

Abstract
Remyelination is a major therapeutic goal in human myelin disorders, serving to restore function to demyelinated axons and providing neuroprotection. The target disorders that might be amenable to the promotion of this repair process are diverse and increasing in number. They range primarily from those of genetic, inflammatory to toxic origin. In order to apply remyelinating strategies to these disorders, it is essential to know whether the myelin damage results from a primary attack on myelin or the oligodendrocyte or both, and whether indeed these lead to myelin breakdown and demyelination. In some disorders, myelin sheath abnormalities are prominent but demyelination does not occur. This review explores the range of human and animal disorders where myelin pathology exists and focusses on defining the myelin changes in each and their cause, to help define whether they are targets for myelin repair therapy.
Keywords: Myelin vacuolation, Demyelination, Leukodystrophy, Multiple sclerosis, Toxin, Deficiency
Highlights
-
•
We reviewed myelin disorders of the CNS in humans and animals.
-
•
Myelin damage results from primary attack on the oligodendrocyte or myelin sheath.
-
•
All major categories of disease can affect CNS myelin.
-
•
Myelin vacuolation is common, yet does not always result in demyelination.
1. Introduction
Disorders of myelin of the central nervous system (CNS) form a large and growing list of neurological disorders in humans, ranging from the most common myelin disease, multiple sclerosis (MS) to rare genetic disorders. The increase is mainly due to detailed investigation of uncharacterized disorders of the CNS using contemporary imaging or gene sequencing but also to the finding of new white matter diseases. There are also many spontaneous inherited disorders of myelin in animals (the myelin mutants) and those generated in mice by genetic manipulation (transgenic mice). Some of these animals have mutations in myelin or myelin-associated genes implicated in the human myelin genetic disorders thus are models of these diseases. There is also an increasing recognition of myelin disorders where the candidate gene is novel and apparently unrelated to myelin or oligodendrocyte (OL) function and affecting cells other than OLs (Nave and Werner, 2014). Acquired myelin disorders have also been reported in animals. This special issue is directed toward remyelination or repair of CNS myelin of these disorders. In order to approach this topic and potentially devise therapeutic strategies to treat the myriad of disorders that affect myelin, a basic understanding of the development and maintenance of the mature myelin sheath is important. The origin of the OLs in the brain and spinal cord and their differentiation have been the subject of intense research and publication and have been reviewed in detail in this issue and elsewhere (Bradl and Lassmann, 2010, Richardson et al., 2006). Here a brief overview of the OL and the developing myelin sheath will lead into the discussion of myelin pathology in individual myelin disorders.
Myelin loss can occur as a result of direct damage to the myelin sheath or indirectly through a primary genetic disorder or attack on the OL during inflammation or toxic exposure. In addition, axon degeneration will lead to secondary loss of the myelin sheath (as in Wallerian degeneration). In genetic disease in which there is a failure of normal myelination, the CNS white matter is either hypomyelinated or dysmyelinated. In the former, axons are either myelinated with a thin myelin sheath or non-myelinated. Those disorders classified as dysmyelinating will show a greater deficit in myelination than hypomyelinating disorders and marked or subtle changes in the ultrastructure of the myelin present. An alternative definition of dysmyelination however is that it includes all disorders with minor disruption of the myelin sheath such as in Plp1 knockout, thus the term may have different interpretation. Genetic disorders can also result in myelin breakdown and demyelination at a variable time after birth. Acquired disorders of myelin result in damage to the myelin sheath that usually, but not always, result in demyelination. In this review we will discuss the disorders that affect myelin and in select cases present what is known about the mechanism of damage to either the myelin sheath or OL. Examples will be presented of the genetic and acquired disorders and note will be made if axon loss occurs as this will clearly affect the extent of remyelination. The goal is to provide the framework to the subject of remyelination or myelin repair, and the target disorders where this repair process may be of functional and clinical importance.
2. The OL and its myelin sheath
The OL is a remarkable cell that during development of the myelin sheath is required to produce vast quantities of lipids and proteins (Baron and Hoekstra, 2010, Pfeiffer et al., 1993). Lipid production however is much greater than protein, with lipids representing 70% of the dry weight of myelin membranes (Chrast et al., 2011). Indeed this high lipid content is greater than other membranes in the body and provides the insulation function of myelin required for saltatory conduction along myelinated axons that is essential for normal neurologic function. Unlike myelin proteins however, there are no myelin-specific lipids (Chrast et al., 2011). While the myelin proteins form a lesser component of myelin, they are critical for myelin production and maintenance and more recently have been found to be important for glial–axonal interactions and axon health (reviewed by Simons and Nave (2015)). In order for myelin to be synthesized normally, expression of the genes that encode for the production of enzymes responsible for lipid synthesis and the myelin-specific proteins, must be highly synchronized. It has been estimated that during development, the myelin membrane surface area of a single OL expands at a rate of 5–50 × 103 μm2/day compared to ~ 0.3 × 103 μm2 of the surface area of the cell soma (Chrast et al., 2011). This corresponds to as much as 5000 μm2 of myelin surface per day and ~ 105 myelin protein molecules per minute (Pfeiffer et al., 1993). Thus from a developmental standpoint, minor disruptions in this highly orchestrated myelinating program have significant consequences for myelin production and can result in severe lack of myelin formation that varies between humans and animals.
In addition to this critical time period in development, there is a slow turnover of lipid and protein in mature myelin that requires normal OL function though myelin is remarkably metabolically stable (Benjamins and Smith, 1984, Morell et al., 1994). Myelin components, including lipid and protein exhibit a great heterogeneity of metabolic turnover (Chrast et al., 2011). The intense biochemical activity during myelin development and to a lesser extent in maturity, is reflected by the structure of the OL and its organelle constituents (Fig. 1 ). At the ultrastructural level, Mori and Leblond (1970) pioneered studies that identified the stages of the OL lineage at which cell division ceased and axon ensheathment and myelination ensued. They identified three types of OLs as seen on EM. Light and medium OLs (terms used principally to describe the cytoplasmic contrast) are still mitotic and represent the earliest, likely pre-myelinating stages that are initiating contact with axons, though medium OLs may at late stage become early myelinating cells (Fig. 1). The third stage, or dark OLs are taking part in active myelination and as such have copious cytoplasm enriched with organelles required for lipid and protein production. While each organelle plays a critical yet unique role in myelin production, they all can be a target or the site of dysfunction that leads to a failure in myelin production or its loss. These organelles and dysfunction in each that affects myelination are as follows.
-
1)
Nucleus: Genetic disorders resulting in disturbance of gene transcription, gene deletion, gene dosage or involvement or modification by modifier genes with resultant failure or disruption of myelin production.
-
2)
Rough endoplasmic reticulum (RER): As the RER is the site of synthesis of certain key myelin proteins and lipid enzymes, mutations can result in a failure of translation of these myelin constituents and the generation of misfolded proteins that accumulate and distend the RER (Mayer et al., 2015). This may result in an unfolded protein response that can lead to OL apoptosis (Southwood et al., 2002). Examples are those humans and animals that have a mutation of the major CNS myelin protein, proteolipid protein (PLP) gene (Duncan, 1995). A block or delay in transport of PLP can lead to the failure of development of the myelin sheath or in hypomyelination.
-
3)
Smooth endoplasmic reticulum: This organelle is rarely seen in OLs (Peters et al., 1991). While smooth ER is the site of production of certain lipids in eukaryotic cells, it may be that lipid synthesis is restricted to the RER of OLs. Lipids are certainly present in the Golgi apparatus and lipid rafts associate with certain proteins such as PLP prior to incorporation in the myelin membrane (Baron and Hoekstra, 2010). We have identified smooth ER in OLs in the myelin mutant taiep rat where it associates with accumulating microtubules.
-
4)
Golgi apparatus: This prominent organelle is involved in vesicular transport and trafficking and the site of post translational modifications of proteins such as PLP and myelin-associated glycoprotein (MAG). The rat mutant, vacuole formation (vf) may be the only myelin mutant with a specific defect in Golgi (see later).
-
5)
Microtubules: Microtubules are essential to the intracellular transport of protein and certain mRNAs (MBP and MAG mRNA) to the cell membrane and developing myelin sheath. The taiep rat has a specific defect in OL microtubules that result in both the initial failure of normal myelin production but also in its subsequent loss (see later).
-
6)
Lysosomes: This organelle is responsible for the intracellular degradation of effete organelles and extracellular materials through the action of hydrolytic enzymes that generate an acidic environment within the lysosome. A specific class of disorders, the lysosomal storage disorders result from a mutation in one of these enzymes that results in a build-up of the respective substrates. Two classic childhood lysosomal storage disorders, Krabbe's disease and metachromatic leukodystrophy result in severe CNS and peripheral nervous system (PNS) demyelination.
-
7)
Peroxisomes: They are involved in the metabolism of lipids in particular the β-oxidation of very long chain fatty acids. The primary example of a white matter disorder resulting from peroxisomal dysfunction is adrenoleukodystrophy.
Fig. 1.
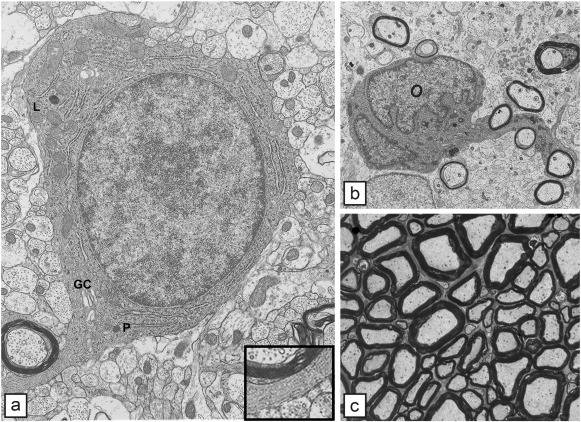
The normal oligodendrocyte and the development of myelin. a) A single oligodendrocyte, early in development has all the organelles required for lipid and protein production. Cisternae of rough endoplasmic reticulum are seen throughout the cell; the Golgi complex (GC) is present though not copious. Scattered mitochondria can be seen while a single lysosome (L) and peroxisome (P) are present. In the cytoplasmic tongue adjacent to the myelin sheath, microtubules (inset) are prominent. b) Most oligodendrocytes myelinate multiple axons as seen here in the developing cat spinal cord where the OL is in contact with and likely myelinates four axons. In a) and b) it is clear that myelination occurs first around larger diameter axons. c) In the mature white matter, myelinated axons are tightly packed. The correlation between axon diameter and myelin sheath thickness is obvious.
Fig. 1a reproduced from Lunn et al. (1997), J. Neurocytol, 26, p. 267 — with permission.
3. Evaluating changes in myelin
Unequivocal proof of myelin pathology and demyelination in the CNS requires definitive morphologic evidence that frequently can be hard to acquire, especially in human tissue. Tissue artifact following immersion fixation, poor perfusion (animals) or a delay in fixation after death is common and can lead to a de-compaction of myelin and misinterpretation of these and other changes. Paradoxically, the best fixation of CNS myelin is achieved where the density and thickness of myelin is less, i.e. in the genetic disorders. It is harder to achieve good fixation of myelin in the normal CNS because of the density and thickness of myelin sheaths perhaps leading to poor penetration of fixatives. The best fixation is achieved by cardiac or aortic perfusion with glutaraldehyde or Karnovsky fixative. In larger animal models, perfusion through the ascending and descending aorta is optimal. Using these methods and fixatives, tissue is processed for embedding in plastic resin that allows both light microscopy (1 μm, toluidine blue stained sections) or electron microscopy (ultrathin sections) to be performed. These sections provide superior resolution and quality to other histologic techniques and are the gold standard in myelin evaluation. Most recently, high pressure freezing of the CNS to avoid aldehyde fixation artifact has provided an alternative method to provide high quality ultrastructural resolution (Mobius et al., 2016).
Previously, a loss or lack of myelin has been evaluated primarily in paraffin-embedded tissue stained with Luxol fast blue, or in immunolabeled sections using antibodies primarily to the major myelin proteins, MBP or PLP. However, secondary loss of myelin following axonal loss cannot be distinguished from primary loss using these methods. In addition, staining results can be variable hence the interpretation of a primary myelin pathology can be difficult. However, evaluation of myelin abnormalities in large tissue blocks such as the whole brain can only be performed in paraffin embedded or cryostat sectioned tissue, as plastic embedding for 1 μm sections can only be carried out in relatively small pieces of tissue (maximum is around 2–4 mm2). Other less used stains for the demonstration of myelin in the brain include the Gallyas silver stain and the Heidenhain stain. These stains can provide a first indication of myelin absence or loss.
Finally, the area of the CNS sampled to evaluate changes in myelin is important. In cases where there are global abnormalities, the spinal cord and optic nerves are best as they provide myelinated fiber tracts that can be sampled on cross section and contain all diameters of axons (the optic nerve however has mainly small/medium diameter). In the brain, myelin pathology is most often evaluated in the corpus callosum as it also allows study of axons in transverse section. It is the target site for studying demyelination and remyelination in cuprizone toxicity, but is one of the few sites in the CNS, affected by this toxin. However, regional variations in axon caliber in the genu, body and splenium of the corpus callosum, along with normally thinner myelin sheaths seen in this structure (Stidworthy et al., 2003), can make quantitation of pathology and repair more challenging. In other major white matter tracts of the brain such as the internal capsule, cerebellar medulla and sub-cortical areas, axons are not aligned in parallel hence quantitation of myelin changes is difficult.
4. Pathology of demyelination
Myelin is lost either because of, 1) a direct attack on the myelin sheath, 2) disruption or death of OLs. This clear difference in the cause of demyelination may not be obvious in some instances and in some cases, damage to both may occur. While the end-stage result, i.e. demyelination is the same, there are various causes of damage to myelin or the OL. The etiology of myelin loss includes immune-mediated, viral, metabolic (deficiency or mineral or vitamin), toxic, genetic, traumatic and neoplastic disease. The immune-mediated diseases are highlighted by MS, and the possible causes of demyelination in MS and its animal model counterpart, experimental autoimmune-mediated encephalomyelitis (EAE) have been reviewed in great detail elsewhere (see later). These disorders are characterized by the invasion of the CNS by T and B lymphocytes and macrophages, and the production of many cytokines, both pro- and anti-inflammatory, some of which may directly damage myelin (Frohman et al., 2006, Lassmann, 2014, Lucchinetti et al., 1996).
There is a wide variety of toxins that result in demyelination, some that kill OLs and others that directly target myelin. Other toxins such as lysolecithin and ethidium bromide can produce different changes in myelin sheath, though these are not diagnostic.
5. Myelin vacuolation
Vacuolation of myelin is the most common pathologic alteration of the myelin sheath. When severe and involving all or large areas of the CNS, it is called status spongiosus though it should be distinguished from neuronal disorders such as the transmissible encephalopathies where vacuolation of neurons occurs. Myelin vacuolation has been described in many species, including humans, cow, pig, goat, fox, dog, cat, hedgehog and bird. Thus it is a shared response in diverse species and disorders that affect myelin and OLs.
Frequently, myelin vacuolation is thought to be synonymous with myelin degeneration and is called spongiform degeneration, but this may be inaccurate. Thus diseases such as Canavan's disease and in certain mitochondrial disorders such as the Kearns Sayre syndrome, have profound myelin vacuolation but little to no demyelination as has been documented (Fig. 2 ). To advance from myelin vacuolation to demyelination may depend on whether the primary target is the myelin sheath or the OL. The actual mechanism of myelin vacuolation likewise may vary between different myelin disorders though there may be some shared mechanisms. It is seen in many myelin disorders including those of genetic, toxic and inflammatory origin thus it clearly lacks specificity. Additionally, in some instances it may not be cell autonomous as disorders of astrocytes may result in ionic imbalance in the neuropil resulting in fluid accumulation within the myelin sheath (Baslow, 2003). This fluid accumulation is predominately seen in the intraperiod line of the myelin sheath, i.e. the extracellular space. Disorders where myelin vacuolation is seen will be presented later.
Fig. 2.
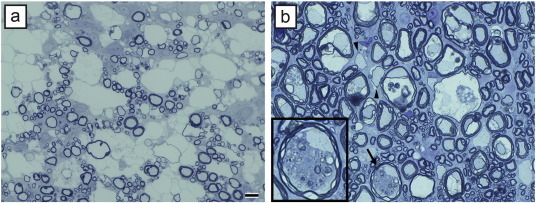
Myelin vacuolation does not always lead to myelin degeneration. a) In a case of canine mitochondrial DNA mutation, there is extensive vacuolation but no evidence of myelin breakdown or demyelination. b) In contrast in a feline model (FIDID), there is severe vacuolation, with evidence of degeneration of the inside loops of myelin adjacent to intact axons (arrow and enlarged in inset), and scattered demyelination and remyelination (arrowheads). Scale bar: 20 μm.
6. Genetic disorders of myelin
6.1. Pelizaeus Merzbacher disease (PMD) and the plp1 mutants
PMD is a rare X-linked disorder of humans that presents as a heterogeneous disease, has been classified into three sub-types, 1) connatal PMD, the most severe form with death usually before 10 years, 2) classical PMD, the most common form and though severe, affected males can survive into the 6th decade, and 3) spastic paraplegia Type 2 (SPG2) the mildest form (reviewed in (Mayer et al., 2015). The disease results from mutations, though more commonly, duplications of the proteolipid protein—PLP gene (Plp1). A separate group of disorders, named the PMD-like diseases has also been reported, notably those with mutations in the connexin 47 gene (Hobson and Garbern, 2012). PMD is the archetypic disorder of failure of development of myelin in the CNS, that in both PMD and its animal models, results in the gross absence of myelin (Fig. 3A). There are a large number of mutations of the Plp1 gene in animals ranging in species from mouse, rat, rabbit, pig to the dog (Duncan, 1995, Griffiths et al., 1998, Werner et al., 1998). Like the human disease, these mutants form a heterogeneous group with respect to their phenotype and variable deficiency in myelination. The mildest myelin deficiency is seen in the rumpshaker mouse (rsh), while the most severe is a transgenic rat model that overexpresses plp1 (Mayer et al.), Fig. 3). With the exception of the rsh mouse, all the mutant rodent models are similar to connatal disease, i.e. they die young with severe deficiency in myelin. The canine model, the shaking pup (shp) is the only mutant that models classical PMD (Mayer et al., 2015). In the spinal cord of the shp, myelin formation increases with time and by two years of age many myelinated axons are present, though with thin myelin sheaths (Fig. 3B), correlating with an increase in mature OLs that occurs with aging in the spinal cord (Mayer et al., 2015). In contrast, the brain and optic nerves remain severely affected throughout life (Mayer et al., 2015).
Fig. 3.
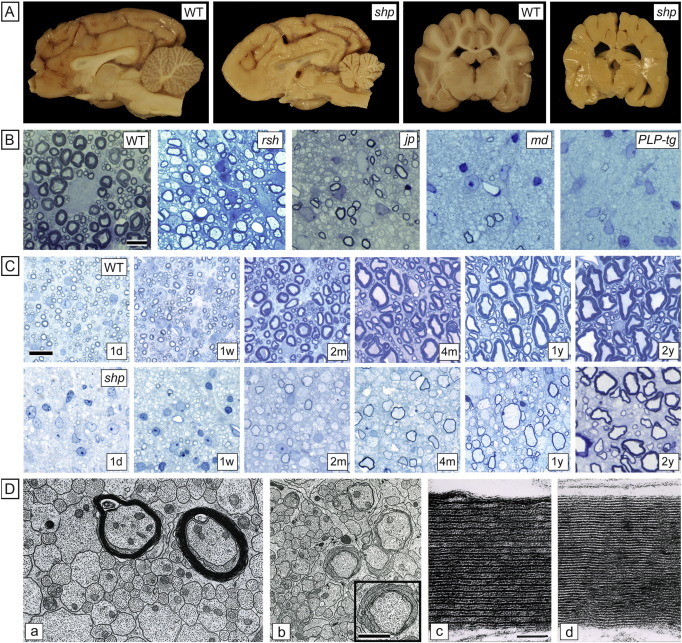
PLP mutations result in severe but variable deficiency in myelination. A. Sagittal and coronal sections of the brain of mature control and canine shp brains (over 18 months) show the marked deficiency in myelination of the mutant brain. B. In the rodent PLP mutants at 17–20 days of age compared to wildtype, there is a clear range of myelination from the mild hypomyelination seen in the mouse jprsh to the almost total absence of myelin in the PLP1 transgenic. In both the md rat and jp mouse the scattered myelin sheaths can be normal thickness and well compacted. C. In 1 μm sections of the spinal cord ventral column of the shp from 1 day to 2 years shows that the mutant lacks myelin at 1 day compared to control but myelin gradually increases with time. D.a. In the 80 day old long-lived md rat the majority of axons are non-myelinated but two larger ones have well compacted sheaths (D.a), while in other areas, sheaths are poorly compacted (D.b). On high power, while well compacted the mutant sheath in the md rat lacks an intraperiod line (D.d) compared to control (D.c). Wild type (WT), rumpshaker (jprsh), jimpy (jp), myelin deficient rat (md), PLP transgenic rat (PLP-tg). Scale bar: 20 μm (B), 10 μm (C).
Panel C reproduced from Mayer et al. (2015), Neurobiol Dis, 75, 118; Panel D.a, b Duncan et al. (1995), J. Neurocytol, 24, 745, c, d, Duncan et al. (1987), PNAS, 84, 6287. All with permission.
In the rodent models, their short lifespan allows observations on myelination/glia over 3–4 weeks only with the exception of a long-lived strain of the myelin deficient (md) rat (Duncan et al., 1995). In the jimpy mouse (jp) and md rat, the majority of axons remain non-myelinated, though focal ‘clusters’ of myelinated axons can be seen, likely associated with a common OL (Duncan et al., 1995). This suggests that some OLs escape the cell death fate that is common in these mutants. In addition, these myelin sheaths often appear quite thick and well compacted (Fig. 3). Immunolabeling of such sheaths in the md rat and jp mouse shows that they lack PLP but are MBP positive and on EM they lack an intraperiod line, the site of localization of PLP (Duncan et al., 1989, Duncan et al., 1987). It is commonly reported that myelin sheaths are poorly compacted in the PLP mutants and indeed this can be seen in the long-lived md rat strain (Fig. 3C). The failure of myelin development in PMD and its models relates to mutations in plp1 or to its overexpression in those with duplications of the gene. Accumulation of a misfolded or excess PLP in the RER and possibly Golgi leads to distension of RER, most definitively seen in the md rat and shp. This has been postulated to lead to an unfolded protein response and OL death (Southwood et al., 2002). However in the shp where distended RER is most frequent, it rarely leads to cell death (Mayer et al., 2015). The slow yet progressive endogenous increase in myelination of the spinal cord in the shp suggests that promotion of host myelination might have therapeutic significance and might be a simpler alternative to cell replacement therapy (Duncan et al., 2011, Gupta et al., 2012).
6.2. Canavan's disease
Canavan's disease (CD) is a rare disorder of the CNS caused by mutations in the aspartoacylase gene (ASPA) that results in accumulation of N-acetyl-aspartic acid (NAA) in the brain and in body fluids (Kaul et al., 1993). It is the archetypical example of myelin vacuolation and status spongiosus of the white matter. The disease has an early onset, between 2 and 6 months of age with symptoms that include, lethargy, mental retardation, blindness and others, and affected children usually die by 5 years. Affected children frequently have macrocephaly. The prevalence of the disease is much higher in children of Ashkenazi Jewish origin.
ASPA is highly expressed in OLs where it hydrolyzes NAA to acetate and aspartic acid. The build up of NA in CD leads to its excretion in the urine and this can be diagnostic. While pathologic examination of the CNS has identified the extensive white matter vacuolation which has been referred to as myelin (spongy) degeneration, in fact the evidence for myelin breakdown and demyelination in CD is very limited. Neuropathological studies were primarily carried in the 1960–70s and although some studies reported EM findings (e.g. Adachi et al., 1973; Jellinger and Seitelberger, 1970), not all contemporary technical approaches to studying myelin pathology have been carried out. Myelin vacuolation is seen throughout the WM of the brain and spinal cord, though variable in its severity in different parts of the CNS. Definitive proof of demyelinated axons (i.e. 1 μm sections or on EM) is lacking and Jellinger and Seitelberger (1970) noted that there was a lack of myelin breakdown products and OLs appear intact. Both of these studies noted that astrocytes had swollen, ‘watery’ cytoplasm with large, abnormal mitochondria (Adachi et al., 1973, Jellinger and Seitelberger, 1970). However, these tissues were immersion fixed some time after death and artifact needs to be considered.
The understanding of the pathophysiology of CD and its possible therapy has been advanced by the generation of mutant mice, knock-outs, knock-in and ENU-induced (Nur7) mutations affecting the ASPA gene and a spontaneous mutant rat (Kitada et al., 2000). The cause of myelin vacuolation in CD has been debated and is still unclear as discussed by Hoshino and Kubota (2014). The two most discussed hypotheses have been the role of accumulating NAA and a decrease in acetate and aspartate leading to a decrease in myelin-associated lipids, and secondly the role that NAA may play as a molecular water pump (Baslow, 2003). Support for the second hypothesis comes from the demonstration of the alteration of distribution of aquaporin-4 to solely astrocyte end feet (Clarner et al., 2014). In addition, three other studies have challenged the lipid hypothesis. Traka et al. (2008) showed that reduction of cerebroside in the ASPAnur7 mouse did not worsen the CNS pathology. Using the same double transgenic, (Guo et al., 2015) and (Maier et al., 2015) showed that ablating NAA synthesis prevented the development of myelin vacuolation, though the survival time of the mice was not prolonged (Maier et al., 2015) and other similar studies confirm this (Guo et al., 2015). It can be concluded that to repair the myelin vacuolation in CD, a complete understanding of the pathophysiology is important as is definitive evidence that demyelination occurs. Gene therapy may prove to be successful (Guo et al., 2015) even if the mode of action on vacuolated myelin is not known.
6.3. Alexander's disease (AD)
This is a rare disorder of the CNS that presents most commonly in infants (0–2 years) though it also can have a juvenile (2–12 years) or adult onset (> 12 years) (Messing et al., 2012). It is classified as leukodystrophy based on an abnormality of WM, however the causative mutation is in the astrocyte GFAP gene (Messing et al., 2012). This makes Alexander's disease of particular interest and importance as it would be a proven example of a primary abnormality in astrocytes resulting in a disorder of myelin though this is debated. Most patients have de novo mutations in GFAP, though observations from adults with AD who have offspring demonstrate that it is inherited as an autosomal dominant trait (Messing et al., 2012).
The pathological hallmark of AD is the presence of Rosenthal fibers within abnormal astrocytes (Herndon et al., 1970). Rosenthal fibers are eosinophilic inclusions in H&E stained sections, that on EM appear osmophilic and contain αB-crystallin and the heat shock protein 27 (Messing et al., 2012, Mignot et al., 2004). There is massive gliosis throughout the brain. The myelin abnormalities which are identified in life by MRI Van der Knaap and Valk (2005) are more variable and in many ways, remain a conundrum. In infantile AD it has been proposed that there is hypomyelination of frontal white matter of the brain (Mignot et al., 2004). Little to no evidence of macrophage activity or microglial activation are seen suggesting lack of myelin breakdown and the greater likelihood a developmental disorder of myelin (Mignot et al., 2004). In adults, where hypomyelination is not as severe, it has been proposed that demyelination occurs. There may also be some loss of axons that may be severe, and though it has been suggested that there is some OL loss, this has been disputed (Mignot et al., 2004). In some cases there is little or no demyelination and this has raised the question as to whether AD is really a leukodystrophy (Mignot et al., 2004). However, Herndon et al. (1970) described two patients where EM of tissue obtained at biopsy and post-mortem showed notable lack of myelin, and reviewed ten other patients where myelin pathology was seen.
Clarification of this issue could have come from the mouse models of, yet in GFAP overexpressing mice which died of encephalopathy and where abnormal astrocytes were seen with profuse Rosenthal fibers, no myelin changes were present (Messing et al., 1998). In AD, myelin abnormalities, particularly hypomyelination could result from an interference with the astrocyte-OL that are required during development (Messing et al., 2012, Mignot et al., 2004). What appears to be missing in the understanding of myelin changes in AD are contemporary studies of the neuropathology. Efforts should be made to fix tissue appropriately for plastic embedding, 1 μm sectioning and ultrastructural examination. Such studies would define whether there is truly hypomyelination or demyelination or a mixture of the two and will also define the presence or loss of axons.
6.4. Adrenoleukodystrophy (ALD)
X-linked adrenoleukodystrophy is the most common inherited peroxisomal defect, affecting 1:1500 Caucasian males (Berger et al., 2014). It has a heterogeneous presentation, even within the same family (Brown et al., 1993, Moser, 1997, Powers and Moser, 1998). The best known form is the childhood ALD (cALD), made famous in the movie, “Lorenzo's Oil.” Boys with cALD become symptomatic at 5–7 years of age and usually die within 2–3 years from a progressive, unrelenting neurologic dysfunction. The disease also affects the adrenal glands and the testes though this can be variable. The second major presentation of the disease is known as adrenomyeloneuropathy (AMN) which develops in the teens and twenties, primarily as a degenerative disease of long tracts (distal axonopathy) of the spinal cord (Powers et al., 2000). Peripheral nerve may also be affected and around 30% of AMN patients develop cerebral disease in their 30–40s. Female carriers (heterozygotes) are also at risk of developing AMN, though later in life and less severely.
The primary biochemical defect in ALD is the accumulation of very long-chain fatty acids (VLFCA), usually C22–26 in plasma (Kemp and Wanders, 2010). Until the ALD gene was discovered it was assumed that the primary defect was a mutation of the VLFCA-CoA synthetase gene. However, positional cloning identified a mutation in the ATP-binding cassette (ABC) transporter subfamily D member 1 (ABCD1) gene (Moser, 1997), and now over 643 different mutations have been identified (Berger et al., 2014). These mutations are presumed to affect the import of VLCFA-CoA synthetase into peroxisomes resulting in the accumulation of VLCFAs.
Pathologically, cALD is characterized by progressive demyelination of the brain, beginning in the corpus callosum with progression outward to result in confluent demyelination of the hemispheres (Fig. 4B) (Dubois-Dalcq et al., 1999). This progression is monitored by MRI (Fig. 4A) and gadolinium enhancement later in the disease results from disruption of the blood-brain barrier. Migration of leucocytes occurs resulting in a later, and marked inflammatory stage involving both T and B cells, with microglial activation and macrophage recruitment. Most recently, the mechanism of BBB disruption and brain inflammation in ALD has been explored (Musolino et al., 2015). This study showed that ABCD1 is highly expressed in endothelial cells and RNAI silencing leads to increase in MMP9 and an increase in transmigration of monocytes.
Fig. 4.
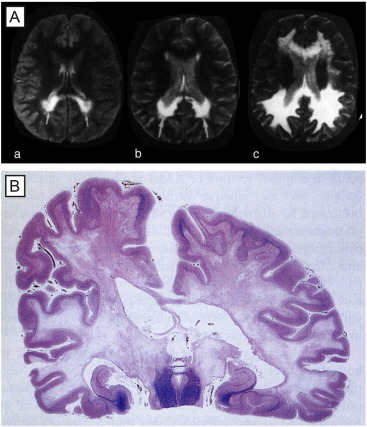
Myelin loss in adrenoleukodystrophy. (A) Magnetic resonance imaging (MRI) scan showing the progression of demyelination in the parieto-occipital white matter over a period of three years in a child. At first, the patient had only neuropsychological deficits [(a), age seven; (b), age eight)], then showed a marked neurological deterioration at nine years of age (c). Note the progression of demyelinating lesions toward frontal lobes from (a) to (c). (B) ALD. Confluent demyelination of parietal and temporal white matter and posterior limb with some arcuate sparing in most superior parietal gyri. (Luxol fast blue-periodic acid Schiff, × 1).
Reproduced from Powers et al. (2000), J Neuropathol Exp Neurol, 59, 715 — with permission).
Despite our increased understanding of the pathophysiology of cALD and AMN, the reason for myelin breakdown is still debated (Berger et al., 2014). Accumulation of VLCFA in myelin may lead to its instability and demyelination, and these precede the inflammatory phase of the disease hence inflammation is unlikely the primary cause of myelin breakdown (Berger et al., 2014, Dubois-Dalcq et al., 1999). OL death occurs but is perhaps not by apoptosis. Further progress in our understanding may depend on the generation of an animal model with cerebral demyelination and inflammation. To-date, the mouse models of ALD lack these changes though they do present with a phenotype similar to AMN as they age (Pujol et al., 2002). Inactivation of peroxisomes has been achieved in a conditional knockout of the mouse peroxin-5 gene (Pex5) in OLs (Kassmann et al., 2007), that developed a progressive neurologic disease from around 6 months with death at 12 months. These mice have both MRI then pathologic evidence of sub-cortical demyelination, axon death and marked inflammation. While the pattern of demyelination did not match that seen in cALD and OLs were preserved, this model has similarities to cALD that may be instructive (Aubourg, 2007). A potential alternative to the current mouse models is to study mutant OLs derived from fibroblasts then induced pluripotent stem cells (iPSCs) from cALD patients (Jang et al., 2011, Wang et al., 2012). These studies have shown that OLs differentiate normally and on differentiation begin to express high levels of VLFCAs. However, an in vitro approach alone may be limited in proving what happens to OLs in vivo and why myelin degenerates.
6.5. Lysosomal storage disorders (LSDs)
Mutations that result in the deficiency of lysosomal catalytic enzymes lead to an increase in the enzyme's substrate and result in a lysosomal storage disorder (LSD) (Jeyakumar et al., 2005). LSDs are usually inherited as autosomal recessive traits. While increase in substrate can affect all tissue, they have their greatest effect on the nervous system. However the majority of LSDs result in storage in neurons leading to neuronal dysfunction, there are two classical diseases that affect white matter, 1) Krabbe's disease (globoid cell leukodystrophy), and 2) metachromatic leukodystrophy (MLD). A major difference between these two diseases and other myelin disorders discussed in this review is that they affect both the CNS and PNS, and demyelination of the PNS may dominate the clinical presentation.
6.6. Krabbe's disease — globoid cell leukodystrophy
Krabbe's disease is a lysosomal storage disorder caused by mutations in galactosylceramidase (Galc) gene (Wenger et al., 2000). Loss of function of the enzyme leads to accumulation of the substrates, galactocerebroside and psychosine. Accumulation of psychosine is thought to play the more significant role in the degeneration of OLs resulting in severe demyelination. Clinically, the infantile form, which is most common, is apparent from early life, with pronounced and progressive neurologic dysfunction leading to death in 1–2 years. Later onset infantile and adult-onset forms of the disease also occur. Considerable advances in our understanding of the disease comes from the study of naturally-occurring models especially the twitcher (twi) mouse (Suzuki, 1994). GCLD is also found in Rhesus monkeys, and Cairn and West Highland terriers (Wenger, 2000).
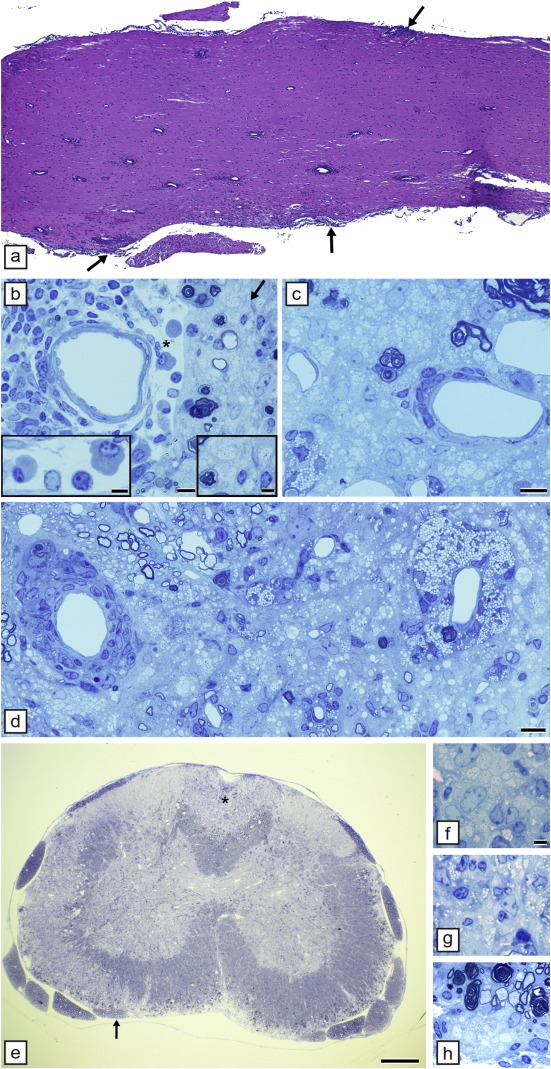
In the twi mouse, both the CNS and PNS develop profound demyelination, as they do in the other Krabbe's models. Demyelination begins in the peripheral nerve before the spinal cord or brain (Fig. 5a, b). In general, the pathologic onset of disease is seen first in the earliest myelinating structures of the CNS (Taniike and Suzuki, 1994). While myelination precedes normally in the twi spinal cord (10 days of age), axons are hypomyelinated prior to the onset of demyelination (Suzuki and Taniike, 1995). The primary reason for demyelination is the apoptotic death of OLs thought to be due to psychosine accumulation (Taniike et al., 1999). A unique feature of the neuropathology of twi and Krabbe's disease and the other models is the presence of multinucleated macrophages (globoid cells) throughout the white matter (Fig. 5e, f). These cells may not only participate in myelin debris removal but may be part of the demyelination process, migrating into the white matter prior to demyelination and extending processes to myelin sheaths where they may be involved in myelin stripping (Suzuki and Taniike, 1995). On EM, globoid cells are noted to contain cytoplasmic inclusions, also seen in OLs and Schwann cells, that are diagnostic for GCLD. We have shown however that globoid cells may be beneficial as in a double mutant mouse, twitcher/osteopetrotic (twi/op), the macrophage deficiency in op results in the double mutant presenting with a more severe phenotype than age-matched twi mice (Kondo et al., 2011). It was proposed that this worsening related to a reduction in remyelination in the double mutant (Kondo et al., 2011). This result differs from a study that showed that a reduction of MHCII class molecule expression in twi led to an improved phenotype with less severe demyelination in the brain yet not the spinal cord (Matsushima et al., 1994). There are differences in the literature on whether axons are lost in the twi and by extrapolation in Krabbe's disease. Careful morphometric analysis by Suzuki and colleagues concluded there was no axon loss (Suzuki and Taniike, 1995) but more recent studies using more contemporary techniques have detected axonal swellings in the CNS and PNS though it is unclear if this lead to axon loss (Castelvetri et al., 2011, Teixeira et al., 2014).
Fig. 5.
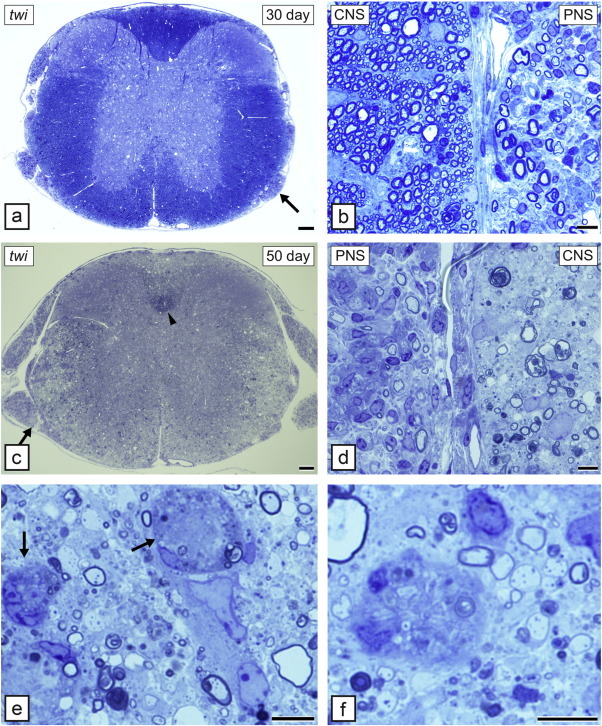
The twitcher (twi) mouse develops progressive demyelination of the spinal cord and peripheral nerve. At 30 days of age the spinal cord appears normal (a) but a nerve root (arrow) seen at higher power (b) shows that demyelination has begun in the PNS but not CNS. By 50 days however the spinal cord is extensively demyelinated (c) with only the cortico-spinal tract (arrowhead) appearing to have retained myelin. In the area marked in (c) (arrow) there is clear demyelination of both the PNS and CNS (d). In the CNS, the white matter is invaded by unique, large macrophages known as globoid cells. These are seen in areas of severe demyelination (e and f). In some cases they are close to thinly demyelinated and myelinated axons that may be remyelinated or hypomyelinated (f). Toluidine blue. Scale bar: 0.2 mm (a, c), 20 μm (b, d–f).
6.7. Metachromatic leukodystrophy (MLD)
Like Krabbe's disease, MLD is an autosomal recessive disease caused by a mutation in the lysosomal enzyme, aryl-sulfatase A (ASA) that results in profound demyelination of both the CNS and PNS (Von Figura et al., 2001). The disease is manifest in late infancy (12–20 months of age) with progressive neurologic deterioration and eventual death. It also can initially present at juvenile and adult time points. The mutations in the ASA gene result in an increase in sulfatide storage though this occurs slowly hence the later onset of the infantile disease in contrast to Krabbe's disease (Ramakrishnan et al., 2007). Unlike Krabbe's disease, there are no spontaneous animal models of MLD and this has affected progress in the developing treatments.
Studies on the pathophysiology and potential therapies of MLD have relied on transgenic mouse models. Initially, the first ASA knockout mouse was shown to have an increase in sulfatide in the CNS but little to no pathology at early time points. By 2 years of age there was evidence of modest gliosis and microglial activation in the CNS but no demyelination (Hess et al., 1996). Myelination may be delayed however in the absence of ASA (Yaghootfam et al., 2005). In attempts to create a mouse with a more meaningful phenotype, Ramakrishnan et al. (2007) generated a galactose-3-O-sulfotransferase-1 double transgenic mouse that lacked ASA and overexpressed the sulfatide synthesizing enzyme in OLs, targeted to myelinating cells of the CNS and PNS. By one year of age, the double transgenic mouse developed severe behavioral abnormalities and pathological studies showed notable changes in the PNS though less severe myelin changes in the CNS. Peripheral nerves were larger than normal, axons and myelin sheaths and some Schwann cells contained storage material, yet there was no obvious demyelination. Though this mouse is more representative of what is seen in MLD, the less generalized and less severe involvement of the CNS suggest that additional models may be required.
6.8. Vanishing white matter disease (VWM disease)
Vanishing white matter disease or childhood ataxia with central hypomyelination (CACH) as it was previously known, has an evocative name that describes the dramatic loss of myelin, the hallmark of this disease (Schiffmann et al., 1994, van der Knaap et al., 1997). It is an autosomal recessive disorder that primarily affects young children (2–6 years of age) but can also have an adult onset. The disease onset is associated with sensitivity to febrile infections, minor head trauma and fright (Labauge et al., 2009, van der Knaap et al., 2006). Vacuolation of white matter is a common finding though whether this leads to demyelination is not clear. The original title of the disease, CACH implied that hypomyelination occurs and this was reported by Schiffmann et al. (1994) and Francalanci et al. (2001). However the definitive presence of hypomyelination or demyelination remains to be confirmed quantitatively. Despite an increase in OL number (Bruck et al., 2001, Rodriguez et al., 1999) apoptosis of OLs is also seen. The presence of ‘foamy’ vacuoles is a hallmark abnormality of OLs (Schiffmann et al., 1994, van der Knaap et al., 2006). Astrocytes may be reduced in number which is unusual given their usual response to WM changes, especially with axon loss which is variable but is present.
The disease is caused by mutations in the eukaryotic translation initiation factor 2B genes (E1F2B 1–5) (van der Knaap et al., 2002). These genes are known to be involved in protein translation, and it is unclear why a mutation in a housekeeping gene leads to myelin loss. Further exploration of the pathophysiology of the disease was reported by Bugiani et al. (2013) who examined WM lesions in VWM patients and showed an increase in hyaluronan, a known inhibiter of astrocyte and OL progenitor cell maturation.
To approach these questions, two transgenic mice have been generated. In the first, introduction of a point mutation in the Eif2b5 gene locus resulted in a delay in development of myelin and an increased in oligodendrocytes and astrocytes though with no abnormal phenotype (Geva et al., 2010). In the second mouse, the temporal activation of PERK (pancreatic endoplasmic reticulum kinase) which is enhanced in oligodendrocytes in VWM disease, many of the characteristic features of the disease were found, including foamy OLs and apoptosis (Lin et al., 2014).
This is a complicated disease and it is clear that we lack a complete understanding of the changes reported in WM. Most recently, M. van der Knaap has studied double knock in mice (mutations in two of the EIF2B genese) which there were severe changes in myelin and OL (ULF meeting, Omaha, Nebraska 2015) and these mice may be instructive in unraveling the VWM disease ‘mystery’.
6.9. Mitochondrial mutations
Mitochondrial DNA (mtDNA) mutations result in a diverse group of severe and often fatal neurologic disorders in humans and animals (Oldfors and Tulinius, 2003). While many tissues can be involved in mtDNA mutations, their greatest effect is seen in the CNS and skeletal muscle. The neuropathologic effects in the brain are also diverse and non-specific including neuronal degeneration, widespread gliosis and sometimes necrosis. Vacuolation of white matter is also common and has been described in Kearns-Sayre (KSS) syndrome (Oldfors et al., 1990, Oldfors and Tulinius, 2003, Sparaco et al., 1993) and also in Leber's optic neuropathy (Kovacs et al., 2005) and Leigh syndrome (Kimura et al., 1991, Lake et al., 2015).
However, in KSS and other mtDNA mutations, it is not clear whether this leads to significant demyelination. We have described a mitochondrial disorder in two breeds of dog (Li et al., 2006). Affected dogs developed a tremor at 2–9 weeks of age of varying severity and progression, typical of the clinical heterogeneity of maternally inherited disorders. A mutation in cytochrome b in the OXPHOS pathway was found by Li et al. (2006). There was widespread vacuolation of the brain and spinal cord of affected dogs but little demyelination (Fig. 6 ). Interestingly, conditional knockout of the Cox10 gene leading to failure of assembly of stable mitochondrial cytochrome c oxidase, resulting in peripheral neuropathy but no CNS WM changes (Funfschilling et al., 2012). While the mechanism of myelin vacuolation in mtDNA disorders remains unknown (Morato et al., 2014), it is possible that vacuolation may be connected to astrocyte mitochondrial dysfunction. The apparent lack of demyelination and in the human mitochondrial disorders noted above, suggests that therapeutic approaches to myelin repair in these disorders might be aimed at resolving the myelin vacuolation with subsequent myelin recompaction.
Fig. 6.
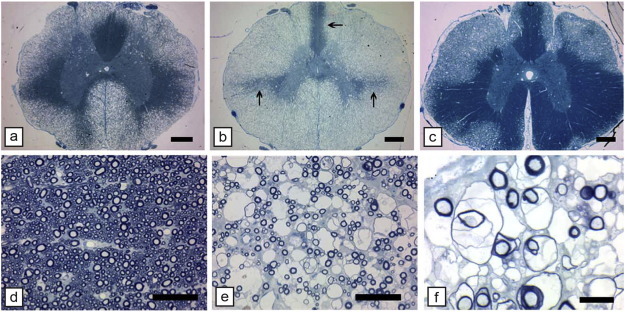
Myelin vacuolation of the CNS in mitochondrial encephalomyelopathy. The spinal cord of a 4-week-old (a, b) and 15-week-old (c) affected dog shows variable but extensive myelin vacuolation, the severity of which is unrelated to age. In the 4-week-old dog, the myelin vacuolation is more severe in the thoracic cord (b) than the cervical cord (a); in panel b, there is little normal myelin (arrows). In the 15-week-old affected dog's thoracic cord, areas of myelin vacuolation occupy about 50% of the white matter (c). On higher power of adjacent normal (d) and vacuolated (e) areas of myelin, the splitting and vacuolation of myelin sheaths can be seen (toluidine blue). Higher power (f) of a vacuolated area clearly shows that the spongiform change is due to splitting of the myelin sheaths. Scale bar: a–c, 500 μm; d, e, 50 μm; f, 20 μm.
Reproduced from Li et al. (2006), Neurobiol Dis, 21, 35 — with permission.
6.10. Mutation of the hyccin gene with leukodystrophy
An unusual autosomal recessive disorder with hypomyelination of both the CNS and PNS, in association with development of congenital cataracts has been reported in five unrelated families (Biancheri et al., 2007, Zara et al., 2006). Hypomyelination of the brain was noted on MRI and on peripheral nerve biopsy (Biancheri et al., 2007). While hyccin may be primarily expressed in neurons (Gazzerro et al., 2012), more recently it has been shown to play a role in the synthesis of phosphatidylinositol 4-phosphate, a determinant of plasma membrane identity, hence its deficiency may impair plasma membrane expansion and oligodendrocyte function (Baskin et al., 2016).
6.11. Mutations of the myelin basic protein (MBP) gene
Two mouse mutants that are allelic, the shiverer (shi) and myelin deficient (mld) mice were first described in the 1970s (Mikoshiba et al., 1992). In the rat, an MBP mutant, the Long Evans Shaker (les), was reported in 1995 (Delaney et al., 1995). In all of these mutants the trait is autosomal recessive though the mutations in each of these differs as well as the phenotype and the development of myelin.
-
i)
Shiverer (shi) mouse: This mutant was discovered in 1973 (Biddle et al., 1973) and was the subject of intense morphologic, biochemical and molecular investigation in the 1970′s and 80′s (Campagnoni and Skoff, 2001). Since that time there have been many fewer publications on the shi mouse with the exception of its use as a recipient of transplanted glial cells. Affected mice have a severe tremor and die around 140 days. The shi mutation is a deletion of exons 3–7 of the MBP gene. In the spinal cord, there is a paucity of myelinated axons and the myelin present is poorly compacted and lacks major dense line (site of localization of MBP). Biochemically and immunohistochemically, there is a global lack of MBP. Despite this, there is a gradual increase in the number of myelinated fibers over time; in the fasciculus gracilis at 2 weeks of age 55.8% of axons are myelinated compared to 77.4% at 20 weeks (Inoue et al., 1983).
-
ii)
Myelin deficient (shi mld) mouse: This mouse has a unique mutation involving the 3′ portion of the gene with a duplication that generates two tandem genes, the first that contains an extensive inversion. This results in the formation of an antisense MBP mRNA that interrupts transcription of MBP with a severe reduction in MBP and generation of myelin (Matthieu et al., 1984, Popko, 1988) that lacks a major dense line. However, with time more MBP is detected in shi mld (from 30 to 135 days) with an increase in the number of myelinated axons that coincided with the appearance of major dense lines (Matthieu et al., 1984).
-
iii)
Long Evans Shaker (les) rat: This mutation was reported in 1995 and was found to have a similar phenotype to the mouse MBP mutants. It is inherited as an autosomal recessive trait, develops a tremor at 10–14 days and seizures around 4–5 weeks (Delaney et al., 1995). However in contrast to shi and shi mld, the les rat can survive for up to 9–12 months, though retaining significant neurologic deficits. The mutation in les is an insertion of an endogenous retrotransposon into intron 3 of the MBP gene that leads to a failure of transcription and subsequent absence of MBP protein (O'Connor et al., 1999) (Fig. 7A). In contrast to both shi and shi mld, a substantial number of axons are myelinated (≈ 47% of large diameter axons) in the spinal cord at 2 weeks, but by 3 months < 1% of these axons have retained their sheaths (Smith et al., 2013a). The myelin that is formed is abnormal however, and is poorly compacted with no major dense line (Kwiecien et al., 1998, Smith et al., 2013a) (Fig. 7C). During this time, OLs accumulate multiple organelles and undergo autophagy (Smith et al., 2013b). Like shi (Bu et al., 2004), there is an increase in OL number possibly as a result of increased progenitor division in response to the global absence of myelin (Kwiecien et al., 1998, Smith et al., 2013b).
Fig. 7.
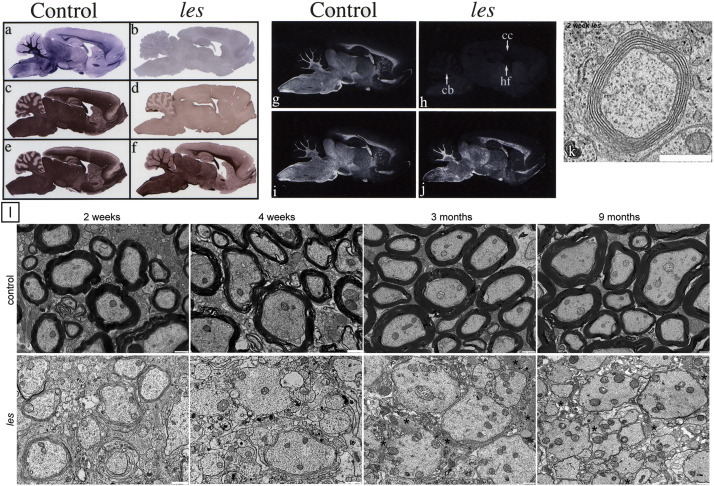
Mutation of the MBP gene in the Long Evans shaker (les) rat results in profound lack of myelin in the brain and loss of rudimentary myelin in the spinal cord. Staining of the brain of the les (b) rat fails to demonstrate myelin compared to controls (a). No detectable MBP immunostaining was evident throughout the CNS of the les rat (c, d), whereas levels of PLP appear to be only modestly reduced compared with that in controls (e, f) (44 day old les rats and controls). No MBP mRNA was detected throughout the les brain, whereas the expression and distribution of PLP mRNA appear similar to that in the control. g–i), Dark-field photomicrographs of 44-d-old sagittal brain sections from les (h, j) and control (g, i) rats hybridized with 35S-labeled probe for MBP (g–h) and PLP (i–j) mRNA. Robust expression of PLP mRNA in les verifies that normal numbers of mature oligodendrocytes are present in the mutant CNS. cb, Cerebellum; cc, corpus callosum; hf, hippocampal fimbria. k. An example of a myelinated axon in les. Although it is uncompact, the membrane sheaths in les form an organized multilamellar structure that is characteristic of a myelin sheath. Scale bar, 1 μm. l. Electron micrographs of the spinal cord from 2 week, 4 week, 3 month, and 9 month control and les animals. At 2 weeks, control rats begin to develop myelin sheaths that persist as the animal ages. In contrast, at 2 weeks, les animals develop thin, uncompact myelin sheaths. However, most of this attempt at myelination is lost by 4 weeks, and at later ages there is practically no myelin in the CNS. Scale bar, 1 μm.
Panels a–j reproduced from O'Connor et al. (1999), J Neurosci, 19, 3044; k–l reproduced from Smith et al. (2013a), J Neurosci, 33, 2719 — with permission.
6.12. The taiep rat
The taiep rat mutant was discovered serendipitously by Ruth and Bjorn Holmgren in Puebla, Mexico in 1989 as a spontaneous mutation in a colony of Sprague Dawley rats (Holmgren et al., 1989). By careful back crossing, they identified it as an autosomal recessive mutation and made the important observation that the brains of mutant rats were lighter in weight than control littermates though there were no gross abnormalities suggesting a lack of myelin. Taiep is a unique myelin mutant in that it initially demonstrates hypomyelination of the CNS then develops severe demyelination of the brain and optic nerves, fasciculus gracilis and corticospinal tract of the spinal cord (Duncan et al., 1992, Lunn et al., 1997, O'Connor et al., 2000) (Fig. 8a, b–f, i–p). This leads to a progressive neurologic disease though affected rats can live up to two years. We and others have proposed that these defects in myelin relate to the progressive accumulation of microtubules in OLs (Couve et al., 1997, Song et al., 2003, Song et al., 1999). Ultrastructural examination showed a marked progressive accumulation of microtubules in the OL cell soma and its processes (Fig. 8g–l).
Fig. 8.
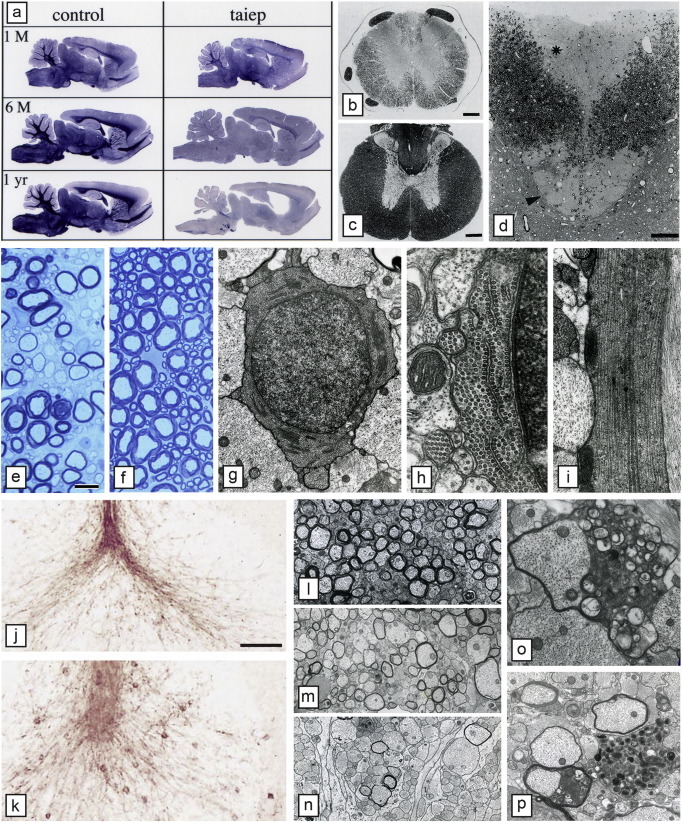
Hypomyelination then demyelination in the taiep rat result from microtubule accumulation in oligodendrocytes. a) Sagittal sections of the brain of control and taiep rats at 1, 6 and 12 months show that the taiep rat has myelin at 1 month though less than control but myelin is gradually lost throughout the brain and brainstem. The spinal cord area of the taiep cord at 6 months (b) is less than control (a). This is most obvious in the dorsal column where by 12 months the fasciculus gracilis (asterisk) and corticospinal tract (arrowhead) have no obvious myelin (d). At 4 months, the ventral column of the taiep spinal cord (e) contains non-myelinated and hypomyelinated axons compared to controls (f). EM of the 4 month taiep spinal cord shows accumulation of microtubules in the cytoplasm of an OL (g) that are forming arrays, aligned with smooth ER (h). Processes of OLs contained densely packed microtubules (i). Accumulation of microtubules leads to failure of transport of certain proteins such as MAG that accumulate around the cell body of OLs in the taiep cerebellum (k) not seen in controls (j). Myelin is gradually lost in the optic nerve of taiep from 3 months (l), 6 months (m) and 12 months (n). Occasional examples of myelin breakdown are seen (o) and rare degenerating axons (p). Scale bar: 20 μm (e, f), 100 μm (j, k).
Panel a, reproduced from O'Connor et al. (2000), MCN, 16, 396; b–d, reproduced from Lunn et al. (1995), Microsc. Res. Tech., 32, 183; o, reproduced from Duncan et al. (1992), J. Neurocytol, 21, 870. All with permission.
Cultured taiep OLs recapitulated the microtubule defect and showed clear differences in protein transport in taiep OLs (Song et al., 2003). In addition it was shown that the transport of MBP mRNA was compromised in mutant cells that on EM were shown to have an increase in microtubules with an abnormality in their polarity (Song et al., 1999). In conclusion it appears that the accumulation of microtubules creates a physical barrier to the transport of myelin components that use this network. This initially results in hypomyelination and later demyelination, especially of small axons. The taiep gene has been mapped to rat chromosome 9 (Li et al., 2003) but the mutation is unknown.
6.13. Other rat myelin mutants
In addition to the md, taiep, and les spontaneous rat mutants, a number of other mutants of autosomal recessive origin have been described and investigated in Japan, in particular by Kuwamura and colleagues. The genes involved in these mutations are diverse and apparently unlikely candidates in myelin disease, yet this reflects the broad range of genetic disorders that affect myelin formation and maintenance. These are shown below.
6.13.1. Demyelination mutant (dmy) rat
This mutation was first described in 1996 in a Sprague Dawley colony (Kuramoto et al., 1996). Homozygous rat pups appear normal until around 6 weeks of age when they develop hindlimb ataxia with later paresis then paralysis (Kuwamura et al., 2004). If hand reared however, rats can survive for up to two years (Kuramoto et al., 1996). Myelination commences normally in dmy but at the time of onset of clinical signs, marked vacuolation of the lateral and ventral columns of the thoracic spinal cord develop yet the dorsal column is unaffected. Similar lesions are seen throughout the major white matter tracts of the brain. Myelin vacuolation leads to extensive demyelination and there is a notable decrease in the number of mature OLs. Interestingly, there is a marked accumulation of iron in astrocytes which was suggested to result in oxidative stress in OLs. Positional cloning identified a mutation in the mitochondrial Mg2 + transporter gene (Mrs2), resulting in mitochondrial dysfunction of OLs leading to demyelination (Kuramoto et al., 2011). Transgenic rescue in two lines of rats expressing Mrs2 wild-type cDNA confirmed this as the causative mutation. Thus the dmy rat provides significant opportunities to study mitochondrial dysfunction and myelin breakdown.
6.13.2. Vacuole formation (vf) rat
This mutation was found in an inbred strain of WTC-tm rats (Tanaka et al., 2012). A mild tremor is prominent at 4–8 weeks of age then disappears with time. There is moderate myelin vacuolation in the ventral and lateral columns of the spinal cord and in certain areas of the brain (Tanaka et al., 2012). These areas are hypomyelinated throughout life (up to 80 weeks) though the vacuolation subsides by 20 weeks of age. The likely causative gene is Dopey1, a member of the Dopey leucine zipper-like family (Tanaka et al., 2014). Dopey1 is expressed mainly in OLs and neurons (Tanaka et al., 2014). The DOPEY1 protein is localized to the Golgi apparatus in OLs and is involved in endosome transport in Golgi. Dilation of Golgi in the vf rat OL might reflect the accumulation of myelin proteins including PLP and MAG as demonstrated immunocytochemically (Tanaka et al., 2012). The potential primary involvement of the Golgi apparatus makes vf a unique myelin mutant.
6.13.3. Myelin vacuolation (mv) rat
This mutation was also found in Sprague Dawley rats (Kuwamura et al., 2002). Homozygous rats develop a tremor at around 3 weeks of age that progresses and leads to occasional seizures though most rats lived at least 25 weeks of age. Hypomyelination is seen in the spinal cord from 2 weeks and becomes more prominent with time as does myelin vacuolation. No abnormalities are seen in OLs nor are they increased in number, though gliosis is prominent as microglial activation. The mutation in the mv rat is in the attractin (Atrn gene). Atrn is expressed in OLs (Izawa et al., 2008) though in the mv rat and there is a complete lack of Atrn positive cells. Attractin is a glycoprotein with multiple functions, yet the mechanism for OL dysfunction in this mutation is unknown (Izawa et al., 2008).
6.13.4. The zitter (zi) rat
Zitter (zi) was also found as an autosomal recessive mutation in the Sprague Dawley rat and is an allele of the myelin vacuolation (mv) rat (Kuramoto et al., 2001). It develops a fine tremor at 3 weeks of age and paresis of the hind limbs at 6 months (Rehm et al., 1982). At 3 weeks of age there are clearly fewer myelinated axons in the optic nerve and spinal cord than controls, though myelin sheath thickness appears normal (Kondo et al., 1992), suggesting that there may be an impairment of differentiation of OLs (Sakakibara et al., 2008). It has been proposed that this delay in differentiation is related to an increase in macrophage/microglial activation, with increased expression of certain cytokines (Sakakibara et al., 2008). As an allele of the mv rat, zi has also been found to have a mutation in the Atrn gene (Kuramoto et al., 2001). Likewise, a mutation in Atrn has been found in a Syrian hamster (Kuramoto et al., 2002).
6.14. The quaking (qk) mouse
Quaking is an autosomal recessive murine mutation also known as quaking viable (qk v), that results in marked delay and hypomyelination of the CNS in the early stages of development (reviewed by Nagara and Suzuki, 1981). In essence, qk v OLs generate compact myelin at a slower rate than controls and though myelin sheath thickness increases with time, at four months, hypomyelination appears to persist (Nagara and Suzuki, 1981). qk v has more OLs than controls in early myelination, and these cells frequently contain abnormal vesicles and cisterns. It was the first murine mutant to be identified in which the mutation did not involve a myelin protein gene. The genetic defect in the qk v mouse arises from a large deletion on chromosome 17. The mutation results in a deletion in the enhancer of the qkI gene resulting in diminished transcription of qkI (Ebersole et al., 1996), one of three isoforms of the QKI selective RNA-binding proteins expressed in glia. While other genes have been implicated as potential causative genes such as parkin, rescue of the qk v phenotype by a QKI-6 transgene expressed specifically in OLs confirms the role of QKI in this mutant (Zhao et al., 2006).
7. Acquired disorders of myelin
7.1. Multiple sclerosis (MS)
Multiple sclerosis is the archetypal demyelinating disorder of humans affecting 2.5 million people world-wide and being the most common debilitating neurologic disorder of young adults Compston et al. (2006). It is more common in women than men and this gender difference may be increasing. It is most frequently diagnosed in the 2nd–3rd decade and 80–85% of those patients will develop relapsing-remitting disease with the majority developing secondary progressive disease at a later time point. Around 15% of patients diagnosed with MS will have primary progressive disease in which no remissions are seen (Compston et al., 2006). Despite being first described in 1838, the cause of MS remains unknown and there is no cure, yet major advances have been made in its treatment, primarily through immunomodulatory therapies. MS is an inflammatory disease of the CNS and is likely to be immune mediated (Lassmann, 2014). It is clear from the voluminous descriptions of the pathology and immunopathology of MS that it is not a purely demyelinating disease of the WM and that axons are lost both in acute and chronic lesions (Lassmann et al., 2007) with neural loss and significant sub-cortical demyelination (Frohman et al., 2006, Lassmann, 2014, Stadelmann et al., 2011). These latter changes have highlighted the significance of cognitive abnormalities in many MS patients. In addition to demyelination and axon and neural loss, there is a notable astrocyte response and chronic lesions are characterized by severe gliosis (Fig. 9 ).
Fig. 9.
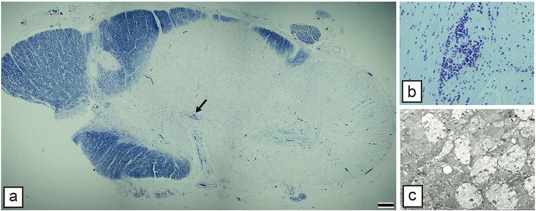
Myelin loss in the MS spinal cord can be severe. In the spinal cord from a patient with secondary progressive MS, almost two thirds of the cervical spinal cord is demyelinated. Perivascular cuffs of T cells (arrow in a) and in inset b) persist though inflammation is less likely than earlier in disease. c) EM of an area from a similar large plaque shows demyelinated axons embedded in gliotic scar. Scale bar: 1 mm (a).
Given the complexity of the neuropathology of MS, strenuous efforts have been made to classify lesion types to help the understanding of the pathophysiology of this disorder and how myelin damage occurs. In the 1990′s Luchinetti and Lassman and colleagues (Lucchinetti et al., 2000) defined four pathological sub-types of the disease. Patterns I and II showed demyelination, frequently periventricular, with preservation of OLs. In Pattern II, there was deposition of IgG and activation of complement. Patterns III and IV showed demyelination unrelated to the vasculature, with T cell and macrophage infiltration. In addition there was marked OL loss in both III and IV and in Pattern III, a preferential loss of the myelin-associated glycoprotein suggestion a dying-back oligodendrogliopathy (Lucchinetti et al., 2000). These patterns were thought to be patient-specific, with homogeneity of pattern type in patients examined at autopsy, though heterogeneous between patients.
This lesion classification has been the topic of much discussion (see Stadelmann et al., 2011) since that time, particularly with regard to the homogeneity of lesions in individual patients. In a challenge to the dogma, Barnett and Prineas (2004) suggested that OL death by apoptosis might precede the inflammatory response, based on observations of lesions in patients who had died shortly after the onset of a relapse. The issue of OL death by apoptosis is controversial however as others have found no evidence of apoptotic OLs in MS lesions (Bonetti and Raine, 1997, Breij et al., 2008). In addition, experimental induction of primary OL death in a mouse model, did not result in CNS inflammation (Locatelli et al., 2012). However, this result has recently been challenged in an alternative model of conditional OL ablation, in which mice developed a secondary bout of disease with activation of CD4 + T cells and late-onset demyelination (Traka et al., 2015). These different outcomes were attributed to differences in the models and the early death of mice in the models of Locatelli et al (Traka et al., 2015). In addition to the ongoing debate on OL death and inflammation and which comes first, the basic tenant that MS is an autoimmune, inflammatory disease of the CNS has been challenged and an alternative hypothesis is that it may be a neurodegenerative disorder with secondary inflammation (Stys et al., 2012, Trapp and Nave, 2008). This ‘inside-out’ theory where axonal changes are the primary event has also been postulated to occur in virus-induced demyelination (Tsunoda and Fujinami, 2002). At present these two hypotheses remain on the table though the majority view remains that inflammation drives the disease.
In regard to the current issue of myelin damage discussed here, does this extensive background suggest a mechanism for myelin damage and loss in MS? Actual damage to myelin sheaths in MS has been summarized by Lucchinetti et al. (1996) to be represented by four patterns, 1) Receptor-mediated phagocytosis of myelin. This is mediated through the interaction of coated pits and vesicles on macrophages with myelin sheaths (Prineas, 1985), 2) Myelin stripping. Lymphocytes and macrophages can be seen to invade myelin sheaths and disrupt the normal myelin, myelin-axon structure, 3) Vesicular disruption of myelin. This is seen mainly in acute lesions where there is notable vesiculation of the myelin sheath and vacuole formation (Frohman et al., 2006, Prineas, 1985), and 4) Dying back oligodendrogliopathy. This is the hallmark change seen in Pattern III lesions in MS (Lucchinetti et al., 2000) and myelin damage is the result of primary OL dysfunction.
7.2. Experimental autoimmune encephalomyelitis (EAE)
This is undoubtedly the most commonly used model in studying the pathophysiology of MS. EAE is an experimental model in which an immune response against myelin and OLs is generated by immunization of a variety of animal species including mice, rats and non-human primates with whole spinal cord, myelin proteins, or defined peptides, usually in complete Freund's adjuvant (Brok et al., 2001, Jagessar et al., 2010, Merkler et al., 2006, Raine et al., 1999, Ransohoff, 2012). The subsequent disease has immunological and pathological similarities to MS and in some cases, a relapsing–remitting clinical course. However, there are significant differences and EAE is not identical to MS. Despite the differences it has been extremely useful in helping dissect the immunopathology of MS and in the pursuit of MS therapies, and if utilized appropriately, EAE remains the closest model to MS (Denic et al., 2011, Gold et al., 2006, Ransohoff, 2012, Steinman and Zamvil, 2006).
The pathologic hallmark of EAE is inflammation and indeed many of the original protocols to generate EAE, resulted solely in inflammation and axon degeneration and little or no demyelination. Presently, the most commonly used EAE protocol uses C57BL/6 mice immunized with MOG peptide (residues 35–55) emulsified with Freund's adjuvant supplemented with Mycobacterium tuberculosis extract (Ransohoff, 2012). Like most EAE models, this results in primarily spinal cord disease. In general, involvement of the brain and optic nerves is less common in many EAE models. An alternative rodent model in which there is marked demyelination in the immunization of dark agouti (DA) rats with MOG peptide (Storch et al., 1998). A similar immunization protocol in marmosets, also produces significant spinal cord demyelination and optic nerve lesions (Jagessar et al., 2010).
We have utilized an alternative model of EAE in Biozzi (ABH) mice that are immunized with whole spinal cord homogenate with Freund's adjuvant (Fig. 10 ) (Baker et al., 1990). This is a very useful model as unlike many EAE models where there is a monophasic illnesses, this model relapse and remits spontaneously, sometimes with up to three relapses, hence it may mimic the course of relapsing-remitting MS. While there is significant demyelination in the spinal cord associated with perivascular cuffing and infiltration of the CNS with T cells, B cells, and macrophages, axonal degeneration also occurs and this is cumulative (Fig. 10). Indeed the progressive neurologic worsening seen after successive relapses correlates well with progressive axon loss (Hampton et al., 2008, Jackson et al., 2009). This correlation has also been found in the DA rat/MOG model (Papadopoulos et al., 2006).
Fig. 10.
EAE is an inflammatory, demyelinating disease with extensive cell infiltrate of T cells, B cells and macrophages from the periphery. All sections from relapsing–remitting EAE in the Biozzi mouse. a) Longitudinal section of the thoracic spinal cord showing both sub-pial inflammation (arrows) and perivascular cuffing of all vessels. b) There is clear cuffing of the ventral spinal artery of the spinal cord with cells migrating (*) toward the spinal cord. Inset of the area marked shows both plasma cells and T lymphocytes. A small area of demyelination (arrow) is enlarged in the right inset. c) Demyelination is extensive adjacent to a partially cuffed vessel and extends into the neuropil. d) Macrophages cuffing a vessel and migrating into the CNS on the left while those that have engulfed myelin are lipid filled and are surrounding a vessel on right, likely prior to returning to the circulation. e) In a Biozzi mouse in the third relapse, the loss of myelin is uneven with the left side of the cord and part of the dorsal column being severely affected. Three areas of the spinal cord (inset) are enlarged in f–h. The dorsal column (e-asterisk) contains both areas of demyelination (f) and adjacent areas with marked axon loss (g). In the ventral cord (↑) the hallmarks of EAE are seen (h) with demyelination, remyelination and axon degeneration. Scale bar: 10 μm (b-inset, f–h), 20 μm (b, c, d), 0.5 mm (e).
As EAE lesions are found scattered randomly along the spinal cord, Kerschensteiner et al. (2004) devised a focal EAE model that was reproducible and could result in quantitatable motor deficits. They induced sub-clinical EAE with low-dose MOG (AA1–125) in incomplete Freund's adjuvant then injected a cocktail of cytokines (TNFα and IFNγ) into the dorsal column of the spinal cord, targeting the cortico-spinal tract. This resulted in large but focal lesions of the dorsal column. However, this model has not been used extensively by others, perhaps relating to a strain specificity of the Lewis rats used. More recently, focal hemispheric lesions were produced in a similar fashion in marmosets (Stassart et al., 2015). Sasaki et al. (2010) injected vascular endothelial growth factor into Lewis rats, sub-clinically immunized with MOG in attempts to create focal breaches of the blood brain barrier. This succeeded in producing quite extensive demyelination at the injection site.
It is likely that EAE will remain as the primary experimental model of MS though it has to be remembered that there are differences between the two. Attempts to study remyelination in EAE however are futile given disease variability, timing and location, etc. (Ransohoff, 2012), hence alternative models must be sought to study myelin repair.
7.3. Other human inflammatory demyelinating diseases
-
1)
Acute disseminated encephalomyelitis (ADEM)
This disorder is characterized by a monophasic illness that can be difficult to differentiate from MS, especially in children who are most frequently affected by ADEM and present with symptoms of encephalitis. It is usually associated with a viral or bacterial infection or rarely, following vaccination. The brain, spinal cord, and occasionally the optic nerves are involved. It is characterized pathologically by perivenous inflammation with T cells and macrophages with associated demyelination (Kuhlmann et al., 2008). This perivenous demyelination has been proposed to distinguish ADEM from MS where larger, confluent areas of demyelination are seen (Young et al., 2010).
-
2)
Neuromyelitis optica (Devic's disease)
Neuromyelitis optica (NMO) was previously thought to be part of the MS spectrum, but the discovery a serum autoantibody in NMO patients, resulted in its nosologic separation from MS. Lennon et al. (2004) first described the finding of an antibody (NMO-IgG) in NMO patients that was later determined to target the astrocytic water channel aquaporin-4 (Lennon et al., 2005). In NMO the primary target sites in the CNS are the cervical spinal cord and optic nerves. Acute lesions are characterized by perivenous inflammation that differs from MS with neutrophils and eosinophils as well as T and B cells (Lucchinetti et al., 2014). In addition there is prominent macrophage infiltration, severe axon loss and pronounced loss of astrocytes (Kuhlmann et al., 2008). In some cases there is pronounced vacuolation of myelin with little demyelination (Lucchinetti et al., 2014). In general, lesions in NMO are more destructive than in MS and display a wide spectrum of pathologic changes (Misu et al., 2013). While it was thought initially that lesions were seen only in the spinal cord and optic nerve, it is now clear that the brain an especially the brain stem have MRI and pathologic evidence of disease (Lucchinetti et al., 2014).
8. Infectious disorders of myelin
8.1. Viral demyelination
The potential viral involvement in the etiology of MS has led to considerable interest in viruses that cause demyelination as models to explore the pathophysiology of MS. A prevailing view derived from the epidemiology of MS is that viral exposure, early in life in those with a specific genetic background predisposes to a later autoimmune-mediated attack on the CNS (Dal Canto and Rabinowitz, 1982, Denic et al., 2011, Stohlman and Hinton, 2001). Currently, the Epstein Barr virus is the prime candidate as the possible prior infection. Theiler's murine encephalomyelitis virus (TMEV) and mouse hepatitis virus (MHV) are the most explored viruses associated with experimental CNS demyelination.
8.2. Theiler's murine encephalomyelitis (TMEV)
Mice are infected intracerebrally with TMEV and can develop a fatal encephalitis with a virulent virus strain or a less severe mono or biphasic disease with less virulent strains of TMEV. In both models the strain and dose of virus used and the host mouse strain chosen will influence the disease, its course and immunopathology. Transient inflammation of the CNS develops shortly after infection and clears in around 3 weeks. Thereafter, all mice develop a chronic demyelinating disorder that like EAE, primarily affects the spinal cord. These mice develop severe inflammation, with the invasion of T and B cells and macrophages and the activation of microglia (Denic et al., 2011, Mecha et al., 2013) that precedes demyelination. The mechanism of myelin damage and loss in TMEV remains debated however. The proposed mechanisms include, 1) TMEV-specific immune destruction of infected OLs, 2) direct viral cytopathic effects on OLs, 3) direct phagocytosis by macrophages or myelin damage due to cytokines, complement, nitric oxide metabolites and reactive oxygen species produced by these cells, 4) epitope spreading, 5) molecular mimicry, 6) primary damage to axons resulting in the “inside-out” loss of myelin, and 7) excitotoxicity (Denic et al., 2011, Mecha et al., 2013).
8.3. Mouse hepatitis (MHV)
Mouse hepatitis virus (MHV) is a coronavirus that when injected into the brain produces a monophasic CNS infection characterized by encephalomyelitis and acute demyelination unlike TMEV where no myelin is lost in the acute stage. The most commonly used MHV strain is JHMV in either Swiss mice or C57BL/6. There is predominant macrophage infiltration and microglial activation though some B cell infiltration can be seen in chronic spinal cord disease. The demyelination seen in MHV is predominantly a result of OL cell death both by necrosis and apoptosis, although only the latter is seen during chronic demyelination (Dal Canto and Rabinowitz, 1982, Erlich and Fleming, 1987, Stohlman and Hinton, 2001).
While mice are predominately used to study viral demyelination, other models do exist including caning distemper virus infection (CDV1). Demyelination is thought to precede OL loss (Schobesberger et al., 2002). However, it has also been suggested that axonal loss may precede demyelination thus invoking the ‘inside out’ theory that suggests that myelin damage is secondary (Seehusen and Baumgartner, 2010).
In humans, many other viruses are associated with disseminated encephalopathies and demyelination. These include AIDS and progressive multifocal leukodystrophies. The latter is caused by reactivation of the JC virus in immunocompromised patients ore in some MS patients treated with drugs that have profound immunosuppressive effects. In addition, a number of other viruses such as the Visna virus and measles virus can be associated with severe encephalitis and demyelination (Stohlman and Hinton, 2001).
9. Myelinotoxic/glia-toxic chemicals
There is a broad range of chemicals that either damage myelin or OLs or both. While the majority are only used experimentally, some were originally discovered after outbreaks of neurologic disease following human exposure. In experimental animals they are either delivered orally (cuprizone), by direct injection into the CNS (lysolecithin, ethidium bromide) or by absorption from the skin (hexachlorophene).
9.1. Hexachlorophene (HCP)
Hexachlorophene (HCP) is a compound previously used to control staphylococcal infections of the skin in newborn infants (Towfighi and Gonatas, 1976). However a number of reports identified its CNS toxicity, with widespread white matter vacuolation of the CNS and PNS. This clinical disorder can be modeled in newborn rats by rubbing 3% HCP on their skin for 3 weeks or by feeding a diet containing HCP for up to a month (Towfighi et al., 1974). Both newborn rats and those aged 1–3 months were found to be susceptible to this treatment. Similar work on baboons has confirmed that non-human primates also develop white matter vacuolation on HCP exposure (Tripier et al., 1981) as well as numerous other species. White matter vacuolation results from splitting of myelin at the intraperiod line. Both the CNS and PNS can be severely vacuolated and most areas of WM of the CNS including optic nerves are affected. Interestingly, discontinuation of HCP exposure leads to a significant resolution of the vacuolation (Fig. 11 ) with only a few vacuoles remaining (Towfighi et al., 1974). The key question is whether this is a result of resorption of the fluid within vacuolated myelin sheaths or from remyelination. Even in severe disease, it appears that demyelination is relatively rare. In the PNS there are occasional examples of thin myelin sheaths seen on longitudinal sections that imply remyelination (Towfighi and Gonatas, 1976). The degree of WM vacuolation varies throughout the CNS and in the optic nerves, the sub-pial areas are affected while the core of the nerve is not (Towfighi et al., 1974). In general, the larger diameter fibers appear to be more affected in both the CNS and PNS. Despite profound WM pathology, axons are largely preserved except for the optic nerve where prolonged exposure to HCP leads to axon loss and gliosis (Towfighi et al., 1974). This is likely due to compression of the edematous nerve in the optic canal.
Fig. 11.
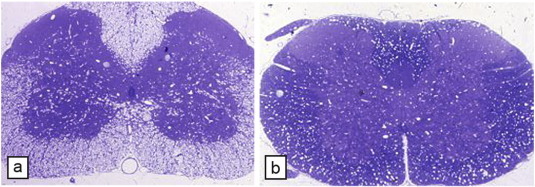
Topical hexachlorophene in the rat pup results in status spongiosus of the white matter. Four weeks after topical administration of hexachlorophene, the entire WM is vacuolated (a) but 2 weeks after stopping treatment (b), myelin appears to have recompacted and only scattered vacuoles remain.
The mechanism of HCP-induced myelin vacuolation has been debated based on its biochemical properties, yet is not resolved. HCP binds to myelin membranes and certain other plasma membranes including certain cells of the retina (Towfighi and Gonatas, 1976). It is a potent uncoupler of oxidative phosphorylation hence it may affect ionic balance in the neuropil. The fact that discontinuation of HCP treatment results in a major decrease in vacuolation may be instructive to myelin repair possibilities in other vacuolating disorders such as Canavan's disease. A better understanding of how vacuolation resolves and whether myelin sheaths can simply ‘recompact’ is required.
9.2. Triethyl tin (TET)
Like HCP, triethyl tin intoxication (TET) was first discovered as a WM toxicant after an outbreak of organotin poisoning in France in 1950 (reviewed in Duncan, 1985). Myelin vacuolation has been produced in rats and is similar to that seen following HCP exposure though the PNS is less severely affected. TET given intravenously to rats produces an acute onset of myelin vacuolation within the CNS (Watanabe, 1980). Termination of TET treatment leads to a resolution of the vacuolation, again like HCP. While the exact mechanism of action is not known, it is thought to be a direct consequence of TET binding to myelin and not through a primary involvement of astrocytes (Jacobs et al., 1977) as been suggested by others. Vacuolation induced by TET may share similar mechanisms to HCP-induced myelin edema.
9.2.1. Lysolecithin (lysophosphatidylcholine)
Lysolecithin (LPC) has been extensively used to create focal demyelinating lesions in the spinal cord and brain of a number of species as demonstrated by Blakemore and colleagues in many publications (e.g. Blakemore, 1978, Jeffery and Blakemore, 1995). Direct injection of lysolecithin into the CNS creates focal areas of myelin loss that are subsequently remyelinated by endogenous cells. LPC acts directly on the myelin sheath as a detergent disrupting myelin membrane and leading to the formation of micelles. Vesicular disruption of myelin occurs but there is no evidence of vacuolation as occurs with HCP and TET. Injection of LPC into the nervous system to produce demyelination was first reported in the mouse sciatic nerve (Hall and Gregson, 1971) and then in the spinal cord (Hall, 1972). Myelin breakdown occurs almost immediately on injection into the CNS, with myelin debris being phagocytosed shortly thereafter. The rapid macrophage recruitment into lesions may be driven by a brief influx of T cells (Ghasemlou et al., 2007). There is little evidence that LPC kills either OLs or astrocytes hence it can be viewed as a primary myelinotoxic compound. Focal lesions with LPC have mainly been performed in the spinal cord (of rodents and cats) but also can be used focally in the brain in the corpus callosum, cerebellar peduncle and optic nerves. EM evidence of remyelination can be seen as early as three days after lesion induction.
9.3. Ethidium bromide (EtBr)
Ethidium bromide (EtBr) is a potent nucleic acid intercalating agent and inhibitor of mitochondrial RNA, DNA and protein synthesis in mammalian cells. Yajima and Suzuki (1979a) injected EtBr intracisternally in 3 week old rats and demonstrated severe status spongiosus of the brain stem at 3 days after injection, with myelin splitting at the intraperiod line (Yajima and Suzuki, 1979b). There was extensive degeneration of OLs that preceded by accumulation of abnormal concentric scrolls of RER prior to death. Six days after injection of EtBr, the majority of axons at the center of the lesion were demyelinated, and there was notable loss of OLs and astrocytes. Similar lesions have been described following injection of EtBr in the cat spinal cord, though the lesions have more prolonged demyelination and subsequent remyelination (Blakemore, 1982). The concentration of EtBr used for focal injection is critical in determining the extent of demyelination and axon loss (Kuypers et al., 2013). There are also significant differences between the mouse and rat in terms of dosage needed to create pure demyelinating disease versus significant axon loss. Myelin vacuolation is present in EtBr lesions, unlike in LPC-induced myelin loss.
9.4. Zymosan
A much less commonly used myelin toxin is zymosan, a yeast cell wall preparation that is a potent activator of microglia/macrophages (Popovich et al., 2002, Schonberg et al., 2007). Zymosan is a TLR2 agonist and has been used to create focal inflammatory lesions in the rat spinal cord characterized by patchy demyelination, marked inflammation, astrocytosis and some axon loss. The myelin breakdown may be associated with myelin vacuolation. Unlike the other myelinotoxic chemicals used to study myelin breakdown, the zymosan lesion is driven by the inflammatory response which is more severe than that seen in other models. We have used the lesion to study migration of potential remyelinating cells toward focal inflammatory demyelinated lesions (Piao et al., 2013, Wang et al., 2011), but the limited demyelination seen in these lesions and the difficulty in reproducing comparable myelin damage is a limitation to its future use.
9.5. Cuprizone
Cuprizone is a copper chelating agent that when fed to mice results in OL cell death with subsequent demyelination of a limited number of areas of the brain. Its neurotoxicity was first recorded by Carlton (reviewed by Gudi et al., 2014) who described the development of hydrocephalus, status spongiosus and demyelination in mice fed cuprizone. Blakemore (1972) described the death of OLs in the brain white matter and noted that remyelination occurred after cessation of the cuprizone diet. Ludwin (1980) developed a chronic model of cuprizone toxicity and showed that remyelination was impaired in contrast to the more acute cuprizone exposure. Thereafter, a gap of almost 18 years occurred before Matsushima and Morell (2001) re-evaluated the cuprizone model from a cellular and molecular basis. Since that time, cuprizone has become the de facto model in which to study demyelination and remyelination of the CNS.
Given its prominence as a model of demyelination and remyelination, several extensive previous (Matsushima and Morell, 2001) and more recent reviews (Gudi et al., 2014, Kipp et al., 2009) have discussed cuprizone toxicity in great detail. Therefore, only a summary of the model will be presented here along with some key facts about its use. In order to establish a reproducible model system, which is a key factor in its wide usage, certain important issues have to be considered. These include, 1) concentration of cuprizone in the food and dosage regimen, 2) the age and strain of the mouse to be used, and 3) the anatomical structure/s in the brain to be studied. The most commonly used model system uses 6–9 week old mice fed a diet containing 0.2–0.3% cuprizone. This concentration must be increased to produce demyelination in older mice. Such a diet will produce extensive demyelination after 4–5 weeks of treatment and extensive remyelination after 3–4 weeks off cuprizone treatment. In Blakemore (1973) initial studies, he chose the rostral cerebellar peduncle as the most reliable tract in which to study myelin breakdown and repair. However, the corpus callosum is also a target of cuprizone and has now become the first choice as the site to study. Other anatomic sites can also be affected by cuprizone, such as the cerebral and cerebellar cortex (Skripuletz et al., 2010, Skripuletz et al., 2008), though definitive structural evidence of demyelination may be more difficult to identify in these sites. It is also known that axons can be damaged by cuprizone exposure especially after long-term dosage (Lindner et al., 2009) and in older mice (Irvine and Blakemore, 2006), though some of this axon death may be reversible (Xie et al., 2010).
Cuprizone has the significant advantage over the other myelinotoxic and gliotoxic chemicals as it can be given orally thus avoiding the need for direct injection into the CNS (as with lysolecithin and ethidium bromide). However, there are some caveats to its use that must be considered. Cuprizone is not only a neurotoxicant but it is hepatoxotic and this may have secondary effects on the CNS. It only affects limited areas in the CNS and not the optic nerves or spinal cord which are much easier structures to study and quantitate demyelination and remyelination. While the corpus callosum is straightforward to sample and analyze, it contains only small to medium caliber myelinated axons hence it is not representative of the entire white matter. In addition, care must be exercised in sampling the callosum as the genu, body and splenium have different myelinated fiber spectrum and the myelin sheath thickness may have the appearance of remyelinated axons, i.e. thin myelin that make quantitation of repair difficult (Stidworthy et al., 2003). There appears to be a clear rostro-caudal gradient of demyelination in the corpus callosum (Steelman et al., 2012, Wu et al., 2008, Xie et al., 2010). Finally, the mechanism of OL death in cuprizone is not fully understood (Gudi et al., 2014, Kipp et al., 2009). This concern also exists for some other models and does not dismiss the usefulness of the study of subsequent events, i.e. remyelination. The reason that cuprizone does not affect OLs globally is worthy of consideration.
There will likely be continued modifications to the model that will enhance its use. For example, in order to explore the AKT/mTOR signaling pathway in myelination, Sachs et al. (2014), injected rapamycin, a potent inhibitor of mTOR into mice on a cuprizone supplemented diet. This resulted in more complete demyelination and slower remyelination. Models such as this as well as those where cuprizone treatment is combined with specific genetic manipulation, will add to usefulness of cuprizone model as a model in studying myelin loss and repair.
9.6. Feline irradiated diet-induced demyelination (FIDID)
There have been three reports of this enigmatic demyelinating disorder in cats fed a diet irradiated at 30 kGy or more (Caulfield et al., 2009, Duncan et al., 2009). Four to six months after feeding the irradiated food, cats develop a progressive neurologic disease with hind limb ataxia and paresis. Over 2–5 weeks this worsens and cats may become paraplegic with urinary incontinence. Eventually the fore limbs may develop paresis. The cranial nerves are primarily uninvolved although vision can be reduced or lost (optic nerve) on rare occasions.
In early, acute disease there is striking vacuolation of the spinal cord though the dorsal column is less severely affected than ventral and lateral columns (Figs. 12a and 11c). Many myelin sheaths are vacuolated yet axons are intact. This vacuolation leads to myelin degeneration and demyelination though subsequent remyelination occurs simultaneously but is incomplete (Fig. 12). In contrast to the spinal cord, the optic nerves can be totally demyelinated (Fig. 12f) with only sparse evidence of remyelination. If cats are returned to a non-irradiated diet at this stage, there is slow neurologic recovery and by 6–8 weeks, neurologic function can be normal. Evaluation of the CNS at this stage shows complete remyelination of the spinal cord and optic nerve. The brain shows more scattered vacuolation though it can be seen in practically all white matter tracts. The cause of FIDID remains an enigma as the reason that irradiated food causes CNS demyelination remains a mystery. While the cause of the myelin sheath vacuolation and degeneration in FIDID may have similarities to other vacuolar myelin disorders, it may be unique to the cat. Nonetheless, the global demyelination/remyelination scene sets it apart from other animal models and makes it a valuable model in which to explore remyelination and how it can be promoted.
Fig. 12.
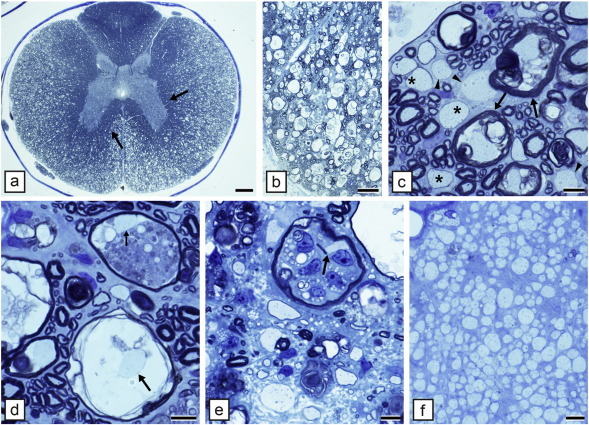
The myelin changes in acute feline irradiated diet induced demyelination (FIDID). a) In early disease there is severe vacuolation of the ventral and lateral columns of the spinal cord, though not in the dorsal column and deep white matter adjacent to the grey matter (arrows). b) Higher power of the mid-line ventral column confirms the generalized vacuolation. c) Details of the cellular changes in b). Myelin breakdown with intact axons is widespread (arrows) as are demyelinated (arrowheads) and remyelinated axons (asterisks). d) One axon is present in a vacuolated myelin sheath (arrow) and in another (small arrow), the axon is bordered by myelin debris. e) Within the myelin debris, numerous macrophages are seen. f) The optic nerve of an affected cat is completely demyelinated. Toluidine blue 1 μm sections. Scale bar: 1 mm (a), 200 μm (b), 20 μm (c–f).
9.7. Myelin vacuolation disorders (acquired and genetic)
In addition to Canavan's disease, there is a wide range of disorders where myelin vacuolation is also seen, either at a mild level or reflected by spongiosus of the entire white matter. These include the following.
-
1)
Toxins: Experimentally there are a great number of chemicals that produce myelin vacuolation, including cycloleucine (Greco et al., 1980), sodium cyanate (Tellez et al., 1979), actinomycin D (Rizzuto and Gambetti, 1976), isoniazid (Blakemore et al., 1972), 6-amino-recotinamide (O'Sullivan and Blakemore, 1980). In domestic animals, status spongiosus has been ascribed to poisoning by a grass-like plant Stypandra imbricata (Huxtable et al., 1980) and to the antiparasitic drug Closantel (van der Lugt and Venter, 2007). Though vacuolation can be severe, in the majority there is no evidence of demyelination. Both copper deficiency (humans and sheep) and copper toxicity result in a vacuolation of WM.
-
2)
Deficiency:
-
i)
Vitamin B12. Deficiency of this vitamin is the cause of sub-acute combined degeneration — SCD which is perhaps the ‘purest’ demyelinating disease of humans (McCaddon, 2013, Miller et al., 2005). Treatment with vitamin B12 early in the course of disease results in complete recovery, likely through remyelination. There are models of SCD, including B12 deficiency in Rhesus monkey (Agamanolis et al., 1978) and fruit bat (Green et al., 1975). While the disorder in monkeys clearly results in demyelination and remyelination, it requires 3 years of B12 deficiency to develop. A third proposed model of in rats in which gastrectomy (to remove the site of production of intrinsic factor required for B12 absorption) is performed, produces extensive vacuolation of the CNS but no definitive evidence of demyelination (Scalabrino et al., 1995).
-
ii)
Pontine myelinolysis. Rapid correction of hyponatremia in patients can result in severe vacuolation of the pons (central pontine myelinolysis). This is a rare and sometimes fatal condition, yet its anatomic location and pathophysiology are poorly understood. A rat model of pontine myelinolysis has been reported (Rojiani et al., 1994). Observation on the pons from patients with CPM suggests that there is focal apoptosis of OLs (DeLuca et al., 2002).
-
3)
Naturally occurring disease — unknown origin:
-
i)
The best described is a WM disorder of the CNS in Norwegian silver foxes where there is vacuolation of both myelin and OLs (Hagen and Bjerkas, 1990, Hagen et al., 1990) with demyelination then spontaneous remyelination. Whether this results from a toxin or is familial is not known.
-
ii)
A progressive neurologic disease was described in two Labrador retrievers, beginning at 4–6 months of age. There was widespread myelin vacuolation throughout the CNS but no myelin breakdown. Abnormal mitochondria were found in astrocytes raising the possibility that these were involved in the myelin vacuolation (Zachary and O'Brien, 1985).
-
iii)
Vacuolar myelinopathy has been reported in Bald Eagles, American Coots, and Red-Tailed Hawks. It appears that the disease arose in Coots due to an environmental toxin (not identified) and was transmitted to Bald Eagles who ate Coots and in Hawks who experimentally fed affected Coot carcasses (Fischer et al., 2003, Thomas et al., 1998).
-
iv)
A myelin vacuolating disease has been reported in hedgehogs in both North America (atelerix) and in Europe (erinaceus sp) (Graesser et al., 2006, Palmer et al., 1998). There is progressive ataxia and paresis associated with myelin vacuolation in the brain and spinal cord. There was little to no evidence of demyelination (Palmer et al., 1998).
-
4)
Genetically induced myelin vacuolation:
-
i)
Connexins: Deletion of the astrocytic connexins Cx30 or Cx40 in the mouse results in both OL and myelin vacuolation (Lutz et al., 2009). The loss of gap junctions in astrocytes and subsequent white matter changes confirm the potential role that astrocytes can have in myelin and OL vacuolation. Deletion of the OL connexins Cx32 and Cx47 also resulted in myelin vacuolation that was most severe in the double knockout (Menichella et al., 2006). These studies confirm the importance of gap junction connections between OLs and astrocytes and K+ buffering.
-
ii)
Galactocerebroside and sulfatide: Knockout of the enzyme UDP-galactose: ceramide galactosyltransferase in mice results in the failure of expression of these two lipids in myelin (Coetzee et al., 1996). These mice developed a tremor at 12–14 days with a progressive loss of function of the hind legs. However, myelin was formed normally in both the CNS and PNS, but there was a gradual yet regional formation of myelin vacuoles that suggest an instability of myelin in the absence of the lipids though this did not lead to demyelination (Coetzee et al., 1996).
-
iii)
Lamin B1: An adult onset, fatal leukodystrophy has been associated with a duplication of the lamin B1 gene (LMNB1) (Padiath et al., 2006). Overexpression of this gene in a transgenic mouse resulted in marked myelin vacuolation of the lateral and ventral columns of the spinal cord at late time points only (13 months) (Rolyan et al., 2015). Analysis of genes associated with lipid synthesis and myelin lipid quantitation showed marked age-dependent reductions. These results imply that overexpression of LMNB1 results in lipid dysregulation in OLs. It may be that this results primarily in hypomyelination however (Rolyan et al. Fig. 4E) and that the myelin sheaths seen at 13 months (Fig. 4C) do not represent remyelination as was suggested.
Other causes of myelin damage in humans and animals
-
1)
Trauma: It has long been known that demyelination occurs at the periphery of lesions following spinal cord injury (SCI) as has been described in both humans and animal SCI models (Smith and Jeffery, 2006), reviewed in Plemel et al. (2014)). Demyelination is likely driven by OL death (McTigue and Tripathi, 2008). As axon degeneration is the primary injury in SCI with the development of an inflammatory milieu, debate remains about the functional significance of demyelination in SCI and whether the promotion of remyelination has therapeutic value. Transplantation of OPCs into acute SCI in rats resulted in functional improvement though it is uncertain whether this relates to remyelination (Plemel et al., 2014). It has been shown that the generation of OPCs occurs for up to 3 months after SCI (Hesp et al., 2015), hence enhancing this response may be important for myelin repair. Traumatic brain injury (TBI) similarly results in OL loss and demyelination (Flygt et al., 2013, Mierzwa et al., 2015). Certainly OLs die by apoptosis and OPCs respond by dividing. Myelin loss has been noted so like SCI, the question arises where remyelination will have functional significance.
It is likely that in both SCI and traumatic brain injury, that damage to myelin is due to ischemic effects on OLs. This is thought primarily due to excitotoxicity and glutamate elevation (Almad et al., 2011, Tsutsui and Stys, 2013). Similarly in ischemic white matter stroke, OL death and myelin damage are found and in newborns who suffer from periventricular leukomalacia (Back and Rosenberg, 2014), this also occurs. Most recently investigation of the pathophysiology of ischemic white matter damage has identified transient receptor potential (TRPA1) as the key to the entry of Ca2 + into OLs and their subsequent death (Hamilton et al., 2016). As for traumatic CNS injury, whether myelin repair here will be of functional significance remains to be seen.
-
2)
Radiation: Radiation therapy is frequently used in treating brain cancer but has also been associated with late-stage necrosis that has been the subject of intense research. Radiation will kill dividing cells including OPCs and there is evidence that suggests that this can result in demyelination (Panagiotakos et al., 2007). Most recently it has been reported that transplantation of OPCs into the irradiated rat brain results in remyelination and functional recovery (Piao et al., 2015). Treatment of radiation damage to the brain is a very important clinical issue, but many questions remain that must be addressed before such therapies will become the standard of care.
10. Conclusions
The range of disorders in which myelin is damaged is almost bewildering and is increasing as new and uncharacterized disorders of white matter are being investigated using contemporary imaging and next generation sequencing to identify the mutant gene. While we have covered most of the known disorders in humans and animals, others have been reported that have not been included here. This includes, for example, a number of inborn errors of metabolism such as maple syrup urine disease which is found in humans and cattle (Harper et al., 1986) and a knockout mouse with disruption of the chloride channel, ClC-2 (Blanz et al., 2007). We have attempted to differentiate those disorders in which there is proven demyelination in contrast to those where changes such as myelin breakdown does not occur, or there is a failure of myelin development. Applying remyelinating strategies should be guided by this knowledge and will be helped if strenuous attempts are made to define the pathologic basis of known and newly recognized myelin diseases, where contemporary pathologic techniques have not been applied. The challenge in human disease is to get access to tissue in the appropriate time and apply fixation, histologic and quantitative tissue analysis that are required. Where possible, we have identified whether each disorder discussed results from disruption of the myelin sheath or alternatively the oligodendrocyte. In some this is not known or it may be a combination of the two. Definition of this will have therapeutic significance in regard to whether OLs need replaced or a genetic deficit corrected or whether remaining normal OLs need to be recruited to replace lost myelin sheaths.
We have not presented details here on the possibility that myelin and OL dysfunction may be important contributors to neurodegenerative and psychiatric disorders. Evidence for this has come from MRI suggesting white matter involvement, differential gene expression showing reduction of oligodendroglial gene transcripts and neuropathological studies showing evidence of WM abnormalities (Haroutunian et al., 2014, Nave and Ehrenreich, 2014). Experimentally, the generation of a transgenic mouse expressing mutant huntingtin (Huntington's disease) specifically in OLs results in progressive neurologic phenotype associated with demyelination and axonal degeneration (Huang et al., 2015). Most recently, analysis of the transcriptomes from Down's syndrome brains has revealed dysregulation of genes associated with OL differentiation, and hypomyelination in a trisomic mouse model (Olmos-Serrano et al., 2016). This paradigm shift may indeed become the new frontier in myelin biology and disease though the causality of these findings remains to be determined. This issue is complex and the next decade may see a resolution of this challenging question.
Finally, while this review has focused on myelin and OL changes in human and animal WM disorders, it is clear from ongoing studies that there is an intricate connection between myelin, the OL and associated axons (Morrison et al., 2013, Nave, 2010). This is critical to restoring lost myelin sheaths that will not occur if there is axon loss for example in MS and certain genetic disorders of myelin proteins (Gruenenfelder et al., 2011, Nave, 2010). However, axon degeneration and loss does not always follow chronic loss of myelin or long-term dysmyelination (Mayer et al., 2015), hence this needs to be determined in each disorder prior to making this assumption. Despite this caution, we need to develop a complete understanding of axo-glial-myelin interactions in health and disease as the background to myelin repair and remyelination.
Acknowledgements
Many people have contributed to studies cited from this lab especially Drs. Y. Kondo, L. O′Connor, K. Lunn, F-Y Li, S. Jackson, J. Song, J. Mayer and C. Smith. Our work has been supported by grants from the NIH (NS32361, NS055816), NMSS (RG-1501-02876), the Boespflug Foundation, MS Hope for a Cure, and the Oscar Rennebohm Foundation.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.expneurol.2016.04.002.
Appendix A. Supplementary data
The following is the supplementary data related to this article.
Supplementary Fig. 1.
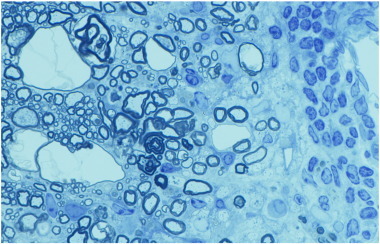
Myelin vacuolation is non-disease specific. In a mouse with EAE, myelin vacuoles are seen close to a vessel with perivascular cuffing and adjacent demyelination. Scale bar: 20 μm.
References
- Adachi M., Schneck L., Cara J., Volk B.W. Spongy degeneration of the central nervous system (van Bogaert and Bertrand type; Canavan's disease). A review. Hum. Pathol. 1973;4:331–347. doi: 10.1016/s0046-8177(73)80098-x. [DOI] [PubMed] [Google Scholar]
- Agamanolis D.P., Victor M., Harris J.W., Hines J.D., Chester E.M., Kark J.A. An ultrastructural study of subacute combined degeneration of the spinal cord in vitamin B12-deficient rhesus monkeys. J. Neuropathol. Exp. Neurol. 1978;37:273–299. doi: 10.1097/00005072-197805000-00006. [DOI] [PubMed] [Google Scholar]
- Almad A., Sahinkaya F.R., McTigue D.M. Oligodendrocyte fate after spinal cord injury. Neurotherapeutics. 2011;8:262–273. doi: 10.1007/s13311-011-0033-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubourg P. Axons need glial peroxisomes. Nat. Genet. 2007;39:936–938. doi: 10.1038/ng0807-936. [DOI] [PubMed] [Google Scholar]
- Back S.A., Rosenberg P.A. Pathophysiology of glia in perinatal white matter injury. Glia. 2014;62:1790–1815. doi: 10.1002/glia.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D., O'Neill J.K., Gschmeissner S.E., Wilcox C.E., Butter C., Turk J.L. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J. Neuroimmunol. 1990;28:261–270. doi: 10.1016/0165-5728(90)90019-j. [DOI] [PubMed] [Google Scholar]
- Barnett M.H., Prineas J.W. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Baron W., Hoekstra D. On the biogenesis of myelin membranes: sorting, trafficking and cell polarity. FEBS Lett. 2010;584:1760–1770. doi: 10.1016/j.febslet.2009.10.085. [DOI] [PubMed] [Google Scholar]
- Baskin J.M., Wu X., Christiano R., Oh M.S., Schauder C.M., Gazzerro E., Messa M., Baldassari S., Assereto S., Biancheri R., Zara F., Minetti C., Raimondi A., Simons M., Walther T.C., Reinisch K.M., De Camilli P. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol. 2016;18:132–138. doi: 10.1038/ncb3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslow M.H. Brain N-acetylaspartate as a molecular water pump and its role in the etiology of Canavan disease — a mechanistic explanation. J. Mol. Neurosci. 2003;21:185–189. doi: 10.1385/jmn:21:3:185. [DOI] [PubMed] [Google Scholar]
- Benjamins J.A., Smith M.E. Metabolism of myelin. In: Morell P., editor. Myelin. second ed. Plenum Press; New York: 1984. pp. 225–258. [Google Scholar]
- Berger J., Forss-Petter S., Eichler F.S. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie. 2014;98:135–142. doi: 10.1016/j.biochi.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancheri R., Zara F., Bruno C., Rossi A., Bordo L., Gazzerro E., Sotgia F., Pedemonte M., Scapolan S., Bado M., Uziel G., Bugiani M., Lamba L.D., Costa V., Schenone A., Rozemuller A.J., Tortori-Donati P., Lisanti M.P., van der Knaap M.S., Minetti C. Phenotypic characterization of hypomyelination and congenital cataract. Ann. Neurol. 2007;62:121–127. doi: 10.1002/ana.21175. [DOI] [PubMed] [Google Scholar]
- Biddle F., March E., Miller J.R. Research news. Mouse News Lett. 1973;48:24. [Google Scholar]
- Blakemore W.F. Observations on oligodendrocyte degeneration, the resolution of status spongiosus and remyelination in cuprizone intoxication in mice. J. Neurocytol. 1972;1:413–426. doi: 10.1007/BF01102943. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. J. Neurol. Sci. 1973;20:63–72. doi: 10.1016/0022-510x(73)90118-4. [DOI] [PubMed] [Google Scholar]
- Blakemore W. Observations on remyelination in the rabbit spinal cord following demyelination induced by lysolecithin. Neuropathol. Appl. Neurobiol. 1978;4:47–59. doi: 10.1111/j.1365-2990.1978.tb00528.x. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F. Ethidium bromide induced demyelination in the spinal cord of the cat. Neuropathol. Appl. Neurobiol. 1982;8:365–375. doi: 10.1111/j.1365-2990.1982.tb00305.x. [DOI] [PubMed] [Google Scholar]
- Blakemore W.F., Palmer A.C., Noel P.R. Ultrastructural changes in isoniazid-induced brain oedema in the dog. J. Neurocytol. 1972;1:263–278. doi: 10.1007/BF01099938. [DOI] [PubMed] [Google Scholar]
- Blanz J., Schweizer M., Auberson M., Maier H., Muenscher A., Hubner C.A., Jentsch T.J. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 2007;27:6581–6589. doi: 10.1523/JNEUROSCI.0338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti B., Raine C.S. Multiple sclerosis: oligodendrocytes display cell death-related molecules in situ but do not undergo apoptosis. Ann. Neurol. 1997;42:74–84. doi: 10.1002/ana.410420113. [DOI] [PubMed] [Google Scholar]
- Bradl M., Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breij E.C., Brink B.P., Veerhuis R., van den B.C., Vloet R., Yan R., Dijkstra C.D., van d.V., Bo L. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann. Neurol. 2008;63:16–25. doi: 10.1002/ana.21311. [DOI] [PubMed] [Google Scholar]
- Brok H.P.M., Bauer J., Jonker M., Blezer E., Amor S., Bontrop R.E., Laman J.D., t Hart B.A. Non-human primate models of multiple sclerosis. Immunol. Rev. 2001;183:173–185. doi: 10.1034/j.1600-065x.2001.1830114.x. [DOI] [PubMed] [Google Scholar]
- Brown F.R., III, Voigt R., Singh A.K., Singh I. Peroxisomal disorders. Neurodevelopmental and biochemical aspects. Am. J. Dis. Child. 1993;147:617–626. doi: 10.1001/archpedi.1993.02160300023015. [DOI] [PubMed] [Google Scholar]
- Bruck W., Herms J., Brockmann K., Schulz-Schaeffer W., Hanefeld F. Myelinopathia centralis diffusa (vanishing white matter disease): evidence of apoptotic oligodendrocyte degeneration in early lesion development. Ann. Neurol. 2001;50:532–536. doi: 10.1002/ana.1227. [DOI] [PubMed] [Google Scholar]
- Bu J., Banki A., Wu Q., Nishiyama A. Increased NG2 + glial cell proliferation and oligodendrocyte generation in the hypomyelinating mutant shiverer. Glia. 2004;48:51–63. doi: 10.1002/glia.20055. [DOI] [PubMed] [Google Scholar]
- Bugiani M., Postma N., Polder E., Dieleman N., Scheffer P.G., Sim F.J., van der Knaap M.S., Boor I. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain. 2013;136:209–222. doi: 10.1093/brain/aws320. [DOI] [PubMed] [Google Scholar]
- Campagnoni A.T., Skoff R.P. The pathobiology of myelin mutants reveal novel biological functions of the MBP and PLP genes. Brain Pathol. 2001;11:74–91. doi: 10.1111/j.1750-3639.2001.tb00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelvetri L.C., Givogri M.I., Zhu H., Smith B., Lopez-Rosas A., Qiu X., van Breemen R., Bongarzone E.R. Axonopathy is a compounding factor in the pathogenesis of Krabbe disease. Acta Neuropathol. 2011;122:35–48. doi: 10.1007/s00401-011-0814-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield C.D., Kelly J.P., Jones B.R., Worrall S., Conlon L., Palmer A.C., Cassidy J.P. The experimental induction of leukoencephalomyelopathy in cats. Vet. Pathol. 2009;46:1258–1269. doi: 10.1354/vp.08-VP-0336-C-FL. [DOI] [PubMed] [Google Scholar]
- Chrast R., Saher G., Nave K.A., Verheijen M.H. Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J. Lipid Res. 2011;52:419–434. doi: 10.1194/jlr.R009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarner T., Wieczorek N., Krauspe B., Jansen K., Beyer C., Kipp M. Astroglial redistribution of aquaporin 4 during spongy degeneration in a Canavan disease mouse model. J. Mol. Neurosci. 2014;53:22–30. doi: 10.1007/s12031-013-0184-4. [DOI] [PubMed] [Google Scholar]
- Coetzee T., Fujita N., Dupree J., Shi R., Blight A., Suzuki K., Suzuki K., Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Compston A., McDonald I., Noseworthy J., Lassmann H., Miller D., Smith K., Wekerle H., Confavreux C. Churchill Livingstone Elsevier; 2006. McAlpine's Multiple Sclerosis. [Google Scholar]
- Couve E., Cabello J.F., Krsulovic J., Roncagliolo M. Binding of microtubules to transitional elements in oligodendrocytes of the myelin mutant taiep rat. J. Neurosci. Res. 1997;47:573–581. [PubMed] [Google Scholar]
- Dal Canto M.C., Rabinowitz S. Experimental models of virus-induced demyelination of the central nervous system. Ann. Neurol. 1982;11:109–127. doi: 10.1002/ana.410110202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney K.H., Kwiecien J., Wegiel J., Wisniewski H.M., Percy D.H., Fletch A.L. Familial dysmyelination in a Long Evans rat mutant. Lab. Anim. Sci. 1995;45:547–553. [PubMed] [Google Scholar]
- DeLuca G.C., Nagy Z., Esiri M.M., Davey P. Evidence for a role for apoptosis in central pontine myelinolysis. Acta Neuropathol. (Berl.) 2002;103:590–598. doi: 10.1007/s00401-001-0508-2. [DOI] [PubMed] [Google Scholar]
- Denic A., Johnson A.J., Bieber A.J., Warrington A.E., Rodriguez M., Pirko I. The relevance of animal models in multiple sclerosis research. Pathophysiology. 2011;18:21–29. doi: 10.1016/j.pathophys.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois-Dalcq M., Feigenbaum V., Aubourg P. The neurobiology of X-linked adrenoleukodystrophy, a demyelinating peroxisomal disorder. Trends Neurosci. 1999;22:4–12. doi: 10.1016/s0166-2236(98)01319-8. [DOI] [PubMed] [Google Scholar]
- Duncan I.D. Toxic Myelinopathies. In: O'Donoghue J.L., editor. Neurotoxicity of Industrial and Commercial Chemicals. vol. 1, 2. CRC Press; Boca Raton: 1985. p. 222 S. [Google Scholar]
- Duncan I.D. Inherited disorders of myelination of the central nervous system. In: Ransom B.R., Kettenmann H.R., editors. Neuroglial Cells. Oxford University Press; Cambridge: 1995. pp. 990–1009. [Google Scholar]
- Duncan I.D., Brower A., Kondo Y., Curlee J.F., Jr., Schultz R.D. Extensive remyelination of the CNS leads to functional recovery. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6832–6836. doi: 10.1073/pnas.0812500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan I.D., Hammang J.P., Goda S., Quarles R.H. Myelination in the jimpy mouse in the absence of PLP. Glia. 1989;2:148–154. doi: 10.1002/glia.440020303. [DOI] [PubMed] [Google Scholar]
- Duncan I.D., Hammang J.P., Trapp B.D. Abnormal compact myelin in the myelin-deficient rat: absence of proteolipid protein correlates with a defect in the intraperiod line. Proc. Natl. Acad. Sci. U. S. A. 1987;84:6287–6291. doi: 10.1073/pnas.84.17.6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan I.D., Kondo Y., Zhang S.C. The myelin mutants as models to study myelin repair in the leukodystrophies. Neurotherapeutics. 2011;8:607–624. doi: 10.1007/s13311-011-0080-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan I.D., Lunn K.F., Holmgren B., Urba-Holmgren R., Brignolo-Holmes L. The taiep rat: a myelin mutant with an associated oligodendrocyte microtubular defect. J. Neurocytol. 1992;21:870–884. doi: 10.1007/BF01191684. [DOI] [PubMed] [Google Scholar]
- Duncan I.D., Nadon N.L., Hoffman R.L., Lunn K.F., Csiza C.K., Wells M.R. Oligodendrocyte survival and function in the long-lived strain of the myelin deficient rat. J. Neurocytol. 1995;24:745–762. doi: 10.1007/BF01191211. [DOI] [PubMed] [Google Scholar]
- Ebersole T.A., Chen Q., Justice M.J., Artzt K. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat. Genet. 1996;12:260–265. doi: 10.1038/ng0396-260. [DOI] [PubMed] [Google Scholar]
- Erlich S.S., Fleming J.O. Experimental neuropathology of chronic demyelination induced by a JHM virus variant (DS) Arch. Neurol. 1987;44:839–842. doi: 10.1001/archneur.1987.00520200043016. [DOI] [PubMed] [Google Scholar]
- Fischer J.R., Lewis-Weis L.A., Tate C.M. Experimental vacuolar myelinopathy in red-tailed hawks. J. Wildl. Dis. 2003;39:400–406. doi: 10.7589/0090-3558-39.2.400. [DOI] [PubMed] [Google Scholar]
- Flygt J., Djupsjo A., Lenne F., Marklund N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur. J. Neurosci. 2013;38:2153–2165. doi: 10.1111/ejn.12179. [DOI] [PubMed] [Google Scholar]
- Francalanci P., Eymard-Pierre E., Dionisi-Vici C., Boldrini R., Piemonte F., Virgili R., Fariello G., Bosman C., Santorelli F.M., Boespflug-Tanguy O., Bertini E. Fatal infantile leukodystrophy — a severe variant of CACH/VWM syndrome, allelic to chromosome 3q27. Neurology. 2001;57:265–270. doi: 10.1212/wnl.57.2.265. [DOI] [PubMed] [Google Scholar]
- Frohman E.M., Racke M.K., Raine C.S. Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Funfschilling U., Supplie L.M., Mahad D., Boretius S., Saab A.S., Edgar J., Brinkmann B.G., Kassmann C.M., Tzvetanova I.D., Mobius W., Diaz F., Meijer D., Suter U., Hamprecht B., Sereda M.W., Moraes C.T., Frahm J., Goebbels S., Nave K.A. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzerro E., Baldassari S., Giacomini C., Musante V., Fruscione F., La Padula V., Biancheri R., Scarfi S., Prada V., Sotgia F., Duncan I.D., Zara F., Werner H.B., Lisanti M.P., Nobbio L., Corradi A., Minetti C. Hyccin, the molecule mutated in the leukodystrophy hypomyelination and congenital cataract (HCC), is a neuronal protein. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva M., Cabilly Y., Assaf Y., Mindroul N., Marom L., Raini G., Pinchasi D., Elroy-Stein O. A mouse model for eukaryotic translation initiation factor 2B-leucodystrophy reveals abnormal development of brain white matter. Brain. 2010;133:2448–2461. doi: 10.1093/brain/awq180. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N., Jeong S.Y., Lacroix S., David S. T cells contribute to lysophosphatidylcholine-induced macrophage activation and demyelination in the CNS. Glia. 2007;55:294–302. doi: 10.1002/glia.20449. [DOI] [PubMed] [Google Scholar]
- Gold R., Linington C., Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006 doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Graesser D., Spraker T.R., Dressen P., Garner M.M., T Raymond J., Terwilliger G., Kim J., Madri J.A. Wobbly Hedgehog Syndrome in African pygmy hedgehogs (Atelerix spp.) J. Exot. Pet. Med. 2006;15:59–65. [Google Scholar]
- Greco C.M., Powell H.C., Garrett R.S., Lampert P.W. Cycloleucine encephalopathy. Neuropathol. Appl. Neurobiol. 1980;6:349–360. doi: 10.1111/j.1365-2990.1980.tb00671.x. [DOI] [PubMed] [Google Scholar]
- Green R., Van Tonder S.V., Oettle G.J., Cole G., Metz J. Neurological changes in fruit bats deficient in vitamin B12. Nature. 1975;254:148–150. doi: 10.1038/254148a0. [DOI] [PubMed] [Google Scholar]
- Griffiths I.R., Klugmann M., Anderson T., Thomson C., Vouyiouklis D.A., Nave K.A. Current concepts of PLP and its role in the nervous system. Microsc. Res. Tech. 1998;41:344–358. doi: 10.1002/(SICI)1097-0029(19980601)41:5<344::AID-JEMT2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Gruenenfelder F.I., Thomson G., Penderis J., Edgar J.M. Axon–glial interaction in the CNS: what we have learned from mouse models of Pelizaeus-Merzbacher disease. J. Anat. 2011;219:33–43. doi: 10.1111/j.1469-7580.2011.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi V., Gingele S., Skripuletz T., Stangel M. Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front. Cell. Neurosci. 2014;8:73. doi: 10.3389/fncel.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F., Bannerman P., Mills Ko E., Miers L., Xu J., Burns T., Li S., Freeman E., McDonough J.A., Pleasure D. Ablating N-acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann. Neurol. 2015;77:884–888. doi: 10.1002/ana.24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N., Henry R.G., Strober J., Kang S.M., Lim D.A., Bucci M., Caverzasi E., Gaetano L., Mandelli M.L., Ryan T., Perry R., Farrell J., Jeremy R.J., Ulman M., Huhn S.L., Barkovich A.J., Rowitch D.H. Neural stem cell engraftment and myelination in the human brain. Sci. Transl. Med. 2012;4:155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen G., Bjerkas I. Spongy degeneration of white matter in the central nervous system of silver foxes (Vulpes vulpes) Vet. Pathol. 1990;27:187–193. doi: 10.1177/030098589002700306. [DOI] [PubMed] [Google Scholar]
- Hagen G., Blakemore W.F., Bjerkas I. Ultrastructural findings in spongy degeneration of white matter in silver foxes (Vulpes vulpes). A naturally occurring demyelinating disease with oligodendrocyte vacuolation. Acta Neuropathol. 1990;80:590–596. doi: 10.1007/BF00307625. [DOI] [PubMed] [Google Scholar]
- Hall S.M. The effect of injections of lysophosphatidyl choline into white metter of the adult mouse spinal cord. J. Cell Sci. 1972;10:535–546. doi: 10.1242/jcs.10.2.535. [DOI] [PubMed] [Google Scholar]
- Hall S.M., Gregson N.A. The in vivo and ultrastructural effects of injection of lysophosphatidyl choline into myelinated peripheral nerve fibres of the adult mouse. J. Cell Sci. 1971;9:769–789. doi: 10.1242/jcs.9.3.769. [DOI] [PubMed] [Google Scholar]
- Hamilton N.B., Kolodziejczyk K., Kougioumtzidou E., Attwell D. Proton-gated Ca2 +-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature. 2016;529:523–527. doi: 10.1038/nature16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton D.W., Anderson J., Pryce G., Irvine K.A., Giovannoni G., Fawcett J.W., Compston A., Franklin R.J., Baker D., Chandran S. An experimental model of secondary progressive multiple sclerosis that shows regional variation in gliosis, remyelination, axonal and neuronal loss. J. Neuroimmunol. 2008;201-202:200–211. doi: 10.1016/j.jneuroim.2008.05.034. [DOI] [PubMed] [Google Scholar]
- Haroutunian V., Katsel P., Roussos P., Davis K.L., Altshuler L.L., Bartzokis G. Myelination, oligodendrocytes, and serious mental illness. Glia. 2014;62:1856–1877. doi: 10.1002/glia.22716. [DOI] [PubMed] [Google Scholar]
- Harper P.A., Healy P.J., Dennis J.A. Ultrastructural findings in maple syrup urine disease in Poll Hereford calves. Acta Neuropathol. 1986;71:316–320. doi: 10.1007/BF00688055. [DOI] [PubMed] [Google Scholar]
- Herndon R.M., Rubinstein L.J., Freeman J.M., Mathieson G. Light and electron microscopic observations on Rosenthal fibers in Alexander's disease and in multiple sclerosis. J. Neuropathol. Exp. Neurol. 1970;29:524–551. doi: 10.1097/00005072-197010000-00002. [DOI] [PubMed] [Google Scholar]
- Hesp Z.C., Goldstein E.Z., Miranda C.J., Kaspar B.K., McTigue D.M. Chronic oligodendrogenesis and remyelination after spinal cord injury in mice and rats. J. Neurosci. 2015;35:1274–1290. doi: 10.1523/JNEUROSCI.2568-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Saftig P., Hartmann D., Coenen R., Lullmann-Rauch R., Goebel H.H., Evers M., von Figura K., D'Hooge R., Nagels G., De Deyn P., Peters C., Gieselmann V. Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc. Natl. Acad. Sci. U. S. A. 1996;93 doi: 10.1073/pnas.93.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson G.M., Garbern J.Y. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin. Neurol. 2012;32:62–67. doi: 10.1055/s-0032-1306388. [DOI] [PubMed] [Google Scholar]
- Holmgren B., Urba-Holmgren R., Riboni L., Vega-SaenzdeMiera E.C. Sprague Dawley rat mutant with tremor, ataxia, tonic immobility episodes, pilepsy and paralysis. Lab. Anim. Sci. 1989;39:226–228. [PubMed] [Google Scholar]
- Hoshino H., Kubota M. Canavan disease: clinical features and recent advances in research. Pediatr. Int. 2014;56:477–483. doi: 10.1111/ped.12422. [DOI] [PubMed] [Google Scholar]
- Huang B., Wei W.J., Wang G.H., Gaertig M.A., Feng Y., Wang W., Li X.J., Li S.H. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron. 2015;85:1212–1226. doi: 10.1016/j.neuron.2015.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable C.R., Dorling P.R., Slatter D.H. Myelin oedema, optic neuropathy and retinopathy in experimental Stypandra imbricata toxicosis. Neuropathol. Appl. Neurobiol. 1980;6:221–232. doi: 10.1111/j.1365-2990.1980.tb00292.x. [DOI] [PubMed] [Google Scholar]
- Inoue Y., Inoue K., Terashima T., Mikoshiba K., Tsukada Y. Developmental changes of oligodendroglia in the posterior funiculus of “shiverer” mutant mouse spinal cord, with special reference to myelin formation. Anat. Embryol. 1983;168:159–171. doi: 10.1007/BF00315814. [DOI] [PubMed] [Google Scholar]
- Irvine K.A., Blakemore W.F. Age increases axon loss associated with primary demyelination in cuprizone-induced demyelination in C57BL/6 mice. J. Neuroimmunol. 2006;175:69–76. doi: 10.1016/j.jneuroim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Izawa T., Takenaka S., Ihara H., Kotani T., Yamate J., Franklin R.J., Kuwamura M. Cellular responses in the spinal cord during development of hypomyelination in the mv rat. Brain Res. 2008;1195:120–129. doi: 10.1016/j.brainres.2007.12.022. [DOI] [PubMed] [Google Scholar]
- Jackson S.J., Lee J., Nikodemova M., Fabry Z., Duncan I.D. Quantification of myelin and axon pathology during relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. J. Neuropathol. Exp. Neurol. 2009;68:616–625. doi: 10.1097/NEN.0b013e3181a41d23. [DOI] [PubMed] [Google Scholar]
- Jacobs J.M., Cremer J.E., Cavanagh J.B. Acute effects of triethyl tin on the rat myelin sheath. Neuropathol. Appl. Neurobiol. 1977;3:169–181. [Google Scholar]
- Jagessar S.A., Kap Y.S., Heijmans N., van Driel N., van Straalen L., Bajramovic J.J., Brok H.P., Blezer E.L., Bauer J., Laman J.D., t Hart B.A. Induction of progressive demyelinating autoimmune encephalomyelitis in common marmoset monkeys using MOG34-56 peptide in incomplete freund adjuvant. J. Neuropathol. Exp. Neurol. 2010;69:372–385. doi: 10.1097/NEN.0b013e3181d5d053. [DOI] [PubMed] [Google Scholar]
- Jang J., Kang H.C., Kim H.S., Kim J.Y., Huh Y.J., Kim D.S., Yoo J.E., Lee J.A., Lim B., Lee J., Yoon T.M., Park I.H., Hwang D.Y., Daley G.Q., Kim D.W. Induced pluripotent stem cell models from X-linked adrenoleukodystrophy patients. Ann. Neurol. 2011;70:402–409. doi: 10.1002/ana.22486. [DOI] [PubMed] [Google Scholar]
- Jeffery N.D., Blakemore W.F. Remyelination of mouse spinal cord axons demyelinated by local injection of lysolecithin. J. Neurocytol. 1995;24:775–781. doi: 10.1007/BF01191213. [DOI] [PubMed] [Google Scholar]
- Jellinger K., Seitelberger F. Spongy degeneration of the central nervous system in infancy. Curr. Top. Pathol. 1970;53:90–160. doi: 10.1007/978-3-662-30514-0_2. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M., Dwek R.A., Butters T.D., Platt F.M. Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- Kassmann C.M., Lappe-Siefke C., Baes M., Brugger B., Mildner A., Werner H.B., Natt O., Michaelis T., Prinz M., Frahm J., Nave K.A. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat. Genet. 2007;39:969–976. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- Kaul R., Gao G.P., Balamurugan K., Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat. Genet. 1993;5:118–123. doi: 10.1038/ng1093-118. [DOI] [PubMed] [Google Scholar]
- Kemp S., Wanders R. Biochemical aspects of X-linked adrenoleukodystrophy. Brain Pathol. 2010;20:831–837. doi: 10.1111/j.1750-3639.2010.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M., Stadelmann C., Buddeberg B.S., Merkler D., Bareyre F.M., Anthony D.C., Linington C., Brück W., Schwab M.E. Targeting experimental autoimmune encephalomyelitis lesions to a predetermined axonal tract system allows for refined behavioral testing in an animal model of multiple sclerosis. Am. J. Pathol. 2004;164:1455–1469. doi: 10.1016/S0002-9440(10)63232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Kobayashi T., Amemiya F. Myelin splitting in the spongy lesion in Leigh encephalopathy. Pediatr. Neurol. 1991;7:56–58. doi: 10.1016/0887-8994(91)90108-w. [DOI] [PubMed] [Google Scholar]
- Kipp M., Clarner T., Dang J., Copray S., Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118:723–736. doi: 10.1007/s00401-009-0591-3. [DOI] [PubMed] [Google Scholar]
- Kitada K., Akimitsu T., Shigematsu Y., Kondo A., Maihara T., Yokoi N., Kuramoto T., Sasa M., Serikawa T. Accumulation of N-acetyl-l-aspartate in the brain of the tremor rat, a mutant exhibiting absence-like seizure and spongiform degeneration in the central nervous system. J. Neurochem. 2000;74:2512–2519. doi: 10.1046/j.1471-4159.2000.0742512.x. [DOI] [PubMed] [Google Scholar]
- van der Knaap M.S., Barth P.G., Gabreëls F.J.M., Franzoni E., Begeer J.H., Stroink H., Rotteveel J.J., Valk J. A new leukoencephalopathy with vanishing white matter. Neurology. 1997:845–855. doi: 10.1212/wnl.48.4.845. [DOI] [PubMed] [Google Scholar]
- van der Knaap M.S., Leegwater P.A.J., Könst A.A.M., Visser A., Naidu S., Oudejans C.B.M., Schutgens R.B.H., Pronk J.C. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol. 2002;51:264–270. doi: 10.1002/ana.10112. [DOI] [PubMed] [Google Scholar]
- van der Knaap M., Pronk J., Scheper G. Vanishing white matter disease. Lancet Neurol. 2006;5:413–423. doi: 10.1016/S1474-4422(06)70440-9. [DOI] [PubMed] [Google Scholar]
- Kondo Y., Adams J.M., Vanier M.T., Duncan I.D. Macrophages counteract demyelination in a mouse model of globoid cell leukodystrophy. J. Neurosci. 2011;31:3610–3624. doi: 10.1523/JNEUROSCI.6344-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo A., Sendoh S., Akazawa K., Sato Y., Nagara H. Early myelination in zitter rat: morphological, immunocytochemical and morphometric studies. Dev. Brain Res. 1992;67:217–228. doi: 10.1016/0165-3806(92)90222-i. [DOI] [PubMed] [Google Scholar]
- Kovacs G.G., Hoftberger R., Majtenyi K., Horvath R., Barsi P., Komoly S., Lassmann H., Budka H., Jakab G. Neuropathology of white matter disease in Leber's hereditary optic neuropathy. Brain. 2005;128:35–41. doi: 10.1093/brain/awh310. [DOI] [PubMed] [Google Scholar]
- Kuhlmann T., Lassmann H., Bruck W. Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol. 2008;115:275–287. doi: 10.1007/s00401-007-0320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto T., Kitada K., Inui T., Sasaki Y., Ito K., Hase T., Kawagachi S., Ogawa Y., Nakao K., Barsh G.S., Nagao M., Ushijima T., Serikawa T. Attractin/mahogany/zitter plays a critical role in myelination of the central nervous system. Proc. Natl. Acad. Sci. U. S. A. 2001;98:559–564. doi: 10.1073/pnas.98.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto T., Kuwamura M., Tokuda S., Izawa T., Nakane Y., Kitada K., Akao M., Guenet J.L., Serikawa T. A mutation in the gene encoding mitochondrial Mg2 + channel MRS2 results in demyelination in the rat. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto T., Nomoto T., Fujiwara A., Mizutani M., Sugimura T., Ushijima T. Insertional mutation of the Attractin gene in the black tremor hamster. Mamm. Genome. 2002;13:36–40. doi: 10.1007/s00335-001-2116-9. [DOI] [PubMed] [Google Scholar]
- Kuramoto T., Sotelo C., Yokoi N., Serikawa T., Gonalons Sintes E., Canto Martorell J., Guenet J.L. A rat mutation producing demyelination (dmy) maps to chromosome 17. Mamm. Genome. 1996;7:890–894. doi: 10.1007/s003359900263. [DOI] [PubMed] [Google Scholar]
- Kuwamura M., Kanehara T., Tokuda S., Kumagai D., Yamate J., Kotani T., Nakane Y., Kuramoto T., Serikawa T. Immunohistochemical and morphometrical studies on myelin breakdown in the demyelination (dmy) mutant rat. Brain Res. 2004;1022:110–116. doi: 10.1016/j.brainres.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Kuwamura M., Maeda M., Kuramoto T., Kitada K., Kanehara T., Moriyama M., Nakane Y., Yamate J., Ushijima T., Kotani T., Serikawa T. The myelin vacuolation (mv) rat with a null mutation in the attractin gene. Lab. Investig. 2002;82:1279–1286. doi: 10.1097/01.lab.0000032375.70196.26. [DOI] [PubMed] [Google Scholar]
- Kuypers N.J., James K.T., Enzmann G.U., Magnuson D.S., Whittemore S.R. Functional consequences of ethidium bromide demyelination of the mouse ventral spinal cord. Exp. Neurol. 2013;247:615–622. doi: 10.1016/j.expneurol.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiecien J.M., O'Connor L.T., Goetz B.D., Delaney K.H., Fletch A.L., Duncan I.D. Morphological and morphometric studies of the dysmyelinating mutant, the Long Evans shaker rat. J. Neurocytol. 1998;27:581–591. doi: 10.1023/a:1006922227791. [DOI] [PubMed] [Google Scholar]
- Labauge P., Horzinski L., Ayrignac X., Blanc P., Vukusic S., Rodriguez D., Mauguiere F., Peter L., Goizet C., Bouhour F., Denier C., Confavreux C., Obadia M., Blanc F., de Seze J., Fogli A., Boespflug-Tanguy O. Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases. Brain. 2009;132:2161–2169. doi: 10.1093/brain/awp171. [DOI] [PubMed] [Google Scholar]
- Lake N.J., Bird M.J., Isohanni P., Paetau A. Leigh syndrome: neuropathology and pathogenesis. J. Neuropathol. Exp. Neurol. 2015;74:482–492. doi: 10.1097/NEN.0000000000000195. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Mechanisms of white matter damage in multiple sclerosis. Glia. 2014;62:1816–1830. doi: 10.1002/glia.22597. [DOI] [PubMed] [Google Scholar]
- Lassmann H., Brück W., Lucchinetti C.F. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17:210–218. doi: 10.1111/j.1750-3639.2007.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon V.A., Kryzer T.J., Pittock S.J., Verkman A.S., Hinson S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon V.A., Wingerchuk D.M., Kryzer T.J., Pittock S.J., Lucchinetti C.F., Fujihara K., Nakashima I., Weinshenker B.G. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Li F.Y., Cuddon P.A., Song J., Wood S.L., Patterson J.S., Shelton G.D., Duncan I.D. Canine spongiform leukoencephalomyelopathy is associated with a missense mutation in cytochrome b. Neurobiol. Dis. 2006;21:35–42. doi: 10.1016/j.nbd.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Li F.Y., Song J., Duncan I.D. Mapping of taiep rat phenotype to rat chromosome 9. Mamm. Genome. 2003;14:703–705. doi: 10.1007/s00335-003-2302-z. [DOI] [PubMed] [Google Scholar]
- Lin Y., Pang X., Huang G., Jamison S., Fang J., Harding H.P., Ron D., Lin W. Impaired eukaryotic translation initiation factor 2B activity specifically in oligodendrocytes reproduces the pathology of vanishing white matter disease in mice. J. Neurosci. 2014;34:12182–12191. doi: 10.1523/JNEUROSCI.1373-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner M., Fokuhl J., Linsmeier F., Trebst C., Stangel M. Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci. Lett. 2009;453:120–125. doi: 10.1016/j.neulet.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Locatelli G., Wortge S., Buch T., Ingold B., Frommer F., Sobottka B., Kruger M., Karram K., Buhlmann C., Bechmann I., Heppner F.L., Waisman A., Becher B. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat. Neurosci. 2012;15:543–550. doi: 10.1038/nn.3062. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C., Brück W., Parisi J., Scheithauer B., Rodrigues M., Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C.F., Brück W., Rodriguez M., Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity in pathogenesis. Brain Pathol. 1996;6:259–274. doi: 10.1111/j.1750-3639.1996.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C.F., Guo Y., Popescu B.F., Fujihara K., Itoyama Y., Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. 2014;24:83–97. doi: 10.1111/bpa.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwin S.K. Chronic demyelination inhibits remyelination in the central nervous system. Lab. Investig. 1980;43:382–387. [PubMed] [Google Scholar]
- van der Lugt J.J., Venter I. Myelin vacuolation, optic neuropathy and retinal degeneration after closantel overdosage in sheep and in a goat. J. Comp. Pathol. 2007;136:87–95. doi: 10.1016/j.jcpa.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Lunn K.F., Fanarraga M., Duncan I.D. Myelin mutants: new models and new observations. Microsc. Res. Tech. 1995;183–203 doi: 10.1002/jemt.1070320303. [DOI] [PubMed] [Google Scholar]
- Lunn K.F., Clayton M.K., Duncan I.D. The temporal progression of the myelination defect in the taiep rat. J. Neurocytol. 1997;26:267–281. doi: 10.1023/a:1018548400536. [DOI] [PubMed] [Google Scholar]
- Lutz S.E., Zhao Y., Gulinello M., Lee S.C., Raine C.S., Brosnan C.F. Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J. Neurosci. 2009;29:7743–7752. doi: 10.1523/JNEUROSCI.0341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier H., Wang-Eckhardt L., Hartmann D., Gieselmann V., Eckhardt M. N-acetylaspartate synthase deficiency Corrects the myelin phenotype in a Canavan disease mouse model but does not affect survival time. J. Neurosci. 2015;35:14501–14516. doi: 10.1523/JNEUROSCI.1056-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima G.K., Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima G.K., Taniike M., Glimcher L.H., Grusby M.J., Frelinger J.A., Suzuki K., Ting J.P.Y. Absence of MHC class II molecules reduces CNS demyelination, microglial/macrophage infiltration, and twitching in murine globoid cell leukodystrophy. Cell. 1994;78:645–656. doi: 10.1016/0092-8674(94)90529-0. [DOI] [PubMed] [Google Scholar]
- Matthieu J.M., Omlin F.X., Ginalski-Winkelmann H., Cooper B.J. Myelination in the CNS of mld mutant mice: comparison between composition and structure. Brain Res. 1984;315:149–158. doi: 10.1016/0165-3806(84)90086-5. [DOI] [PubMed] [Google Scholar]
- Mayer J.A., Griffiths I.R., Goldman J.E., Smith C.M., Cooksey E., Radcliff A.B., Duncan I.D. Modeling the natural history of Pelizaeus-Merzbacher disease. Neurobiol. Dis. 2015;75:115–130. doi: 10.1016/j.nbd.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaddon A. Vitamin B12 in neurology and ageing; clinical and genetic aspects. Biochimie. 2013;95:1066–1076. doi: 10.1016/j.biochi.2012.11.017. [DOI] [PubMed] [Google Scholar]
- McTigue D.M., Tripathi R.B. The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Mecha M., Carrillo-Salinas F.J., Mestre L., Feliu A., Guaza C. Viral models of multiple sclerosis: neurodegeneration and demyelination in mice infected with Theiler's virus. Prog. Neurobiol. 2013;101-102:46–64. doi: 10.1016/j.pneurobio.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella D.M., Majdan M., Awatramani R., Goodenough D.A., Sirkowski E., Scherer S.S., Paul D.L. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 2006;26:10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkler D., Schmelting B., Czeh B., Fuchs E., Stadelmann C., Bruck W. Myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis in the common marmoset reflects the immunopathology of pattern II multiple sclerosis lesions. Mult. Scler. 2006;12:369–374. doi: 10.1191/1352458506ms1290oa. [DOI] [PubMed] [Google Scholar]
- Messing A., Brenner M., Feany M.B., Nedergaard M., Goldman J.E. Alexander disease. J. Neurosci. 2012;32:5017–5023. doi: 10.1523/JNEUROSCI.5384-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A., Head M.W., Galles K., Galbreath E., Goldman J.E., Brenner M. Fatal encephalopathy with astroyte inclusions in GFAP transgenic mice. Am. J. Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- Mierzwa A.J., Marion C.M., Sullivan G.M., McDaniel D.P., Armstrong R.C. Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 2015;74:218–232. doi: 10.1097/NEN.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C., Boespflug-Tanguy O., Gelot A., Dautigny A., Pham-Dinh D., Rodriguez D. Alexander disease: putative mechanisms of an astrocytic encephalopathy. Cell. Mol. Life Sci. 2004;61:369–385. doi: 10.1007/s00018-003-3143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikoshiba K., Aruga J., Ikenaka K., Okano H. Shiverer and allelic mutant MLD mice. In: RE M., editor. Myelin: Biology and Chemistry. CRC Press; Boca Raton, FL: 1992. pp. 723–744. [Google Scholar]
- Miller A., Korem M., Almog R., Galboiz Y. Vitamin B12, demyelination, remyelination and repair in multiple sclerosis. J. Neurol. Sci. 2005;233:93–97. doi: 10.1016/j.jns.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Misu T., Hoftberger R., Fujihara K., Wimmer I., Takai Y., Nishiyama S., Nakashima I., Konno H., Bradl M., Garzuly F., Itoyama Y., Aoki M., Lassmann H. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125:815–827. doi: 10.1007/s00401-013-1116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobius W., Nave K.A., Werner H.B. Electron microscopy of myelin: structure preservation by high-pressure freezing. Brain Res. 2016 doi: 10.1016/j.brainres.2016.02.027. [DOI] [PubMed] [Google Scholar]
- Morato L., Bertini E., Verrigni D., Ardissone A., Ruiz M., Ferrer I., Uziel G., Pujol A. Mitochondrial dysfunction in central nervous system white matter disorders. Glia. 2014;62:1878–1894. doi: 10.1002/glia.22670. [DOI] [PubMed] [Google Scholar]
- Morell P., Quarles R.H., Norton W.T. Myelin formation, structure, and biochemistry. In: Siegel G.J., editor. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. fifth ed. Raven Press; New York: 1994. pp. 117–143. [Google Scholar]
- Mori S., Leblond C.P. Electron microscopic identification of three classes of oligodendrocytes and a preliminary study of their activity in the corpus callosum of young rats. J. Comp. Neurol. 1970;139:1–30. doi: 10.1002/cne.901390102. [DOI] [PubMed] [Google Scholar]
- Morrison B.M., Lee Y., Rothstein J.D. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 2013;23:644–651. doi: 10.1016/j.tcb.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser H.W. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120:1485–1508. doi: 10.1093/brain/120.8.1485. [DOI] [PubMed] [Google Scholar]
- Musolino P.L., Gong Y., Snyder J.M., Jimenez S., Lok J., Lo E.H., Moser A.B., Grabowski E.F., Frosch M.P., Eichler F.S. Brain endothelial dysfunction in cerebral adrenoleukodystrophy. Brain. 2015;138:3206–3220. doi: 10.1093/brain/awv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagara H., Suzuki K. Chronological study of oligodendroglial alterations and myelination in quaking mice. Neuropathol. Appl. Neurobiol. 1981;7:135–149. doi: 10.1111/j.1365-2990.1981.tb00083.x. [DOI] [PubMed] [Google Scholar]
- Nave K.A. Myelination and support of axonal integrity by glia. Nature. 2010;468:244–252. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Nave K.A., Ehrenreich H. Myelination and oligodendrocyte functions in psychiatric diseases. JAMA Psychiatry. 2014;71:582–584. doi: 10.1001/jamapsychiatry.2014.189. [DOI] [PubMed] [Google Scholar]
- Nave K.A., Werner H.B. Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014;30:503–533. doi: 10.1146/annurev-cellbio-100913-013101. [DOI] [PubMed] [Google Scholar]
- O'Connor L.T., Goetz B.D., Couve E., Song J., Duncan I.D. Intracellular distribution of myelin protein gene products is altered in oligodendrocytes of the taiep rat. Mol. Cell. Neurosci. 2000;16:396–407. doi: 10.1006/mcne.2000.0889. [DOI] [PubMed] [Google Scholar]
- O'Connor L.T., Goetz B.D., Kwiecien J.M., Delaney K.H., Fletch A.L., Duncan I.D. Insertion of a retrotransposon in Mbp disrupts mRNA splicing and myelination in a new mutant rat. J. Neurosci. 1999;19:3404–3413. doi: 10.1523/JNEUROSCI.19-09-03404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfors A., Tulinius M. Mitochondrial encephalomyopathies. J. Neuropathol. Exp. Neurol. 2003;62:217–227. doi: 10.1093/jnen/62.3.217. [DOI] [PubMed] [Google Scholar]
- Oldfors A., Fyhr I.M., Holme E., Larsson N.G., Tulinius M. Neuropathology in Kearns–Sayre syndrome. Acta Neuropathol. 1990;80:541–546. doi: 10.1007/BF00294616. [DOI] [PubMed] [Google Scholar]
- Olmos-Serrano J.L., Kang H.J., Tyler W.A., Silbereis J.C., Cheng F., Zhu Y., Pletikos M., Jankovic-Rapan L., Cramer N.P., Galdzicki Z., Goodliffe J., Peters A., Sethares C., Delalle I., Golden J.A., Haydar T.F., Sestan N. Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination. Neuron. 2016;89:1208–1222. doi: 10.1016/j.neuron.2016.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan B.M., Blakemore W.F. Acute nicotinamide deficiency in the pig induced by 6-aminonicotinamide. Vet. Pathol. 1980;17:748–758. doi: 10.1177/030098588001700610. [DOI] [PubMed] [Google Scholar]
- Padiath Q.S., Saigoh K., Schiffmann R., Asahara H., Yamada T., Koeppen A., Hogan K., Ptacek L.J., Fu Y.H. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- Palmer A.C., Blakemore W.F., Franklin R.J., Frost L.M., Gough R.E., Lewis J.C., Macdougall D.F., O'Leary M.T., Stocker L.R. Paralysis in hedgehogs (Erinaceus europaeus) associated with demyelination. Vet. Rec. 1998;143:550–552. doi: 10.1136/vr.143.20.550. [DOI] [PubMed] [Google Scholar]
- Panagiotakos G., Alshamy G., Chan B., Abrams R., Greenberg E., Saxena A., Bradbury M., Edgar M., Gutin P., Tabar V. Long-term Impact of radiation on the stem cell and oligodendrocyte precursors in the brain. PLoS One. 2007;2 doi: 10.1371/journal.pone.0000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos D., Pham-Dinh D., Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp. Neurol. 2006;197:373–385. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Peters A., Palay S.L., Webster H.d. third ed. Oxford Univ. Press; New York U.A.: 1991. The Fine Structure of the Nervous System: Neurons and their Supporting Cells. [Google Scholar]
- Pfeiffer S.E., Warrington A.E., Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- Piao J., Major T., Auyeung G., Policarpio E., Menon J., Droms L., Gutin P., Uryu K., Tchieu J., Soulet D., Tabar V. Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell. 2015;16:198–210. doi: 10.1016/j.stem.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao J.H., Wang Y., Duncan I.D. CD44 is required for the migration of transplanted oligodendrocyte progenitor cells to focal inflammatory demyelinating lesions in the spinal cord. Glia. 2013;61:361–367. doi: 10.1002/glia.22438. [DOI] [PubMed] [Google Scholar]
- Plemel J.R., Keough M.B., Duncan G.J., Sparling J.S., Yong V.W., Stys P.K., Tetzlaff W. Remyelination after spinal cord injury: is it a target for repair? Prog. Neurobiol. 2014;117:54–72. doi: 10.1016/j.pneurobio.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Popko B. A novel mutation in myelin-deficient mice results in unstable myelin basic protein gene transcripts. Neuron. 1988;1:221–225. doi: 10.1016/0896-6273(88)90142-0. [DOI] [PubMed] [Google Scholar]
- Popovich P.G., Guan Z., McGaughy V., Fisher L., Hickey W.F., Basso D.M. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J. Neuropathol. Exp. Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- Powers J.M., Moser H.W. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions and pathogenesis. Brain Pathol. 1998;8:101–120. doi: 10.1111/j.1750-3639.1998.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers J.M., DeCiero D.P., Ito M., Moser A.B., Moser H.W. Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J. Neuropathol. Exp. Neurol. 2000;59:89–102. doi: 10.1093/jnen/59.2.89. [DOI] [PubMed] [Google Scholar]
- Prineas J. The neuropathology of multiple sclerosis. In: Vinken P.J., Bruyn G.W., Klawans H.L., Koetsier J.C., editors. Handbook of Clinical Neurology/Vol. 47, Demyelinating Diseases/Co-edited by Johan C. Koetsier. Elsevier; Amsterdam: 1985. p. xii. (660 pages) [Google Scholar]
- Pujol A., Hindelang C., Callizot N., Bartsch U., Schachner M., Mandel J.L. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum. Mol. Genet. 2002;11:499–505. doi: 10.1093/hmg/11.5.499. [DOI] [PubMed] [Google Scholar]
- Raine C.S., Cannella B., Hauser S.L., Genain C.P. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: a case for antigen-specific antibody mediation. Ann. Neurol. 1999;46:144–160. doi: 10.1002/1531-8249(199908)46:2<144::aid-ana3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan H., Hedayati K.K., Lullmann-Rauch R., Wessig C., Fewou S.N., Maier H., Goebel H.H., Gieselmann V., Eckhardt M. Increasing sulfatide synthesis in myelin-forming cells of arylsulfatase A-deficient mice causes demyelination and neurological symptoms reminiscent of human metachromatic leukodystrophy. J. Neurosci. 2007;27:9482–9490. doi: 10.1523/JNEUROSCI.2287-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff R.M. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat. Neurosci. 2012;15:1074–1077. doi: 10.1038/nn.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm S., Mehraein P., Anzil A.P., Deerberg F. A new rat mutant with defective overhairs and spongy degeneration of the central nervous system: clinical and pathologic studies. Lab. Anim. Sci. 1982;32:70–73. [PubMed] [Google Scholar]
- Richardson W.D., Kessaris N., Pringle N. Oligodendrocyte wars. Nat. Rev. Neurosci. 2006;7:11–18. doi: 10.1038/nrn1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto N., Gambetti P.L. Status spongiosus of rat central nervous system induced by actinomycin D. Acta Neuropathol. 1976;36:21–30. doi: 10.1007/BF00685144. [DOI] [PubMed] [Google Scholar]
- Rodriguez D., Gelot A., Della Gaspera B., Robain O., Ponsot G., SarliŠve L.L., Ghandour S., Pompidou A., Dautigny A., Aubourg P., Pham-Dinh D. Increased density of oligodendrocytes in childhood ataxia with diffuse central hypomyelination (CACH) syndrome: neuropathological and biochemical study of two cases. Acta Neuropathol. (Berl) 1999;97:469–480. doi: 10.1007/s004010051016. [DOI] [PubMed] [Google Scholar]
- Rojiani A.M., Prineas J.W., Cho E.S. Electrolyte-induced demyelination in rats. 1. Role of the blood–brain barrier and edema. Acta Neuropathol. (Berl) 1994;88:287–292. doi: 10.1007/BF00310371. [DOI] [PubMed] [Google Scholar]
- Rolyan H., Tyurina Y.Y., Hernandez M., Amoscato A.A., Sparvero L.J., Nmezi B.C., Lu Y., Estecio M.R., Lin K., Chen J., He R.R., Gong P., Rigatti L.H., Dupree J., Bayir H., Kagan V.E., Casaccia P., Padiath Q.S. Defects of lipid synthesis are linked to the age-dependent demyelination caused by Lamin B1 overexpression. J. Neurosci. 2015;35:12002–12017. doi: 10.1523/JNEUROSCI.1668-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs H.H., Bercury K.K., Popescu D.C., Narayanan S.P., Macklin W.B. A new model of cuprizone-mediated demyelination/remyelination. ASN Neuro. 2014;6 doi: 10.1177/1759091414551955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara S., Nakadate K., Ookawara S., Ueda S. Non-cell autonomous impairment of oligodendrocyte differentiation precedes CNS degeneration in the Zitter rat: implications of macrophage/microglial activation in the pathogenesis. BMC Neurosci. 2008;9:35. doi: 10.1186/1471-2202-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M., Lankford K.L., Brown R.J., Ruddle N.H., Kocsis J.D. Focal experimental autoimmune encephalomyelitis in the Lewis rat induced by immunization with myelin oligodendrocyte glycoprotein and intraspinal injection of vascular endothelial growth factor. Glia. 2010;58:1523–1531. doi: 10.1002/glia.21026. [DOI] [PubMed] [Google Scholar]
- Scalabrino G., Lorenzini E.C., Monzio-Compagnoni B., Colombi R.P., Chiodini E., Buccellato F.R. Subacute combined degeneration in the spinal cords of totally gastrectomized rats. Ornithine decarboxylase induction, cobalamin status, and astroglial reaction. Lab. Investig. 1995;72:114–123. [PubMed] [Google Scholar]
- Schiffmann R., Moller J.R., Trapp B.D., Shih H.H.L., Farrer R.G., Katz D.A., Alger J.R., Parker C.C., Hauer P.E., Kaneshi C.R., Heiss J.D., Kaye E.M., Quarles R.H., Brady R.O., Barton N.W. Childhood ataxia with diffuse central nervous system hypomyelination. Ann. Neurol. 1994;35:331–340. doi: 10.1002/ana.410350314. [DOI] [PubMed] [Google Scholar]
- Schobesberger M., Zurbriggen A., Doherr M.G., Weissenb”ck H., Vandevelde M., Lassmann H., Griot C. Demyelination precedes oligodendrocyte loss in canine distemper virus-induced encephalitis. Acta Neuropathol. (Berl.) 2002;103:11–19. doi: 10.1007/s004010100427. [DOI] [PubMed] [Google Scholar]
- Schonberg D.L., Popovich P.G., McTigue D.M. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. J. Neuropathol. Exp. Neurol. 2007;66:1124–1135. doi: 10.1097/nen.0b013e31815c2530. [DOI] [PubMed] [Google Scholar]
- Seehusen F., Baumgartner W. Axonal pathology and loss precede demyelination and accompany chronic lesions in a spontaneously occurring animal model of multiple sclerosis. Brain Pathol. 2010;20:551–559. doi: 10.1111/j.1750-3639.2009.00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M., Nave K.A. Oligodendrocytes: myelination and axonal support. Cold Spring Harb. Perspect. Biol. 2015;8 doi: 10.1101/cshperspect.a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripuletz T., Bussmann J.H., Gudi V., Koutsoudaki P.N., Pul R., Moharregh-Khiabani D., Lindner M., Stangel M. Cerebellar cortical demyelination in the murine cuprizone model. Brain Pathol. 2010;20:301–312. doi: 10.1111/j.1750-3639.2009.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripuletz T., Lindner M., Kotsiari A., Garde N., Fokuhl J., Linsmeier F., Trebst C., Stangel M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am. J. Pathol. 2008;172:1053–1061. doi: 10.2353/ajpath.2008.070850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.M., Jeffery N.D. Histological and ultrastructural analysis of white matter damage after naturally-occurring spinal cord injury. Brain Pathol. 2006;16:99–109. doi: 10.1111/j.1750-3639.2006.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.M., Cooksey E., Duncan I.D. Myelin loss does not lead to axonal degeneration in a long-lived model of chronic demyelination. J. Neurosci. 2013;33:2718–2727. doi: 10.1523/JNEUROSCI.4627-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.M., Mayer J.A., Duncan I.D. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J. Neurosci. 2013;33:8088–8100. doi: 10.1523/JNEUROSCI.0233-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Carson J.H., Barbarese E., Li F.Y., Duncan I.D. RNA transport in oligodendrocytes from the taiep mutant rat. Mol. Cell. Neurosci. 2003;24:926–938. doi: 10.1016/s1044-7431(03)00254-9. [DOI] [PubMed] [Google Scholar]
- Song J., O'Connor L.T., Yu W., Baas P.W., Duncan I.D. Microtubule alterations in cultured taiep rat oligodendrocytes lead to deficits in myelin membrane formation. J. Neurocytol. 1999;28:671–683. doi: 10.1023/a:1007060832459. [DOI] [PubMed] [Google Scholar]
- Southwood C.M., Garbern J., Jiang W., Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparaco M., Bonilla E., Dimauro S., Powers J.M. Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defects. J. Neuropathol. Exp. Neurol. 1993;52:1–10. doi: 10.1097/00005072-199301000-00001. [DOI] [PubMed] [Google Scholar]
- Stadelmann C., Wegner C., Bruck W. Inflammation, demyelination, and degeneration — recent insights from MS pathology. Biochim. Biophys. Acta. 2011;1812:275–282. doi: 10.1016/j.bbadis.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Stassart R.M., Helms G., Garea-Rodriguez E., Nessler S., Hayardeny L., Wegner C., Schlumbohm C., Fuchs E., Bruck W. A new targeted model of experimental autoimmune encephalomyelitis in the common marmoset. Brain Pathol. 2015 doi: 10.1111/bpa.12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steelman A.J., Thompson J.P., Li J. Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci. Res. 2012;72:32–42. doi: 10.1016/j.neures.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L., Zamvil S.S. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Stidworthy M.F., Genoud S., Suter U., Mantei N., Franklin R.J.M. Quantifying the early stages of remyelination following cuprizone-induced demyelination. Brain Pathol. 2003;13:329–339. doi: 10.1111/j.1750-3639.2003.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohlman S.A., Hinton D.R. Viral induced demyelination. Brain Pathol. 2001;11:92–106. doi: 10.1111/j.1750-3639.2001.tb00384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch M.K., Stefferl A., Brehm U., Weissert R., Wallstr”m E., Kerschensteiner M., Olsson T., Linington C., Lassmann H. Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol. 1998;8:681–694. doi: 10.1111/j.1750-3639.1998.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys P.K., Zamponi G.W., van Minnen J., Geurts J.J. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012;13:507–514. doi: 10.1038/nrn3275. [DOI] [PubMed] [Google Scholar]
- Suzuki K. A genetic demyelinating disease globoid cell leukodystrophy: studies with animal models. J. Neuropathol. Exp. Neurol. 1994;53:359–363. doi: 10.1097/00005072-199407000-00006. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Taniike R. Murine model of genetic demyelinating disease: The twitcher mouse. Microsc. Res. Tech. 1995;32:204–214. doi: 10.1002/jemt.1070320304. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Izawa T., Yamate J., Franklin R.J., Kuramoto T., Serikawa T., Kuwamura M. The VF rat with abnormal myelinogenesis has a mutation in Dopey1. Glia. 2014;62:1530–1542. doi: 10.1002/glia.22698. [DOI] [PubMed] [Google Scholar]
- Tanaka M., Soma K., Izawa T., Yamate J., Franklin R.J., Kuramoto T., Serikawa T., Kuwamura M. Abnormal myelinogenesis in the central nervous system of the VF mutant rat with recoverable tremor. Brain Res. 2012;1488:104–112. doi: 10.1016/j.brainres.2012.09.037. [DOI] [PubMed] [Google Scholar]
- Taniike M., Suzuki K. Spacio-temporal progression of demyelination in twitcher mouse: with clinico-pathological correlation. Acta Neuropathol. (Berl) 1994;88:228–236. doi: 10.1007/BF00293398. [DOI] [PubMed] [Google Scholar]
- Taniike M., Mohri I., Eguchi N., Irikura D., Urade Y., Okada S., Suzuki K. An apoptotic depletion of oligodendrocytes in the twitcher, a murine model of globoid cell leukodystrophy. J. Neuropathol. Exp. Neurol. 1999;58:644–653. doi: 10.1097/00005072-199906000-00009. [DOI] [PubMed] [Google Scholar]
- Teixeira C.A., Miranda C.O., Sousa V.F., Santos T.E., Malheiro A.R., Solomon M., Maegawa G.H., Brites P., Sousa M.M. Early axonal loss accompanied by impaired endocytosis, abnormal axonal transport, and decreased microtubule stability occur in the model of Krabbe's disease. Neurobiol. Dis. 2014;66:92–103. doi: 10.1016/j.nbd.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez I., Johnson D., Nagel R.L., Cerami A. Neurotoxicity of sodium cyanate. New pathological and ultrastructural observations in Maccaca nemestrina. Acta Neuropathol. 1979;47:75–79. doi: 10.1007/BF00698277. [DOI] [PubMed] [Google Scholar]
- Thomas N.J., Meteyer C.U., Sileo L. Epizootic vacuolar myelinopathy of the central nervous system of bald eagles (Haliaeetus leucocephalus) and American coots (Fulica americana) Vet. Pathol. 1998;35:479–487. doi: 10.1177/030098589803500602. [DOI] [PubMed] [Google Scholar]
- Towfighi J., Gonatas N.K. Hexachlorophene and the nervous system. In: Zimmerman H.M., editor. Progress in Neuropathology. vol. 3. Grune and Stratton; New York; London: 1976. p. xii. (495 pages) [Google Scholar]
- Towfighi J., Gonatas N.K., McCree L. Hexachlorophene-induced changes in central and peripheral myelinated axons of developing and adult rats. Lab. Investig. 1974;31:712–721. [PubMed] [Google Scholar]
- Traka M., Podojil J.R., McCarthy D.P., Miller S.D., Popko B. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat. Neurosci. 2015 doi: 10.1038/nn.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M., Wollmann R.L., Cerda S.R., Dugas J., Barres B.A., Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J. Neurosci. 2008;28:11537–11549. doi: 10.1523/JNEUROSCI.1490-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp B.D., Nave K.A. Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Tripier M.F., Berard M., Toga M., Martin-Bouyer G., Le Breton R., Garat J. Hexachlorophene and the central nervous system. Toxic effects in mice and baboons. Acta Neuropathol. 1981;53:65–74. doi: 10.1007/BF00697186. [DOI] [PubMed] [Google Scholar]
- Tsunoda I., Fujinami R.S. Inside-out versus outside-in model for virus induced demyelination: axonal damage triggering demyelination. Springer Semin. Immunopathol. 2002;24:105–125. doi: 10.1007/s00281-002-0105-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui S., Stys P.K. Metabolic injury to axons and myelin. Exp. Neurol. 2013;246:26–34. doi: 10.1016/j.expneurol.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Van der Knaap M.S., Valk J. Alexander disease. In: Van der Knaap M.S., Valk J., editors. Magnetic Resonance of Myelination and Myelin Disorders. third ed. Springer-Verlag Berlin and Heidelberg & Co. KG; Dordrecht: 2005. (p. 1 online resource 1096 pages) [Google Scholar]
- Von Figura K., Gieselmann V., Jaeken J. Metachromatic leukodystrophy. In: Scriver C.R., Beaudet A.L., Valle D., Sly W.S., editors. The Metabolic and Molecular Bases of Inherited Disease. eighth ed. ed. McGraw-Hill; New York; Madrid; Milan etc.: 2001. pp. 3695–3724. [Google Scholar]
- Wang Y., Piao J.H., Larsen E.C., Kondo Y., Duncan I.D. Migration and remyelination by oligodendrocyte progenitor cells transplanted adjacent to focal areas of spinal cord inflammation. J. Neurosci. Res. 2011 doi: 10.1002/jnr.22716. [DOI] [PubMed] [Google Scholar]
- Wang X.M., Yik W.Y., Zhang P., Lu W., Dranchak P.K., Shibata D., Steinberg S.J., Hacia J.G. The gene expression profiles of induced pluripotent stem cells from individuals with childhood cerebral adrenoleukodystrophy are consistent with proposed mechanisms of pathogenesis. Stem Cell. Res. Ther. 2012;3:39. doi: 10.1186/scrt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe I. Organotin (triethyltin) In: Spencer P.S., Schaumburg H.H., editors. Experimental and Clinical Neurotoxicology. Williams & Wilkins; Baltimore: 1980. pp. 545–557. [Google Scholar]
- Wenger D.A. Murine, canine and non-human primate models of Krabbe disease. Mol. Med. Today. 2000;6:449–451. doi: 10.1016/s1357-4310(00)01800-1. [DOI] [PubMed] [Google Scholar]
- Wenger D.A., Rafi M.A., Luzi P., Datto J., Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol. Genet. Metab. 2000;70:1–9. doi: 10.1006/mgme.2000.2990. [DOI] [PubMed] [Google Scholar]
- Werner H., Jung M., Klugmann M., Sereda M., Griffiths I.R., Nave K.A. Mouse models of myelin diseases. Brain Pathol. 1998;8:771–793. doi: 10.1111/j.1750-3639.1998.tb00200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.Z., Yang Q., Cate H.S., Kemper D., Binder M., Wang H.X., Fang K., Quick M.J., Marriott M., Kilpatrick T.J., Egan G.F. MRI identification of the rostral-caudal pattern of pathology within the corpus callosum in the cuprizone mouse model. J. Magn. Reson. Imaging. 2008;27:446–453. doi: 10.1002/jmri.21111. [DOI] [PubMed] [Google Scholar]
- Xie M., Tobin J.E., Budde M.D., Chen C.I., Trinkaus K., Cross A.H., McDaniel D.P., Song S.K., Armstrong R.C. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J. Neuropathol. Exp. Neurol. 2010;69:704–716. doi: 10.1097/NEN.0b013e3181e3de90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaghootfam A., Gieselmann V., Eckhardt M. Delay of myelin formation in arylsulphatase A-deficient mice. Eur. J. Neurosci. 2005;21:711–720. doi: 10.1111/j.1460-9568.2005.03891.x. [DOI] [PubMed] [Google Scholar]
- Yajima K., Suzuki K. Demyelination and remyelination in the rat central nervous system following ethidium bromide injection. Lab. Investig. 1979:385–392. [PubMed] [Google Scholar]
- Yajima K., Suzuki K. Ultrastructural changes of oligodendroglia and myelin sheaths induced by ethidium bromide. Neuropathol. Appl. Neurobiol. 1979;5:49–62. doi: 10.1111/j.1365-2990.1979.tb00613.x. [DOI] [PubMed] [Google Scholar]
- Young N.P., Weinshenker B.G., Parisi J.E., Scheithauer B., Giannini C., Roemer S.F., Thomsen K.M., Mandrekar J.N., Erickson B.J., Lucchinetti C.F. Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain. 2010;133:333–348. doi: 10.1093/brain/awp321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary J.F., O'Brien D.P. Spongy degeneration of the central nervous system in two canine littermates. Vet. Pathol. 1985;22:561–571. doi: 10.1177/030098588502200609. [DOI] [PubMed] [Google Scholar]
- Zara F., Biancheri R., Bruno C., Bordo L., Assereto S., Gazzerro E., Sotgia F., Wang X.B., Gianotti S., Stringara S., Pedemonte M., Uziel G., Rossi A., Schenone A., Tortori-Donati P., van der Knaap M.S., Lisanti M.P., Minetti C. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat. Genet. 2006;38:1111–1113. doi: 10.1038/ng1870. [DOI] [PubMed] [Google Scholar]
- Zhao L., Tian D., Xia M., Macklin W.B., Feng Y. Rescuing qkV dysmyelination by a single isoform of the selective RNA-binding protein QKI. J. Neurosci. 2006;26:11278–11286. doi: 10.1523/JNEUROSCI.2677-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]