

Abstract
Despite the figure of complete bivalve mitochondrial genomes keeps growing, an assessment of the general features of these genomes in a phylogenetic framework is still lacking, despite the fact that bivalve mitochondrial genomes are unusual under different aspects. In this work, we constructed a dataset of one hundred mitochondrial genomes of bivalves to perform the first systematic comparative mitogenomic analysis, developing a phylogenetic background to scaffold the evolutionary history of the class' mitochondrial genomes. Highly conserved domains were identified in all protein coding genes; however, four genes (namely, atp6, nad2, nad4L, and nad6) were found to be very divergent for many respects, notwithstanding the overall purifying selection working on those genomes. Moreover, the atp8 gene was newly annotated in 20 mitochondrial genomes, where it was previously declared as lacking or only signaled. Supernumerary mitochondrial proteins were compared, but it was possible to find homologies only among strictly related species. The rearrangement rate on the molecule is too high to be used as a phylogenetic marker, but here we demonstrate for the first time in mollusks that there is correlation between rearrangement rates and evolutionary rates. We also developed a new index (HERMES) to estimate the amount of mitochondrial evolution. Many genomic features are phylogenetically congruent and this allowed us to highlight three main phases in bivalve history: the origin, the branching of palaeoheterodonts, and the second radiation leading to the present-day biodiversity.
Keywords: mitochondrial genomics, comparative mitogenomics, bivalves, phylogeny, HERMES
Introduction
Mitochondria are key eukaryotic organelles involved not only in the well-known synthesis of ATP through oxidative phosphorylation (OXPHOS; Mitchell 1961; Wallace 2013), but also in many other biological functions, such as intracellular signaling, cell differentiation, programmed cellular death, fertilization, and aging (Scheffler 2008; Tait and Green 2010; Van Blerkom 2011; Lane and Martin 2012; López-Otín et al. 2013; Sousa et al. 2013; Chandel 2014). Currently, it is widely accepted that mitochondria originated from a single endosymbiotic event that took place over a billion of years ago (Gray et al. 1999, 2001; Sicheritz-Ponten and Andersson 2001; Gray 2012). It has been proposed that the putative ancestor of all mitochondria was an alpha-proteobacterium (Müller and Martin 1999; Gray et al. 2001; Fitzpatrick et al. 2006; Williams et al. 2007; Atteia et al. 2009; Abhishek et al. 2011; Thrash et al. 2011; Degli Esposti et al. 2014).
The following evolution of mitochondria is still unclear and a matter of debate. In animals, mitochondrial DNA (mtDNA) is a small (∼16 kb) and compact circular molecule that typically contains 13 OXPHOS-related genes, 2 rRNAs encoding for the two subunits of mitochondrial ribosomes, and an array of tRNAs used for translation within the organelle (Boore 1999; Breton et al. 2014). Most genes of the original endosymbiont were therefore lost or have been transferred to the nucleus during a process called Genome Reductive Evolution (GRE) (Andersson and Kurland 1998; Khachane et al. 2007; Ghiselli et al. 2013; Kannan et al. 2014). As claimed by Meisinger et al. (2008), in mammals no less than 1,500 proteins are needed to keep mitochondria alive and working. Why then only a small cluster of protein coding genes (PCGs) was spared by GRE—the typical 13 in metazoans, and even down to 3 in apicomplexans (Feagin 1994; Rehkopf et al. 2000)?
Many tentative answers were proposed to this question. It is well known, for example, that mitochondria employ a genetic code slightly different from that of the nucleus, which would possibly lead to erroneous translations of some genes if transferred to nucleus (Adams and Palmer 2003); however, this is true for animals, but not for plants (Jukes and Osawa 1990), which also underwent mitochondrial GRE.
The high hydrophobicity of some of these proteins would hamper the import from the cytosol as well (von Heijne 1986; Popot and de Vitry 1990; Claros et al. 1995; Pérez-Martínez et al. 2000, 2001; Daley et al. 2002; Funes et al. 2002; Adams and Palmer 2003). Martin and Schnarrenberger (1997) also claimed that certain gene products are toxic in the cytoplasm. Finally, it has been proposed that the genes still encoded by mtDNAs must be efficiently and directly regulated by mitochondrial redox conditions, so they need mandatory colocation for redox regulation (CoRR) (Race et al. 1999; Allen 2003a, 2003b; Lane 2007).
The sequencing and characterization of several complete mitochondrial genomes is a key to address GRE of mitochondria. Pioneering comparative works of large mtDNA datasets could recently, e.g., shed light on the reduction and ultimate loss in animals’ mitochondria of ribosomal protein genes (Maier et al. 2013) and establish that the mitochondrion-encoded genes of almost all eukaryotes are different subsets of the mitochondrial gene complement of jakobids, a group of free-living, heterotrophic flagellates (Kannan et al. 2014).
At the same time, mitochondrial genomes of single metazoan groups often exhibit peculiar evolution that deserves a careful examination. In this regard, bivalves (Mollusca, Bivalvia) are among the most notable animal taxa, because of several interesting features. Most strikingly, some bivalves follow a non-canonical way of mitochondrial inheritance, the Doubly Uniparental Inheritance (DUI; Skibinski et al. 1994a, 1994b; Zouros et al. 1994a, 1994b). In these species, two separate sex-linked mitochondrial lineages (and their respective mtDNAs) are present, namely, the F and the M (Breton et al. 2007; Passamonti and Ghiselli 2009; Zouros 2013; Breton et al. 2014). F mitochondria are passed from the mother to the complete offspring, and they are typically found in the soma and in the female germline (eggs); the M mitochondria are passed from the father to the male offspring, and they are typically found in the male germline (sperms). However, these general DUI rules may hold imperfectly, thus resulting in a leakage of the M mitochondrial lineage in somatic cells of either sex (Chakrabarti et al. 2006; Batista et al. 2010; Kyriakou et al. 2010; Ghiselli et al. 2011; Obata et al. 2011). DUI represents a case of a triple genomic conflict (nucleus vs. F mtDNA; nucleus vs. M mtDNA; F vs. M mtDNAs) (Passamonti and Ghiselli 2009; Breton et al. 2014).
There are more outstanding features of bivalves mtDNA that are generally shared across the entire class. In fact, bivalve mitochondrial genomes are often very large, up to the 46,985 bp of Scapharca broughtonii (Liu et al. 2013); they present many putative unassigned regions (URs) (Ghiselli et al. 2013); they may encode for supernumerary open reading frames (ORFans) (Breton et al. 2009; Milani et al. 2013); they show an unpaired degree of gene rearrangement (Vallès and Boore 2006; Simison and Boore 2008; Plazzi et al. 2013); they exhibit strong differences in strand usage (see Additional file 6 in Plazzi et al. 2013).
All this considered, the interest of bivalves in the wider picture of the evolution of animal mitochondria is self-evident. In this work, we present the first meta-analysis of 100 bivalve mtDNAs, corresponding to all the species whose mtDNA was available in GenBank in August, 2014.
The polyphyly of bivalves in molecular phylogenetic reconstructions is a well-known artifact (Giribet and Wheeler 2002; Giribet and Distel 2003; Passamaneck et al. 2004; Giribet et al. 2006; Sharma et al. 2012; Plazzi et al. 2013; Stöger and Schrödl 2013; Bieler et al. 2014) and it is probably related to the fact that a small group of bivalves (the protobranchs) separated very early and, regarding mitochondrial markers, before the burst of genomic novelties described above (Plazzi et al. 2013). In the present work, we do not address the issue of bivalve monophyly: therefore, the only protobranch bivalve whose complete mitochondrial genome is available in GenBank in August, 2014 is used as outgroup for phylogenetic analyses.
The comparative mitogenomics of bivalves is critically explored to (i) achieve a deeper knowledge about factors that shaped mitochondrial evolution in these metazoans, and (ii) focus on the specific features of bivalve mitochondrial PCGs.
Materials and Methods
The Database
Hundred bivalve complete mitochondrial genomes (supplementary file S1, Supplementary Material online) were downloaded from GenBank in August, 2014 using CLC Sequence Viewer 7.5 (Qiagen A/S, Aarhus). Annotations and sequences were imported and managed in Microsoft Excel® 2007 through custom functions and VBA macros that were also used to output files in the correct format for downstream analyses. In August, 2014 the annotation of Tegillarca granosa (Sun et al. 2015) was already published, but the sequence was not available yet; the sequence of Nucula nucleus (GenBank accession number EF211991) is still incomplete and unpublished: as a consequence, these species were included in the overall analysis, but not in alignments presented in this paper.
Alignment Algorithms
The first step of most analyses was a structural alignment and data masking: to this purpose, a custom tool was written in bash and R (R Development Core Team 2008) environments, loading the package seqinr (Charif and Lobry 2007). The atp8 gene and tRNAs were excluded from alignments beacuse of many uncertainties in annotations.
This tool was called masking_package and is available with a brief tutorial from the GitHub repository https://github.com/mozoo/masking_package.git. It performs:
structural alignment using T-Coffee (Notredame et al. 2000) and nested packages PSI-BLAST (Altschul et al. 1997), Muscle (Edgar 2004), ProbconsRNA (Do et al. 2005), RNAplfold (Lorenz et al. 2011), and MAFFT (Katoh and Standley 2013), using the option series PSI-Coffee > Expresso > accurate for Protein Coding Gene (PCG) amino acids and the MR-Coffee mode for rRNA nucleotides;
alignment masking in order to eliminate phylogenetic noise using the four softwares Aliscore 2.0 (Misof and Misof 2009), BMGE 1.1 (Criscuolo and Gribaldo 2010), Gblocks 0.91b (Castresana 2000), and Noisy (Dress et al. 2008), setting adequate options for distantly related sequences;
comparison of outputs from different masking strategies in terms of number of sites and percentage of removed sites and output of a final concatenated alignment where only sites kept by at least k softwares are present (where k is up to the user and was set to 2, 3, or 4);
computation of the number of sites selected by all the possible combinations of the four softwares.
All the masking_package scripts adapted for amino acid (PCGs) and nucleotide (rRNAs) alignments of the present study are available as supplementary files S2 and S3, Supplementary Material online, respectively.
Split rrnL genes of ostreids were concatenated prior to alignment. Moreover, all Crassostrea species (with the exception of C. virginica) have a duplicated rrnS gene. In these cases, the two copies were aligned with MR-Coffee, a consensus sequence was computed using seqinr and the result was inserted into the final alignment.
Genomic Features
Five large-scale features of bivalve mtDNAs were extracted: length, percentage of URs, number of genes (including newly annotated ones; see below), and A-T and G-C skew following Reyes et al. (1998). Instead of a typical ANOVA, because of non-normality and non-homoscedasticity of data, differences between subclasses had to be tested using a non-parametric alternative: we used the Kruskal–Wallis test (Kruskal and Wallis 1952), while relative pairwise comparisons were carried out using the Dunn's test (Dunn 1964). Given the poor sample size of Anomalodesmata (N = 1) and Opponobranchia (N = 2), these subclasses were excluded from these preliminary analyses.
Afterwards, single alignments were used as input for several custom bash and R scripts that were used to:
subsample the original dataset, producing subsets corresponding to largest families (Mytilidae, Ostreidae, Pectinidae, Unionidae, Veneridae) and subclasses (Heterodonta, Palaeoheterodonta, Pteriomoprhia) to be aligned and masked;
compute the percentage of cases in which each site of each gene was kept after the masking phase, using the agreement of at least 3 out of 4 masking softwares (across the five families, the three subclasses, and in the complete-dataset analysis);
comparing masking results using a one-sided t-test;
back-translate the original, unmasked amino acid alignment into nucleotides;
use EMBOSS (Rice et al. 2000) to compute different metrics of pairwise and overall distances, both on total gene length (using distmat) and over a sliding window (using plotcon);
compare nucleotide and amino acid distances through the Kolmogorov–Smirnov test;
test alignments for saturation by plotting uncorrected p-distance over the Jin–Nei distance (for nucleotides; Jin and Nei 1990) or the Kimura distance (for amino acids; Kimura 1983);
perform a Principal Component Analysis (PCA) on concatenated distance matrices, using the FactoMineR (Lê et al. 2008) package for computations and ggplot2 (Wickham 2010) for graphics.
The ratio of non-synonymous vs. synonymous substitutions (dN/dS) was computed for back-translated alignments using PAML 4.8a (Yang 1997, 2007); dN/dS was computed both for each pair of sequences and along the phylogenetic tree (see below). In the latter case, we used the Likelihood Ratio Test (LRT) to compare the fitting of single dN/dS for the complete tree with the use of 12 different dN/dS, allowing different dN/dS ratios for different clades. All LRTs were carried out in the R environment; to be conservative, we used a chi-square distribution with 11 degrees of freedom (Wong et al. 2004).
Phylogenetic Analyses
Using the original annotations available in GenBank, phylogenetic analysis was carried out on PCGs and rRNAs; given many uncertainties in annotation and orthology, the atp8 gene and tRNAs were excluded from the analysis. The best-fitting partitioning scheme and molecular evolution models were estimated using PartitionFinderProtein and PartitionFinder 1.1.0 (Lanfear et al. 2012) under the Bayesian Information Criterion and a greedy approach. Gaps were coded following the simple indel method of Simmons and Ochoterena (2000) as implemented in GapCoder (Young and Healy 2003). As a result, the final alignment spanned over 14 genes and three types of data: amino acids for PCGs, nucleotides for rRNAs, and binary data for coded gaps.
The software RAxML 8.2.0 (Stamatakis 2014) was used to infer Maximum Likelihood (ML) phylogeny. The strand usage is variable across different bivalve mtDNAs: while in most cases all genes locate to the same strand, genes are evenly distributed on both strand in N. nucleus, S. velum, and Unionidae. As a strand usage bias can be associated with a compositional bias, which may in turn lead to phylogenetic artifacts, preliminary analyses with 500 bootstrap replicates were separately conducted on those genes mapping on the “+” and on the “−” strand of unionids. If a significant compositional bias is working, we expect to detect possible phylogenetic artifacts by recovering different topologies from the two analyses.
The final tree was computed as follows; explicit RAxML commands are listed in supplementary file S4, Supplementary Material online.
five randomized maximum parsimony (MP) starting trees were generated;
a maximum likelihood (ML) tree was inferred from each MP starting tree with a fixed rearrangement radius of 10 and with an automatically determined one, choosing the setting yielding the best results;
a ML tree was inferred from each MP starting tree with the selected initial rearrangement option under different number of rate categories (10, 25, 40, or 55) for the CAT model accounting for evolutionary rate heterogeneity (Stamatakis 2006), choosing the setting yielding the best results;
the best-known likelihood tree was inferred from the original alignment under the selected rearrangement and rate categories options calling 10 runs from 10 randomized MP starting trees;
500 bootstrap replicates were run; and
confidence values were computed from bootstrap replicates and annotated on the best-known likelihood tree.
The software MrBayes 3.2.1 (Ronquist et al. 2012) was used to carry out Bayesian Inference (BI) using 2 separate runs, 4 chains, and 10,000,000 generations of MC3, sampling every 100 trees. Convergence between runs and burn-in were estimated looking to standard deviation of average split frequencies sampled every 1,000 generation and to Potential Scale Reduction Factor (PSRF) (Gelman and Rubin 1992).
The best-known likelihood tree computed by RAxML was used to obtain a time-scaled chronogram using r8s 1.70 (Sanderson 2003) and the r8s bootstrap kit by Torsten Eriksson (downloaded from https://github.com/TorstenEriksson/r8s-bootstrap in February, 2015). First appearance data for six calibration point were downloaded from the Paleobiology Database (http://fossilworks.org) in February, 2015: the root of all bivalves, Mytilidae, Ostreidae, Pectinidae, Unionidae, and Veneridae (M'Coy 1847; Tillyard and Dunstan 1916; Cromptok and Parrington 1955; Drysdall and Kitching 1963; Nakazawa and Newell 1968; Brasier and Hewitt 1978; Baird and Brett 1983; Grasso 1986; Brett et al. 1991; Mergl and Massa 1992; Kříž 2008; Nesbit et al. 2010). We used the Penalized Likelihood method and the truncated Newton algorithm; the cross-validation approach allowed us to estimate the best smoothing parameter up to the sixth decimal digit and five restarts and five guesses were used each round.
Following the r8s bootstrap kit procedure, 163 bootstrap replicates of the original alignment were generated and the original tree was optimized on each of them; node ages were estimated under the same r8s setting, estimating the best smoothing parameter up to the second decimal digit. The 99% confidence intervals of each node age were computed using custom R script loading the package ape (Paradis et al. 2004) and following methodological recommendations of Hyndman and Fan (1996), i.e., type = 8. Trees were graphically edited using the software PhyloWidget (Jordan and Piel 2008) and Dendroscope 3.3.2 (Huson and Scornavacca 2012).
Phylogenetic Informativeness (PI) was investigated on the amino acid dataset using the PhyDesign portal (López-Giráldez and Townsend 2011) and Rate4Site (Pupko et al. 2002) to estimate site-specific evolutionary rates. Best-fitting amino acid evolutionary models were selected using ProtTest 3.4 (Darriba et al. 2011) and PhyML (Guindon and Gascuel 2003). PI was computed over five different epochs, whose boundaries were taken from the International Stratigraphic Chart v2015/01 (Cohen et al. 2013): Quaternary, “Cenozoic” (Paleogene + Neogene), Mesozoic, “Paleozoic” (from Ordovician to Permian), and “Cambrian” (from 520 to 485.4 Mya). Following Townsend et al. (2012) and Simmons et al. (2004), the phylogenetic signal and noise analysis was carried out in a state space of five.
Finally, we investigated the correlation between gene rearrangements and substitution rates, which has been demonstrated for insects by Shao et al. (2003) and hypothesized for mollusks by Stöger and Schrödl (2013). However, the RGR test of Dowton (2004) and its modified version of Xu et al. (2006) cannot be useful in the case of bivalves to describe gene arrangement variability because it is a relative rate test that involves a comparison with the ancestral gene arrangement. As detailed in Plazzi et al. (2013), this is most likely that of the chiton Katharina tunicata, but, with the notable exception of Solemya velum and Nucula nucleus (Plazzi et al. 2013), most bivalve gene orders are not comparable with each other and seem to be equally very different from that. Therefore, while there is no remarkable difference in terms of distance from the ancestral state, higher rates of mitochondrial gene rearrangement are straightforward in some clusters with respect to others, but this would not be detected by RGR: for this reason, we quantified rates of mitochondrial rearrangement in the clades obtained in the best-known likelihood tree using the architecture rate (AR) as introduced by Gissi et al. (2008). This is given by
where NGA is the number of different gene arrangements found in a clade and NOTU is its number of OTUs. In fact, this index conservatively estimates the number of gene rearrangement events along a clade's evolutionary history, as it must be at least as large as the number of different gene arrangements found in extant taxa, and normalizes it with the number of species, so that clades of different sizes are comparable. AR rate ranges from 0 (the whole clade shares the same gene arrangement) to 1 (each OTU shows a different gene arrangement). The sum of branch lengths for a given clade was computed with the software Phylocom 4.2 (Webb et al. 2008), thus estimating the total number of expected substitution per site in a given cluster; this was divided by the root age in millions of years (see above). The correlation of AR rate and substitution rate was assessed with the Spearman's rho and Kendall's tau tests using R.
Annotation of the atp8 Gene and of ORFans
The EMBOSS suite was used to find all the possible Open Reading Frames (ORFs) in all the complete mitochondrial genomes, using all the six possible frames. A Hidden Markov Model (HMM) was constructed for each ORF using HHblits 2.0 (Remmert et al. 2012) and the latest PDB70 release. All the HMMs were merged in a custom database, which was called Biv_mtDNA_ORFs.
All the annotated atp8 genes were aligned using the T-Coffee (again following the PSI-Coffee > Expresso > accurate option series). A Hidden Markov Model (HMM) was constructed using HHblits 2.0 and the latest Uniprot release and a HMM-HMM alignment was run against the Biv_mtDNA_ORFs database to check for homology with atp8. Positive results were manually screened and original annotations were consequently updated.
Bivalve supernumerary mitochondrial ORFs do not have known homologs and are therefore considered as ORFans (Fischer and Eisenberg 1999). However, it is possible to find homologies at least within Bivalvia. All the published supernumerary ORFs (Breton et al. 2009; Milani et al. 2013; Plazzi et al. 2013; Bettinazzi et al. 2016; GenBank accession numbers HM856636, HQ283344, KC848655, KF030963, NC_015310, NC_015476, NC_015477, NC_015479, NC_015481, NC_015483, NC_018763, NC_022803, NC_023250, NC_023942) were annotated; a HHblits database of all the known ORFans was created as above using Biv_mtDNA_ORFs and was called Biv_mtDNA_ORFans; finally, HHblits was used for each ORFan against this custom database to investigate putative relationships between known ORFans.
Summarizing Data
A PCA was carried out as above using many different parameters (supplementary file S5, Supplementary Material online): nucleotide composition; AT content; A-T and G-C skews; use of between-genes overlapping nucleotides; length; percentage of Unassigned Regions (URs); total number of genes; the strand usage skew (SU skew); number of truncated T–/TA- stop codons in the genome; the Amount of Mitochondrial Identical Gene Arrangements (AMIGA). SU skew and AMIGA were defined as follows.
where H is the number of genes on the H strand and L is the number of genes on the L strand. If the strand usage is perfectly balanced (i.e., H = L), the SU skew is equal to 0; a negative SU skew indicates a bias towards the L strand, while a positive SU skew indicates a bias towards the H strand.
where NIGA is the number of taxa in the analyzed sample that share an Identical Gene Arrangement and N is the total number of taxa. As a consequence, unique gene arrangements will have an AMIGA score equal to 0. Conversely, when all the taxa in a given sample share the same gene arrangement, each AMIGA score will be equal to 1. An intermediate value expresses an intermediate value of conservation. Given many uncertainties in annotations, rRNAs and tRNAs were excluded from the analysis, therefore the AMIGA index relies solely on PCGs.
The HERMES Index
A widespread method of quantifying molecular evolution of mitochondrial genomes in different species and clusters is still lacking for mitogenomic analyses. We developed a new index in this regard, which relies on maximum likelihood factor analysis to summarize different measures that are typically found to be linked with evolutionary rates; it is intended to be computed a posteriori, i.e. after the phylogenetic and genomic analysis. As different empirical measures are merged together in a single score, this is a “hyper-empirical” index. Moreover, a taxon retaining most genomic plesiomorphies of the group (at least following state-of-art knowledge) is selected as a benchmark: thus, it is a relative measure, and that taxon was in our case N. nucleus. All this considered, the index was called Hyper-Empirical Relative Mitochondrial Evolutionary Speed (HERMES) index.
We explored the use of different subsets of the following variables to compute the HERMES index:
the AT content;
the genome length;
the number of (annotated) genes;
the percentage of URs;
the absolute value of SU skew;
AMIGA;
the root-to-tip distance computed on the best-known likelihood tree using Phylocom 4.2;
the ML distance from N. nucleus computed with RAxML specifying the model as above.
The factor analysis was carried out using the psych (Revelle 2014) package of R; the plot was prepared using the ggplot2 package. Normalization and varimax rotation were used, factor scores were found using correlation preserving, and correlations were found using the Pearson method; given the possible presence of a missing value (as T. granosa was not included in the phylogeny and we were therefore unable to compute either the root-to-tip and the ML distance), missing data were set to be imputed using the median. All the variables were pooled together for each species into the value of a single loading: we define this score as the HERMES score of a given species.
The best-performing variable set and the goodness-of-fit of the analysis was assessed following the recommendations of Hu and Bentler (1999): Tucker–Lewis Index (TLI) (Tucker and Lewis 1973) > 0.95; root mean square of the residuals (SRMR) < 0.08; root mean squared error of approximation (RMSEA) < 0.06; moreover, the Kaiser–Meyer–Olkin index (KMO) (Kaiser 1970) was taken into account on this regard.
A Python script was written to compute the HERMES scores of a given assemblage of species, providing the GenBank annotation, gene alignments and a phylogenetic tree; this software can be downloaded from the GitHub repository at the URL https://github.com/mozoo/HERMES.git, along with sample data and a tutorial.
Results
Overall Genomic Features
The 100 mitochondrial genomes (mtDNAs) of the present study range from 14,622 (Lanternula elliptica) to 46,985 (Scapharca broughtonii) bp in length. Excluding the abnormally large genomes of Arcidae, the longest mtDNA is that of Placopecten magellanicus (32,115), followed by the female genome of Venerupis philippinarum (22,676 bp). Conversely, the proportion of putatively Unstranslated Regions (URs) span from 1.44% (L. elliptica) and 2.13% (S. velum) to the high scores of Pectinidae (29.86–51.04%) and Arcidae (52.30–70.76%). The number of annotated genes on the molecule is much more stable: exception made for Scapharca kagoshimensis and S. broughtonii (55 and 54, respectively), this number span from 30 (Mizuhopecten yessoensis) to 46 (P. magellanicus), with a mean value of 38.11 ± 3.20.
As summarized in figure 1, mtDNA length and %UR increases in the order Palaeoheterodonta < Pteriomorphia < Heterodonta. Palaeoheterodonta are in both cases significantly different from either Pteriomorphia or Heterodonta (P < 0.001***; supplementary file S6, Supplementary Material online). The Kruskal–Wallis test detected a significant location difference also for the number of annotated genes, which is due to the Pteriomorphia/Heterodonta comparison only (P < 0.05*; supplementary file S6, Supplementary Material online). Finally, each subclass was significantly different from each other when considering A-T and G-C skews—again, comparisons involving Palaeoheterodonta showed the highest significance values (P < 0.001***; supplementary file S6, Supplementary Material online). Single-species data used in this study are extensively listed in supplementary file S5, Supplementary Material online.
Fig. 1.—
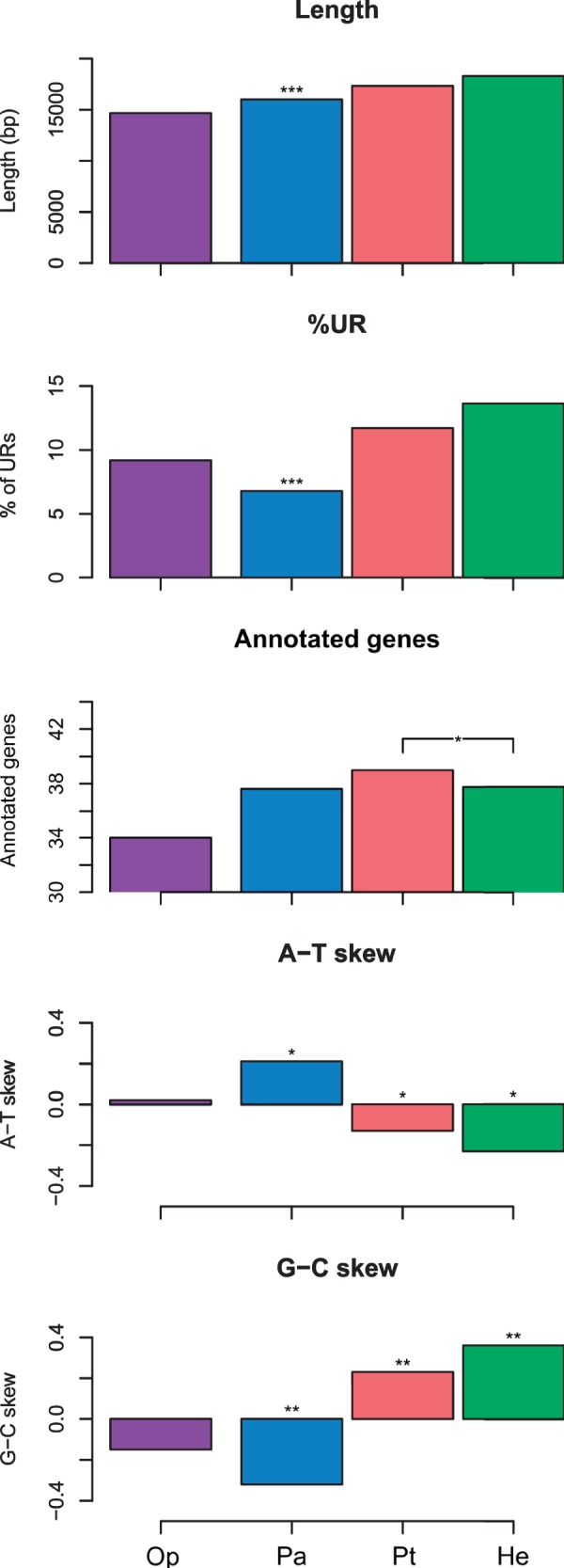
Main features of mitochondrial genomes. Five large-scale mitochondrial features are compared across four bivalvian subclasses (the fifth subclass, Anomalodesmata, was excluded because only one mitogenome was available): Op, Opponobranchia (purple); Pa, Palaeoheterodonta (blue); Pt, Pteriomorphia (red); He, Heterodonta (green). Length, median length of the genome; %UR, median percentage of Untranslated Regions; Annotated genes, mean number of annotated genes; A-T skew, median A-T skew; G-C skew, median G-C skew. Differences between subclasses were tested using the Krukal–Wallis and the Dunn's test; Anomaldesmata and Opponobranchia were excluded because of low sample size (N = 1 and N = 2, respectively). Asterisks above columns refer to levels of significance in both pairwise comparisons or in a single one (bracketed); *P < 0.05; **P < 0.01, ***P < 0.001.
Notwithstanding the large variability, especially in length and %UR, the genome content is quite stable: if we do not take tRNAs into account, all the mtDNAs share the same gene content and few duplication events are present. The most evident duplication is that of the rrnS gene in all species of the genus Crassostrea (with the exception of C. virginica), whose genes show a divergence between 0.11% (C. iredalei) and 5.05% (C. ariakensis), as estimated using uncorrected p-distance (mean = 2.92%). Moreover, as already signaled (Milbury and Gaffney 2005; Ren et al. 2010), in all Ostreidae species the rrnL gene is split in two separate fragments. Finally, a duplication of cox2 was already signaled in Musculista senhousia, male type (Passamonti et al. 2011), and V. philippinarum, female type (GenBank Accession Number AB065375).
Alignments
Protein Coding Genes (PCGs) alignment lengths range from 186 amino acids of nad3 to 1,313 amino acids of cox2 (before masking). The amount and percentage of sites selected by at least 2, 3, or 4 softwares is detailed in supplementary file S7, Supplementary Material online. The use of three masking softwares out of four is a good compromise between the elimination of noise and the elimination of apparently noisy useful sites, therefore this was taken as our preferred setting. All alignments are available from FP upon request.
The masking step affected different genes differently: cox1 and cytb are the genes that were least affected during masking phase (68.63% and 60.13% of their sites were kept, respectively), while cox2, nad4L, and nad6 alignments were heavily reduced after this phase (to the 16.45%, 18.32%, and 21.73% of the original size, respectively). As expected, rRNA alignments are much longer (4,210 and 2,891 bp for rrnL and rrnS) and much more shortened after the masking phase (to the 14.06% and 11.45% of the original size, respectively).
The amount of shortening was not reduced when smaller, more-related subsets were analyzed (supplementary file S8, Supplementary Material online). Differences in the percentages of kept sites among different subsets were never significantly higher when moving towards a smaller, nested taxon, but rather in some cases they were significantly lower (Bivalvia > Pteriomorphia, P < 0.001***; Palaeoheterodonta > Unionidae, P < 0.05*; Pteriomorphia > Pectinidae, P < 0.01**).
Actually, in all the cases, phylogenetically-informative sites are almost the same, being the dataset a single family, a subclass, or the whole class (fig. 2). The longest conserved domains are those of cox1 and cytb, even if also nad genes (with the exception of nad4L and nad6) show extended well-aligned regions. Interestingly, nad4 is the only case of a conserved domain that appears with reduced and less variable alignments: about 120 amino acids at the N-terminus of the protein are not conserved when the complete dataset is considered, but are increasingly more conserved at the subclass and family level (fig. 2). The similarity measure of plotcon over a sliding window show highest values for the most conserved regions, as expected (supplementary files S9 and S10, Supplementary Material online).
Fig. 2.—

Conserved sites of protein coding genes. Single genes were aligned, masked using four different approaches, and finally only sites selected as phylogenetically informative by at least three softwares were kept. Whenever possible in terms of sample size, the same procedure was carried out for reduced datasets, corresponding to single subclasses (3 datasets) and single families (5 families). Each chart shows the proportion of datasets keeping single sites: this is 0 (discarded site) or 100 (kept site) at the class level, and ranges from 0 to 100 at the subclass and family level, because more datasets are available.
Uncorrected (p-) distances are slightly, but significantly (P = 0), higher for amino acids than for nucleotides; however, when a more complex model is used to account for multiple substitution events at a single site, the situation is the opposite, and nucleotide distances are significantly (P = 0) higher than amino acid ones (fig. 3). Moreover, with the exception of the low-scoring cox1, all uncorrected distances are comparable among genes, while using the Jin–Nei/Kimura method (for nucleotides/amino acids, respectively), some genes (namely, atp6, nad2, nad4L, and nad6) have higher distance values than others (fig. 3). A similar scenario is retrieved through saturation plots. The uncorrected distance was plotted on Jin–Nei/Kimura distance: while in many cases uncorrected distances tend to increase with the other model, the same four genes show a plateau, indicating that p-distance is not able to uncover multiple hits and that a significant degree of saturation is present. As an example, cytb and nad6 plots are shown in figure 4, while all genes are detailed in supplementary file S11, Supplementary Material online. Expectedly, aminocid (aa) plot is farther from the plateau than nucleotide (nt) plot in all cases.
Fig. 3.—

Nucleotide and amino acid genetic distances. The mean genetic distance across the complete dataset ± one standard deviation is shown for each PCG; brown, nucleotides; green, amino acids. Differences in distance distributions were tested using the Kolmogorov–Smirnov test; ***P < 0.001.
Fig. 4.—

Example of four saturation plots. Pairwise p-distance is plotted over Jin–Nei/Kimura distance for nucleotides (above)/amino acids (below), respectively; nt, nucleotides; aa, amino acids. All saturation plots are available as supplementary file S11, Supplementary Material online.
A PCA was carried out using pairwise nucleotide and amino acids distances as variables for each gene; the result is shown in figure 5. Taken together, the first two principal components explain the 85.64% of the variance, but it has to be noted that the first component alone explains the 77.62%. The PCA (and specifically the first component) clearly separates atp6, nad2, nad4L, and nad6 from other genes.
Fig. 5.—

Distance PCA. Single-gene pairwise distance matrices were used as input data in order to investigate differences between PCGs.
Finally, the dN/dS ratios for each gene are given in table 1. In all cases, the null hypothesis that a single dN/dS applies to all the tree branches was not rejected by the LRT (P = 1); the dN/dS value computed along the entire tree is one or two orders of magnitude higher than the median of pairwise comparisons (table 1). The highest values are shown by nad6 (dN/dS = 0.344; median of pairwise comparisons = 0.045), while the lowest are shown by cox1 (dN/dS = 0.080; median of pairwise comparisons = 0.004). However, namely in atp6, nad4L, and nad6, a quite large number of high (i.e., greater than 10) pairwise dN/dS values were computed (table 1). In the vast majority of cases, these values expectedly come from pairs of distantly related species.
Table 1.
Values of dN/dS
| Genes | dN/dS | Pairwise median | dN/dS > 10 |
|---|---|---|---|
| atp6 | 0.255 | 0.023 ± 6.030 | 41 |
| cox1 | 0.080 | 0.004 ± 1.436 | 1 |
| cox2 | 0.222 | 0.008 ± 1.436 | 1 |
| cox3 | 0.155 | 0.009 ± 0.027 | 0 |
| cytb | 0.164 | 0.008 ± 0.017 | 0 |
| nad1 | 0.162 | 0.009 ± 0.026 | 0 |
| nad2 | 0.310 | 0.019 ± 2.949 | 9 |
| nad3 | 0.216 | 0.009 ± 1.461 | 2 |
| nad4 | 0.230 | 0.010 ± 0.022 | 0 |
| nad4L | 0.312 | 0.032 ± 8.441 | 67 |
| nad5 | 0.252 | 0.008 ± 0.020 | 0 |
| nad6 | 0.344 | 0.045 ± 11.223 | 103 |
dN/dS, value of dN/dS computed by PAML using the best-known likelihood tree (see text for details) as the given phylogeny; pairwise median, median of all pairwise comparisons ± standard deviation; dN/dS > 10, number of pairwise dN/dS ratios greater than 10.
Phylogenetic Analysis
The two subsets of markers (i.e., genes that are on the “+” strand and genes that are on the “−” strand of Unionoidea) yielded the same topology in the preliminary analyses, therefore the final tree was computed on the complete set of genes. The best partitioning scheme selected by PartitionFinder separated atp6 + nad genes, cytochrome subunit genes, and rRNAs (which were taken together; see supplementary file S12, Supplementary Material online, for details).
The best-known likelihood tree annotated with support values is shown in figure 6 over a geological timescale, while original phylograms are shown in supplementary files S13, Supplementary Material online (best-known likelihood tree with bootstrap proportions) and 14 (Bayesian consensus tree). Both Bootstrap Proportions (BPs) and Bayesian Posterior Probabilities (PPs) are generally high: as BP is typically lower than PP, we conservatively show the consensus tree after collapsing nodes with BP > 70. After the separation of the outgroup S. velum, the tree is divided into two major branches: Palaeoheterodonta (BP = 100, PP = 1.00) and Amarsipobranchia sensu Plazzi et al. (2011) (BP = 97, PP = 1.00). Within Palaeoheterodonta, M and F genomes (if non-DUI species genomes are considered as F ones) cluster separately, both with BP = 100/PP = 1.00; the branching order is also the same. Within Amarsipobranchia, relationships between L. elliptica (Anomalodesmata), Pteriomoprhia (BP = 91, PP = 1.00), and Heterodonta (BP = 73, PP = 1.00) are not resolved. Within Pteriomorphia, Mytilidae (BP = 100, PP = 1.00) are the sister group of all remaining pteriomorphians (BP = 79, PP = 1.00). With the exception of M. californianus M, DUI genomes of Mytilus spp. cluster by sex, as in Palaeoheterodonta, while M. senhousia genomes cluster by species. Within Heterodonta, Lucinindae (Loripes lacteus + Lucinella divaricata, BP = 100, PP = 1.00) are the sister group of all remaining heterodonts (BP = 100, PP = 1.00). All heterodont DUI genomes (i.e., M. lamarckii and V. philippinarum) cluster by species.
Fig. 6.—

The mitochondrial phylogeny of bivalves. Ultrametric tree of 98 bivalve species computed using r8s starting from the best-known likelihood tree computed with RAxML and the consensus Bayesian tree. RAxML bootstrap values/MrBayes posterior probabilities are printed at each node (an asterisk means a bootstrap of 100 and a posterior probability of 1.00). Below the tree the phylogenetic informativeness for each PCG is shown. The geological timescale in Mya follows Cohen et al. (2013).
The main split of the class (Palaeoheterodonta, one side, and Amarsipobranchia, the other side) took place about 500 million years ago (Mya), with a confidence interval (CI) of about 4.5 million years (My). The origin of pteriomorphians is dated at 484 mya (CI = 6 My), while the origin of heterodonts is dated slightly later, 454 mya (CI = 16 My). Oldest families are those of mytilids (421 Mya), pectinids (384 Mya), and venerids (336 Mya). All details about time calibration, including node names, mean estimates across bootstrap replicates, and CIs are shown in supplementary files S15 and S16, Supplementary Material online.
Different genes have different Phylogenetic Informativeness (PI) scores for different periods (fig. 6). Most nad genes and the cytb gene reach their peak in the Cenozoic, while cox genes and atp6 reach it in the Cretaceous. Notably, the peak of cox1 informativeness is shifted back to the Jurassic-Cretaceous limit and decreases slowly towards the deeper past. In noise vs. signal analysis (supplementary file S17, Supplementary Material online) cox1, cytb, nad1, and nad5 typically outperformed all the other markers, although the complete dataset always shows much higher values than single genes.
The AR and substitution rates (supplementary file S18, Supplementary Material online) showed a good correlation when six clades were used, corresponding to the major clades of the tree (Palaeoheterodonta F, Palaeoheterodonta M, Mytilidae, other Pteriomorphia, Lucinidae, other Heterodonta). The value of Spearman's rho was 0.90 (P < 0.01**) and that of Kendall's tau was 0.83 (P < 0.05*).
Annotation of the atp8 Gene and of ORFans
Twenty atp8 genes were annotated using the HHblits approach; they are listed in table 2. All the newly annotated atp8 had a homology probability > 95%, E-value < 0.01, and P-value < 1 × 10−6. The only exceptions to these are the atp8s in Paphia euglypta (Prob. = 92.7%, E-value = 0.071, P-value = 4.8 × 10−6), Acanthocardia tuberculata (Prob. = 86.6%, E-value = 0.46, P-value = 3.1 × 10−5), and Musculista senhousia M (Prob. = 80.6%, E-value = 0.74, P-value = 5.0 × 10−5): as the atp8 gene was not annotated in these species, it is highly probable that the homology is correct, but they anyway should be regarded as only tentatively annotated; the complete list of HHblits scores is available as supplementary file S19, Supplementary Material online. The annotation of atp8 led us to make small changes to the original GenBank annotations; these, along with other corrections of minor flaws that we detected during the analyses, are listed in supplementary file S20, Supplementary Material online, where the complete, updated annotations of all the present mtDNAs are given.
Table 2.
Newly annotated atp8 genes
| Species | Start | Stop | Length | aa |
|---|---|---|---|---|
| Mytilus galloprovincialis M | 8,530 | 8,871 | 345 | 115 |
| Mytilus edulis M | 9,789 | 10,133 | 345 | 115 |
| Mytilus californianus F | 8,735 | 9,037 | 303 | 101 |
| Mytilus galloprovincialis F | 8,802 | 9,062 | 264 | 88 |
| Mytilus edulis F | 10,396 | 10,656 | 264 | 88 |
| Mytilus californianus M | 8,262 | 8,570 | 312 | 104 |
| Ruditapes philippinarum M | 4,630 | 4,752 | 126 | 42 |
| Arctica islandica | 10,343 | 10,493 | 151 | 50 |
| Semele scabra | 11,969 | 12,097 | 129 | 43 |
| Solecurtus divaricata | 11,321 | 11,452 | 135 | 45 |
| Nuttallia olivacea | 12,930 | 13,058 | 132 | 44 |
| Soletellina diphos | 11,214 | 11,342 | 132 | 44 |
| Mimachlamys nobilis | 7,937 | 8,086 | 153 | 51 |
| Moerella iredescens | 11,625 | 11,753 | 132 | 44 |
| Meretrix petechialis | 8,532 | 8,669 | 141 | 47 |
| Meretrix meretrix | 8,532 | 8,669 | 141 | 47 |
| Ruditapes philippinarum F | 5,968 | 6,084 | 120 | 40 |
| Paphia euglypta | 12,994 | 13,107 | 117 | 39 |
| Acanthocardia tuberculata | 12,546 | 12,648 | 103 | 34 |
| Musculista senhousia M | 7,403 | 7,591 | 192 | 64 |
Start and stop refer to the complete mitochondrial genome as referenced in supplementary file S1, Supplementary Material online; length, nucleotide length without stop codon; aa, amino acid length. All newly annotated atp8 are on the “+” strand; ORFs are listed in order of hit, as in supplementary file S19, Supplementary Material online.
Conversely, ORFans seem to be poorly connectible across the entire class: the complete list of HHblits clusters of homologs shows that any given ORFan shows sharp similarities only with ORFans of strictly related species (supplementary file S21, Supplementary Material online). Indeed, ORFans from Unionidae are phylogenetically clumped in the same clusters that appear in the tree (fig. 6) and, notably, F and M ORFans are never intermingled. The same apply for F ORFans of mytilids, but not for M ORFans, whose homology scores are always unsignificant for all entries of the database. The only exception to this is given by Musculista senhousia. Three ORFans were described on the F mtDNA of this species (Guerra et al. 2014): while no significant hits were retrieved for one of these, the other two and the single M ORFan are clearly homolog (supplementary file S21, Supplementary Material online; see also Guerra et al. 2014). Finally, S. velum ORFans did not show significant homologies with other ORFans.
Summarizing Data: PCA and HERMES Score
All bivalve mitogenomic features are listed in supplementary file S5, Supplementary Material online. The resulting PCA is shown in figure 7; the first two principal components account for the 66.61% of the dataset variability. In the PCA plot, larger taxonomic assemblages are easy identified: Palaeoheterodonta on one side and the wide cluster of Amarsipobranchia on the other side. Most families create also a cluster, like Pectinidae, Ostreidae, and Margaritiferidae. Contrastingly, some points are sharply separated from others: N. nucleus, S. velum, Scapharca spp., P. magellanicus, and L. elliptica.
Fig. 7.—

Bivalve mitogenomics PCA. Input data are shown in supplementary file S5, Supplementary Material online (see text for details). Different subclasses are indicated with different shades: shades of blue, Palaeoheterodonta; shades of violet, Opponobranchia; shades of red, Pteriomorphia; shades of green, Heterodonta + Anomalodesmata; species of the same family are indicated with the same color.
The best-performing variable set to compute the HERMES score was the following:
percentage of URs;
absolute value of SU skew;
AMIGA;
root-to-tip distance; and
ML distance from N. nucleus.
For example, when inserting also the AT content, the goodness-of-fit parameters become only slightly better, but AT content was given a communality of 0.25%; therefore, we may conclude that this variable is not highly linked with the other five and thus it not significant in quantifying molecular evolution of mitochondrial genomes.
The HERMES factor analysis index (fig. 8) shows good levels of correlation between the selected variables: TLI = 0.965, SRMR = 0.061, RMSEA 95% CI = 0.025-0.233, KMO = 0.764, which are all within boundaries suggested by Hu and Bentler (1999). The lowest communality was scored by %UR (13.20%), while the highest was scored by the ML distance from N. nucleus (98.66%); the mean communality of the model was 60.62%, meaning that the HERMES index accounts for the 60.62% of the total variability of the source matrix.
Fig. 8.—

The HERMES index. To highlight HERMES differences between subclasses, species are horizontally listed by subclass and then in alphabetical order.
Discussion
To the best of our knowledge, the present study is the first overall and detailed appraisal to the mitogenomics of bivalve mollusks. Many similar studies have been published in the past on similar topics, but generally they focused on a single family, like Mytilidae (Breton et al. 2006), Unionidae (Breton et al. 2009), Pectinidae (Wu et al. 2009), or on the DUI phenomenon (Doucet-Beaupré et al. 2010). As stated in the introduction, however, large comparative studies are essential to understand the main pathways followed by the evolution of mitochondrial genomes.
At the class level, we decided to especially concentrate on PCGs, as annotations are more trustworthy, homology is certain, and the same set of genes is present in all genomes under study. Actually, we find only two duplication events (cox2 in M. senhousia M and V. philippinarum F) and we were able to detect the atp8 gene in 20 species where this gene was originally described as missing (table 2): some of these atp8 genes were already signaled (Stöger and Schrödl 2013), namely those of A. tuberculata, H. arctica, and V. philippinarum (Dreyer and Steiner 2006); Mytilus spp. (Breton et al. 2010; Smietanka et al. 2010); Unionidae (Doucet-Beaupré et al. 2010). After our analysis, 71 species out of 100 have an annotated atp8. The nucleotide/amino acid sequence of this gene is scarcely conserved, and this probably led to annotation flaws, as already stated (Plazzi et al. 2013; Zouros 2013; Bettinazzi et al. 2016; Gaitán-Espitia et al. 2016). Structural analyses are needed to recover some homology with known atp8, and we may suggest the importance of such analyses in other groups where atp8 is reported as missing, like nematodes (Okimoto et al. 1992), platyhelminths (Le et al. 2000), and chaetognaths (Boore et al. 2004).
Contrastingly, the GenBank annotation of rRNAs is still commonly obtained by the boundaries of the upstream and downstream genes, despite accurate ongoing work of re-annotation (Bernt et al. 2013; Stöger and Schrödl 2013). The use of these genes is therefore problematic for analyses like nucleotide composition, skews, or biases. However, phylogenetic methods should be robust enough to overcome this issue (but a masking phase is required): for this reason, we decided to insert rRNAs in the phylogenetic analysis anyway.
Finally, tRNAs are extremely prone to gene rearrangement (Boore 1999; Vallès and Boore 2006; Xu et al. 2006; Gissi et al. 2008) and recruitment (Lavrov and Lang 2005; Wu, Li, Li, Xu, et al. 2012; Wu, Li, Li, Yu, et al. 2012), and therefore they have to be assessed at a lower taxonomical level (Wu et al. 2014).
PCGs in Bivalve Mitochondrial Genomes
The distance analyses reveal a high degree of divergence for single genes. p-Distance, which is an underestimation of the true distance in that it does not account for multiple substitution events, is generally comprised between 40% and 60% (fig. 3). However, when the correct model of molecular evolution is used, it is clear that most of this variability is found in synonymous mutations (fig. 3), which is clearly demonstrated by the low values of dN/dS (table 1).
Pairwise dN/dS have very low medians, but a very high standard deviations, probably due to high dN/dS computed for very distantly related species; therefore, we consider as the best estimation of dN/dS in our dataset the value computed along the phylogenetic tree, which is comprised between 0.080 (cox1) and 0.344 (nad6; table 1). Such a pattern of overall negative selection was already detected for the species of genus Mytilus (Zbawicka et al. 2014; Gaitán-Espitia et al. 2016).
However, conservation and variability should not be assessed at the general gene level, because our results clearly indicate a sharp contrast between strongly conserved domains and highly variable regions. Those domains are shown in figure 2 (see also supplementary files S9 and S10, Supplementary Material online). Concluding, some domains of the mitochondrial PCGs are currently under severe purifying selection in bivalves, while, in most cases, variability is due to indel events typical of some species (see, e.g., the cox2 gene).
If the general pattern is the conservation of specific domains in each PCG, some genes seem to follow different evolutionary pathways. In most analyses, atp6, nad2, nad4L, and nad6 behave differently from other PCGs: they are more heavily affected by masking phase (supplementary file S7, Supplementary Material online), show higher dN/dS values (table 1), are heavily saturated (fig. 4 and supplementary file S11, Supplementary Material online), and are much more variable (fig. 3). In a nutshell, these genes are driven by different evolutionary constraints with respect to the others (fig. 5); although the second principal component accounts for only the 8.02% of the variability, this may indicate that, furthermore, each of these genes follows its own evolutionary pathway.
History of Bivalve Mitochondria
The present phylogeny (fig. 6) corroborates a view of bivalve evolution where most extant families were essentially already present in the Lower Ordovician (Mondal and Harries 2016b). Expectedly, the mitogenomic differentiation of the main clades of extant bivalves predates the paleontological evidences of the well-known Ordovician bivalve radiation (Cope 1996; Fang 2006; Sánchez 2008; Fang and Sánchez 2012; Polechová 2015; Mondal and Harries 2016b). Conversely, the root of most families is placed after the Ordovician, probably because of limited taxon sampling: the same would hold for Palaeoheterodonta, but they appear to be much more recent (Early Triassic) since only members of the superfamily Unionoidea were actually inserted in this phylogenetic analysis.
The topology of our phylogenetic tree is in perfect agreement with our previous results (Plazzi and Passamonti 2010; Plazzi et al. 2011). It is particularly noteworthy the presence of the Amarsipobranchia clade, i.e., Pteriomorphia + Heterodonta. Our previous analyses were based on four mitochondrial genes (cox1, cytb, rrnL, and rrnS), and the use of the complete mitochondrial gene array led to the same result, even with a larger sample. Furthermore, the signal vs. noise analysis indicates that the phylogenetic signal of all genes is suitable (supplementary file S17, Supplementary Material online), and it becomes even stronger for the complete, concatenated dataset. Indeed, the same Amarsipobranchia clade was also retrieved by other studies using mitochondrial markers (Giribet and Distel 2003; Doucet-Beaupré et al. 2010; Stöger and Schrödl 2013).
On the other side, the use of morphological characters leads generally to the Heteroconchia sensu Waller (1998) clade (i.e., Palaeoheterodonta + Heterodonta; Giribet and Distel 2003; Bieler et al. 2014)—however, some morphological analyses retrieved instead the Amarsipobranchia clade (Cope 1996). Recent analyses based on nuclear genes and transcriptomes (Kocot et al. 2011; Smith et al. 2011; Sharma et al. 2012; González et al. 2015) also recovered the Heteroconchia clade.
Thus, the discrepancy between phylogenies based on morphology/nuclear genes vs. mitochondrial genes still holds. The amount of data in mitochondrial genomes is clearly restricted with respect to nuclear genomes. It is also possible that specific mitochondrial features may lead to a wrong relationship between heterodonts and pteriomorphians, thus disrupting Heteroconchia. Nucleotide composition may be one of these features: Amarsipobranchia have all negative A-T and positive G-C skews, while the situation is reversed for Palaeoheterodonta (supplementary file S5, Supplementary Material online). Another feature which is correlated with nucleotide composition is the substitution rate (Kowalczuk et al. 2001; Siepel and Haussler 2004; Gowri-Shankar and Rattray 2006; Hobolth et al. 2006), which we show is linked with AR rate (supplementary file S18, Supplementary Material online), as already hypothesized by Stöger and Schrödl (2013).
However, our HERMES factor analysis demonstrates that the AT content practically does not explain other phylogenetic parameters like root-to-tip distance and ML distance from N. nucleus, and the protobranch Solemya velum show the same situation of Palaeoheterodonta, corroborating the idea that this is the plesiomorphic condition of bivalves. The same can be seen from the general PCA (fig. 7): Amarsipobranchia and Palaeoheterodonta are separated, many features are considered (not only nucleotide composition), and Opponobranchia are shifted towards Palaeoheterodonta.
Conclusions
The HERMES Index: Tempo and Mode of Mitochondrial Evolution
The use of a single score to quantify mitochondrial evolution is a complex task, as many different parameters (that, furthermore, are linked together to different extents) can be considered and have to be summarized into a single number. High goodness-of-fit test results (given the complexity of the dataset) show that HERMES (fig. 8) is indeed a suitable measure of this evolution. We are planning to apply the HERMES index in other taxa, and this will lead us to the identification of taxon-specific mitochondrial features that are suitable to measure molecular evolution; a Python script was written and made publicly available for this purpose (available at https://github.com/mozoo/HERMES.git).
Concerning bivalves, there is consistence between all the aforementioned data and the HERMES measure: S. velum is the slowest-evolving mitogenome, followed by those of Palaeoheterodonta. Amarsipobranchia have similar HERMES scores, typically higher than Palaeoheterodonta; finally, the position of L. elliptica is intriguing and deserves further investigation. Within Palaeoheterodonta, F genomes have HERMES scores that are sharply lower than the M counterparts. The very early onset of DUI in Palaeoheterodonta led to the highest inter-sex distance values for DUI species (Bettinazzi et al. 2016) and to the well-known gender-joining pattern in phylogenetic trees (Curole and Kocher 2005; Doucet-Beaupré et al. 2010; Zouros 2013; fig. 6). The divergence of the two lineages started at least from the origin of Unionidae, ∼250 Mya (Tillyard and Dunstan 1916; Cromptok and Parrington 1955; Drysdall and Kitching 1963; Nesbitt et al. 2010; fig. 6; supplementary file S16, Supplementary Material online): F and M genomes are separately evolving since then. Recall that the organization of the ancestral bivalve mitochondrial genome should have resembled that of Solemya and Nucula (Plazzi et al. 2013), we can conclude that, while somehow the F mtDNAs of unionids retain much of this original condition, the M mtDNAs seem to have diverged more quickly on their own. DUI in other bivalve lineages appears to be a much more recent phenomenon (fig. 6), or masked by masculinization role reversals (Hoeh et al. 1997; Quesada et al. 1999; Zouros 2013), therefore the gender-joining pattern of unionids remains essentially unique across the phylogeny of the entire class.
It is tempting to explore links between the molecular evolution of bivalve mitochondrial genomes as depicted by the HERMES score (and the mitogenomic feature-based PCA as well; fig. 7) and the fossil evidences of the class. First known bivalves originated in the Early-Middle Cambrian and slowly faded away during the Late Cambrian; no Cambrian species are known from the Ordovician (Fang 2006; Sánchez 2008; Fang and Sánchez 2012; Cope and Kříž 2013; Polechová 2015; Mondal and Harries 2016a).
However, some of them gave rise to extant bivalve clades in the Ordovician period (Cope 2002; Sánchez 2008; Polechová 2015). Protobranch forms were the first branching clade (Morton 1996; Cope and Babin 1999; Kocot et al. 2011; Plazzi et al. 2011; Smith et al. 2011; Fang and Sánchez 2012; Sharma et al. 2012; Bieler et al. 2014; González et al. 2015; and reference therein) before the evolution of the true feeding gill of all remaining autobranchs. The strict similarity of the mitochondrial genome of Solemya (and Nucula as well) with some gastropods (Plazzi et al. 2013) strengthens this hypothesis and allows to set a direction in the evolution of bivalve mtDNAs after the original appearance of extant bivalves; the HERMES index detects two main phases of this evolution (fig. 8).
According to the HERMES pattern, a first phase was the split of palaeoheterodonts from Amarsipobranchia. Again, fossil data may shed light on this issue. The most ancient families of Ordovician bivalves (dating to the Lower Ordovician, upper Tremadoc, ∼480 Mya) are Ucumariidae, Modiolopsidae (Goniophorinidae), and Lipanellidae (Sánchez 2006; Fang and Sánchez 2012): following current revised bivalve systematics, they should be classified within Heterodonta, Pteriomorphia, and Heterodonta, respectively (Carter et al. 2011).
However, the state-of-art phylogenetic reconstruction is Lipanellidae + (Modiolopsidae + Ucumariidae) (Sánchez 2006, p. 117; Fang and Sánchez 2012, p. 12), and Heterodonta should be therefore considered as polyphyletic at their root. Moreover, Ucumariidae are interpreted as connected to extant anamalodesmatans (Sánchez 2006; Fang and Sánchez 2012), which are currently considered as nested within heterodonts (Giribet and Wheeler 2002; Dreyer et al. 2003; Giribet and Distel 2003; Harper et al. 2006; Taylor et al. 2007, 2009; Sharma et al. 2012; Bieler et al. 2014; González et al. 2015).
Indeed, our phylogenetic reconstruction and the Amarsipobranchia hypothesis retrieve the same clade; namely, in the present phylogenetic tree it exhibits a basal tritomy (Heterodonta + Pteriomorphia + Anomalodesmata; fig. 6). Furthermore, it is worth recalling the fossil record of the family Thoraliidae, which is also known from the Lower Ordovician (Morris 1980; Sánchez and Babin 2003; Cope and Kříž 2013) and it is currently classified within Palaeoheterodonta (Carter et al. 2011). Thus, in the Lower Ordovician a monophyletic Amarsipobranchia-like clade is hypothesized, while the first palaeoheterodonts have been found from the same epoch: concluding, phylogenetic paleontological reconstructions are not discordant with our molecular phylogentic tree or with the HERMES pattern.
Eventually, the second phase of bivalve mitochondrial evolution as depicted by the HERMES score is the diversification of Amarsipobranchia, which definitely lost most plesiomorphic mitogenomic features (fig. 7): all genes, with rarest exceptions, migrated on the same coding strand; an increase in length was coupled to an increase of the genomic regions not assigned to canonical genes; there was the inversion in A-T and G-C skews (fig. 1; supplementary file S5, Supplementary Material online); most strikingly, the gene rearrangement rate was given an unprecedented boost (supplementary file S18, Supplementary Material online).
Supplementary Material
Supplementary file S1–S21 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
We would like to thank Fabrizio Ghiselli, Liliana Milani, Stefano Bettinazzi, and Mariangela Iannello for suggestions and stimulating discussion. Thanks are also due to Davide Guerra for his unvaluable guide to the taming of R and to Liliana Silva for having introduced us to the basics of factor analysis. The original manuscript was greatly improved by comments and suggestions by two anonymous reviewers. This work was financed by the “Canziani Bequest” fund (University of Bologna, grant number A.31.CANZELSEW).
Literature Cited
- Abhishek A, Bavishi A, Bavishi A, Choudhary M. 2011. Bacterial genome chimaerism and the origin of mitochondria. Can J Microbiol. 57:49. 46. [DOI] [PubMed] [Google Scholar]
- Adams KL, Palmer JD. 2003. Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol. 29:380–395. [DOI] [PubMed] [Google Scholar]
- Allen JF. 2003a. The function of genomes in bioenergetic organelles. Philos Trans R Soc Lond B Biol Sci. 358:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JF. 2003b. Why chloroplasts and mitochondria contain genomes. Comp Funct Genomics 4:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG. 1998. Reductive evolution of resident genomes. Trends Microbiol. 6:263–268. [DOI] [PubMed] [Google Scholar]
- Atteia A, et al. 2009. A proteomic survey of Chlamydomonas reinhardtii mitochondria sheds new light on the metabolic plasticity of the organelle and on the nature of the alpha-proteobacterial mitochondrial ancestor. Mol Biol Evol. 26:1533–1548. [DOI] [PubMed] [Google Scholar]
- Baird GC, Brett CE. 1983. Regional variation and paleontology of two coral beds in the Middle Devonian Hamilton Group of Western New York. J Paleontol 57:417–446. [Google Scholar]
- Batista FM, Lallias D, Taris N, Guerdes-Pinto H, Beaumont AR. 2010. Relative quantification of the M and F mitochondrial DNA types in the blue mussel Mytilus edulis by real-time PCR. J Molluscan Stud 71:24–29. [Google Scholar]
- Bernt M, et al. 2013. A comprehensive analysis of metazoan mitochondrial genomes and animal phylogeny. Mol Phylogenet Evol. 69:352–364. [DOI] [PubMed] [Google Scholar]
- Bettinazzi S, Plazzi F, Passamonti M. 2016. The Complete Female- and Male-Transmitted Mitochondrial Genome of Meretrix lamarckii. PLoS One 11:e0153631.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieler R, et al. 2014. Investigating the Bivalve Tree of Life—an exemplar-based approach combining molecular and novel morphological characters. Invertebr Syst 28:32. 115. [Google Scholar]
- Boore JL, Medina M, Rosenberg LA. 2004. Complete sequences of the highly rearranged molluscan mitochondrial genomes of the scaphopod Graptacme eborea and the bivalve Mytilus edulis. Mol Biol Evol. 21:1492–1503. [DOI] [PubMed] [Google Scholar]
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27:1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier MD, Hewitt RA. 1978. On the late precambrian- early cambrian Hartshill formation of warwickshire. Geol Mag 115:21–36. [Google Scholar]
- Breton S, et al. 2009. Comparative mitochondrial genomics of freshwater mussels (Bivalvia: Unionoida) with doubly uniparental inheritance of mtDNA: gender-specific open reading frames and putative origins of replication. Genetics 183:1575–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Burger G, Stewart DT, Blier PU. 2006. Comparative analysis of gender-associated complete mitochondrial genomes in marine mussels (Mytilus spp.). Genetics 172:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton S, Doucet-Beaupré H, Stewart DT, Hoeh WR, Blier PU. 2007. The unusual system of doubly uniparental inheritance of mtDNA: isn't one enough? Trends Genet. 23:465–474. [DOI] [PubMed] [Google Scholar]
- Breton S, et al. 2014. A resourceful genome: updating the functional repertoire and evolutionary role of animal mitochondrial DNAs. Trends Genet. 30:555–564. [DOI] [PubMed] [Google Scholar]
- Breton S, Stewart DT, Hoeh WR. 2010. Characterization of a mitochondrial ORF from the gender-associated mtDNAs of Mytilus spp. (Bivalvia: Mytilidae): identification of the “missing” ATPase 8 gene. Mar Genomics 3:11–18. [DOI] [PubMed] [Google Scholar]
- Brett CE, Dick VB, Baird GC. 1991. Comparative taphonomy and paleoecology of middle Devonian dark gray and black shale facies from western New York. State Mus Bull. 469:5–36. [Google Scholar]
- Carter JG, et al. 2011. A Synoptical Classification of the Bivalvia (Mollusca). The University of Kansas Paleontological Contributions 4:1–47. [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, et al. 2006. Presence of a unique male-specific extension of C-terminus to the cytochrome c oxidase subunit II protein coded by the male-transmitted mitochondrial genome of Venustaconcha ellipsiformis (Bivalvia: Unionoidea). FEBS Lett. 580:862–866. [DOI] [PubMed] [Google Scholar]
- Chandel NS. 2014. Mitochondria as signaling organelles. BMC Biol. 12:34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charif D, Lobry JR. 2007. SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis In: Bastolla U, Porto M, Roman HE, Vendruscolo M, editors. Structural approaches to sequence evolution: Molecules, networks, populations. New York: Springer Verlag; p. 207–232. [Google Scholar]
- Claros MG, et al. 1995. Limitations to in vivo import of hydrophobic proteins into yeast mitochondria. The case of a cytoplasmically synthesized apocytochrome b. Eur J Biochem. 228:762–771. [PubMed] [Google Scholar]
- Cohen KM, Finney SC, Gibbard PL, Fan J-X. 2013. The ICS International Chronostratigraphic Chart. Episodes 36:199–204. [Google Scholar]
- Cope JCW, Babin C. 1999. Diversification of bivalves in the Ordovician. Geobios 32:175–185. [Google Scholar]
- Cope JCW, Kříž J. 2013. The Lower Palaeozoic palaeobiogeography of Bivalvia. Geol Soc Lond Mem 38:221–241. [Google Scholar]
- Cope JCW. 1996. The early evolution of the Bivalvia In: Taylor JD, editor. Origin and Evolutionary Radiation of the Mollusca. Oxford: Oxford University Press; p. 361–370. [Google Scholar]
- Cope JCW. 2002. Diversification and biogeography of bivalves during the Ordovician Period In: Crame JA, Owen AW, editors. Palaeobiogeography and Biodiversity Change: the Ordovician and Mesozoic-Cenozoic Radiations. London: Geological Society; p. 25–52. [Google Scholar]
- Criscuolo A, Gribaldo S. 2010. BMGE (Block Mapping and Gathering with Entropy): selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 10:210.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromptok AW, Parrington FR. 1955. On some Triassic cynodonts from Tanganyika. J Zool 125:617–669. [Google Scholar]
- Curole JP, Kocher TD. 2005. Evolution of a unique mitotype-specific protein-coding extension of the cytochrome c oxidase II gene in freshwater mussels (Bivalvia: Unionida). J Mol Evol. 61:381–389. [DOI] [PubMed] [Google Scholar]
- Daley DO, Clifton R, Whelan J. 2002. Intracellular gene transfer: reduced hydrophobicity facilitates gene transfer for subunit 2 of cytochrome c oxidase. Proc Natl Acad Sci U S A. 99:10510–10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti M, et al. 2014. Evolution of mitochondria reconstructed from the energy metabolism of living bacteria. PLoS One 9:e96566.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S. 2005. PROBCONS: probabilistic consistency-based multiple sequence alignment. Genome Res. 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet-Beaupré H, et al. 2010. Mitochondrial phylogenomics of the Bivalvia (Mollusca): searching for the origin and mitogenomic correlates of doubly uniparental inheritance of mtDNA. BMC Evol Biol. 10:50.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M. 2004. Assessing the relative rate of (mitochondrial) genomic change. Genetics 167:1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dress AWM, et al. 2008. Noisy: identification of problematic columns in multiple sequence alignments. Algorithm Mol Biol. 3:7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer H, Steiner G. 2006. The complete sequences and gene organization of the mitochondrial genomes of the heterodont bivalves Acanthocardia tubercolata and Hiatella arctica and the first record for a putative Atpase subunit 8 gene in marine bivalves. Front Zool 3:13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer H, Steiner G, Harper EM. 2003. Molecular phylogeny of Anomalodesmata (Mollusca: Bivalvia) inferred from 18S rRNA sequences. Zool J Linn Soc 139:229–246. [Google Scholar]
- Drysdall AR, Kitching JW. 1963. A re-examination of the Karroo succession and fossil localities of part of the Upper Luangwa Valley. Memoir of the Geological Survey of Northern Rhodesia 1:1–62. [Google Scholar]
- Dunn OJ. 1964. Multiple comparisons using rank sums. Technometrics 6:241–252. [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z-J, Sánchez TM. 2012. Part N, revised, volume 1, chapter 16: origin and early evolution of the Bivalvia. Treatise Online 43:1–21 [Google Scholar]
- Fang Z-J. 2006. An introduction to Ordovician bivalves of southern China, with a discussion of the early evolution of the Bivalvia. Geo J. 41:303–328. [Google Scholar]
- Feagin JE. 1994. The extrachromosomal DNAs of apicomplexan parasites. Annu Rev Microbiol. 48:81–104. [DOI] [PubMed] [Google Scholar]
- Fischer D, Eisenberg D. 1999. Finding families for genomic ORFans. Bioinformatics 15:759–762. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DA, Creevey CJ, McInerney JO. 2006. Genome phylogenies indicate a meaningful alpha-proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales. Mol Biol Evol. 23:74–85. [DOI] [PubMed] [Google Scholar]
- Funes S, et al. 2002. The typically mitochondrial DNA-encoded ATP6 subunit of the F1F0-ATPase is encoded by a nuclear gene in Chlamydomonas reinhardtii. J Biol Chem. 277:6051–6058. [DOI] [PubMed] [Google Scholar]
- Gaitán-Espitia JD, Quintero-Galvis JF, Mesas A, D'Elía G. 2016. Mitogenomics of southern hemisphere blue mussels (Bivalvia: Pteriomorphia): insights into the evolutionary characteristics of the Mytilus edulis complex. Sci Rep 6:26853.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Rubin DB. 1992. Inference from iterative simulation using multiple sequences. Stat Sci. 7:457–511. [Google Scholar]
- Ghiselli F, et al. 2013. Structure, transcription, and variability of metazoan mitochondrial genome: perspectives from an unusual mitochondrial inheritance system. Genome Biol Evol. 5:1535–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiselli F, Milani L, Passamonti M. 2011. Strict sex-specific mtDNA segregation in the germ line of the DUI species Venerupis philippinarum (Bivalvia: Veneridae). Mol Biol Evol. 28:949–961. [DOI] [PubMed] [Google Scholar]
- Giribet G, et al. 2006. Evidence for a clade composed of molluscs with serially repeated structures: monoplacophorans are related to chitons. Proc Natl Acad Sci U S A. 103:7723–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giribet G, Distel DL. 2003. Bivalve phylogeny and molecular data In: Lydeard C, Lindberg D, editors. Molecular Systematics and Phylogeography of Mollusks. Washington, DC: Smithsonian Institution Press; p. 45–90. [Google Scholar]
- Giribet G, Wheeler WC. 2002. On bivalve phylogeny: a high-level analysis of the Bivalvia (Mollusca) based on combined morphology and DNA sequence data. Invertebr Biol. 121:271–324. [Google Scholar]
- Gissi C, Iannelli F, Pesole G. 2008. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101:301–320. [DOI] [PubMed] [Google Scholar]
- González VL, et al. 2015. A phylogenetic backbone for Bivalvia: an RNA-seq approach. Proc R Soc B Biol Sci. 282:20142332.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowri-Shankar V, Rattray M. 2006. On the correlation between composition and site-specific evolutionary rate: implications for phylogenetic inference. Mol Biol Evol. 23:352–364. [DOI] [PubMed] [Google Scholar]
- Grasso TX. 1986. Redefinition, stratigraphy and depostional environments of the mottville member (hamilton group) in central and eastern New York. N Y State Mus Bull. 457:5–31. [Google Scholar]
- Gray MW, Burger G, Lang BF. 1999. Mitochondrial evolution. Science 283:1476–1481. [DOI] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. 2001. The origin and early evolution of mitochondria. Genome Biol. 2:1018.1–1018.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW. 2012. Mitochondrial evolution. Cold Spring Harb Perspect Biol. 4:a011403.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra D, Ghiselli F, Passamonti M. 2014. The largest unassigned regions of the male- and female-transmitted mitochondrial DNAs in Musculista senhousia (Bivalvia Mytilidae). Gene 536:316–325. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. [DOI] [PubMed] [Google Scholar]
- Harper EM, Dreyer H, Steiner G. 2006. Reconstructing the Anomalodesmata (Mollusca: Bivalvia): morphology and molecules. Zool J Linn Soc 148:395–420. [Google Scholar]
- Hobolth A, Nielsen R, Wang Y, Wu F, Tanksley SD. 2006. CpG + CpNpG analysis of protein-coding sequences from tomato. Mol Biol Evol. 23 1318–1323. [DOI] [PubMed] [Google Scholar]
- Hoeh WR, Stewart DT, Saavedra C, Sutherland BW, Zouros E. 1997. Phylogenetic evidence for role-reversals of gender-associated mitochondrial DNA genomes in Mytilus (Bivalvia: Mytilidae). Mol Biol Evol. 14:959–967. [DOI] [PubMed] [Google Scholar]
- Hu L-T, Bentler PM. 1999. Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Struct Equat Model Multidiscipl J. 6:1–55. [Google Scholar]
- Huson DH, Scornavacca C. 2012. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 61 1061–1067. [DOI] [PubMed] [Google Scholar]
- Hyndman RJ, Fan Y. 1996. Sample quantiles in statistical packages. Am Stat 50:361–365. [Google Scholar]
- Jin L, Nei M. 1990. Limitations of the evolutionary parsimony method of phylogenetic analysis. Mol Biol Evol. 7:82–102. [DOI] [PubMed] [Google Scholar]
- Jordan GE, Piel WH. 2008. PhyloWidget: web-based visualizations for the tree of life. Bioinformatics 24:1641–1642. [DOI] [PubMed] [Google Scholar]
- Jukes TH, Osawa S. 1990. The genetic code in mitochondria and chloroplasts. Experientia 46:1117–1126. [DOI] [PubMed] [Google Scholar]
- Kaiser HF. 1970. A second generation little jiffy. Psychometrika 35:401–415. [Google Scholar]
- Kannan S, Rogozin IB, Koonin EV. 2014. MitoCOGs: clusters of orthologous genes from mitochondria and implications for the evolution of eukaryotes. BMC Evol Biol. 14:237.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachane AN, Timmis KN, Martins dos Santos VA. 2007. Dynamics of reductive genome evolution in mitochondria and obligate intracellular microbes. Mol Biol Evol. 24:449–456. [DOI] [PubMed] [Google Scholar]
- Kimura M. 1983. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge University Press. [Google Scholar]
- Kocot KM, et al. 2011. Phylogenomics reveals deep molluscan relationships. Nature 477:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczuk M, et al. 2001. High correlation between the turnover of nucleotides under High correlation between the turnover of nucleotides under. BMC Evol Biol. 1:13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kříž J. 2008. A new bivalve community from the lower Ludlow of the Prague Basin (Perunica, Bohemia). Bull Geosci 83:237–280. [Google Scholar]
- Kruskal WH, Wallis A. 1952. Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47:583–621. [Google Scholar]
- Kyriakou E, Zouros E, Rodakis GC. 2010. The atypical presence of the paternal mitochondrial DNA in somatic tissues of male and female individuals of the blue mussel species Mytilus galloprovincialis. BMC Res Notes 3:222.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, Martin WF. 2012. The origin of membrane bioenergetics. Cell 151:1406–1416. [DOI] [PubMed] [Google Scholar]
- Lane N. 2007. Mitochondria: key to complexity In: Martin WF, Müller M, editors. Origin of Mitochondria and Hydrogenosomes. Berlin-Heidelberg: Springer-Verlag; p. 13–38. [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701. [DOI] [PubMed] [Google Scholar]
- Lavrov DV, Lang BF. 2005. Transfer RNA gene recruitment in mitochondrial DNA. Trends Genet. 21:129–133. [DOI] [PubMed] [Google Scholar]
- Lê S, Josse J, Husson F. 2008. FactoMineR: an R package for multivariate analysis. J Stat Softw 25:1–18. [Google Scholar]
- Le TH, et al. 2000. Phylogenies inferred from mitochondrial gene orders—a cautionary tale from the parasitic flatworms. Mol Biol Evol. 17:1123–1125. [DOI] [PubMed] [Google Scholar]
- Liu YG, Kurokawa T, Sekino M, Tanabe T, Watanabe K. 2013. Complete mitochondrial DNA sequence of the ark shell Scapharca broughtonii: an ultra-large metazoan mitochondrial genome. Comp Biochem Physiol D Genomics Proteomics 8:72–81. [DOI] [PubMed] [Google Scholar]
- López-Giráldez F, Townsend JP. 2011. PhyDesign: an online application for profiling phylogenetic informativeness. BMC Evol Biol. 11:152.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153:19994–21217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, et al. 2011. ViennaRNA Package 2.0. Algorithm Mol Biol. 6:26.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier UG, et al. 2013. Massively convergent evolution for ribosomal protein gene content in plastid and mitochondrial genomes. Genome Biol Evol. 5:2318–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Schnarrenberger C. 1997. The evolution of the Calvin cycle from prokaryotic to eukaryotic chromosomes: a case study of functional redundancy in ancient pathways through endosymbiosis. Curr Genet. 32:1–18. [DOI] [PubMed] [Google Scholar]
- M'Coy F. 1847. XXI. On the fossil botany and zoology of the rocks associated with the coal of Australia. Ann Mag Nat History 20:226–236. [Google Scholar]
- Meisinger C, Sickmann A, Pfanner N. 2008. The mitochondrial proteome: from inventory to function. Cell 134:22–24. [DOI] [PubMed] [Google Scholar]
- Mergl M, Massa D. 1992. Devonian and Lower Carboniferous brachiopods and bivalves from western Libya. Biostratigraphie Du Paleozoique 12:1–115. [Google Scholar]
- Milani L, Ghiselli F, Guerra D, Breton S, Passamonti M. 2013. A comparative analysis of Mitochondrial ORFans: new clues on their origin and role in species with Doubly Uniparental Inheritance. Genome Biol Evol. 5:1408–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbury CA, Gaffney PM. 2005. Complete mitochondrial DNA sequence of the eastern oyster Crassostrea virginica. Mar Biotechnol 7:697–712. [DOI] [PubMed] [Google Scholar]
- Misof B, Misof K. 2009. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst Biol. 58:21–34. [DOI] [PubMed] [Google Scholar]
- Mitchell P. 1961. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191:144–148. [DOI] [PubMed] [Google Scholar]
- Mondal S, Harries PJ. 2016a. Phanerozoic trends in ecospace utilization: the bivalve perspective. Earth Sci Rev 152:106–118. [Google Scholar]
- Mondal S, Harries PJ. 2016b. The effect of taxonomic corrections on phanerozoic generic richness trends in marine bivalves with a discussion on the Clade’s overall history. Paleobiology 42:157–171. [Google Scholar]
- Morris NJ. 1980. A new Lower Ordovician bivalve family, the Thoraliidae (? Nuculoida), intepreted as actinodont deposit feeders. Bull Br Mus Nat History (Geology) 34:265–272. [Google Scholar]
- Morton B. 1996. The evolutionary history of the Bivalvia In: Taylor JD, editor. Origin and Evolutionary Radiation of the Mollusca. Oxford: Oxford University Press; p. 337–359. [Google Scholar]
- Müller M, Martin W. 1999. The genome of Rickettsia prowazekii and some thoughts on the origin of mitochondria and hydrogenosomes. Bioessays 21:377–381. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Newell ND. 1968. Permian bivalves of Japan. Memoirs of the Faculty of Science, Kyoto University. Ser Geol Mineral 35:1–108. [Google Scholar]
- Nesbitt SJ, et al. 2010. Ecologically distinct dinosaurian sister group shows early diversification of Ornithodira. Nature 464:95–98. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 302:205–217. [DOI] [PubMed] [Google Scholar]
- Obata M, Sano N, Komaru A. 2011. Different transcriptional ratios of male and female transmitted mitochondrial DNA and tissue-specific expression patterns in the blue mussel, Mytilus galloprovincialis. Dev Growth Diff 53:878–886. [DOI] [PubMed] [Google Scholar]
- Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR. 1992. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics 130:471–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. [DOI] [PubMed] [Google Scholar]
- Passamaneck YJ, Schander C, Halanych KM. 2004. Investigation of molluscan phylogeny using large-subunit and small-subunit nuclear rRNA sequences. Mol Phylogenet Evol. 32:25–38. [DOI] [PubMed] [Google Scholar]
- Passamonti M, Ghiselli F. 2009. Doubly uniparental inheritance: two mitochondrial genomes, one precious model for organelle DNA inheritance and evolution. DNA Cell Biol. 28:1–10. [DOI] [PubMed] [Google Scholar]
- Passamonti M, Ricci A, Milani L, Ghiselli F. 2011. Mitochondrial genomes and Doubly Uniparental Inheritance: new insights from Musculista senhousia sex-linked mitochondrial DNAs (Bivalvia Mytilidae). BMC Genomics 12:442.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Martínez X, et al. 2001. Subunit II of cytochrome c oxidase in chlamydomonad algae is a heterodimer encoded by two independent nuclear genes. J Biol Chem. 276:11302–11309. [DOI] [PubMed] [Google Scholar]
- Pérez-Martínez X, et al. 2000. Unusual Location of a Mitochondrial Gene. Subunit III of cytochrome c oxidase is encoded in the nucleus of chlamydomonad algae. J Biol Chem. 275:30144–30152. [DOI] [PubMed] [Google Scholar]
- Plazzi F, Ceregato A, Taviani M, Passamonti M. 2011. A molecular phylogeny of bivalve mollusks: ancient radiations and divergences as revealed by mitochondrial genes. PLoS One 6:e27174.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plazzi F, Passamonti M. 2010. Towards a molecular phylogeny of Mollusks: Bivalves’ early evolution as revealed by mitochondrial genes. Mol Phylogenet Evol. 57:641–657. [DOI] [PubMed] [Google Scholar]
- Plazzi F, Ribani A, Passamonti M. 2013. The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics 14:409.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polechová M. 2015. The bivalve fauna from the Fezouata Formation (Lower Ordovician) of Morocco and its significance for palaeobiogeography, palaeoecology and early diversification of bivalves. Paleogeogr Paleoclimatol Paleoecol. http://dx.doi.org/10.1016/j.palaeo.2015.12.016. [Google Scholar]
- Popot JL, de Vitry C. 1990. On the microassembly of integral membrane proteins. Annu Rev Biophys Biophys Chem. 19:369–403. [DOI] [PubMed] [Google Scholar]
- Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N. 2002. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18:S71–S77. [DOI] [PubMed] [Google Scholar]
- Quesada H, Wenne R, Skibinski DOF. 1999. Interspecies transfer of female mitochondrial DNA is coupled with role-reversals and departure from neutrality in the mussel Mytilus trossulus. Mol Biol Evol. 16:655–665. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2008. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing.
- Race HL, Herrmann RG, Martin W. 1999. Why have organelles retained genomes? Trends Genet. 15:364–370. [DOI] [PubMed] [Google Scholar]
- Rehkopf DH, Gillespie DE, Harrell MI, Feagin JE. 2000. Transcriptional mapping and RNA processing of the Plasmodium falciparum mitochondrial mRNAs. Mol Biochem Parasitol. 105:91–103. [DOI] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Hauser A, Söding J. 2012. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 9:173–175. [DOI] [PubMed] [Google Scholar]
- Ren J, Liu X, Jiang F, Guo X, Liu B. 2010. Unusual conservation of mitochondrial gene order in Crassostrea oysters: evidence for recent speciation in Asia. BMC Evol Biol. 10:394.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelle W. 2014. psych: Procedures for Personality and Psychological Research. Evanston: Northwestern University. R package version 1.4.12.
- Reyes A, Gissi C, Pesole G, Saccone C. 1998. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 15:957–966. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Ronquist F, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez TM, Babin C. 2003. Distribution paléogéographique des mollusques bivalves durant l'Ordovicien. Géodiversitas 25:243–259. [Google Scholar]
- Sánchez TM. 2006. Taxonomic position and phylogenetic relationships of the bivalve Goniophorina Isberg and related genera from the Early Ordovician of northwestern Argentina. Ameghiniana 43:113–122. [Google Scholar]
- Sánchez TM. 2008. The early bivalve radiation in the Ordovician Gondwanan basins of Argentina. Alcheringa 32:223–246. [Google Scholar]
- Sanderson MJ. 2003. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19:301–302. [DOI] [PubMed] [Google Scholar]
- Scheffler I. 2008. Mitochondria. New York: J. Wiley and Sons. [Google Scholar]
- Shao R, Dowton M, Murrell A, Barker SC. 2003. Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol Biol Evol. 20:1612–1619. [DOI] [PubMed] [Google Scholar]
- Sharma PP, et al. 2012. Phylogenetic analysis of four nuclear protein-encoding genes largely corroborates the traditional classification of Bivalvia (Mollusca). Mol Phylogenet Evol. 65:64–74. [DOI] [PubMed] [Google Scholar]
- Sicheritz-Ponten T, Andersson SG. 2001. A phylogenomic approach to microbial evolution. Nucleic Acids Res. 29:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. 2004. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol. 21:468–488. [DOI] [PubMed] [Google Scholar]
- Simison WB, Boore JL. 2008. Molluscan evolutionary genomics In: Ponder W, Lindberg DR, editors. Phylogeny and Evolution of the Mollusca. Berkeley: University of California Press; p. 447–461. [Google Scholar]
- Simmons MP, Carr TG, O’Neill K. 2004. Relative character-state space, amount of potential phylogenetic information, and heterogeneity of nucleotide and amino acid characters. Mol Phylogenet Evol. 32:913–926. [DOI] [PubMed] [Google Scholar]
- Simmons MP, Ochoterena H. 2000. Gaps as characters in sequence-based phylogenetic analyses. Syst Biol. 49:369–381. [PubMed] [Google Scholar]
- Skibinski DOF, Gallagher C, Beynon CM. 1994a. Mitochondrial DNA inheritance. Nature 368:817–818. [DOI] [PubMed] [Google Scholar]
- Skibinski DOF, Gallagher C, Beynon CM. 1994b. Sex limited mitochondrial DNA transmission in the marine mussel Mytilus edulis. Genetics 138:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smietanka B, Burzyński A, Wenne R. 2010. Comparative genomics of marine mussels (Mytilus spp.) gender associated mtDNA: rapidly evolving atp8. J Mol Evol. 71:385–400. [DOI] [PubMed] [Google Scholar]
- Smith SA, et al. 2011. Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature 480:364–367. [DOI] [PubMed] [Google Scholar]
- Sousa FL, et al. 2013. Early bioenergetic evolution. Philos Trans R Soc Lond B Biol Sci. 368:20130088.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. Phylogenetic Models of Rate Heterogeneity: A High Performance Computing Perspective. In: Proceedings 20th IEEE International Parallel & Distributed Processing Symposium. Washington: IEEE Computer Society Press. p. 278–286.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöger I, Schrödl M. 2013. Mitogenomics does not resolve deep molluscan relationships (yet?). Mol Phylogenet Evol. 69:376–392. [DOI] [PubMed] [Google Scholar]
- Sun S, Kong L, Yu H, Li Q. 2015. The complete mitochondrial DNA of Tegillarca granosa and comparative mitogenomic analyses of three Arcidae species. Gene 557:61–70. [DOI] [PubMed] [Google Scholar]
- Tait SW, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 11:621–632. [DOI] [PubMed] [Google Scholar]
- Taylor JD, Glover EA, Williams ST. 2009. Phylogenetic position of the bivalve family Cyrenoididae—removal from (and further dismantling of) the superfamily Lucinoidea. Nautilus 123:9–13. [Google Scholar]
- Taylor JD, Williams ST, Glover EA, Dyal P. 2007. A molecular phylogeny of heterodont bivalves (Mollusca: Bivalvia: Heterodonta): new analyses of 18S and 28S rRNA genes. Zool Scr 36:587–606. [Google Scholar]
- Thrash JC, et al. 2011. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci Rep 1:13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillyard RJ, Dunstan B. 1916. Mesozoic and tertiary insects of Queensland and New South Wales. Descriptions of the fossil Insects and stratigraphical features. Queensl Geol Survey 253:1–63. [Google Scholar]
- Townsend JP, Su Z, Tekle YI. 2012. Phylogenetic signal and noise: predicting the power of a data set to resolve phylogeny. Syst Biol. 61:835–849. [DOI] [PubMed] [Google Scholar]
- Tucker LR, Lewis C. 1973. A reliability coefficient for maximum likelihood factor analysis. Psychometrika 38:1–10. [Google Scholar]
- Vallès Y, Boore JL. 2006. Lophotrochozoan mitochondrial genomes. Integr Comp Biol. 46:544–557. [DOI] [PubMed] [Google Scholar]
- Van Blerkom J. 2011. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion. 11 797–813. [DOI] [PubMed] [Google Scholar]
- von Heijne G. 1986. Why mitochondria need a genome. FEBS Lett. 198:1–4. [DOI] [PubMed] [Google Scholar]
- Wallace DC. 2013. Bioenergetics in human evolution and disease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos Trans R Soc Lond B Biol Sci. 368:20120267.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller TR. 1998. Origin of the molluscan class Bivalvia and a phylogeny of major groups In: Johnston PA, Haggart JW, editors. Bivalves: An Eon of Evolution. Calgary: University of Calgary Press; p. 1–45. [Google Scholar]
- Webb CO, Ackerly DD, Kembel SW. 2008. Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 24:2098–2100. [DOI] [PubMed] [Google Scholar]
- Wickham H. 2010. ggplot2 Elegant Graphics for Data Analysis. New York: Springer. [Google Scholar]
- Williams KP, Sobral BW, Dickerman AW. 2007. A robust species tree for the alphaproteobacteria. J Bacteriol 189:4578–4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WSW, Yang Z, Goldman N, Nielsen R. 2004. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168:1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li X, Li L, Xu X, et al. 2012. New features of Asian Crassostrea oyster mitochondrial genomes: a novel alloacceptor tRNA gene recruitment and two novel ORFs. Gene 507:112–118. [DOI] [PubMed] [Google Scholar]
- Wu X, Li X, Li L, Yu Z. 2012. A unique tRNA gene family and a novel, highly expressed ORF in the mitochondrial genome of the silver-lip pearl oyster, Pinctada maxima (Bivalvia: Pteriidae). Gene 510:22–31. [DOI] [PubMed] [Google Scholar]
- Wu X, et al. 2014. Evolution of the tRNA gene family in mitochondrial genomes of five Meretrix clams (Bivalvia, Veneridae). Gene 533:439–446. [DOI] [PubMed] [Google Scholar]
- Wu X, Xu X, Yu Z, Kong X. 2009. Comparative mitogenomic analyses of three scallops (Bivalvia: Pectinidae) reveal high level variation of genomic organization and a diversity of transfer RNA gene sets. BMC Research Notes 2:69.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Jameson D, Tang B, Higgs PG. 2006. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 63:375–392. [DOI] [PubMed] [Google Scholar]
- Yang Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555–556. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Young ND, Healy J. 2003. GapCoder automates the use of indel characters in phylogenetic analysis. BMC Bioinformatics 4:6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbawicka M, Wenne R, Burzyński A. 2014. Mitogenomics of recombinant mitochondrial genomes of Baltic Sea Mytilus mussels. Mol Genet Genomics 289:1275–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E, Oberhauser Ball A, Saavedra C, Freeman KR. 1994a. An unusual type of mitochondrial DNA inheritance in the blue mussel Mytilus. Proc Natl Acad Sci U S A. 91:7463–7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E, Oberhauser Ball A, Saavedra C, Freeman KR. 1994b. Mitochondrial DNA inheritance—reply. Nature 368:818.. [DOI] [PubMed] [Google Scholar]
- Zouros E. 2013. Biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol Biol. 40:1–31. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.