

Abstract
We propose a probabilistic framework to infer autosomal and sex-linked genes from RNA-seq data of a cross for any sex chromosome type (XY, ZW, and UV). Sex chromosomes (especially the non-recombining and repeat-dense Y, W, U, and V) are notoriously difficult to sequence. Strategies have been developed to obtain partially assembled sex chromosome sequences. Most of them remain difficult to apply to numerous non-model organisms, either because they require a reference genome, or because they are designed for evolutionarily old systems. Sequencing a cross (parents and progeny) by RNA-seq to study the segregation of alleles and infer sex-linked genes is a cost-efficient strategy, which also provides expression level estimates. However, the lack of a proper statistical framework has limited a broader application of this approach. Tests on empirical Silene data show that our method identifies 20–35% more sex-linked genes than existing pipelines, while making reliable inferences for downstream analyses. Approximately 12 individuals are needed for optimal results based on simulations. For species with an unknown sex-determination system, the method can assess the presence and type (XY vs. ZW) of sex chromosomes through a model comparison strategy. The method is particularly well optimized for sex chromosomes of young or intermediate age, which are expected in thousands of yet unstudied lineages. Any organisms, including non-model ones for which nothing is known a priori, that can be bred in the lab, are suitable for our method. SEX-DETector and its implementation in a Galaxy workflow are made freely available.
Keywords: XY, ZW, UV, sex-linked genes, RNA-seq, Galaxy workflow
Introduction
Species with separate sexes (males and females) represent ∼95% of animals (Weeks 2012). In angiosperms, although rarer, separated sexes (dioecy) can be found in ∼15,000 species (Renner 2014). Approximately 20% of the crops (e.g. papaya, grapevine, strawberries, kiwi, and spinach) are dioecious or derive from a dioecious progenitor (Ming et al. 2011). However, the mechanisms for sex determination remain unknown for most plant species and a number of animal species (Bachtrog et al. 2014). In numerous cases, it is even unknown whether sex chromosomes are present. In angiosperms, dioecy has evolved from an ancestral hermaphrodite state 871–5000 times independently (Renner 2014), but less than 40 sex chromosomes have been reported so far (Ming et al. 2011). This suggests that sex determination is unknown in 95–99% of the dioecious angiosperm species. The situation is even worse in other plants where only a handful of sex chromosomes have been described among the ∼6000 dioecious liverworts, ∼7250 dioecious leafy mosses and ∼381 dioecious gymnosperms (Ming et al. 2011). Precise estimates of the frequency of dioecy in brown and green algae are currently missing and very few sex chromosomes have been described in those groups. Consequently, further research is required to describe the diversity of sex determination and sex chromosomes in non-model organisms (Bachtrog et al. 2014).
Sex chromosomes were originally a normal pair of autosomes that, after acquiring sex-determining genes, stopped recombining and diverged from one another (Bachtrog 2013). In male heterogametic systems, males are XY and females XX whereas in female heterogametic systems, females are ZW and males ZZ. In species with a haplodiploid life cycle, sex can be expressed at the haploid phase with U females and V males and diploid individuals are heterogametic UV (Bachtrog et al. 2011). Y, W, U, and V chromosomes all have a non-recombining region that can be small or spread to most of the chromosome. Sex chromosomes with a small non-recombining region are homomorphic (X and Y of similar size), which makes their identification through cytology difficult. And yet this type of sex chromosomes is probably frequent in groups such as angiosperms where many dioecious species have evolved recently and must have young sex chromosomes (Ming et al. 2011), in groups such as fish where sex chromosome turnover is high (Mank and Avise 2009), or in groups such as amphibians where occasional recombination limits sex chromosome divergence (Stock et al. 2013). In such cases, sequences are required to identify sex chromosomes.
However, obtaining well assembled sequences of sex chromosomes is extremely difficult due to the repeats that accumulate in their non-recombining regions (Charlesworth et al. 1994; Gaut et al. 2007). Only the costly use of BAC clones organized in a physical map makes it possible to completely assemble DNA sequences from non-recombining regions (Hughes and Rozen 2012). This is why only a handful of non-recombining sex chromosomes (Y, W, U, or V) have been fully sequenced and assembled to date (<15). This includes eight mammalian Y chromosomes (Bellott et al. 2014), and other species which have small non-recombining regions: the liverwort Marchantia (Yamato et al. 2007), the fish medaka (Kondo et al. 2006), the green alga Volvox (Ferris et al. 2010), the tree papaya (Wang et al. 2012), and the brown alga Ectocarpus (Ahmed et al. 2014). The scarcity of assembled Y is true even for well studied groups such as Drosophila. Only 10% of the Y chromosome has so far been sequenced in the Drosophila melanogaster genome release 6 (Bachtrog 2013; Hoskins et al. 2015), and the Drosophila miranda neo-Y chromosome is a draft (Zhou and Bachtrog 2012). However, producing high-quality assemblies is not always necessary and alternative, less expensive strategies have been recently developed for identifying sex chromosome sequences based on next-generation sequencing (NGS) data (reviewed in Muyle et al. 2016).
A first category of approaches relies on the comparison of female and male DNA-seq (DNA sequencing using NGS) data (Vicoso and Bachtrog 2011; Carvalho and Clark 2013; Vicoso, Emerson et al. 2013; Vicoso, Kaiser et al. 2013; Akagi et al. 2014; Cortez et al. 2014). As these methods require a reference genome (either from the studied species or a close relative), they will be difficult to apply to non-model organisms because reference genomes are lacking and/or genomes are large and complex. Another method uses the ploidy of SNPs in order to identify sex chromosome sequences (Gautier 2014) but requires the sequencing of a 100 individuals, which, depending on the sequencing method, could be too expensive for non-model organisms. Long-read sequencing such as PacBio can improve these approaches by providing larger Y scaffolds as recently shown in malaria mosquitoes (Hall et al. 2016) or in gorilla (Tomaszkiewicz et al. 2016). Using long-read sequencing is more affordable than the use of BAC clones, but remains quite expensive for organisms with large genomes even when the Y chromosome can be isolated (Tomaszkiewicz et al. 2016). The methods cited thus far require that X and Y sequences be divergent enough not to co-assemble or map onto one another. This means that they will work well in old systems but will probably fail with recently evolved sex chromosomes. Other approaches work well on young sex chromosomes, such as the use of sex markers (inferred from polymorphism data or genetic maps) to identify scaffolds belonging to sex chromosomes in a genome assembly (Al-Dous et al. 2011; Picq et al. 2014; Hou et al. 2015). However, the need for a reference genome can again be a hindrance for many non-model organisms, especially those with large genomes. In such cases, studying the transcriptome rather than the genome can be a very effective cost saving measure. RNA-seq gives direct access to gene sequences and their expression levels, which can be valuable for various biological analyses. Identifying which genes in a transcriptome are sex-linked (i.e., located on the non-recombining region of sex chromosomes) can be done through the sequencing of males and females and the analysis of SNPs. For instance, brothers and sisters from an inbred line can be sequenced and used to infer sex-linked genes (Muyle et al. 2012). But inbred lines are unlikely to be available in most non-model organisms. A strategy relying on the sequencing of a cross (parents and progeny of each sex) by RNA-seq has proved very successful in the identification of sex-linked genes through the study of allele segregation (fig. 1). This strategy requires that X and Y copies co-assemble and map onto one another in order to identify X/Y genes using X/Y SNPs (fig. 1b). X copies can also be identified on their own if the Y copy is absent because of degeneration or if X and Y were too diverged to co-assemble (fig. 1c). Therefore, this strategy is better suited for sex chromosomes that have either a low or intermediate level of divergence. However, it will still provide X copies of sex-linked genes in old systems, as long as appropriate SNPs are present in the dataset (fig. 1c). Crosses can be obtained in any organism that can be grown in controlled conditions and are a common resource because they are needed for building genetic maps. Hundreds of sex-linked genes were identified this way in species with unavailable fully sequenced genomes such as Silene latifolia (Bergero and Charlesworth 2011; Chibalina and Filatov 2011) or in species without any genomic resource such as Rumex hastatulus (Hough et al. 2014) and Rumex acetosa (Michalovova et al. 2015).
Fig. 1.—

Examples for the three segregation types: (a) autosomal, (b) X/Y, and (c) X-hemizygous (when the Y copy was lost or was assembled in a separate contig). TFG stands for true heterogametic parent genotype and TMG for true homogametic parent genotype.
Although this RNA-seq cross-based strategy is very promising for studying sex chromosomes in non-model organisms, the existing approaches have a number of limitations due to the fact that inference of sex-linkage was done with empirical filters and without a statistical framework. Once RNA-seq reads have been mapped to a reference transcriptome, individuals need to be genotyped in order to study allele segregation in the cross (fig. 1). Genotyping was either done by filtering the number of reads at each locus with fixed thresholds (Bergero and Charlesworth 2011; Chibalina and Filatov 2011) or using genotypers designed for DNA-seq data (Hough et al. 2014; Michalovova et al. 2015). This is problematic in the case of sex-linked genes where the Y allele is frequently less expressed than the X (reviewed in Bachtrog 2013) and can be confounded with a sequencing error in RNA-seq data. Read number thresholds determined empirically for a given dataset can be sub-optimal for another dataset with different sequencing depth. In many cases, progeny individuals were pooled separately for each sex for sequencing in order to lower costs (Bergero and Charlesworth 2011; Chibalina and Filatov 2011; Michalovova et al. 2015). However, this makes it impossible to differentiate between all individuals of the pool or only a few being heterozygous, a criterion that is crucial to disentangle sex-linked genes from autosomal ones (fig. 1). Finally, sex-linked genes were filtered for having more than a given number of sex-linked SNPs (Chibalina and Filatov 2011; Hough et al. 2014), and for not having any autosomal SNPs (Bergero and Charlesworth 2011; Michalovova et al. 2015). These arbitrary filters clearly limit the application of this strategy to only specific datasets and probably prevent the detection of many true sex-linked genes. Also, a method allowing the study of UV systems is currently lacking.
Here, we propose a probabilistic method called SEX-DETector that solves the caveats of previous RNA-seq cross-based approaches and works on any sex chromosome type (XY, ZW, and UV). The method was designed to discover as many sex-linked genes as possible from the data, whereas keeping inferences extremely reliable for downstream biological analyses. The pipeline, implemented in a galaxy workflow, was tested on empirical and simulated data and proved very promising for the discovery of many sex chromosomes and sex-linked genes in non-model organisms, especially in young systems.
Materials and Methods
The Probabilistic Model
Observed and Hidden Data
The data consist of genotypes in a cross (parents and progeny of each sex), at each position of every contig and can typically be obtained from RNA-seq experiments. The model aims to describe the transmission of alleles from parents to progeny, in the given cross, in order to infer whether a gene is sex-linked, i.e., if it is located in the non-recombining region of sex-chromosomes (fig. 1). The observed data, denoted by G, consist of the observed genotypes of the parents and the progeny, and we suppose that the probabilities of observing these genotypes depend on unknown information or hidden variables that we want to recover.
The segregation type S describes whether the studied locus is autosomal or sex-linked, which influences allele transmission from parents to progeny. There are three segregation types j: autosomal (j = 1), X/Y (or Z/W, j = 2) and X-hemizygous when the Y allele is absent (or Z-hemizygous when the W allele is absent, j = 3). The probability of segregation type j for position t in contig k is .
The true homogametic parent genotype TMG (for true mother genotype in the case of male heterogamety) is introduced to account for genotyping errors that can cause the observed genotype to differ from the true genotype. There are ten possible genotypes m for the homogametic parent: AA, AC, AG, AT, CC, CG, CT, GG, GT, and TT. The probability for the true homogametic parent genotype of being m at position t of contig k, given segregation type j is: . It is assumed that the true mother genotype frequencies do not differ between autosomal and sex-linked loci, so that parameter α is independent from segregation type j.
The true heterogametic parent genotype TFG (for true father genotype in the case of male heterogamety). The possible genotypes n for the heterogametic parent depend on the segregation type of the studied locus: there are ten possibilities for an autosomal segregation type (AA, AC, AG, AT, CC, CG, CT, GG, GT, and TT), twelve for an X/Y (or Z/W) segregation type (XY, XY, XY, XY, XY, XY, XY, XY, XY, XY, XY, and XY) and four for an X-hemizygous (or Z-hemizygous) segregation type (X, X, X, and X). Given the segregation type j, the probability for the true heterogametic parent genotype of being n at position t of contig k, is: . It is also assumed that the true parental genotype frequencies do not differ for autosomal loci ().
Genotyping error (GE). This variable describes whether there has been a genotyping error made for the studied individual i. It depends on the segregation type and the true parental genotypes of the studied locus. The probability for a genotyping error of having occured for individual i at position t in contig k, given the segregation type j and the true parental genotypes m and n is: . It is assumed that this parameter is fixed for all contigs and contig positions.
Y (or W) genotyping error YGE. This variable accounts for the fact that genotyping errors are more common for Y and W alleles due to degeneration and lower expression. A Y or W genotyping error can only occur in a heterogametic individual and in a X/Y segregation type (j = 2). Similarly to the genotyping error , it depends on the true parental genotypes. The probability for a Y or W genotyping error of having occured for individual i of sex r at position t in contig k, given the segregation type j and the true parental genotypes m and n is: . pjr is equal to zero for homogametic individuals r = hom in any segregation type. pjr is also equal to zero for heterogametic individuals r = het in autosomal and X-hemizygous segregation types (). It is assumed that this parameter is fixed for all contigs and contig positions.
The probabilities of observing the parent and offspring genotypes can be defined when conditioned by all the hidden data of the model. The probability of observing , the genotype of offspring individual i of sex r at position t of contig k, given the segregation type j, the true parental genotypes m and n, the genotyping error h (either with an error or without error ) and the Y genotyping error d (either with an error or without error ) is . And similarly the probability of observing , the genotype of parent individual i of sex r at position t of contig k, given all the hidden data is , where w is the true genotype of the studied individual (either m or n). For instance, in the case of an autosomal segregation type (j = 1), if the heterogametic parent true genotype n is AC and the homogametic parent true genotype m is AA (as shown in fig. 1a) and if no genotyping error has occurred [ and ], then the probability of observing genotype in the offspring is 1/2 for both males and females. However, in the case of X/Y segregation type, as shown in figure 1b, then genotype AC is observed with probability 1 in males and genotype AA with probability 1 in females. Similarly, if the true homogametic parent genotype m is AA and no genotyping error has occurred, then the probability of observing genotype in the homogametic parent is 1, and if a genotyping error occurred for this individual then all other genotypes can be observed with probability 1/9 (as there are nine genotypes other than AA). In the case of an X/Y segregation type with the true heterogametic parent genotype n being XY, the probability of observing genotype AA for this individual is 1 if there has been a Y genotyping error. All values for λ and μ can be found in supplementary table S1, Supplementary Material online.
Inferences
An Expectation Maximization (EM) algorithm is used to estimate the parameter values of the model . Detailed equations of the EM algorithm can be found in supplementary text S1, Supplementary Material online. Once parameters have been estimated, the posterior probabilities of the hidden data are computed given the observed data: is the posterior probability of segregation type j at position t in contig k, given the observed data G. Using Bayes’ rule we get:
with and:
Similar derivations are done for the other posterior probabilities of hidden variables .
Then, the segregation type of each position in the dataset can be inferred using the posterior segregation type . Contigs are attributed to a segregation category using positions that are polymorphic and informative. A position that was inferred as X or Z-hemizygous and that is polymorphic is always informative. A position that was inferred as autosomal or X/Y is considered informative only if the heterogametic parent is heterozygous and has a genotype that is different from the homogametic parent (otherwise it is not possible to differentiate between X/Y and autosomal segregation). The posterior segregation type of the contig is the average of the informative positions in the contig (assumed independent), with the positions being attributed a weight according to the posterior probability of genotyping errors (if a position has high genotyping error posterior probabilities it will be given less weight in the final decision for the contig segregation type):
A contig is attributed to a sex-linked (X/Y or X-hemizygous) segregation type if its posterior probability of being X/Y plus X-hemizygous is higher than a tunable threshold (0.8 by default) and if the contig has at least one X/Y or X-hemizygous SNP without error (a genotyping error is inferred when its posterior probability is higher than 0.5). Similarly, a contig is attributed to an autosomal segregation if its posterior probability of being autosomal is higher than the chosen threshold and if the contig has at least one autosomal SNP without error. The threshold of 0.8 used here was chosen using the tester set (see below), a value of 0.8 provided the best possible specificity. If users have access to a tester set in their species, they can choose the threshold value accordingly, otherwise we suggest to use 0.8 by default.
For each SNP, the true parental genotypes are inferred as the ones that have the highest and probabilities. Expression levels are retrieved using the X and Y (or Z and W) alleles predicted by this method and written in outputs. The model described above is adapted to X/Y and Z/W systems. Another version of the SEX-DETector model was written for U/V systems and can be found in supplementary text S2, Supplementary Material online.
Bayesian Information Criterion Test for the Presence of Sex Chromosomes
The Maximum Likelihood framework of the method allows the use of a model selection strategy to assess the presence of sex-linked genes in the dataset. A model with the three possible segregation types can be compared with a model with only autosomal segregation type using the Bayesian information criterion (BIC), defined such that
Where is the BIC value of model is the likelihood of the model, is the number of free parameters of the model and n is the sample size. The model with the lower BIC value is chosen. It is also possible to test for a X/Y versus a Z/W system by comparing both BIC values. In case a model with sex chromosomes fits best the data but no sex-linked genes are inferred, then it means there are no sex chromosomes in the dataset. This could happen because of the extra Y genotyping error parameter that is specific to the model with sex chromosomes, which could account for mapping and genotyping errors in the data better than the genotyping error parameter ϵ alone. Note that if an X/Y (or Z/W) system fits best the data but only X-hemizygous (or Z-hemizygous) genes are inferred, the system can be X0 (or Z0) or X/Y with fully degenerated Y chromosome and no Y expression (or Z/W with fully degenerated W chromosomes).
Data Analysis
Plant Material and Sequencing
RNA-seq data were generated from a cross in Silene latifolia, a dioecious plant that has sex chromosomes, and from a cross in Silene vulgaris, a gynodioecious plant that does not have sex chromosomes. We used the following RNAseq libraries that were used in previous studies: Leuk144-3, a male from a wild population; U10_37, a female from a ten-generation inbred line (Muyle et al. 2012); and their progeny (C1_01, C1_3, C1_04, C1_05, C1_26, C1_27, C1_29, C1_34). For S. vulgaris, the father came from a wild population (Guard_1), the mother from another wild population (See_02) and had progeny individuals (V1_1, V1_2, V1_4, V1_5, V1_8, V1_9). Individuals were grown in a temperature-controlled greenhouse. The QiagenRNeasy Mini Plant extraction kit was used to extract total RNA two times separately from four flower buds at developmental stages B1–B2 after removing the calyx. Samples were treated additionally with QiagenDNase. RNA quality was assessed with an Aligent Bioanalyzer (RIN larger than 9) and quantity with an Invitrogen Qubit. An intron-spanning PCR product was checked on an agarose gel to exclude the possibility of genomic DNA contamination. Then, the two extractions of the same individual were pooled. Individuals were tagged and then pooled for sequencing. Samples were sequenced by GATC, Konstanz, Germany on an Illumina HiSeq2000 following an Illumina paired-end protocol (fragment lengths 150–250bp, 100 bp sequenced from each end). A normalized 454 library was generated for S. latifolia using bud extracts from four different developmental stages. The RNA-seq data used in this study is available under the European Nucleotide Archive accession number PRJEB14171 (Zemp et al. n.d.).
Assembly
Adaptors, low quality and identical reads were removed. The transcriptome was then assembled using Trinity (Haas et al. 2013) on the combined 10 individuals described previously as well as the six individuals from (Muyle et al. 2012) and the normalized 454 sequencing that was transformed to illumina using 454-to-illumina-transformed-reads (because Trinity cannot take 454 reads as input). Then, isoforms were collapsed using /trinity-plugins/rsem-1.2.0/rsem-prepare-reference. PolyA tails and ribosomal RNAs were removed using ribopicker. ORFs were predicted with Trinity's transcripts_to_best_scoring_ORFs.pl (this step is facultative and SEX-DETector can work on coding or non-coding sequences). In order to increase the probability of assembling X and Y sequences in the same contig, ORFs were further assembled using CAP3 (cap3 -p 70, Version 15 October 2007, Huang and Madan 1999) inside of Trinity components (in Trinity, components can group contigs that are alternative transcripts of the same gene or paralogs). CAP3 was shown to be useful to supplement Illumina read assemblers in order to get a better de novo assembly of a transcriptome in a non-model organism (Cahais et al. 2012).
Mapping, Genotyping, and Segregation Inference
Illumina reads from the 10 individuals of the cross were mapped onto the assembly using BWA (version 0.6.2, bwa aln -n 5 and bwa sample; Li and Durbin 2009). The libraries were then merged using SAMTOOLS (Version 0.1.18; Li et al. 2009). The obtained alignments were locally realigned using IndelRealigner (GATK) (McKenna et al. 2010; DePristo et al. 2011) and were analyzed using Reads2snp (Version 2.0, -fis 0 -model M2 -output_genotype best -multi_alleles acc -min_coverage 3 -par false) (Tsagkogeorga et al. 2012) in order to genotype individuals at each loci while allowing for biases in allele expression and not cleaning for paralogous SNPs as X/Y SNPs tend to be filtered out by paraclean (the program that removes paralogous positions) (Gayral et al. 2013). SEX-DETector was then used to infer contig segregation types after estimation of parameters using an EM algorithm. Posterior segregation types probabilities were filtered to be higher than 0.8. All these steps are implemented in a Galaxy workflow (see pipeline in fig. 2).
Fig. 2.—

The SEX-DETector pipeline: from required data to outputs. The advised number of offspring individual to sequence was determined with simulations. Parameters of the model are written in red: the segregation type π, the parents true genotype frequencies α and β, the genotyping error ϵ and the Y genotyping error p. An X/Y system is represented here but the pipeline is equivalent for a Z/W system. For a U/V system, only two individuals of each sex and one parent are advised and can be sequenced on a single Illumina Hi-seq 2000 lane. The pipeline was implemented in Galaxy for steps following Trinity. Note that different assembler and mapper could be chosen, only the genotyper Reads2snp is required to run SEX-DETector.
The Tester Set, Sensitivity, and Specificity
For various tests, we used 209 genes with previously known segregation type: 129 experimentally known autosomal genes, 31 experimentally known sex-linked genes (X/Y or X-hemizygous) and 49 X-linked CDS from BAC sequences (supplementary table S2, Supplementary Material online). The sequences of these 209 genes were blasted (blast -e 1E-5) (Altschul et al. 1990) onto the de novo assembly in order to find the corresponding ORF of each gene. Blasts were filtered for having a percentage of identity over 90% and an alignment length over 100 bp and manually checked. Multiple RNA-seq contigs were accepted for a single gene if they matched different regions of the gene. If multiple contigs matched the same region of a gene, only the contig with the best identity percentage was kept. The gene was considered inferred as sex-linked if at least one of its matching contig was sex-linked. The inferred status of the genes by SEX-DETector was then used to compute specificity and sensitivity values. The same approach was used to compute sensitivity and specificity values for the three previous studies that inferred S. latifolia RNA-seq contig segregation patterns (Bergero and Charlesworth 2011; Chibalina and Filatov 2011; Muyle et al. 2012).
Sensitivity (or true-positive rate) measures the capacity to detect true positives TP (genes that are sex-linked and inferred as such by the method). False negatives FN are sex-linked genes missed by the method.
Specificity (or true-negative rate) measures the capacity to avoid false positives FP (genes that are not sex-linked but inferred as such by the method). True negatives TN are non-sex-linked genes detected as such by the method.
The false discovery rate measures the proportion of false positives FP among all inferred sex-linked genes.
Simulations
Simulated genotypes were used in order to test the effect of various parameters on the sensitivity and specificity of SEX-DETector. Sequences were first simulated for two parents (or a single parent in the case of a UV system) using the program ms to generate a coalescent tree (Hudson 2002; see supplementary figure S1, Supplementary Material online and then the program seq-gen to generate sequences using the ms tree and molecular evolution parameters (version 1.3.2x, seq-gen -mHKY -l contig_length -f 0.26 0.21 0.23 0.3 -t 2 -s theta) (Rambaut and Grassly 1997). Different types of sequences were generated: either autosomal (ms 4 1 -T) or X/Y (ms 4 1 -T -I 2 3 1 -n 2 0.25 -n 1 0.75 -ej XY_divergence_time 2 1 -eN XY_divergence_time 1) or X-hemizygous (same parameters as X/Y but no Y sequence drawn) or U/V (ms 2 1 -T -I 2 1 1 -n 2 0.5 -n 1 0.5 -ej UV_divergence 2 1 -eN UV_divergence 1). Then, allele segregation was randomly carried on for a given number of progeny of each sex, using the segregation pattern determined when generating sequences with ms and seq-gen (see supplementary table S1, Supplementary Material online for segregation tables). was set to 0.0275 as estimated in S. latifolia by (Qiu et al. 2010). μ was set to , which implies that was equal to ∼70,000. Contig lengths were randomly assigned from the observed distribution of contig lengths of the S. latifolia assembly presented previously. Equilibrium frequencies used for seq-gen were retrieved from SEX-DETector inferences on the observed S. latifolia data. The transition to transversion ratio was set to 2 as inferred by PAML (Yang 2007) on S. latifolia data (Käfer et al. 2013). The rate of genotyping error (ϵ) was set to 0.01 and the rate of Y genotyping error () was set to 0.13 as inferred by SEX-DETector on the observed S. latifolia data. Five types of datasets were simulated, with ten repetitions for each set of parameters and 10,000 contigs simulated for each dataset:
Effect of X–Y divergence: Five different X–Y divergence times in units of generations were tested, either S. latifolia X–Y divergence time (4.5 My) or 10 times or 100 times older or younger. The proportion of X-hemizygous contigs among sex-linked contigs was set accordingly to X–Y divergence time: 0.002, 0.02, 0.2, 0.6, and 1 for 45,000 years, 450,000 years, 4.5 My, 45 My, and 450 My divergence time, respectively. As well as the proportion of Y genotyping error (because Y expression is known to decrease with X–Y divergence): 0, 0.01, 0.13, 0.2, and 1, respectively. Four offspring of each sex were simulated. The proportion of sex-linked contigs was set to 10%.
Effect of the number of sex-linked contigs: Five different proportions of sex-linked contigs (X/Y pairs or X hemizygous) were tested: 30% (3000 sex-linked contigs out of 10,000), 5%, 1%, 0.1%, and 0.01%. Four offspring of each sex were simulated and X–Y divergence was set to 4.5 My.
Effect of theta: Three different (polymorphism) were tested: 0.000275, 0.00275, and 0.0275. Five offspring of each sex were simulated and X–Y divergence was set to 4.5 My, the X–Y divergence time in unit of generations varied accordingly to the value of theta. The proportion of sex-linked contigs was set to 10%.
Effect of the number of individuals in Z/W and X/Y systems: Nine different numbers of offspring individuals of each sex were tested for the X/Y system: 2, 3, 4, 5, 6, 7, 8, 12, or 16 individuals of each sex. Sex chromosome size was set to 10% and X–Y/Z–W divergence to 4.5 My.
Effect of the number of individuals in U/V systems: Eight different numbers of offspring individuals of each sex were tested for the U/V system: 1, 2, 3, 4, 5, 6, 7, or 8 individuals of each sex. Sex chromosome size was set to 10% and U–V divergence to 4.5 My.
For each simulated dataset, segregation types were inferred using SEX-DETector and were compared with the true segregation types in order to compute sensitivity and specificity values.
Implementation and Availability
The SEX-DETector code was written in perl and a Galaxy workflow was also developed (see user guide and source codes at http://lbbe.univ-lyon1.fr/-SEX-DETector-.html, last accessed 30 July 2016).
Results
The SEX-DETector Pipeline
SEX-DETector takes as input file the genotypes of a cross (parents and progeny of each sex) for different contigs of an assembly. These data can typically be obtained from RNA-seq. The output is the inferred segregation type for every SNP and contig of the data (autosomal, X/Y or X-hemizygous, see fig. 1 for an example) along with allelic X and Y (or Z and W or U and V) expression levels. The SEX-DETector pipeline is pictured in figure 2 and has been implemented as a Galaxy workflow. Simulations showed that the sequencing of two parents plus five progeny of each sex (12 individuals in total) is sufficient to obtain good results in an X/Y or Z/W system (see below). The use of RNA-seq lowers the cost, especially for species with large genomes. The pipeline can easily be modified to handle DNA-seq data. In order to obtain sufficient coverage, sequencing 20 to 25 million reads per individual is recommended in the case of RNA-seq (i.e., two Illumina lanes for 12 individuals on a Hiseq 2000). It is also recommended to use RNA extracted from a complex tissue where many genes would be expressed, especially sex-determining genes (e.g., flower buds in plants). The parents should be sampled from two different populations in order to increase the number of SNPs and therefore the power of the method. RNA-seq reads can be assembled into transcripts using Trinity (Haas et al. 2013), although a different assembler could be chosen. An important step after assembly is to further assemble transcripts, for example with CAP3 (Huang and Madan 1999), in order to coassemble X and Y alleles in a single X/Y contig and avoid X/Y contigs to be misinferred as X-hemizygous. After mapping the reads onto the assembly (with any mapper), all individuals can be genotyped. The use of Reads2snp (Tsagkogeorga et al. 2012) is highly recommended as it was designed for RNA-seq data in non-model organisms and allows for allelic expression biases, a key parameter when dealing with sex chromosomes and the poorly expressed Y alleles (Bachtrog 2013). SEX-DETector takes Reads2snp_2.0 output as input.
SEX-DETector uses a probabilistic model to cluster contigs into segregation types. The parameters of the SEX-DETector model are estimated from the data using an EM algorithm (see “Materials and Methods” section for details). Parameter π, for segregation type frequencies, makes it possible to deal with different sex chromosome sizes. Parameters α and β, the parental genotype frequencies, accommodate for the heterozygosity level of the parents as well as the base composition of the species. The probability for a genotyping error to occur ϵ accounts for possible differences between observed and true genotypes (due to sequencing, mapping or genotyping errors). A specific Y genotyping error parameter p fits the high genotyping error rate for Y alleles due to degeneration and lower expression.
Testing Our Pipeline’s Performance Using a Silene latifolia Dataset
The SEX-DETector pipeline (fig. 2) was run on a cross dataset sequenced by RNA-seq in the plant Silene latifolia. This dioecious species was interesting for benchmarking our method/pipeline as its sex chromosomes are well known: they are relatively recent (∼5 My old) (Rautenberg et al. 2010) but old enough to be clearly heteromorphic (the X is 400 Mb and the Y is 550 Mb) (Matsunaga et al. 1994). X–Y synonymous divergence ranges from 5% to 25% (Bergero et al. 2007), S. latifolia thus represents a system of intermediate age. Also, a tester set of 209 genes for which segregation type has been established is available in this species (supplementary table S2, Supplementary Material online). The dataset that was used here consists of a cross (two parents and four offspring of each sex). RNA-seq data were obtained for each of these individuals tagged separately and the reads were assembled using Trinity and then CAP3, the final assembly included 46,178 ORFs (table 1). RNA-seq reads were mapped onto this assembly (see supplementary table S3, Supplementary Material online for library sizes and mapping statistics) and genotyping was done for each individual using Reads2snp. SEX-DETector was run on the genotyping data to infer autosomal and sex-linked genes (table 1). Figure 3 shows examples from the tester set. For some genes, all SNPs show clearly the same correct segregation type (fig. 3A–C), whereas in some genes mixed segregation patterns were inferred, which we attribute to co-assembly of recent paralogs or other assembly/mapping problems (fig. 3D). These mixed cases can be filtered by the user, although they can happen in true sex-linked genes as is the case in figure 3D.
Table 1.
Results of the SEX-DETector Pipeline on the S. latifolia dataset.
| ORF types | Numbers |
|---|---|
| ORFs in final assembly | 46,178 |
| ORFs with enough coverage to be studied | 43,901 |
| ORFs with enough informative SNPs to compute a segregation probability | 17,189 |
| ORFs with posterior segregation probability over 0.8 | 15,164 |
| ORFs assigned to an autosomal segregation type | 13,807 (91%) |
| ORFs assigned to a X/Y segregation type | 1,025 (7%) |
| ORFs assigned to a X-hemizygous segregation type | 332 (2%) |
Fig. 3.—

Results of the SEX-DETector pipeline for known S. latifolia genes. Segregation type posterior probabilities are shown for each informative SNP (see “Materials and Methods” section), see legend on figure for colour code, and inferred number of genotyping errors (see “Materials and Methods” section) are shown inside the bars. (A) SlE72 is known to be autosomal, its weighted autosomal mean probability (see “Materials and Methods” section) is 0.99. (B) SlCypX is known to be X/Y, its weighted sex-linked mean probability is 0.96. (C) WUS1 is known to be X-hemizygous, its weighted sex-linked mean probability is 0.99. (D) BAC284N5-CDS13_SlX6a is known to be sex-linked, its weighted sex-linked mean probability is 0.82.
We used our tester set to measure the performance of our pipeline, i.e., estimate its sensitivity (the capacity to detect true sex-linked genes) and specificity (the capacity not to assign autosomal genes as sex-linked, see “Materials and Methods” section). About 83% of the known sex-linked genes expressed in the RNA-seq data used here (i.e. flower bud) were detected, indicating a high sensitivity. We obtained a specificity of 99% for this dataset as one gene, OxRZn, was supposedly wrongly assigned as a sex-linked gene by SEX-DETector. However, this gene was earlier assessed as autosomal based on the absence of male specific alleles (Marais et al. 2011) and SEX-DETector assigned it to a sex-linked category because out of the four SNPs detected in this gene, all where inferred to be X-hemizygous, and all were without genotyping error. It is therefore very likely that OxRZn is in fact a true positive and more research on that gene is required.
Comparing Our Pipeline to Others Using a S. latifolia Dataset
We compared the performance of our pipeline to those used in previous work on inferring sex-linkage with RNA-seq data in S. latifolia (Bergero and Charlesworth 2011; Chibalina and Filatov 2011; Muyle et al. 2012). Those pipelines differ in many ways, and the data themselves can be different. In previous work, offspring individuals of the same sex were sometimes pooled before sequencing (Bergero and Charlesworth 2011; Chibalina and Filatov 2011). We used again the tester set of 209 S. latifolia genes with known segregation types, which we blasted onto each data set to find the corresponding contigs and their inferred segregation type (for details see supplementary table S2, Supplementary Material online). Because the different pipelines require different types of data (pooled progeny versus individually tagged offspring) and with different read coverages, we computed sensitivity on all known genes (expressed or not). Our pipeline outperformed other pipelines in terms of sensitivity, whereas specificity was comparable (see fig. 4 and supplementary table S4, Supplementary Material online for details). This indicates that SEX-DETector can uncover more sex-linked contigs, without increasing the rate of false positives.
Fig. 4.—
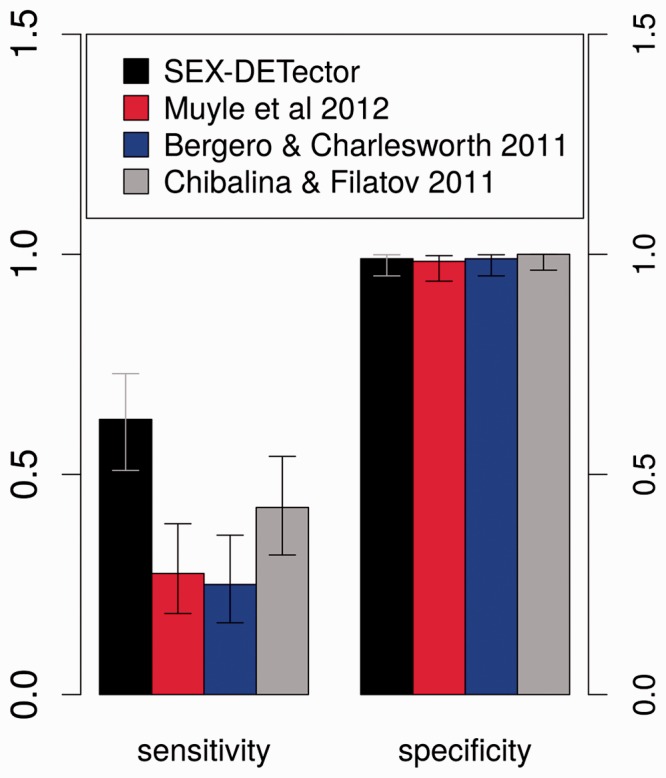
Comparison of SEX-DETector with other methods: sensitivity and specificity values (see “Materials and Methods” section) along with their 95% confidence intervals are shown. Values were obtained using 209 known S. latifolia genes (see supplementary table S2, Supplementary Material online).
As further analysis showed, this was due to overly conservative filtering in previous work. To exclude false positives, genes with at least five sex-linked SNPs were retained in previous studies. More filtering was done by excluding contigs with autosomal SNPs (Bergero and Charlesworth 2011; Hough et al. 2014). As shown in figure 5, keeping only contigs with at least five sex-linked SNPs removes nearly half of the contigs inferred as sex-linked by SEX-DETector, many of which have a high posterior probability. Excluding further those with autosomal SNPs (keeping those with sex-linked SNPs only) removes 74% of the contigs (fig. 5B). Comparatively, SEX-DETector removes 12% of contigs when filtering for a posterior probability higher than 0.8 (table 1), as most genes have a very high posterior segregation type probability which indicates a strong signal in the data and illustrates the benefits of using a model-based approach.
Fig. 5.—

Performance of the method. The number of SNPs without genotyping error was plotted against the posterior segregation type probability for each autosomal (A) and sex-linked (B) contigs of the S. latifolia dataset. The distributions of both variables are shown, and means for each category on the histograms are indicated by red dots. Sex-linked genes that remain after filters that are commonly applied in empirical methods are shown in green (at least five sex-linked SNPs and no autosomal SNPs). SEX-DETector on the other hand filters for a posterior probability above 0.8 (horizontal line on graph) and at least one sex-linked SNP, so that more contigs can be inferred as sex-linked, without increasing the false positive rate compared with other empirical methods (fig. 4).
Simulations Show that SEX-DETector Requires a Modest Experimental Effort and Works On Different Sex Chromosome Systems
We simulated genotypes for a cross (parents and progeny) by generating coalescent trees with either autosomal or sex-linked history (supplementary fig. S1, Supplementary Material online) and generated the parental sequences using these trees and molecular evolution parameters. Progeny genotypes were obtained by random segregation of alleles from the parents and a genotyping error layer was added (see “Materials and Methods” section). About 10,000 contigs were simulated for each dataset. SEX-DETector was run on every dataset (see supplementary table S5, Supplementary Material online for details on the inferences).
In order to know how many offspring of each sex should be sequenced to achieve the best sensitivity and specificity trade-off using SEX-DETector, we varied the number of progeny individuals in the simulations. For an X/Y or Z/W system, optimal results were obtained when sequencing five progeny individuals of each sex (fig. 6A); sequencing more progeny individuals did not improve the results further. This suggests that sequencing 12 individuals (two parents and five progeny individuals of each sex) may be sufficient to achieve optimal performances with SEX-DETector on an X/Y or Z/W system. For a U/V system, two progeny individuals of each sex seems sufficient to obtain optimal SEX-DETector performance (fig. 6B), which suggests that sequencing five individuals (the sporophyte parent and two progeny of each sex) may be enough in the case of a U/V system. Our simulations thus suggest that SEX-DETector requires a modest experimental effort to reliably identify expressed sex-linked genes.
Fig. 6.—

Measure of the effect of the number of sequenced offspring individuals using simulations. ROC curve (true positive rate represented as a function of false positive rate) showing the effect of the number of progeny sequenced on sensitivity (TPR, true positive rate) and specificity (1-FPR, false positive rate) in simulated data. A perfect classification of contigs would lead to a point having TPR equal to one and FPR equal to zero (top left corner of the graph). (A) X/Y or Z/W sex determination system (all points overlap in the top left corner when over five progeny of each sex are used). (B) U/V system (all points overlap in the top left corner when over two progeny of each sex are used).
In order to assess the applicability of SEX-DETector to different types of sex chromosomes (old versus young, homomorphic versus heteromorphic) and species (highly vs. weakly polymorphic), we used the same simulation procedure and tested the effect of one parameter at a time on SEX-DETector sensitivity and specificity. In our simulations, the degree of polymorphism within species had no influence on the performance of our method (supplementary fig. S2A, Supplementary Material online). As for the influence of the size of the non-recombining region (homomorphic or heteromorphic sex chromosomes), it was tested using different % of sex-linked genes in a genome with no effect on the performance of SEX-DETector (supplementary fig. S2B, Supplementary Material online). The limit of detection of a sex-linked contig was reached only when one sex-linked contig out of 10,000 contigs was present. Finally, the simulations indicated that our method is robust to X–Y divergence, as young and old sex chromosomes were evenly detected (supplementary fig. S2C, Supplementary Material online).
SEX-DETector Identifies Unknown Sex Chromosomes Using Model Selection
It is common that the sex determination system is unknown in species with separated sexes, i.e., it is unknown whether they have sex chromosomes and if they do, what the system is (Z/W or X/Y). The likelihood-based framework of SEX-DETector allows us to test for these assumptions by comparing the model fit to the data using the Bayesian Information Criterion (BIC, see “Materials and Methods” section). In species for which sex determination is unknown, it is possible to compare models with and without sex chromosomes, and, if sex chromosomes are detected, it is possible to compare models with X/Y or Z/W system. This model selection strategy was tested on empirical and simulated data.
In the S. latifolia dataset, the best model inferred by SEX-DETector was a model with sex chromosomes as expected, with 1357 sex-linked contigs (which represents 9% of the contigs with a posterior probability higher than 0.8). In the S. vulgaris data set (a species without sex chromosomes), no sex-linked contigs were inferred, the best model fit to the data was thus a model without sex chromosomes as expected (see “Materials and Methods” section).
In order to know from which proportion of sex-linked genes sex chromosomes can be detected, we compared models on simulated data with varying numbers of sex-linked contigs out of 10,000 simulated contigs (table 2 and supplementary table S6, Supplementary Material online). When no sex-linked contigs were simulated, as expected the best model was the one without sex chromosomes. This was also the case when a single sex-linked contig was simulated. In this case, SEX-DETector could not detect it due to lack of information in the dataset. When ten or more sex-linked contigs were simulated, the best model was the one with sex chromosomes as expected. Thus, ten sex-linked contigs out of 10,000 provide sufficient information for SEX-DETector (i.e., one sex-linked gene out of 1000 genes can be detected).
Table 2.
Model comparison using SEX-DETector on empirical datasets in Silene latifolia (with sex chromosomes) and S. vulgaris (without sex chromosomes) and simulated X/Y datasets with varying number of sex-linked contigs out of 10,000 simulated contigs. The best model is chosen as the one having the lowest BIC value (see “Materials and Methods” section and supplementary table S6, Supplementary Material online for details)
| Best model | number of sex-linked genes in the best model | ||
|---|---|---|---|
| Empirical datasets | Silene latifolia (X/Y system) | X/Y | 1357 |
| Silene vulgaris (no sex chromosomes) | Z/W | 0 | |
| Simulated datasets of 10,000 genes with different numbers of sex-linked genes (XY system) | 0 sex-linked genes | no sex chromosomes | 0 |
| 1 sex-linked gene | no sex chromosomes | 0 | |
| 10 sex-linked genes | X/Y | 16–57 | |
| 100 sex-linked genes | X/Y | 156–181 | |
| 500 sex-linked genes | X/Y | 592–624 | |
| 3000 sex-linked genes | X/Y | 3159–3200 |
Once the presence of sex chromosomes has been inferred, it can be tested whether the system is X/Y or Z/W. The model comparison between X/Y and Z/W systems worked on both empirical and simulated data: the best model for S. latifolia was, as expected, the X/Y system (table 2 and supplementary table S6, Supplementary Material online).
Discussion
To summarize, SEX-DETector implements a probabilistic model that is used to compute the posterior probabilities of being autosomal, X/Y and X-hemizygous (X-linked copy only) for each RNA-seq contig in data from a full-sib family. The method is suitable for any sex chromosome type (XY, ZW, and UV). SEX-DETector uses genotypes obtained from a genotyper specifically designed for RNA-seq data (Tsagkogeorga et al. 2012; Gayral et al. 2013). This genotyper takes unequal allelic expression into account, which is particularly important as Y (or W) copies tend to be less expressed than their X (or Z) counterpart (reviewed in Bachtrog 2013). The SEX-DETector model also accounts for genotyping errors. The pipeline that includes steps from assembly to sex-linkage inference (fig. 2) was implemented in Galaxy for easier use. The pipeline was successfully tested on RNA-seq data of a family from Silene latifolia, a dioecious plant with relatively recent but heteromorphic sex chromosomes. Genes have previously been experimentally characterised as autosomal or sex-linked in this species, which made it possible to assess the performances of the method. About 83% of known sex-linked genes that were expressed in the sampled tissue could be identified using the SEX-DETector pipeline. Sensitivity and specificity values were used to compare SEX-DETector to other RNA-seq based approaches that used S. latifolia data sets (Bergero and Charlesworth 2011; Chibalina and Filatov 2011; Muyle et al. 2012). SEX-DETector showed a much higher sensitivity (0.63 compared with 0.25–0.43) while specificity remained close to 1. Thanks to a statistically grounded method, the SEX-DETector pipeline can detect many more genes than previous approaches, while keeping the inferences extremely reliable. The SEX-DETector pipeline was also run on a comparable RNA-seq data from Silene vulgaris (a plant without sex chromosomes), and yielded no sex-linked genes, as expected. We further tested the SEX-DETector method using simulations, which indicated good performance on different sex chromosome systems (old or young and homomorphic or heteromorphic). Simulations also showed that few individuals need to be sequenced for optimal results (under 12 individuals for a ZW or XY system and five individuals for a UV system). This makes the strategy very accessible given the cost of RNA-seq, in particular in species with large genomes. The likelihood framework of SEX-DETector makes it possible to assess the presence and type of sex chromosomes in the data using a model comparison strategy. This procedure proved efficient on empirical and simulated data, provided more than one gene in 10,000 was sex-linked in the data.
The downside of using RNA-seq data is, of course, that only expressed genes can be identified by the SEX-DETector pipeline. This can be overcome by the use of DNA-seq data, or the combination of multiple tissues for RNA-seq data. Moreover, because Y-specific genes cannot be differentiated from autosomal male-specific genes in RNA-seq data, Y genes are not inferred by SEX-DETector unless they coassemble with an X counterpart. This requirement makes the method less adapted to old sex chromosome systems where X and Y copies of a given gene could be too diverged to coassemble. However, X copies can still be identified on their own if the Y copy is absent or did not coassemble with the X (fig. 1c). To try and identify missed Y contigs, X-hemizygous genes can be blasted onto male-specific contigs, which may represent the diverged Y copy. This was done for the 332 inferred X-hemizygous genes in the S. latifolia dataset, and only five of them had a significant match with a male-specific contig. This suggests that very few true X/Y gene pairs were wrongly inferred as X-hemizygous due to a too divergent Y. In S. latifolia, X–Y synonymous divergence ranges from 5% to 25% (Bergero et al. 2007). This is comparable to regions in human sex chromosomes that stopped recombining last: the mean X–Y synonymous divergence for strata 3, 4, and 5 in humans is respectively 30%, 10%, and 5% (Skaletsky et al. 2003). SEX-DETector will therefore perform best in species with sex chromosomes of young or intermediate age, but will also work on recent strata of old systems. A complete list of cases where inferences could be difficult (e.g., presence of pseudoautosomal genes, X chromosome inactivation and imprinting) along with possible solutions is provided in the online SEX-DETector user manual (p. 3–4). A list of cases where SEX-DETector could be applied to detect dominant loci associated with a phenotype, other than sex chromosomes is also provided in the user manual.
Other approaches to identify sequences of sex chromosomes based on female and male DNA-seq data comparison can only detect regions where the X and the Y are divergent enough not to coassemble nor map onto one another (Vicoso and Bachtrog 2011; Vicoso, Emerson et al. 2013; Vicoso, Kaiser et al. 2013; Carvalho and Clark 2013; Akagi et al. 2014; Cortez et al. 2014). These approaches are therefore best suited to old sex chromosome systems. Other methods work on young systems but rely on genome sequencing (Al-Dous et al. 2011; Picq et al. 2014; Hou et al. 2015). Obtaining a reference genome can be difficult for non-model organisms, especially those with large genomes. In such cases, RNA-seq data is a lot cheaper. Therefore, SEX-DETector is a highly promising method for uncovering sex-chromosomes in non-model organisms, and especially those with young sex chromosomes. These types of sex chromosomes are expected in thousands of yet unstudied independent taxa across plants and animals (see “Introduction” section) (Ming et al. 2011; Bachtrog et al. 2014; Renner 2014), and probably many more in all eukaryotes.
Supplementary Material
Supplementary tables S1–S6 and figures S1–S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We thank Alex Widmer for access to the RNA-seq data sets and comments on the manuscript, Nicolas Galtier and Sylvain Glémin (ISEM-Montpellier) for useful discussions about Reads2snp, Vincent Miele (LBBE) for SEX-DETector profiling and advice on code performance, Khalid Belkhir (ISEM-Montpellier) for providing and adapting Galaxy wrappers for analyses used upstream of SEX-DETector (BWA, Reads2snp) and Philippe Veber (LBBE) for help with Galaxy. We thank the editor and three anonymous referees for their helpful comments. This work was supported by Agence Nationale de la Recherche grants to GABM (grants numbers: ANR-11-BSV7-013, ANR-11-BSV7-024; ANR-14-CE19-0021) and SNF project to Alex Widmer (SNF 31003A_141260).
Literature Cited
- Ahmed S, et al. 2014. A haploid system of sex determination in the brown alga Ectocarpus sp. Curr Biol. 24:1945–1957. [DOI] [PubMed] [Google Scholar]
- Akagi T, Henry IM, Tao R, Comai L. 2014. Plant genetics. A Y-chromosome-encoded small RNA acts as a sex determinant in persimmons. Science (New York, N.Y.) 346:646–650. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. 2013. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet. 14:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, et al. 2011. Are all sex chromosomes created equal? Trends in Genet. 27:350–357. [DOI] [PubMed] [Google Scholar]
- Bachtrog D, et al. 2014. Sex determination: why so many ways of doing it? PLoS Biol. 12:e1001899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, et al. 2014. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508:494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergero R, Charlesworth D. 2011. Preservation of the Y transcriptome in a 10-million-year-old plant sex chromosome system. Curr Biol. 21:1470–1474. [DOI] [PubMed] [Google Scholar]
- Bergero R, Forrest A, Kamau E, Charlesworth D. 2007. Evolutionary strata on the X chromosomes of the dioecious plant Silene latifolia: evidence from new sex-linked genes. Genetics 175:1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahais V, et al. 2012. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol Ecol Resources 12:834–845. [DOI] [PubMed] [Google Scholar]
- Carvalho AB, Clark AG. 2013. Efficient identification of Y chromosome sequences in the human and Drosophila genomes. Genome Res. 23:1894–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Sniegowski P, Stephan W. 1994. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 371:215–220. [DOI] [PubMed] [Google Scholar]
- Chibalina MV, Filatov DA. 2011. Plant Y chromosome degeneration is retarded by haploid purifying selection. Curr Biol. 21:1475–1479. [DOI] [PubMed] [Google Scholar]
- Cortez D, et al. 2014. Origins and functional evolution of Y chromosomes across mammals. Nature 508:488–493. [DOI] [PubMed] [Google Scholar]
- DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dous EK, et al. 2011. De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nat Biotechnol 29:521–527. [DOI] [PubMed] [Google Scholar]
- Ferris P, et al. 2010. Evolution of an expanded sex-determining locus in Volvox. Science (New York, N.Y.) 328:351–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, Wright SI, Rizzon C, Dvorak J, Anderson LK. 2007. Recombination: an underappreciated factor in the evolution of plant genomes. Nat Rev Genet. 8:77–84. [DOI] [PubMed] [Google Scholar]
- Gautier M. 2014. Using genotyping data to assign markers to their chromosome type and to infer the sex of individuals: a Bayesian model-based classifier. Mol Ecol Resources 14:1141–1159. [DOI] [PubMed] [Google Scholar]
- Gayral P, et al. 2013. Reference-free population genomics from next-generation transcriptome data and the vertebrate-invertebrate gap. PLoS Genet. 9:e1003457.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, et al. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AB, et al. 2016. Radical remodeling of the Y chromosome in a recent radiation of malaria mosquitoes. Proc Natl Acad Sci USA 113:E2114–E2123. [DOI] [PMC free article] [PubMed]
- Hoskins RA, et al. 2015. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, et al. 2015. Different autosomes evolved into sex chromosomes in the sister genera of Salix and Populus. Sci Rep 5:9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough J, Hollister JD, Wang W, Barrett SCH, Wright SI. 2014. Genetic degeneration of old and young Y chromosomes in the flowering plant Rumex hastatulus. Proc Natl Acad Sci U S A. 111:7713–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Madan A. 1999. CAP3: a DNA sequence assembly program. Genome Res. 9:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics (Oxford, England) 18:337–338. [DOI] [PubMed] [Google Scholar]
- Hughes JF, Rozen S. 2012. Genomics and genetics of human and primate y chromosomes. Annu Rev Genomics Hum Genet. 13:83–108. [DOI] [PubMed] [Google Scholar]
- Käfer J, et al. 2013. Patterns of molecular evolution in dioecious and non-dioecious Silene. J Evol Biol. 26:335–346. [DOI] [PubMed] [Google Scholar]
- Kondo M, et al. 2006. Genomic organization of the sex-determining and adjacent regions of the sex chromosomes of medaka. Genome Res. 16:815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Avise JC. 2009. Evolutionary diversity and turn-over of sex determination in teleost fishes. Sex Dev. 3:60–67. [DOI] [PubMed] [Google Scholar]
- Marais GAB, et al. 2011. Multiple nuclear gene phylogenetic analysis of the evolution of dioecy and sex chromosomes in the genus Silene. PLoS One 6:e21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga S, Hizume M, Kawano S, Kuroiwa T. 1994. Cytological analyses in Melandrium album: genome size, chromosome size and fluorescence in situ hybridization. Cytologia 59:135–141. [Google Scholar]
- McKenna A, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovova M, Kubat Z, Hobza R, Vyskot B, Kejnovsky E. 2015. Fully automated pipeline for detection of sex linked genes using RNA-Seq data. BMC Bioinformatics 16:78.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming R, Bendahmane A, Renner SS. 2011. Sex chromosomes in land plants. Annu Rev Plant Biol. 62:485–514. [DOI] [PubMed] [Google Scholar]
- Muyle A, Shearn R, Marais G. 2016. The evolution of sex chromosomes and dosage compensation in plants. Genome Biol Evol. [DOI] [PMC free article] [PubMed]
- Muyle A, et al. 2012. Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLoS Biol. 10:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picq S, et al. 2014. A small XY chromosomal region explains sex determination in wild dioecious V. vinifera and the reversal to hermaphroditism in domesticated grapevines. BMC Plant Biol. 14:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Bergero R, Forrest A, Kaiser VB, Charlesworth D. 2010. Nucleotide diversity in Silene latifolia autosomal and sex-linked genes. Proc Biol Sci R Soc 277:3283–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Grassly NC. 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput Appl Biosci 13:235–238. [DOI] [PubMed] [Google Scholar]
- Rautenberg A, Hathaway L, Oxelman B, Prentice HC. 2010. Geographic and phylogenetic patterns in Silene section Melandrium (Caryophyllaceae) as inferred from chloroplast and nuclear DNA sequences. Mol Phylogenet Evol. 57:978–991. [DOI] [PubMed] [Google Scholar]
- Renner SS. 2014. The relative and absolute frequencies of angiosperm sexual systems: dioecy, monoecy, gynodioecy, and an updated online database. Am J Bot 101:1588–1596. [DOI] [PubMed] [Google Scholar]
- Skaletsky H, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837. [DOI] [PubMed] [Google Scholar]
- Stock M, et al. 2013. Low rates of X-Y recombination, not turnovers, account for homomorphic sex chromosomes in several diploid species of Palearctic green toads (Bufo viridis subgroup). J Evol Biol. 26:674–682. [DOI] [PubMed] [Google Scholar]
- Tomaszkiewicz M, et al. 2016. A time- and cost-effective strategy to sequence mammalian Y Chromosomes: an application to the de novo assembly of gorilla Y. Genome Res. 26:530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagkogeorga G, Cahais V, Galtier N. 2012. The population genomics of a fast evolver: high levels of diversity, functional constraint, and molecular adaptation in the tunicate Ciona intestinalis. Genome Biol Evol. 4:740–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Bachtrog D. 2011. Lack of global dosage compensation in Schistosoma mansoni, a female-heterogametic parasite. Genome Biol Evol. 3:230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Emerson JJ, Zektser Y, Mahajan S, Bachtrog D. 2013. Comparative sex chromosome genomics in snakes: differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol. 11:e1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Kaiser VB, Bachtrog D. 2013. Sex-biased gene expression at homomorphic sex chromosomes in emus and its implication for sex chromosome evolution. Proc Natl Acad Sci U S A. 110:6453–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, et al. 2012. Sequencing papaya X and Yh chromosomes reveals molecular basis of incipient sex chromosome evolution. Proc Natl Acad Sci U S A. 109:13710–13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks SC. 2012. The role of androdioecy and gynodioecy in mediating evolutionary transitions between dioecy and hermaphroditism in the animalia. Evolution 66:3670–3686. [DOI] [PubMed] [Google Scholar]
- Yamato KT, et al. 2007. Gene organization of the liverwort Y chromosome reveals distinct sex chromosome evolution in a haploid system. Proc Natl Acad Sci U S A. 104:6472–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Zemp N, et al. Regulatory changes in females drive the evolution of sex-biased gene expression.
- Zhou Q, Bachtrog D. 2012. Sex-specific adaptation drives early sex chromosome evolution in Drosophila. Science (New York, N.Y.) 337:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.