (Putnam et al. 2008), there must be many thousands more CNEs. The phylogenetic distance between cephalochordates and vertebrates is evidently too great for most regulatory elements to be readily identified. This is exemplified by comparison of the Hedgehog locus between Branchiostoma and vertebrates, which showed there was virtually no conservation of noncoding DNA sequences. In contrast, comparisons of the Hedgehog locus among three species of Branchiostoma (Branchiostoma lanceolatum, Branchiostoma floridae, and Branchiostoma belcheri) revealed altogether too much conservation of noncoding DNA sequences for regulatory elements to be readily identified (Pascual-Anaya et al. 2008; Irimia et al. 2012a). Moreover, comparison of whole genome sequences of B. belcheri and B. floridae, which diverged about 112 mya (Nohara et al. 2005), revealed up to 180,000 CNEs (Huang et al. 2014). Many of these are likely due to insufficient divergence rather than functional constraints. Thus, the genomic sequences of Branchiostoma species are too close while those of Branchiostoma and vertebrates are too different for ready identification of CNEs and understanding their evolution. Therefore, we compared genomes of Asymmetron lucayanum and B. floridae, which diverged about 120–160 mya. We found that these genomes differ just about the right amount with approximately 113,000 CNEs. Some of these may not be functional regulatory elements. If they were, that would mean about five regulatory elements per gene, which is not altogether unreasonable, as some might be enhancers and others repressors. Even so, included in this set of CNEs were most of those previously identified as being conserved with vertebrates. We performed functional tests for five amphioxus CNEs that are not conserved with vertebrates at the sequence level and confirmed that they are indeed regulatory elements. Interestingly, when expressed in zebrafish, one of these CNEs directed expression to a domain that normally does not express the gene. Therefore, while the function of CNEs may be conserved across wide evolutionary distances, the genes they regulate may not always be conserved.
Materials and Methods
Whole Genome Shotgun Sequencing
We used three adult animals (two males and one female) and 22 larvae (from the cross of two of those three adults) for this study. The adult animals were collected from Bimini, Bahamas and maintained in the laboratory. The DNA of one male was used to make the first two libraries—one short-insert (approximately 300 bp) library and one long-insert (approximately 5,000 bp) library. We denoted these two libraries as Aluca4 and Aluca15, respectively. A moonlight regime (Fishbowl Innovations, Spokane, WA) was used to induce the spawning of another male and a female. The DNA of this pair with 22 of their offspring was used to make an additional pooled library (approximately 300 bp insert size and individually barcoded). We denoted this library as Aluca39. The genomic DNA of all samples was extracted by the DNAEasy kit (Qiagen Inc., Valencia CA, USA) and sequencing libraries were prepared by the Nextera kit (Illumina Inc, San Diego, CA, USA), according to the manufacturers’ protocols. Illumina paired-end sequencing was performed in three separate lanes on the Illumina HiSeq2000 platform. Aluca4 was sequenced at the Human and Molecular Genetics Center, Medical College of Wisconsin (Milwaukee, WI, USA). Auca15 and Aluca39 were sequenced at BGI (Shenzhen, Guangdong, China). The raw sequencing data are available from the NCBI SRA database via NCBI BioProject accession PRJNA280114 and PRJNA301923.
Whole-Genome Shotgun Sequencing Reads Processing and k-Mer Analysis
For each whole genome sequencing (WGS) library, raw reads were trimmed by Trimmomatic (v0.32) (Bolger et al. 2014) and processed by Deconseq (v0.4.3) (Schmieder and Edwards 2011a) to remove potential contaminations. For k-mer analysis, the reads from Aluca4 were counted by Jellyfish (v2.0) (Marçais and Kingsford 2011) using the two-pass method described in its manual. Different values of k (17, 19, 21, 23, and 25) were explored independently. The multiplicity of each k-mer and the number of distinct k-mers given such multiplicity were summarized.
Whole Genome Shotgun Sequencing Data Assembly
We used Platanus (v1.2.1) (Kajitani et al. 2014) to generate a de novo assembly for A. lucayanum with trimmed WGS reads from the Aluca4 and Aluca15 libraries, which were from the same animal. We chose Platanus because it was designed to handle highly heterozygous genomes. The maximum difference for bubble crush (-u) was set as 0.2 following the suggestion of the Platanus user manual for highly heterozygous genomes. The statistical summary for the assembly result was calculated by NGSQCToolkit (v2.3) (Patel and Jain 2012). This genome assembly has been deposited at DDBJ/ENA/GenBank under the accession LZCU00000000. The version described in this article is the version LZCU01000000.
Whole Genome Shotgun Reads Mapping
For each animal sample, the software package Stampy (v1.0.23) (Lunter and Goodson 2011) with “divergence” set to 10% was used to map trimmed reads to the B. floridae reference genome (v2.0) (Putnam et al. 2008). The mapping alignments were further processed with three programs 1) SAMtools (v0.1.19) (Li et al. 2009), 2) Picard-Tools (v1.106) (http://broadinstitute.github.io/picard/, last accessed July 20, 2016), and 3) GATK (v2.8-1) (McKenna et al. 2010). Depth of coverage across the reference genome was calculated by GATK with a mapping quality cutoff of 20.
RNA-Seq Sequencing and Assembly
The sequencing data from four RNA-Seq libraries used for this study included the following: 1) pooled A. lucayanum adults (denoted as asymAD), 2) pooled A. lucayanum 20h-larvae (phylotypic stage) (denoted as asym20h), 3) pooled B. floridae 20h-larvae (phylotypic stage) (denoted as bf20h), and 4) pooled B. floridae adults (denoted as bfAD). Details of reads processing and transcriptome assembly of the two A. lucayanum RNA-Seq libraries were previously described (Yue et al. 2014). The raw reads of these two A. lucayanum RNA-Seq libraries have been deposited in NCBI SRA database via NCBI BioProject accession PRJNA235900. The raw asymAD transcriptome assembly has been deposited at DDBJ/EMBL/GenBank under the accession GESY00000000 and version number GESY01000000. The raw asym20h transcriptome assembly has been deposited at DDBJ/EMBL/GenBank under the accession GETC00000000 and version number GETC01000000. The B. floridae pooled 20h-larvae library was prepared by the RNA-Seq kit (NuGen Inc., San Carlos, CA, USA) and sequenced by the Illumina GAII platform (paired-end 100 bp at the BioGem facility at University of California, San Diego, La Jolla, CA, USA). About 204 million raw reads were obtained for the bf20h library. Raw sequences are in the NCBI SRA database via NCBI BioProject accession PRJNA280115. The B. floridae pooled adults RNA-Seq library was constructed by as described by Fidler et al. (2014). The raw reads were deposited in NCBI SRA database (accession number: PRJNA215261). We processed the bf20h and bfAD RNA-Seq data following the same protocol as for the two Asymmetron libraries: raw reads were processed by Trimmomatic (v0.32) (Bolger et al. 2014), prinseq (v0.20.4) (Schmieder and Edwards 2011b), and Deconseq (v0.4.3) (Schmieder and Edwards 2011a) sequentially to trim the sequence and remove potential contamination; Trinity (r20131110) (Grabherr et al. 2011) and TransDecoder (http://transdecoder.github.io/, last accessed July 20, 2016) were used to generate the transcriptome assembly and infer the likely coding sequences (CDSs). The raw bf20h transcriptome assembly has been deposited at DDBJ/EMBL/GenBank under the accession GESZ00000000 and version number GESZ01000000. The raw bfAD transcriptome assembly has been deposited at DDBJ/EMBL/GenBank under the accession GETA00000000 and version number GETA01000000.
Genome Size Estimation
We used the k-mer-based method to estimate the genome size of A. lucayanum. The k-mer multiplicity distribution curve contains three peaks: the error peak with multiplicity (0-3), the heterozygous peak with multiplicity of dk/2, and the homozygous peak with multiplicity of dk, where dk is the k-mer depth of coverage. We used the 21-mer-multiplicity distribution to estimate the genome size. Assuming that Nk is the total number of non-error k-mers (after excluding the error peak from the k-mer multiplicity distribution), then the genome size G can be estimated by Nk/dk.
Repeat and Coding Sequence Masking for the B. floridae Reference Genome
To facilitate CNE identification, we created a copy of a repeat-and-coding-masked B. floridae genome as follows: we first performed repeat-masking for the reference genome using the software package RepeatMasker (open-v4.0.3) (http://www.repeatmasker.org, last accessed July 20, 2016) together with the B. floridae’s repeats library (retrieved from http://genome.jgi.doe.gov/Brafl1/Brafl1.home.html, last accessed July 20, 2016). Then we masked all the B. floridae CDS regions based on a previously corrected copy of B. floridae gene annotation file (Yue et al. 2014). To exclude potential CDS regions as cleanly as possible, we used Exonerate (v2.2.0) (Slater and Birney 2005) to align the inferred CDSs from our A. lucayanum and B. floridae RNA-Seq assemblies to the B. floridae reference and further masked those matched regions. After masking, the remaining unmasked regions along the B. floridae reference assembly should represent the noncoding and non-repetitive portion of the B. floridae genome.
Identification of CNEs Shared between Asymmetron and Branchiostoma and between Two Branchiostoma Species
Two methods were adopted to identify CNEs shared between Asymmetron and Branchiostoma. The first is a whole-genome-alignment-based (WGA-based) method. In this method, we used the progressive Cactus pipeline (Paten et al. 2011) to align our fragmented genome assembly of A. lucayanum to the B. floridae v2.0 reference assembly. To identify highly conserved regions shared between these two species, the Asymmetron-Branchiostoma whole genome alignments were analyzed by VISTA (v1.4.26) (Frazer et al. 2004) with B. floridae as the reference and the criteria of 45-bp sliding window, 90% identity, and 45-bp minimal length. The intersection of the highly conserved regions identified by VISTA and the repeat-and-coding-masked B. floridae reference genome formed our preliminary set of WGA-based CNEs. The second method is cross-species-reads-mapping-based (CSRM-based). In this method, we took the A. lucayanum-to-B. floridae reads mapping alignment and masked all the regions covered by < 5 reads (mapping quality cutoff = 20) along the B. floridae reference genome. To form our preliminary set of CSRM-based CNEs, the unmasked regions after such mapping-depth masking were used to identify the intersections with the repeat-and-coding-masked B. floridae reference genome.
We annotated the noncoding RNAs (ncRNAs) contained in our preliminary CNE sets with a combination of BLASTN and an Infernal (v1.0.2) (Nawrocki et al. 2009) search using the wrapper script rfam_scan.pl provided by Rfam, against the Rfam database (v11.0) (Gardner et al. 2011). The miRNAs were further annotated by miRBase (Release 21) (Kozomara and Griffiths-Jones 2013). To further exclude potential coding sequences in our preliminary CNE sets, we trained AUGUSTUS (v3.0.2) (Stanke et al. 2004) with the gene annotation information for the B. floridae reference genome and subsequently performed ab initio gene prediction for our preliminary CNE sets. This identified likely CDS sequences that were not previously identified based on the reference B. floridae gene annotation and our four RNA-Seq libraries. After eliminating all annotated ncRNAs and likely CDS regions, we applied a final length cutoff of 45 bp for the remaining CNEs to form the final version of the cephalochordate CNE sets for the WGA-based method and CSRM-based method. Bedtools (v2.22.0) (Quinlan and Hall 2010) was used to take the intersection and union of these two final CNE sets to form another two CNE sets: the CNEs-intersection and CNEs-union sets. Neighboring CNEs that are <10 bp away were rejoined together as a single CNE. The raw sequences of WGA-based CNEs for A. lucayanum–B. floridae comparison were deposited at GitHub (https://github.com/yjx1217/SupplementaryData_for_Asymmetron_CNE_paper_2016.git, last accessed July 20, 2016).
In parallel, we also identified WGA-based CNEs based on the comparison between two Branchiostoma species (B. floridae and B. belcheri) for downstream analysis. The genome assembly and gene annotation information of B. belcheri used for this analysis were retrieved from http://mosas.sysu.edu.cn/genome/download_data.php, last accessed July 20, 2016 (v18h27.r3) (Huang et al. 2014). The raw sequences of WGA-based CNEs for B. floridae–B. belcheri comparison were also deposited at GitHub (https://github.com/yjx1217/SupplementaryData_for_Asymmetron_CNE_paper_2016.git, last accessed July 20, 2016).
Physical Distribution and Functional Association of cephalochordate CNEs
For each cephalochordate CNE, we examined its physical position and distance relative to the nearest B. floridae gene using a 100-kb maximum distance cutoff. This allowed grouping our cephalochordate CNEs into six classes: intronic, 5’-flanking, 3’-flanking, equidistant (nearest CDSs were found in both 5’- and 3’-flanking directions with equal distance), no flanking (CDSs were found >100 kb away in both 5’- and 3’-flanking directions), and undefined (the flanking region of these CNEs encountered the end of the corresponding scaffold before reaching the 100-kb distance cutoff or any CDS). For the cephalochordate CNEs in the first three classes (intronic, 5’-flanking, and 3’-flanking), we examined the functional annotation of their nearest protein coding genes based on BLAST2GO’s annotation (Conesa et al. 2005). We ranked these genes by the number of CNEs that they are associated with and selected the top 5% of this ranked list for examining the enriched gene ontology (GO) terms. To assess statistical significance, Fisher’s exact test (Fisher 1922) with false discovery rate (FDR) correction (Benjamini and Hochberg 1995) was used.
Identification of Enriched Motifs in our CNE Sets
We used the de novo motif discovery tool HOMER (v4.7) (Heinz et al. 2010) to identify the enriched motifs of our CNE sets. Each discovered motif was searched against the known motif database collected by HOMER to identify its best-matched known motifs.
Manual Annotation for Important Cephalochordate Developmental Genes
Based on previous developmental biology studies on cephalochordates, we selected 50 important cephalochordate developmental genes (i.e. 12 Hox genes [Hox1-Hox10 and Hox12-Hox13–Hox11 is partially missing in the B. floridae v2.0 assembly], 5 Parahox genes [EvxA, EvxB, Mox, Cdx, Gsx–Xlox/Pdx is missing in the B. floridae v2.0 assembly], and 33 other genes [ADMP/SPTSSB, Ap2, BMP2/4, Brachyury, Bsx, Chordin, Engrailed, FoxA1, FoxA2/HNF3, FoxD, FoxG1, Gbx, Gli, Hedgehog, Id, Msx, MyoD, Nanos, Nk2.1, Nk2.2, Nk2.3/4/5, Nodal, Otx, Pax1/9, Pax2/5/8, Pax3/7, Pax6, Pitx, SoxE, Tbx1/10, Tbx2/3, Wnt1, and ZNF503/703]). We manually annotated these genes in the B. floridae reference assembly (v2.0) with BLAST. The protein domain composition of our annotated genes was further verified by Pfam v27.0 (http://pfam.xfam.org, last accessed July 20, 2016).
Orthologous CNEs in Vertebrates
To investigate the evolution of cephalochordate CNEs associated with those important amphioxus developmental genes within chordates, for each of the CNEs associated with those 50 genes, we used Lastz (v1.02) (Harris 2007) to conduct genome-wide searches in seven well-annotated vertebrates: elephant shark (Callorhinchus milii), zebrafish (Danio rerio), fugu (Takifugu rubripes), frog (Xenopus tropicalis), chicken (Gallus gallus), mouse (Mus musculus), and human (Homo sapiens). The genome assemblies (repeat soft-masked version) and gene annotations (in the gene transfer format [GTF]) of elephant shark were downloaded from http://esharkgenome.imcb.a-star.edu.sg/ (last accessed July 20, 2016), and those of all the other species were downloaded from Ensembl (Release 74). We masked the CDS regions of these genomes according to the gene annotations described in those GTF files before the search.
We used a combination of rather low thresholds (hspthresh = 1,500, gappedthresh = 2,500) in Lastz to maximize the sensitivity of our search. A following filter of identity ≥ 65% and the entropy score ≥ 1.7 were adopted to filter out most dubious alignments. The entropy score was calculated in the same way as proposed by Hiller et al. (2012):
Equation 1 for calculating sequence alignment entropy based on Hiller et al. 2012
Finally, we applied a stringent syntenic check to filter out those alignments for which the orthology relationships were uncertain. We manually inspected the orthologous relationship for the nearest gene of the cephalochordate query and its Lastz hits in those vertebrate genomes. For each cephalochordate CNE query, we only retained those query-hit pairs that are associated with the same orthologous developmental genes. The true orthologous vertebrate CNE hits and the corresponding cephalochordate CNE query were extracted from their respective genomes with 100-bp flanking regions at both 5’- and 3’-sides. These extracted orthologous cephalochordate-vertebrate CNEs sequences were aligned by MAFFT (v7.0) (Katoh and Standley 2013) using the “L-INS-i” strategy and visualized in Jalview (Waterhouse et al. 2009). The core CNEs were subsequently extracted based on the alignment sequence identity conservation profile provided by Jalview.
Comparison with Previous Studies
Each B. floridae CNE or regulatory element previously identified was searched against the B. floridae v2.0 assembly using Exonerate (v2.2.0) (Slater and Birney 2005). We also built the LiftOver chain file to facilitate automatic genomic coordinate conversion between the B. floridae v1.0 assembly and v2.0 assembly using UCSC’s Kent utilities.
Three-Way VISTA Plot
For the genomic regions around ADMP, BMP2/4, Brachyury, Mox, and Msx genes, we made three-way VISTA plots for B. floridae, B. belcheri, and A. lucayanum with B. floridae as the reference. The genome assembly for B. belcheri was obtained from http://mosas.sysu.edu.cn/genome/download_data.php (last accessed July 20, 2016). We located the genomic coordinates of these five genes in B. floridae based on our manual annotation and found their orthologous counterparts in B. belcheri and A. lucayanum using Exonerate (v2.2.0) (Slater and Birney 2005). The genomic regions were then retrieved with 3–5 kb 5’ and 3’ flanking regions. We aligned the sequences by FSA (v1.15.9) (Bradley et al. 2009) and used VISTA (v1.4.26) (Frazer et al. 2004) to visualize sequence conservation profile based on the criteria of 45-bp sliding window and 90% identity cutoff.
Zebrafish Transgenic Experiment
The CNE sequences were amplified from B. floridae genomic DNA using primers listed in supplementary table S9 and introduced into the ZED vector upstream of the minimal gata2a promoter and EGFP. To control for the efficiency of transgenesis in vivo, the reporter genes contained a second cassette composed of a cardiac actin promoter driving the expression of a red fluorescent protein (DsRed). EGFP and DsRed transcriptional units in the ZED vector are separated by an insulator (Bessa et al. 2009). For transgenesis, the Tol2 transposon/transposase method (Kawakami et al. 2004) was used with minor modifications. A mixture containing 30 ng/μl of transposase mRNA, 30 ng/μl of Qiagen column purified DNA, and 0.05% phenol red was injected in the cell of one-cell stage embryos. Embryos were raised at 28.5°C and staged by hours post fertilization. Embryos selected for imaging were anaesthetised with tricaine and mounted in low-melting agarose. Images were taken on Leica SP5 confocal microscope.
Amphioxus (B. floridae) Transgenic Experiment
The genomic DNA of Florida amphioxus (B. floridae) was isolated by phenol-chloroform extraction from an adult individual cultured in the laboratory. The amphioxus Msx-CNE region was amplified from amphioxus genomic DNA by PCR with FastStart Taq DNA Polymerase, dNTPack (Roche Applied Science, Indianapolis, USA), and cloned between the HindIII site and AsiSI site of the reporter construct derived from the 72-1.27 vector containing the minimal promoter of B. floridae FoxD (Corbo et al. 1997; Yu et al. 2004). Primers were 5’-gggAAGCTTcaatacaaacgcgctctgtaaaggtc-3’ (forward primer) and 5’-tctGCGATCGCcaatagttccaaaacggtggtagag-3’ (reverse primer). This construct contains the amphioxus Msx-CNE region, 593 bp upstream of the ATG start site of amphioxus FoxD including the TATA box, CCAAT box, and GC box elements and the first 15 amino acids of the amphioxus FoxD coding region upstream of the LacZ gene. Methods for microinjection and staining are according to Holland and Yu (2004).
Results
Sequencing, Assembly, and Mapping of the Asymmetron Genome
Illumina sequencing of the three genomic libraries of A. lucayanum yielded about 351 million paired-end 100-bp reads and 294 million paired-end 90-bp reads from the Aluca4 and Aluca15 library, respectively, and another 285 million paired-end 100-bp from the Aluca39 library. The completeness of the sequencing was evaluated by matching our previous A. lucayanum RNA-Seq assemblies to the A. lucayanum WGS reads. Of the two RNA-Seq libraries, about 93.37% and 98.43% of the transcriptome contigs had significant hits (BLASTN, e-value < 1E-6) with the WGS reads, indicating that our WGS represented most of the A. lucayanum genome.
Using the WGS reads from the Aluca4 and Aluca15 libraries, which are from the same individual animal, we attempted de novo whole genome assembly using the software package Platanus (Kajitani et al. 2014). We estimated the genome size of A. lucayanum to be approximately 644.45 mb (supplementary fig. S1). Unfortunately, largely owing to the high polymorphism of the A. lucayanum genome, the assembled contigs/scaffolds were fragmented. A total of 141,535 scaffolds (>1 kb) were obtained, for a combined length of 409.53 mb. The N50 length of these scaffolds is 3,567 bp. Only approximately 60% of Asymmetron reads from our previous RNA-seq study (Yue et al. 2014) could be placed into the Asymmetron scaffolds (58.47% for asymAD and 60.64% for asym20h), indicating many assembly gaps, in contrast to the >90% completeness of raw WGS reads. Nevertheless, this assembly offers a first draft genome sequence of A. lucayanum, which will provide the foundation for more complete assemblies based on additional sequencing.
We next mapped the WGS reads from the three Asymmetron genomic libraries to the B. floridae reference genome (v2.0). The genome-wide average mapping depth was 3.61X with approximately 9.96% of the genome covered by ≥ 5 reads (mapping quality cutoff = 20). For the coding regions (CDS) of the 28,593 B. floridae gene models in the v2.0 reference assembly, the average mapping depth is 19.35 with 48.56% of the CDS regions covered by ≥ 5 reads. Even though the mapping depth was good for the mapped region, only about 20% of the A. lucayanum reads could be mapped to the B. floridae reference genome, which was fairly consistent across all three WGS libraries (Aluca4, Aluca15, and Aluca39). This observation demonstrates considerable divergence between the Asymmetron and Branchiostoma genomes and indicates a high probability that the conserved noncoding regions between the two species were retained owing to functional constraints.
Identification of Cephalochordate CNEs Shared between Asymmetron and Branchiostoma
To identify the CNEs shared between Asymmetron and Branchiostoma, we used two independent approaches: 1) a WGA-based method and 2) a CSRM-based method (fig. 1). The WGA-based method assumes colinearity or syntenic conservation of CNEs between the species compared, whereas this positional information was largely uncaptured in the CSRM-based method. Thus, the WGA-based method is more stringent and well defined but, given the fragmented genome assembly of A. lucayanum, it can miss many CNEs. The CSRM-based method tends to give a more complete result but the conservation levels of those CNEs are less well defined. Not surprisingly, after excluding ncRNAs, the first method yielded fewer CNEs (45,515) than the second (109,410 CNEs) (table 1 and supplementary files S2 and S3). The 45,515 WGA-based CNEs account for 4.02 mb (0.84%) of the B. floridae v2.0 reference genome (480.40 mb after excluding the sequencing and assembly gaps in the B. floridae reference genome), while the 109,410 CSRM-based CNEs covered 15.51 mb (3.23%) of the reference genome (table 1). We generated another two CNE sets by taking the intersection and the union of the CSRM-based and WGA-based CNE sets, respectively (fig. 1). The intersection set comprises 40,957 CNEs with a cumulative length of 3.67 mb, while the union set contains 113,070 CNEs accounting for 3.30% (15.84 mb) of the B. floridae v2.0 reference genome (table 1 and supplementary files S4 and S5). Fig. 2 shows the CNEs around the Msx gene using the B. floridae v2.0 assembly as the genomic coordinate reference. Outside of the coding regions, there are several conserved regions; of particular note is the block downstream of the 3’ untranslated region (3’-UTR), which we experimentally verified to be a functional CNE (see below).
Fig. 1.—
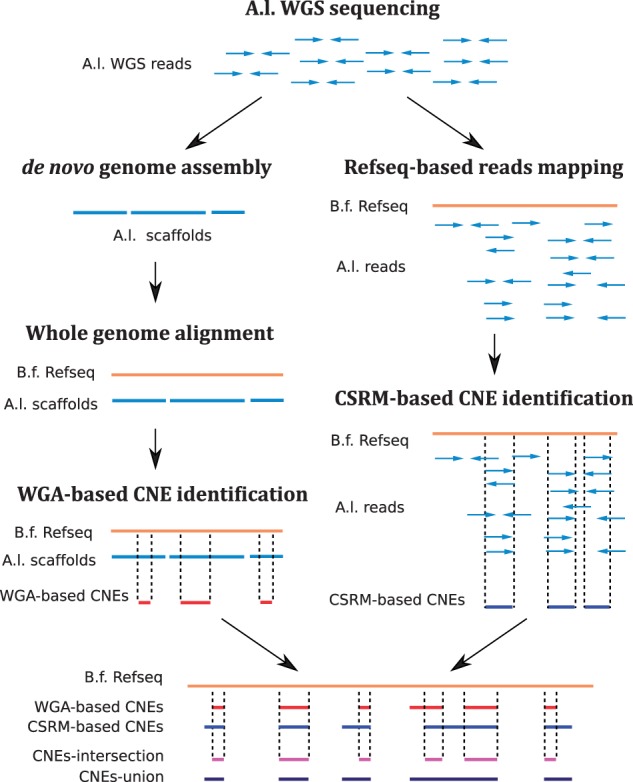
A diagram to show the two parallel strategies used in this study for CNE identification. Starting from the raw reads, a whole genome assembly of A. lucayanum was generated and further aligned to the B. floridae reference genome for CNE identification. We refer CNEs identified by this way as whole-genome alignment-based CNEs (WGA-based CNEs). Alternatively, another CNE set was generated by directly mapping the A. lucyanuam reads to the B. floridae reference genome and we refer these CNEs as cross-species reads mapping-based CNEs (CSRM-based CNEs). The intersection and union of WGA-based CNEs and CSRM-based CNEs sets were also extracted.
Table 1.
Summary of cephalochordate CNEs identified in this study
| CNE set | Sequence number | Accumulated length | Mean length | Median length | Maximum length |
|---|---|---|---|---|---|
| WGA-based CNEs | 45,515 | 4.02 Mb | 88.26 bp | 66.00 bp | 1,524.00 bp |
| CSRM-based CNEs | 109,410 | 15.51 Mb | 141.73 bp | 106.00 bp | 3,161.00 bp |
| CNEs-intersection | 40,957 | 3.67 Mb | 89.50 bp | 67.00 bp | 1,524.00 bp |
| CNEs-union | 113,070 | 15.84 Mb | 140.05 bp | 104.00 bp | 3,161.00 bp |
Fig. 2.—

An example of the cephalochordate CNEs identified in this study. The B. floridae Msx gene (B. floridae gene model ID = 278777). The genomic coordinates on the top of the figure show the location (Bf_V2_40:950,557-959,820) of this region according to the B. floridae reference assembly. Seven tracks are shown underneath: 1) A. lucayanum reads mapping depth, 2) mapped A. lucayanum reads, 3) B. floridae gene model, 4) WGA-based CNEs, 5) CSRM-based CNEs, 6) the intersection of WGA-based and CSRM-based CNEs, and 7) the union of WGA-based and CSRM-based CNEs. On the mapped reads track, each gray block represents a mapped A. lucayanum read with mapping quality ≥20. On the B. floridae gene model track, the orange blocks represent the CDS region and the orange line represents the intronic region, whereas the arrows represent the transcription direction of the corresponding gene. On the cephalochordate CNE tracks, each block represents an individual CNE that we identified in this study.
Cephalochordate CNEs are Enriched in the Proximity of trans-dev Genes
CNEs typically function as cis-regulatory elements and tend to cluster in introns or in the immediate proximity of transcription factors or signaling genes involved in developmental processes (“trans-dev” genes) (Bejerano et al. 2004; Woolfe et al. 2005; Vavouri et al. 2006; Vavouri et al. 2007). Therefore, we investigated the distribution of our identified cephalochordate CNEs as well as their nearest genes (supplementary files S2–S5) within a 100-kb radius using the B. floridae genome as the reference. Because the UTRs of most B. floridae genes are currently unknown, we marked the boundary of each B. floridae gene by its start and stop codons and defined the 100-kb upstream region from the start codon as the 5’-flanking region, and the 100-kb downstream region from the stop codon as 3’-flanking region.
By the above definitions, 41–46% of cephalochordate CNEs are located in introns, while 29–32% and 25–27% are located in 5’ and 3’-flanking regions, respectively (supplementary table S1). Very few (approximately 0.01%) cephalochordate CNEs are equidistant from upstream and downstream neighboring genes. The remaining few CNEs (<1%) are either outside of the 100-kb radius or their neighboring genes cannot be identified because the scaffold boundary was reached before any genes were identified within the 100-kb flanking radius. For those CNEs located in the 5’ and 3’ gene flanking regions, there was a strong pattern of CNE enrichment immediately adjacent the target genes (supplementary figs S2 and S3). Because of their proximity to coding regions, many of the CNEs probably represent cis-regulatory elements, although those within 1 kb of the start and stop codons are likely in 5’- or 3’-UTRs and may include binding sites for proteins initiating transcription or miRNAs in addition to cis-acting ncRNAs (Guil and Esteller 2012). We next asked what types of B. floridae genes are frequently associated with our identified CNEs. We ranked the genes by the number of CNEs they are associated with. The top 5% chiefly included genes involved in regulatory functions and developmental processes (table 2 and supplementary tables S2–S4). In particular, some genes such as those encoding Bruno4/6, Pax2, TIF1-alpha, and Neurexin-1-beta are associated with >100 CNEs, suggesting considerable evolutionary constraints surrounding these genes (table 3 and supplementary table S5–S7).
Table 2.
Enriched Gene Ontology (GO) terms for the genes associated with most CNEs based on the CNEs-union set
| GO-ID | Term | Category | FDR |
|---|---|---|---|
| GO:0048468 | Cell development | P | 1.46E-63 |
| GO:0048869 | Cellular developmental process | P | 2.86E-56 |
| GO:0030154 | Cell differentiation | P | 2.16E-54 |
| GO:0022008 | Neurogenesis | P | 2.49E-51 |
| GO:0048699 | Generation of neurons | P | 1.95E-49 |
| GO:0030182 | Neuron differentiation | P | 1.86E-46 |
| GO:0007399 | Nervous system development | P | 9.71E-46 |
| GO:0009653 | Anatomical structure morphogenesis | P | 1.01E-45 |
| GO:0048856 | Anatomical structure development | P | 1.99E-40 |
| GO:0030030 | Cell projection organization | P | 2.00E-39 |
Note.—The highlighted GO terms are shown as top 10 enriched GO terms based on all of our four CNE sets. For the GO term category column, “C” stands for cellular component, “F” for biological function, and “P” biological process. The statistical significance was assessed by Fisher’s exact test with FDR correction.
Table 3.
Cephalochordate genes associated with most CNEs based on the CNEs-union set
| B. floridae gene ID | CNE count | Gene product description |
|---|---|---|
| 89423 | 190 | Bruno 4/6 |
| 56669 | 173 | Paired box protein Pax-2 |
| 84482 | 171 | Transcription intermediary factor 1-alpha (TIF1-alpha) |
| 96723 | 145 | Unknown |
| 201173 | 137 | Protein CBFA2T1-like |
| 89425 | 126 | UPF0676 protein C1494.01-like |
| 68413 | 125 | Fibrinogen C domain-containing protein 1-like |
| 281312 | 121 | Receptor-type tyrosine-protein phosphatase delta-like |
| 208446 | 119 | Neurexin-1-beta-like |
| 84479 | 119 | Disks large homolog 5-like |
Note.—The highlighted genes are shown as top 10 genes associated with most CNEs based on all of our four CNE sets. The gene descriptions were retrieved based on BLASTP against NCBI nr database.
Enriched Transcription Factor Binding Motifs in Cephalochordate CNEs
After excluding the low-complexity motifs and other potential false positives, comparing the motifs enriched in our cephalochordate CNE sets to previously characterized binding motifs of various transcription factors yielded 29 enriched motifs in the WGA-based CNE set, 32 in the CSRM-based CNE set, 30 in the CNEs-intersection set, and 36 in the CNEs-union set (supplementary table S5–S8). These enriched motifs matched the binding motifs of several transcription factors including homeobox, basic helix-loop-helix, Zinc finger, and basic region-leucine zipper factors. These genes included Atf2, Esrrb, E2f, Ebf1, FoxO3, Nrf, Pbx3, Pit1, Ptf1a, and Yy1 (supplementary table S5–S8). This suggests that many, if not most, of the identified cephalochordate CNEs bind transcription factors.
Highly Conserved Cephalochordate CNEs Shared with Vertebrates
Given that cephalochordates and vertebrates shared a common ancestor >520 mya, we would expect at least some of our identified cephalochordate CNEs to also be conserved in vertebrates. As a proof of concept, for cephalochordate CNEs (from our CNEs-union set) associated with 50 important cephalochordate developmental genes, which include 12 Hox genes (Hox1-Hox10 and Hox12-Hox13—Hox11 is partially missing in the B. floridae v2.0 assembly), 5 Parahox genes (EvxA, EvxB, Mox, Cdx, Gsx—Xlox/Pdx is missing in the B. floridae v2.0 assembly), and 33 other genes (ADMP/SPTSSB, Ap2, BMP2/4, Brachyury, Bsx, Chordin, Engrailed, FoxA1, FoxA2/HNF3, FoxD, FoxG1, Gbx, Gli, Hedgehog, Id, Msx, MyoD, Nanos, Nk2.1, Nk2.2, Nk2.3/4/5, Nodal, Otx, Pax1/9, Pax2/5/8, Pax3/7, Pax6, Pitx, SoxE, Tbx1/10, Tbx2/3, Wnt1, and ZNF503/703), we preformed synteny checks to search for their orthologs in seven vertebrates (elephant shark, zebrafish, fugu, frog, chicken, mouse, and human) (See Methods section). One Hox4-CNE, one Gbx-CNE, one Msx-CNE, two Tbx2/3-CNEs, and three Znf503/703-CNEs were highly conserved with our sampled vertebrates (table 4 and supplementary figs S4–S11). We examined these eight CNEs using Ensembl (http://www.ensembl.org/, last accessed July 20, 2016) and the UCSC genome browser (http://genome.ucsc.edu/index.html, last accessed July 20, 2016) based on the well-annotated human genome (GRCh37/hg19). We found that the Hox4-CNE (Hox4-CNE-1) should be a 5’-UTR, whereas the other CNEs evidently have bona fide cis-regulatory functions, as suggested by several epigenomic signatures annotated by The Encode Project (The ENCODE Project Consortium 2007). Furthermore, the Msx-CNE (Msx-CNE-1), 5’ of the coding sequence, and one ZNF503/703-CNE (ZNF503/703-CNE-1) have been experimentally verified by previous studies (Holland et al. 2008; Hufton et al. 2009; Royo et al. 2011; Clarke et al. 2012). Finally, for all of these eight cephalochordate-vertebrate CNEs, we found that the sequence conservation between the two cephalochordates (A. lucayanum and B. floridae) clearly extends beyond the core CNE regions, echoing the trend previously observed in vertebrates (McEwen et al. 2009; Maeso and Tena 2016) that flanking sequences of ancient CNEs tend to be more conserved between more closely related lineages.
Table 4.
The genomic coordinates of eight cephalochordate CNEs that are also conserved in vertebrates
| CNE | Species | Assembly version | Chromosome/scaffolds | Start | End | Strand |
|---|---|---|---|---|---|---|
| Hox4-CNE-1 | B. floridae | JGI v2.0 | Bf_V2_12 | 935683 | 935734 | + |
| C. milii | v6.1.3 | scaffold_14 | 5156972 | 5157024 | + | |
| C. milii | v6.1.3 | scaffold_79 | 1146070 | 1146121 | + | |
| D. rerio | Zv9 | chr23 | 36182558 | 36182606 | + | |
| T. rubripes | FUGU4 | scaffold_66 | 127864 | 127912 | - | |
| X. tropicalis | JGI v4.2 | GL172692.1 | 1443781 | 1443838 | + | |
| X. tropicalis | JGI v4.2 | GL172862.1 | 463024 | 463072 | + | |
| G. gallus | Galgal4 | chr2 | 32825928 | 32825980 | - | |
| G. gallus | Galgal4 | chr7 | 15779237 | 15779290 | - | |
| M. musculus | GRCm38 | chr2 | 74727226 | 74727278 | + | |
| M. musculus | GRCm38 | chr6 | 52191689 | 52191741 | - | |
| M. musculus | GRCm38 | chr15 | 103034674 | 103034722 | + | |
| H. sapiens | GRCh37 | chr2 | 177016308 | 177016361 | + | |
| H. sapiens | GRCh37 | chr7 | 27170353 | 27170406 | - | |
| H. sapiens | GRCh37 | chr12 | 54447658 | 54447706 | + | |
| Gbx-CNE-1 | B. floridae | JGI v2.0 | Bf_V2_98 | 338705 | 338749 | + |
| D. rerio | Zv9 | chr24 | 35438769 | 35438813 | - | |
| T. rubripes | FUGU4 | scaffold_107 | 110492 | 110536 | + | |
| X. tropicalis | JGI v4.2 | GL172651.1 | 1936474 | 1936517 | - | |
| X. tropicalis | JGI v4.2 | GL172651.1 | 1942293 | 1942336 | + | |
| G. gallus | Galgal4 | chr2 | 173160 | 173204 | + | |
| M. musculus | GRCm38 | chr5 | 24526927 | 24526971 | - | |
| H. sapiens | GRCh37 | chr7 | 150864986 | 150865030 | - | |
| Msx-CNE-1 | B. floridae | JGI v2.0 | Bf_V2_40 | 953325 | 953435 | + |
| C. milii | v6.1.3 | scaffold_3 | 13863219 | 13863320 | - | |
| C. milii | v6.1.3 | scaffold_53 | 4190366 | 4190495 | - | |
| D. rerio | Zv9 | chr14 | 168407 | 168543 | + | |
| T. rubripes | FUGU4 | scaffold_116 | 868022 | 868152 | + | |
| T. rubripes | FUGU4 | scaffold_3613 | 3253 | 3383 | + | |
| X. tropicalis | JGI v4.2 | GL173077.1 | 691910 | 692039 | + | |
| G. gallus | Galgal4 | chr4 | 78386959 | 78387088 | + | |
| M. musculus | GRCm38 | chr5 | 37826828 | 37826956 | - | |
| H. sapiens | GRCh37 | chr4 | 4858647 | 4858775 | + | |
| Tbx2/3-CNE-1 | B. floridae | JGI v2.0 | Bf_V2_147 | 3548171 | 3548268 | + |
| C. milii | v6.1.3 | scaffold_47 | 3346941 | 3347032 | - | |
| D. rerio | Zv9 | chr5 | 57923023 | 57923117 | + | |
| D. rerio | Zv9 | chr5 | 75416575 | 75416677 | - | |
| D. rerio | Zv9 | chr15 | 26713164 | 26713258 | - | |
| T. rubripes | FUGU4 | scaffold_84 | 934772 | 934875 | + | |
| X. tropicalis | JGI v4.2 | GL172708.1 | 2281836 | 2281930 | - | |
| X. tropicalis | JGI v4.2 | GL173091.1 | 420790 | 420895 | - | |
| G. gallus | Galgal4 | chr15 | 12206709 | 12206815 | - | |
| G. gallus | Galgal4 | chr19 | 7638979 | 7639073 | + | |
| M. musculus | GRCm38 | chr5 | 119670890 | 119670994 | - | |
| M. musculus | GRCm38 | chr11 | 85832678 | 85832773 | - | |
| H. sapiens | GRCh37 | chr12 | 115122004 | 115122108 | + | |
| H. sapiens | GRCh37 | chr17 | 59477114 | 59477209 | - | |
| Tbx2/3-CNE-2 | B. floridae | JGI v2.0 | Bf_V2_147 | 3549098 | 3549149 | + |
| D. rerio | Zv9 | chr5 | 57923920 | 57923971 | - | |
| D. rerio | Zv9 | chr15 | 26712039 | 26712091 | - | |
| T. rubripes | FUGU4 | scaffold_300 | 238408 | 238461 | + | |
| T. rubripes | FUGU4 | scaffold_300 | 243979 | 244032 | + | |
| X. tropicalis | JGI v4.2 | GL172708.1 | 2280047 | 2280099 | - | |
| X. tropicalis | JGI v4.2 | GL173091.1 | 419230 | 419282 | - | |
| G. gallus | Galgal4 | chr15 | 12205007 | 12205059 | - | |
| M. musculus | GRCm38 | chr5 | 119669212 | 119669264 | - | |
| H. sapiens | GRCh37 | chr12 | 115123879 | 115123931 | + | |
| Znf503/703-CNE-1 | B. floridae | JGI v2.0 | Bf_V2_167 | 2385480 | 2385629 | + |
| C. milii | v6.1.3 | scaffold_3 | 13121560 | 13121704 | + | |
| D. rerio | Zv9 | chr5 | 26009609 | 26009746 | - | |
| D. rerio | Zv9 | chr13 | 17515432 | 17515575 | + | |
| T. rubripes | FUGU4 | scaffold_7 | 2065877 | 2066001 | - | |
| T. rubripes | FUGU4 | scaffold_86 | 79136 | 79279 | - | |
| X. tropicalis | JGI v4.2 | GL172676.1 | 1844110 | 1844257 | - | |
| X. tropicalis | JGI v4.2 | GL172901.1 | 202622 | 202765 | - | |
| G. gallus | Galgal4 | chr6 | 14130380 | 14130523 | + | |
| M. musculus | GRCm38 | chr8 | 26961370 | 26961507 | + | |
| M. musculus | GRCm38 | chr14 | 21991346 | 21991490 | - | |
| H. sapiens | GRCh37 | chr8 | 37532872 | 37533008 | + | |
| H. sapiens | GRCh37 | chr10 | 77165028 | 77165172 | - | |
| Znf503/703-CNE-2 | B. floridae | JGI v2.0 | Bf_V2_167 | 2387049 | 2387137 | + |
| C. milii | v6.1.3 | scaffold_3 | 13125570 | 13125657 | + | |
| D. rerio | Zv9 | chr13 | 17516606 | 17516696 | + | |
| T. rubripes | FUGU4 | scaffold_86 | 78313 | 78398 | - | |
| X. tropicalis | JGI v4.2 | GL172901.1 | 199576 | 199665 | - | |
| M. musculus | GRCm38 | chr14 | 21988662 | 21988751 | - | |
| H. sapiens | GRCh37 | chr10 | 77162348 | 77162437 | - | |
| Znf503/703-CNE-3 | B. floridae | JGI v2.0 | Bf_V2_167 | 2387253 | 2387382 | + |
| C. milii | v6.1.3 | scaffold_3 | 13125892 | 13126016 | + | |
| D. rerio | Zv9 | chr13 | 17517345 | 17517461 | + | |
| T. rubripes | FUGU4 | scaffold_86 | 77609 | 77725 | - | |
| X. tropicalis | JGI v4.2 | GL172901.1 | 193902 | 194019 | - | |
| M. musculus | GRCm38 | chr14 | 21987870 | 21987987 | - | |
| H. sapiens | GRCh37 | chr10 | 77161518 | 77161635 | - |
Previously Verified Branchiostoma cis-Regulatory Elements Are Largely Conserved with Asymmetron
We compared the CNEs shared between A. lucayanum and B. floridae with the 30 amphioxus (25 in B. floridae and 5 in B. lanceolatum) regulatory elements previously verified in reporter assays (Manzanares et al. 2000; Yu et al. 2004; Wada et al. 2006; Beaster-Jones et al. 2007; Holland et al. 2008; Royo et al. 2011; Clarke et al. 2012; Irimia et al. 2012a; Maeso et al. 2012; Van Otterloo et al. 2012; Acemel et al. 2016). Twenty-six of these 30 can be mapped to the version 2.0 assembly of B. floridae; presumably the absence of the other four is owing to errors in the assembly, which represents a single composite allele, whereas version 1.0 of the B. floridae genome used by Hufton et al. (2009) includes both alleles. Of these 26 functional elements 20 (76.92%) were also present in our WGA-based CNE set and in the CNEs-intersection set, and 24 (92.30%) were in our CSRM-based CNE set and CNEs-union set (table 5). The two CNEs that we missed are one Elav-like CNE at Bf_V2_69:357679-357729 and one Irx-Sowah 9d CNE at Bf_V2_14:300101-300634. However, we did find two other CNEs located close to these genes (3 bp and 27 bp away, respectively), suggesting that we might still recover the potential functionally important regions represented by these two CNEs. For those CNEs matched with previously verified regulatory elements, we found that their sequences tend to be generally longer than those non-matched CNEs (Wilcoxon rank sum test, P-value = 1.849E-3 for WGA-based CNEs and P-value = 5.485E-12 for CSRM-based CNEs), but no statistical difference was found in sequence identity between A. lucayanum and B. floridae (Wilcoxon rank sum test, P-value = 0.9154 for WGA-based CNEs; sequence identity for CSRM-based CNEs is not readily calculable).
Table 5.
Comparison between previously experimentally verified B. floridae CNEs or regulatory elements (REs) and this study
| Functional B. floridae | Genomic coordinate | Literature source | Comparison with this study |
|||
|---|---|---|---|---|---|---|
| CNE/RE name | (in the B. floridae v2.0 assembly) | WGA-based CNEs | CSRM-based CNEs | CNEs- intersection | CNEs- union | |
| Elav-like CNEa | Bf_V2_69:357679-357729 | Hufton et al. (2009) | X | X | X | X |
| Engrailed RE | Bf_V2_9:1267978-1274277 | Beaster-Jones et al. (2007) | ✓ | ✓ | ✓ | ✓ |
| EvxA? RE 2473 | Bf_V2_12:378112-378330 | Acemel et al. (2016) | ✓ | ✓ | ✓ | ✓ |
| FoxD RE | Bf_V2_113:901685-902885 | Yu et al. (2004) | ✓ | ✓ | ✓ | ✓ |
| Hedgehog RE | Bf_V2_205:188300-193172 | Irimia et al. (2012a) | ✓ | ✓ | ✓ | ✓ |
| Hox1A RE | Bf_V2_12:999003-1001503 | Manzanares et al. (2000); Wada et al. (2006) | ✓ | ✓ | ✓ | ✓ |
| Hox2B RE | Bf_V2_12:986507-990914 | Manzanares et al. (2000); Wada et al. (2006) | ✓ | ✓ | ✓ | ✓ |
| Hox2C RE | Bf_V2_12:980069-981704 | Manzanares et al. (2000); Wada et al. (2006) | ✓ | ✓ | ✓ | ✓ |
| Hox3B RE | Bf_V2_12:977525-979277 | Manzanares et al. (2000); Wada et al. (2006) | ✓ | ✓ | ✓ | ✓ |
| Hox1 1655 RE | Bf_V2_12:1119489-1119959 | Acemel et al. (2016) | ✓ | ✓ | ✓ | ✓ |
| Hox1 1739 RE | Bf_V2_12:1060551-1060880 | Acemel et al. (2016) | ✓ | ✓ | ✓ | ✓ |
| Hox1 1784 RE | Bf_V2_12:1018545-1019524 | Acemel et al. (2016) | ✓ | ✓ | ✓ | ✓ |
| Hox1 1801 RE | Bf_V2_12:1002265-1002697 | Acemel et al. (2016) | ✓ | ✓ | ✓ | ✓ |
| Irx-Sowah 1a CNE | Bf_V2_14:276485-278804 | Maeso et al. (2012) | ✓ | ✓ | ✓ | ✓ |
| Irx-Sowah 5b CNE | Bf_V2_14:351877-352857 | Maeso et al. (2012) | X | ✓ | X | ✓ |
| Irx-Sowah 6a CNE | Bf_V2_14:424282-425437 | Maeso et al. (2012) | X | ✓ | X | ✓ |
| Irx-Sowah 6c CNE | Bf_V2_14:315985-317211 | Maeso et al. (2012) | X | ✓ | X | ✓ |
| Irx-Sowah 9d CNEa | Bf_V2_14:300102-300634 | Maeso et al. (2012) | X | X | X | X |
| Irx-Sowah 10b CNE | Bf_V2_14:378423-380890 | Maeso et al. (2012) | X | ✓ | X | ✓ |
| Irx-Sowah 10d CNE | Bf_V2_14:301002-303621 | Maeso et al. (2012) | ✓ | ✓ | ✓ | ✓ |
| Msx CNE | Bf_V2_40:953259-953496 | Hufton et al. (2009); Royo et al. (2011); Clarke et al. (2012) | ✓ | ✓ | ✓ | ✓ |
| Six3/6 CNE | Bf_V2_245:211307-211352 | Hufton et al. (2009); Royo et al. (2011) | ✓ | ✓ | ✓ | ✓ |
| SoxB2 CNE | Bf_V2_196:4387592-4387942 | Hufton et al. (2009); Royo et al. (2011) | ✓ | ✓ | ✓ | ✓ |
| SoxE RE | Bf_V2_174:2742068 2744567 | Van Otterloo et al. (2012) | ✓ | ✓ | ✓ | ✓ |
| Sp5 CNE | Bf_V2_149:861774-862000 | Hufton et al. (2009) | ✓ | ✓ | ✓ | ✓ |
| ZNF503/703 CNE | Bf_V2_167:2385491-2385650 | Holland et al. (2008); Royo et al. (2011); Clarke et al. (2012) | ✓ | ✓ | ✓ | ✓ |
aAlthough there is no direct overlap between our CNEs and these two regions, we found other CNEs in the immediate proximity of these regions.
The Number of CNEs Identified is Highly Dependent on the Method
There are three previous genome-wide studies identifying CNEs shared between Branchiostoma sp. and vertebrates. Two used the B. floridae v1.0 assembly. Putnam et al. (2008) identified 77 CNEs based on B. floridae versus human, while Hufton et al. (2009) identified 1,299 CNEs based on B. floridae versus mouse, Fugu, and zebrafish. After removing redundancies by mapping these CNEs to the B. floridae v2.0 assembly and removing those overlapping with CDS regions or ncRNAs, 54 and 669 CNEs, respectively, were left. However, our CNEs-union set matched only 21 of these 54 CNEs (identified by Putnam et al. 2008) and just 120 of those 669 CNEs (identified by Hufton et al. 2009). Surprisingly, only 6 of the 54 CNEs in the first set are also in the second set, even though both compared cephalochordates and vertebrates. We think this large discrepancy likely comes from the differences in methodology and conservation criteria. In the study by Putnam et al. (2008), a WGA-based method similar to ours was used, with the criterion of 60% nucleotide identity across a 50-bp window, whereas Hufton et al. (2009) used a local-similarity-based method centered on conserved gene families. Also for the Hufton et al. (2009) study, a later review pointed out that the authors might have overestimated the CNEs shared between cephalochordates and vertebrates given that they did not check the detailed position and orientation of those CNEs relative to their respective target genes (Maeso et al. 2013).
The third study compared the Chinese amphioxus, B. belcheri, with B. floridae and vertebrates (human and opossum) using a combination of Lastz-ChainNet-based and BLASTN-based methods. It found at least 135,046 CNEs shared between B. floridae and B. belcheri, with 1,084 also shared with vertebrates (Huang et al. 2014). Because this result is based on their B. belcheri reference coordinate system and not on B. floridae, it was not straightforward to compare their results with ours. Therefore, using the same criteria that we used for the Asymmetron–Branchiostoma comparison, we ran our WGA-based CNE pipeline to generate our own CNE set shared between these two Branchiostoma species. This resulted in 179,224 CNEs with a cumulative length of 16.48 Mb, which is considerably more than the CNEs-union set we obtained for the Asymmetron–Branchiostoma comparison (CNE count: 113,070; cumulative length: 15.84 Mb). Most of the A. lucayanum–B. floridae CNEs (77.32% when calculating based on the CNEs-union set) were recapitulated in the B. floridae–B. belcheri CNE set. Moreover, for these shared CNEs across all three amphioxus species, the comparison between the two Branchiostoma species reveals longer sequence conservation tracts than the comparison between Asymmetron and Branchiostoma under the same criteria (Wilcoxon rank sum test, P-value < 2.2E-16). The same trend holds for those ancient CNEs that are shared between cephalochordate and vertebrates. All of these observations are consistent with what we would expect based on the phylogenetic relationship among these three cephalochordate species. Chances are that many of these 180,000 CNEs shared between B. belcheri and B. floridae are not gene regulatory elements. These two congeners diverged about 100 mya (Nohara et al. 2005). However, as cephalochordates are evolving particularly slowly (Yue et al. 2014), 100 mya appears to be insufficient for meaningful CNEs identification in cephalochordate genomes.
Experimental Verification of in silico CNEs in Zebrafish and Amphioxus
To verify that some of the CNEs that were identified computationally really are functional cis-regulatory elements, we generated Vista alignments between B. floridae and B. belcheri and between B. floridae and A. lucayanum for five genes (ADMP, BMP2/4, Brachyury, Mox, and Msx) (fig. 3). As this figure shows, the Vista alignments for the two Branchiostoma species reveal altogether too much conservation outside the coding regions. Therefore, for each of these genes, we randomly selected a noncoding region conserved between A. lucayanum and B. floridae as well as with B. belcheri (boxed in fig. 3) to test experimentally by linking them to reporter constructs. For ADMP and BMP2/4, so many regions are conserved between the two genera that our selection was somewhat arbitrary. All reporter constructs were injected into zebrafish eggs and, for the Msx 3’ CNE, into B. floridae eggs as well (supplementary table S9; fig. 4). As injections into amphioxus eggs are time-consuming, we did not attempt to express the other constructs in amphioxus. The Msx CNE downstream of the 3’ UTR has a central region of 11 bp that is not conserved; therefore, while it could be considered to be two CNEs in our in silico CNE identification, the entire region was tested in expression assays.
Fig. 3.—
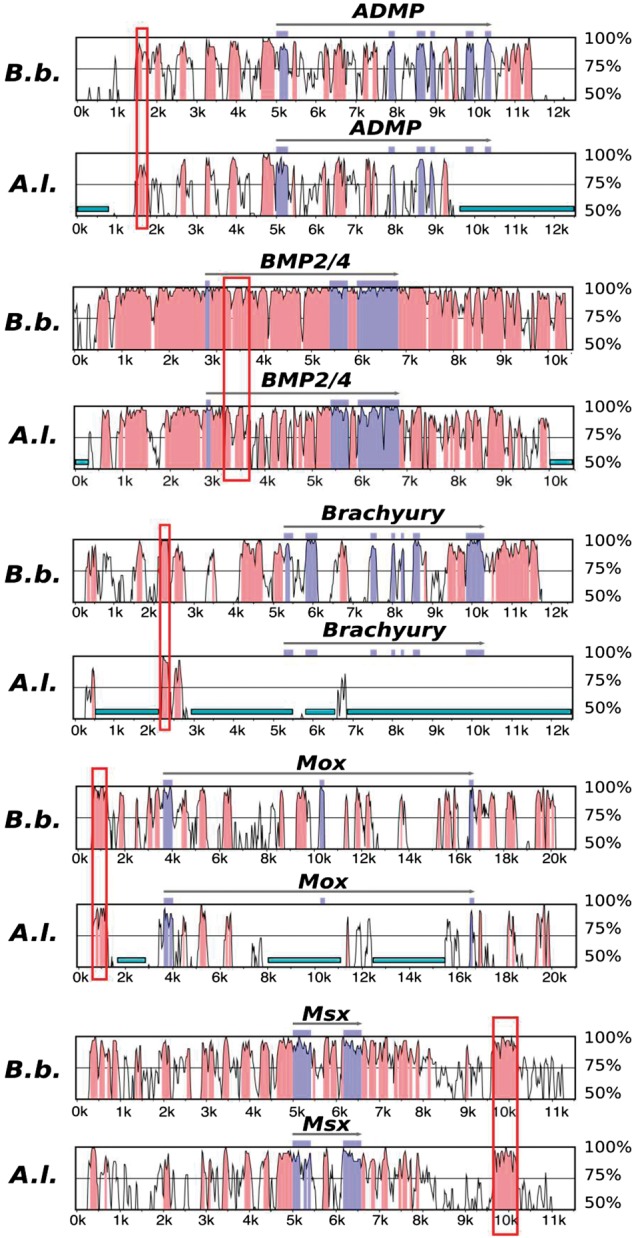
Vistaplots reveal high sequence conservation across cephalochordate genomes. The genomic sequences with >50% sequence identity compared with B. floridae are shown for B. belcheri (denoted as B. b.) and A. lucayanum (denoted as A. l.) around ADMP, BMP2/4, Brachyury, Mox, and Msx genes. The CDS regions are depicted in blue, while CNEs with 90% identity over 45-bp window are depicted in red. The tested CNE for each gene was highlighted by the red boxes. The cyan bars at the bottom indicate assembly gaps in A. lucyanuam.
Fig. 4.—

Amphioxus CNEs identified in silico by comparing cephalochordate genomes are functional enhancers in zebrafish. (A) ADMP CNE drives the expression of enhanced green fluorescent protein (EGFP) into dorsal shield of zebrafish late gastrula. (B) Negative control for A shows no expression. (C) EGFP expression driven by BMP2/4 CNE is present throughout the blastoderm with lower intensity in the shield region at gastrula stage. (D) Negative control for C shows no expression. (E) The activity of the Brachyury CNE at late segmentation period. White arrows indicate expression in the notochord. (F) The Msx CNE directs expression to the muscles. Red marks expression of DSRED directed by a muscle-specific zebrafish enhancer. Yellow shows co-expression directed by both enhancers in the muscles. (G) The Mox CNE from amphioxus directs expression to muscle in the zebrafish. Red marks expression directed by a muscle-specific zebrafish enhancer. Yellow shows co-expression directed by both enhancers in the muscles. (H) Negative control. EGFP expression with no enhancer is nonspecific. Red indicates expression of a co-injected muscle-specific enhancer. (I–N). Amphioxus embryos. Anterior to the left. (I) Mid-neurula. Expression driven by the Msx-CNE is mosaic in muscle. (J) Late-neurula. Expression driven by Msx-CNE is expressed strongly in muscle. (K) Negative control. Early neurula. Expression limited to a single sick cell in the gut lumen. (L) Dorsal view. Expression of the Msx-CNE in muscles on the right side. (M) Dorsal view. Expression driven by the Msx-CNE is in muscles on the left side. (N) Negative control. Expression of the empty vector in a single anterior ectodermal cell and in a single cell in the vicinity of the muscles.
All of the cephalochordate CNEs directed tissue-specific expression in zebrafish, while the 3’ Msx-CNE also directed expression in amphioxus (fig. 4). The amphioxus ADMP-CNE directs expression throughout much of the zebrafish shield at 90% epiboly (fig. 4A). This domain is somewhat broader than expression of the native zebrafish ADMP gene at the same stage (Dickmeis et al. 2001). Expression driven by the BMP2/4 CNE recapitulates the expression pattern of the native genes in both zebrafish and amphioxus at the gastrula stage—broadly in both ectoderm and mesendoderm (fig. 4C). The amphioxus Brachyury CNE recapitulates native expression of the zebrafish Brachyury in the notochord (fig. 4E) (Schulte-Merker et al. 1994). The native Brachyury gene is also expressed in the amphioxus notochord (Holland et al. 1995). The amphioxus Mox CNE directs expression to developing muscle in zebrafish (fig. 4G). Mox is expressed in developing muscle in both amphioxus (Minguillón and Garcia-Fernàndez 2002) and in Xenopus, as well as in other mesodermal tissues (Candia and Wright 1995). Zebrafish Mox orthologs meox2a and meox2 are first expressed at the end of somitogenesis in formed myotomes and later in fin myoblasts and specific muscles of the head (Nguyen et al. 2014). The amphioxus Msx CNE directs expression to developing muscle in both amphioxus and vertebrates (fig. 4I-M), one of the two domains that express the native gene (Sharman et al. 1999). The other domain is in the neural tube. An amphioxus CNE 5’ of the Msx coding region was previously shown to direct expression to the neural tube in zebrafish (Hufton et al. 2009). Interestingly, none of the four zebrafish Msx genes is expressed in muscle (Akimenko et al. 1995). However, mouse Msx1 is expressed in limb muscle precursor cells (Bendall et al. 1999). In the invertebrates Platynereis dumerilli and Drosophila melanogaster, Msx is expressed in presumptive myoblasts that give rise to segmental muscles (Jagla et al. 1999; Ramos and Robert 2005; Saudemont et al. 2008).
These results show that comparisons between Asymmetron and Branchiostoma are highly effective in revealing functional CNEs. Although the ones we tested functionally were also conserved with vertebrates, it is likely that most of those conserved between the two cephalochordate genera but not vertebrates will also prove to be functional enhancers.
Discussion
The Goldilocks Principle in CNE Identification
When CNEs are identified by comparisons of homologous regions of DNA among different species, the sequences being compared have to be conserved to just the right degree. If they are too divergent, regulatory DNA sequences may have moved in relation to the coding sequence; transcription factor binding sites may have shifted position within the regulatory element; or the sequence may have changed considerably. If, on the other hand, the sequences are too highly conserved, regulatory elements cannot be distinguished from the background nonfunctional DNA.
The effective phylogenetic distance for comparisons of genome sequences to reveal meaningful CNEs depends not only on the divergence time but on the rates of evolution as well. For fast-evolving organisms, the phylogenetic distance must be small, while for slow-evolving ones, the distance between the organisms being compared must be much larger. For example, tunicates are evolving much, much faster than vertebrates and cephalochordates (Tsagkogeorga et al. 2010; Yue et al. 2014). Genome sequence alignments between the tunicate Ciona intestinalis and vertebrates revealed few, if any conserved non-coding elements. In contrast, the genetic distance between the tunicates C. intestinalis and Ciona savignyi, which split 3–4 mya, seems to be about the same as that between human and chicken, which split about 310 mya (Furlong 2005; Irvine 2013). Consequently, like comparisons between humans and chickens and/or frogs, comparisons between the two congeners of Ciona have revealed many CNEs (Russo et al. 2004; Irvine 2013). In addition, separate analyses of the genomes of the two Ciona species and six vertebrates, revealed 183 CNEs that are syntenic among vertebrates. However, 182 of them were located in non-syntenic positions in tunicate genomes (Sanges et al. 2013).
In contrast with tunicates, comparisons between fairly distant vertebrates, which are evolving relatively slowly, with agnathans splitting from gnathostomes about 450 mya, and mammals first appearing about 320 mya, have revealed numerous CNEs. Comparisons between human and fugu initially revealed about 1,400 CNEs (Woolfe et al. 2005). In addition, of a set of 1,205 human CNEs distributed across about 1% of the human genome, 1,142 were conserved with chicken, 1,035 with fugu, 789 with elephant shark but only 73 with the lamprey (McEwen et al. 2009). This implies that CNEs conserved between amphioxus and vertebrates are probably performing vital functions. Although cephalochordates and vertebrates diverged >520 mya, because cephalochordates are evolving even more slowly than the slowest evolving vertebrate known, the elephant shark (Venkatesh et al. 2014; Yue et al. 2014), hundreds of CNEs that are shared between amphioxus and vertebrates have been identified (Putnam et al. 2008; Hufton et al. 2009; Huang et al. 2014). Some of them are even conserved across greater evolutionary distance (e.g. also conserved in hemichordates and even protostomes or cnidarians; Royo et al. 2011; Clarke et al. 2012; Simakov et al. 2015). To identify more regulatory elements, especially those cephalochordate-specific ones, we compared the two most phylogenetically distant genera of cephalochordates (Asymmetron and Branchiostoma), which diverged about 120–160 mya. While intra-genus comparisons for very fast-evolving species such as tunicates and relatively fast-evolving ones such as Drosophila (Schmid and Tautz 1997; Makunin et al. 2014), which diverged 30–40 mya, have revealed numerous enhancers (>2,000 for Drosophila), for very slowly evolving species, comparisons over a much wider phylogenic distance are better. Thus, as fig. 3 shows, for cephalochordates, even the 112 million years of separation for Branchiostoma (B. floridae and B. belcheri) estimated from mitochondrial DNA sequences (Nohara et al. 2005) does not suffice to separate the CNEs from background sequences. Levels of conservation for ADMP, BMP2/4, Brachyury, Mox, and Msx in the 3–5 kb up- and downstream of the coding regions and in the introns are high between two Branchiostoma species, revealing only a few regions with ≤50% identity. This widespread conservation in the noncoding regions of Branchiostoma species echoes a previous observation about the Hedgehog locus of Branchiostoma; the noncoding regions of this locus are strikingly similar among the three Branchiostoma species (B. floridae, B. lanceolatum, and B. belcheri; Irimia et al. 2012b). In line with the very slow evolution, conservation between Branchiostoma and Asymmetron is also fairly high given the 120–160 million years since they diverged (Kon et al. 2007; Yue et al. 2014). Although some of the 113,070 CNEs we identified that are conserved between A. lucayanum and B. floridae may not be functional regulatory elements, with 23,000 genes in cephalochordates, one would expect to find a minimum of 50,000 regulatory elements. Therefore, in contrast to the species of Branchiostoma, the Asymmetron versus Branchiostoma comparison seems to be better for identifying functional regulatory elements. Remarkably, these two genera will hybridize and develop at least to the mid-larval stage even though they have different numbers of chromosomes (2n = 38 in B. floridae; 2n = 34 in A. lucayanum) and different sized genomes (480.40 mb after excluding the sequencing and assembly gaps in B. floridae; 645 mb in A. lucayanum) (Holland et al. 2015). Comparisons between these two genera and their hybrids promise to be highly informative for understanding the genetic mechanisms of development, in general, and the construction of gene regulatory networks in particular.
CNE Evolution
Once CNEs have been identified between one or more groups, comparisons with somewhat more distantly related organisms can show how CNEs have evolved as organisms have diverged. For example, comparisons among mammals have revealed loss of many CNEs in one mammalian lineage or another (Hiller et al. 2012; Villar et al. 2015). CNEs that were not lost in any mammalian lineage were, in general, older than those that were lost. Similarly, comparative genomics revealed several possible enhancers near the Shh gene conserved between the coelacanth and some sarcopterygian and actinopterygian fishes and verified in reporter assays (Lang et al. 2010). However, several of these CNEs were missing in more recently evolved sarcopterygian and actinopterygian fishes.
Not only can old enhancers disappear during evolution, if they persist they may retain old functions and/or acquire new functions. Examples of such functional conservation are the Msx CNE in the present study and a CNE near the EBF3 gene in lamprey and human (McEwen et al. 2009). The amphioxus CNE near ZNF503/703 and its two vertebrate homologs show that CNEs can both retain old functions and acquire new ones in evolution. This amphioxus CNE directs expression to the amphioxus notochord and somites but not to the central nervous system (CNS) and to some, but not all, of the domains that the corresponding enhancers adjacent the human ZNF503 and ZNF703 genes direct expression to in the mouse (Holland et al. 2008). Moreover, the amphioxus enhancer directs expression in the mouse to one domain in the eye to which the vertebrate counterparts do not direct expression. Similarly, in vertebrates, some CNEs associated with GLI3, which transduces Shh signaling, show conserved expression in mouse and zebrafish, while one CNE, which directs expression to the limb bud in the mouse and chick, directs expression to the notochord and blood cell precursors, but not to the limb, in the zebrafish (Anwar et al. 2015). In fact, such examples of acquiring new regulatory functions by co-option or modification of preexisting CNEs are prevalent in the evolution of new regulatory elements (Maeso and Tena 2016).
New regulatory elements can also be gained via exaptation of repetitive elements and transposable elements. For example one family of RSR elements that function as transcriptional enhancers in the strongylocentrotid family of sea urchins evolved from repetitive sequence at the base of that family; RSR elements are absent from other sea urchins such as Heliocidaris and Lytechinus (Dayal et al. 2004). The co-option of transposable elements for regulatory elements has been well-documented (Britten 1997). An example of how transposable elements may become candidates for new regulatory elements is shown by the amphioxus FoxD gene (Yu et al. 2004). We noted a transposable element with many TCF binding sites located about 1 kb upstream of the start codon of the FoxD gene in a clone from a genomic library. However, this sequence was not part of the tissue-specific enhancer for this gene and, as it was absent from the same place in the FoxD gene in the genome sequenced from another individual, it had not become fixed in the population.
As more genomes become available, it may be informative to investigate the amphioxus CNEs in the context of hemichordates and early-diverged echinoderms. Two noncoding elements associated with the Pax1/9 gene were found to be conserved among vertebrates, amphioxus and hemichordates (Simakov et al. 2015), raising the question of just when the amphioxus CNEs that we determined in the present study evolved. However, given the different body plans of chordates and ambulacrarians, as well as the rapid turnovers of cis-regulatory elements in general, it may be difficult to trace the origins of most amphioxus CNEs in deuterostome evolution.
Supplementary Material
Supplementary files S1–S5, Figures S1–S11, and Tables S1–S11 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank Dr Michael Hiller for sharing the script for alignment entropy calculation, Dr Michael H. Kohn for helpful suggestions during manuscript writing, Dr Robert A. Cornell, Dr Peter W. H. Holland, Dr Manuel Irimia, Dr Ignacio Maeso, Dr Matthew Blow, Dr Len A. Pennacchio, and Dr Hiroshi Wada and for providing detailed information about the amphioxus regulatory regions that they identified in their previous studies, Dr. Nicholas D. Holland for collecting the specimens of A. lucayanum, Dr Koichi Kawakami and Dr Jose Bessa for the reagents used in zebrafish transgenic experiment, and the anonymous reviewers for their valuable comments that helped us to improve the manuscript. J.X.Y. was supported by a doctoral fellowship from Rice University when conducting this study; he is currently supported by a postdoctoral fellowship from Fondation ARC pour la Recherche sur le Cancer (n°PDF20150602803). L.Z.H. was supported by the grant NSF IOS1353688 from National Science Foundation. I.K. and Z.K. were supported by the grant GC15-21285J from Grantová Agentura České Republiky. J.K.Y. was supported by the Career Development Award AS-98-CDA-L06 from Academia Sinica, Taiwan, and grants 102-2311-B001-011-MY3 and 104-2923-B-001-002-MY3 from the Ministry of Science and Technology (MOST), Taiwan.
Literature Cited
- Acemel RD, et al. 2016. A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat Genet. 48:336–341. [DOI] [PubMed] [Google Scholar]
- Akimenko MA, Johnson SL, Westerfield M, Ekker M. 1995. Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development 121:347–357. [DOI] [PubMed] [Google Scholar]
- Anwar S, et al. 2015. Identification and functional characterization of novel transcriptional enhancers involved in regulating human GLI3 expression during early development. Dev Growth Differ. 57:570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaster-Jones L, Schubert M, Holland LZ. 2007. Cis-regulation of the amphioxus engrailed gene: insights into evolution of a muscle-specific enhancer. Mech Dev. 124:532–542. [DOI] [PubMed] [Google Scholar]
- Bejerano G, et al. 2004. Ultraconserved elements in the human genome. Science 304:1321–1325. [DOI] [PubMed] [Google Scholar]
- Bendall AJ, Ding J, Hu G, Shen MM, Abate-Shen C. 1999. Msx1 antagonizes the myogenic activity of Pax3 in migrating limb muscle precursors. Development 126:4965–4976. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 57:289–300. [Google Scholar]
- Bessa J, et al. 2009. Zebrafish enhancer detection (ZED) vector: a new tool to facilitate transgenesis and the functional analysis of cis-regulatory regions in zebrafish. Dev Dyn. 238:2409–2417. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braasch I, et al. 2016. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat Genet. 48:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RK, et al. 2009. Fast statistical alignment. PLoS Comput Biol. 5:e1000392.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. 1997. Mobile elements inserted in the distant past have taken on important functions. Gene 205:177–182. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candia AF, Wright CV. 1995. The expression pattern of Xenopus Mox-2 implies a role in initial mesodermal differentiation. Mech Dev. 52:27–36. [DOI] [PubMed] [Google Scholar]
- Clarke SL, et al. 2012. Human Developmental enhancers conserved between deuterostomes and protostomes. PLoS Genet. 8:e1002852.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. [DOI] [PubMed] [Google Scholar]
- Corbo JC, Levine M, Zeller RW. 1997. Characterization of a notochord-specific enhancer from the Brachyury promoter region of the ascidian, Ciona intestinalis. Development 124:589–602. [DOI] [PubMed] [Google Scholar]
- Davidson EH. 2011. Evolutionary bioscience as regulatory systems biology. Dev Biol. 357:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal S, et al. 2004. Creation of cis-regulatory elements during sea urchin evolution by co-option and optimization of a repetitive sequence adjacent to the spec2a gene. Dev Biol. 273:436–453. [DOI] [PubMed] [Google Scholar]
- Dickmeis T, et al. 2001. Expression of the anti-dorsalizing morphogenetic protein gene in the zebrafish embryo. Dev Genes Evol. 211:568–572. [DOI] [PubMed] [Google Scholar]
- Dogan N, et al. 2015. Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenetics Chromatin 8:16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doglio L, et al. 2013. Parallel evolution of chordate cis-regulatory code for development. PLoS Genet. 9:e1003904.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. 2014. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 15:7–21. [DOI] [PubMed] [Google Scholar]
- Fidler AL, et al. 2014. A unique covalent bond in basement membrane is a primordial innovation for tissue evolution. Proc Natl Acad Sci U S A. 111:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. 1922. On the interpretation of χ2 from contingency tables, and the calculation of P. J R Stat Soc. 85:87–94. [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. 2004. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32:W273–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong RF. 2005. Insights into vertebrate evolution from the chicken genome sequence. Genome Biol. 6:207.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, et al. 2011. Rfam: Wikipedia, clans and the “decimal” release. Nucleic Acids Res. 39:D141–D145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guil S, Esteller M. 2012. Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol. 19:1068–1075. [DOI] [PubMed] [Google Scholar]
- Harris RS. 2007. Improved pairwise alignment of genomic DNA. Ph.D. dissertation, Pennsylvania State University.
- Hausser J, Zavolan M. 2014. Identification and consequences of miRNA-target interactions - beyond repression of gene expression. Nat Rev Genet. 15:599–612. [DOI] [PubMed] [Google Scholar]
- Heinz S, et al. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemberg M, et al. 2012. Integrated genome analysis suggests that most conserved non-coding sequences are regulatory factor binding sites. Nucleic Acids Res. 40:7858–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Schaar BT, Bejerano G. 2012. Hundreds of conserved non-coding genomic regions are independently lost in mammals. Nucleic Acids Res. 40:11463–11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LZ, et al. 2008. The amphioxus genome illuminates vertebrate origins and cephalochordate biology. Genome Res. 18:1100–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland LZ, Yu JK. 2004. Cephalochordate (amphioxus) embryos: procurement, culture, and basic methods. Methods Cell Biol. 74:195–215. [DOI] [PubMed] [Google Scholar]
- Holland ND, Holland LZ, Heimberg A. 2015. Hybrids between the Florida amphioxus (Branchiostoma floridae) and the Bahamas lancelet (Asymmetron lucayanum): developmental morphology and chromosome counts. Biol Bull. 228:13–24. [DOI] [PubMed] [Google Scholar]
- Holland PW, Koschorz B, Holland LZ, Herrmann BG. 1995. Conservation of Brachyury (T) genes in amphioxus and vertebrates: developmental and evolutionary implications. Development 121:4283–4291. [DOI] [PubMed] [Google Scholar]
- Huang S, et al. 2014. Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes. Nat Commun. 5:5896.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufton AL, et al. 2009. Deeply conserved chordate noncoding sequences preserve genome synteny but do not drive gene duplicate retention. Genome Res. 19:2036–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, et al. 2012a. Comparative genomics of the Hedgehog loci in chordates and the origins of Shh regulatory novelties. Sci Rep. 2:433.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, et al. 2012b. Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Res. 22:2356–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine SQ. 2013. Study of cis-regulatory elements in the ascidian Ciona intestinalis. Curr Genomics. 14:56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi M, Noda AO, Sakate R, Imanishi T. 2012. Evolutionary growth process of highly conserved sequences in vertebrate genomes. Gene 504:1–5. [DOI] [PubMed] [Google Scholar]
- Jagla T, et al. 1999. Plasticity within the lateral somatic mesoderm of Drosophila embryos. Int J Dev Biol. 43:571–573. [PubMed] [Google Scholar]
- Kajitani R, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, et al. 2004. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev Cell 7:133–144. [DOI] [PubMed] [Google Scholar]
- Kon T, et al. 2007. Phylogenetic position of a whale-fall lancelet (Cephalochordata) inferred from whole mitochondrial genome sequences. BMC Evol Biol. 7:127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2013. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M, et al. 2010. Conservation of shh cis-regulatory architecture of the coelacanth is consistent with its ancestral phylogenetic position. Evodevo 1:11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Goodson M. 2011. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21:936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeso I, Irimia M, Tena JJ, Casares F, Gómez-Skarmeta JL. 2013. Deep conservation of cis-regulatory elements in metazoans. Philos Trans R Soc Lond B Biol Sci. 368:20130020.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeso I, et al. 2012. An ancient genomic regulatory block conserved across bilaterians and its dismantling in tetrapods by retrogene replacement. Genome Res. 22:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeso I, Tena JJ. 2016. Favorable genomic environments for cis-regulatory evolution: a novel theoretical framework. Semin Cell Dev Biol. 57:2–10. [DOI] [PubMed] [Google Scholar]
- Makunin IV, Kolesnikova TD, Andreyenkova NG. 2014. Underreplicated regions in D. melanogaster are enriched with fast evolving genes and highly conserved noncoding sequences. Genome Biol Evol. 6:2050–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanares M, et al. 2000. Conservation and elaboration of Hox gene regulation during evolution of the vertebrate head. Nature 408:854–857. [DOI] [PubMed] [Google Scholar]
- Marçais G, Kingsford C. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Morales JR. 2015. Toward understanding the evolution of vertebrate gene regulatory networks: comparative genomics and epigenomic approaches. Brief Funct Genomics 15(4):315–321. [DOI] [PubMed] [Google Scholar]
- McEwen GK, et al. 2009. Early evolution of conserved regulatory sequences associated with development in vertebrates. PLoS Genet. 5:e1000762.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meader S, Ponting CP, Lunter G. 2010. Massive turnover of functional sequence in human and other mammalian genomes. Genome Res. 20:1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguillón C, Garcia-Fernàndez J. 2002. The single amphioxus Mox gene: insights into the functional evolution of Mox genes, somites, and the asymmetry of amphioxus somitogenesis. Dev. Biol. 246:455–465. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AC, Wardle FC. 2013. Conserved non-coding elements and cis regulation: actions speak louder than words. Development 140:1385–1395. [DOI] [PubMed] [Google Scholar]
- Nguyen PD, et al. 2014. Haematopoietic stem cell induction by somite-derived endothelial cells controlled by meox1. Nature 512:314–318. [DOI] [PubMed] [Google Scholar]
- Nohara M, Nishida M, Miya M, Nishikawa T. 2005. Evolution of the mitochondrial genome in cephalochordata as inferred from complete nucleotide sequences from two epigonichthys species. J Mol Evol. 60:526–537. [DOI] [PubMed] [Google Scholar]
- Ono H, Kozmik Z, Yu JK, Wada H. 2014. A novel N-terminal motif is responsible for the evolution of neural crest-specific gene-regulatory activity in vertebrate FoxD3. Dev Biol. 385:396–404. [DOI] [PubMed] [Google Scholar]
- Van Otterloo E, et al. 2012. Novel Tfap2-mediated control of soxE expression facilitated the evolutionary emergence of the neural crest. Development 139:720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker HJ, Sauka-Spengler T, Bronner M, Elgar G. 2014. A reporter assay in lamprey embryos reveals both functional conservation and elaboration of vertebrate enhancers. PLoS One 9:e85492.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Anaya J, D’Aniello S, Garcia-Fernàndez J. 2008. Unexpectedly large number of conserved noncoding regions within the ancestral chordate Hox cluster. Dev Genes Evol. 218:591–597. [DOI] [PubMed] [Google Scholar]
- Patel RK, Jain M. 2012. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, et al. 2011. Cactus: algorithms for genome multiple sequence alignment. Genome Res. 21:1512–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio L, et al. 2006. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444:499–502. [DOI] [PubMed] [Google Scholar]
- Putnam NH, et al. 2008. The amphioxus genome and the evolution of the chordate karyotype. Nature 453:1064–1071. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos C, Robert B. 2005. msh/Msx gene family in neural development. Trends Genet. 21:624–632. [DOI] [PubMed] [Google Scholar]
- Royo JL, et al. 2011. Transphyletic conservation of developmental regulatory state in animal evolution. Proc Natl Acad Sci U S A. 108:14186–14191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MT, et al. 2004. Regulatory elements controlling Ci-msxb tissue-specific expression during Ciona intestinalis embryonic development. Dev Biol. 267:517–528. [DOI] [PubMed] [Google Scholar]
- Sanges R, et al. 2013. Highly conserved elements discovered in vertebrates are present in non-syntenic loci of tunicates, act as enhancers and can be transcribed during development. Nucleic Acids Res. 41:3600–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudemont A, et al. 2008. Complementary striped expression patterns of NK homeobox genes during segment formation in the annelid Platynereis. Dev Biol. 317:430–443. [DOI] [PubMed] [Google Scholar]
- Schmid KJ, Tautz D. 1997. A screen for fast evolving genes from Drosophila. Proc Natl Acad Sci U S A. 94:9746–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011a. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6:e17288.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011b. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Merker S, van Eeden FJ, Halpern ME, Kimmel CB, Nüsslein-Volhard C. 1994. no tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development 120:1009–1015. [DOI] [PubMed] [Google Scholar]
- Sharman AC, Shimeld SM, Holland PWH. 1999. An amphioxus Msx gene expressed predominantly in the dorsal neural tube. Dev Genes Evol. 209:260–263. [DOI] [PubMed] [Google Scholar]
- Simakov O, et al. 2015. Hemichordate genomes and deuterostome origins. Nature 527:459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GSC, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32:W309–W312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, et al. 2014. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagkogeorga G, Turon X, Galtier N, Douzery EJP, Delsuc F. 2010. Accelerated evolutionary rate of housekeeping genes in tunicates. J Mol Evol. 71:153–167. [DOI] [PubMed] [Google Scholar]
- Valouev A, et al. 2008. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods 5:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, McEwen GK, Woolfe A, Gilks WR, Elgar G. 2006. Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet. 22:5–10. [DOI] [PubMed] [Google Scholar]
- Vavouri T, Walter K, Gilks WR, Lehner B, Elgar G. 2007. Parallel evolution of conserved non-coding elements that target a common set of developmental regulatory genes from worms to humans. Genome Biol. 8:R15.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh B, et al. 2014. Elephant shark genome provides unique insights into gnathostome evolution. Nature 505:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Escriva H, Zhang S, Laudet V. 2006. Conserved RARE localization in amphioxus Hox clusters and implications for Hox code evolution in the vertebrate neural crest. Dev Dyn. 235:1522–1531. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe A, et al. 2005. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3:e7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousaf A, Sohail Raza M, Ali Abbasi A. 2015. The evolution of bony vertebrate enhancers at odds with their coding sequence landscape. Genome Biol Evol. 7:2333–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JK, Holland ND, Holland LZ. 2004. Tissue-specific expression of FoxD reporter constructs in amphioxus embryos. Dev Biol. 274:452–461. [DOI] [PubMed] [Google Scholar]
- Yue JX, Yu JK, Putnam NH, Holland LZ. 2014. The transcriptome of an amphioxus, Asymmetron lucayanum, from the Bahamas: a window into chordate evolution. Genome Biol Evol. 6:2681–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.