

Abstract
In order to relieve anxiety and depression accompanying stress, physicians resort to tricyclic antidepressants, such as imipramine. We had previously shown that imipramine reversed stress-induced social avoidance behavior, and down-regulated microglial activation 24 days after stress cessation. To further characterize the effects of imipramine on stress induced neuroimmune dysregulation and associated changes in behavior, the aims of this study were to determine if imipramine 1) ameliorated stress-induced inflammation in the periphery and central nervous system, and 2) prevented stress related anxiety- and depressive-like behaviors. C57BL/6 mice were treated with imipramine (15mg/kg) in their drinking water, and exposed to repeated social defeat (RSD). Imipramine attenuated stress-induced corticosterone and IL-6 responses in plasma. Imipramine decreased the percentage of monocytes and granulocytes in the bone marrow and circulation. However, imipramine did not prevent splenomegaly, stress-related increased percentage of granulocytes in this organ, and the production of pro-inflammatory cytokines in the spleen, following RSD. Moreover, imipramine abrogated the accumulation of macrophages in the brain in mice exposed to RSD. Imipramine blocked neuroinflammatory signaling and prevented stress-related anxiety- and depressive-like behaviors. These data support the notion that pharmacomodulation of the monoaminergic system, besides exerting anxiolytic and antidepressant effects, may have therapeutic effects as a neuroimmunomodulator during stress.
Keywords: psychosocial stress, social defeat, imipramine, anxiety-like behavior, social avoidance, microglia
1. Introduction
The prolonged inflammatory state associated with social stress has the potential to contribute to the etiology of anxiety and depression. Analysis of peripheral inflammatory markers in patients with mood disorders reveals constant elevations in interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor-α (TNF-α) (Russo and Nestler, 2013). There is evidence that activation of microglia in animal models of stress, potentiates hypothalamic-pituitary-adrenal (HPA) axis stimulation through the release of IL-1β within the hypothalamus (Goshen and Yirmiya, 2009), augmenting neuroendocrine outflow that may reinforce stress-associated behaviors. Thus, physiological microglia alterations are likely to contribute to the dysfunctional neurobiological stress interpretation in the CNS.
Repeated social defeat (RSD), a model of psychosocial stress in mice, provides a probe to study the mechanisms leading to stress-related alterations in inflammation in both the periphery and CNS, and associated anxiety- and depressive-like behaviors (Kinsey et al., 2007; Wohleb et al., 2013). RSD causes an increase in pro-inflammatory factors and enhances the capacity of immune cells to enter circulation and organs, such as the spleen and brain. Also, RSD augments levels of corticosterone in circulation following the second cycle of RSD, and peaks at the sixth cycle (Engler et al., 2005).
Mice exposed to RSD display an increase in plasma and tissue catecholamines. This is meaningful, since peripheral immune cells express receptors for norepinephrine, and when these receptors are stimulated, functional responses occur that influence the development and mobility of these cells, as well as their inflammatory phenotype (Powell et al., 2013). For example, repeated activation of the sympathetic nervous system (SNS) causes an increase of norepinephrine in the bone marrow (BM) (Hanke et al., 2012) that promotes a shift in myelopoiesis after RSD exposure. These inflammatory myeloid progenitor cells (MPCs) were found to be glucocorticoid (GC)-resistant and trafficked throughout the body with an increased production of pro-inflammatory cytokines in response to lipopolysaccharide (LPS) stimulation (Avitsur et al., 2001; Bailey et al., 2009).
Microglia isolated from socially defeated mice have a higher mRNA expression of pro-inflammatory cytokines and chemokines compared to home cage controls (Wohleb et al., 2011). Also, microglia from socially defeated mice and cultured ex vivo produced exaggerated levels of IL-6, TNF-α, and CCL-2 following stimulation with LPS (Wohleb et al, 2011), even 24 days after stress cessation (Ramirez et al., 2015). Neuroinflammatory factors such as IL1-β, TNF-α, and IL-6, are associated in the neurobiological changes that reinforce fear/anxiety and threat circuitry (Wohleb et al., 2014), promoting anxiety-like behavior.
Neuronal and microglia activation and production of pro-inflammatory molecules as a result of stress exposure, promote the development of a reactive endothelium (Wohleb et al., 2014). Peripherally derived monocytes differentiate into perivascular and parenchymal macrophages (Wohleb et al., 2014) within the fear, anxiety, and threat appraisal circuitry (Wohleb et al., 2013). The accumulation of macrophages in the CNS elicited by RSD, increases neuroinflammatory signaling.
Clinical and experimental research has demonstrated that antidepressants can also prevent the expression of pro-inflammatory cytokines (Xia et al., 1996; Yirmiya et al., 2001; Castanon et al., 2002; Hashioka et al., 2007; Hwang et al., 2008). In animal models, imipramine and fluoxetine suppressed the production of TNF-α, IL-1β, and IL-6 by glial cells (Lim et al., 2009). Imipramine inhibited interferon (IFN)-γ stimulated microglial production of IL-6 and nitric oxide (Hashioka et al., 2007), and TNF-α production in microglia and astrocyte cultures (Hwang et al., 2008). In patients suffering from acute depression, fluoxetine reduced enhanced plasma levels of IL-6 (Sluzewska et al., 1995).
Recent findings from our laboratory showed RSD promoted long-lasting microglial activation associated with social avoidance behavior, which was maintained for at least 24 days after stress cessation (Wohleb et al. 2013). Imipramine treatment by intraperitoneal (i.p.) injection (20mg/kg) or in the drinking water (15mg/kg) reversed stress-associated social avoidance behavior and stress-induced increased neuroinflammatory signaling at this time point (Ramirez et al., 2015). Moreover, microglia from RSD mice produced exaggerated levels of pro-inflammatory molecules following LPS-stimulation, even 24 days after stress termination, and this was prevented with imipramine treatment.
The mechanism of medicinal action of tricyclic antidepressants such as imipramine in relation to the monoaminergic system has been well established. These drugs inhibit the reuptake of serotonin, norepinephrine, and dopamine by directly blocking neurotransmitter transporters (Eshleman et al., 1999; Zhou et al., 2007). Neurotransmitter transporters for serotonin, norepinephrine, and dopamine in the presynaptic membrane restricts neuronal signal transmission (Glowinski and Axelrod 1964, Iversen et al., 2006) and drugs used to block these systems have been used successfully for the treatment of depression (Klimek et al., 1997; Zhou et l., 2007). However, more research is needed to establish the influence of antidepressants on stress-related catecholaminergic mechanisms, specifically in the context of HPA axis, SNS activation, and cytokine production in both the periphery and central nervous system (CNS). The objective of this study was to further determine if neuroinflammatory signaling, and behavioral alterations after six cycles of RSD, could be reversed with imipramine treatment. Hence, the effect of imipramine on stress-induced shift in myelopoiesis, and trafficking of MPCs to blood, spleen, and brain, and associated anxiety- and depressive like behaviors were studied.
2. Materials and Methods
2.1 Animals
Male C57BL/6 (6–8 weeks old) and CD-1 (12 months old, retired breeders) mice were purchased from Charles River Breeding Laboratories (Wilmington, Massachusetts) and allowed to acclimate to their surroundings for 7–10 days prior to initiation of experiments. C57BL/6 mice were housed in cohorts of three and CD-1 mice were singly housed in 11.5 x 7.5 x 6 inch polypropylene cages. Mice were maintained at 21°C under a 12:12 h light: dark cycle with ad libitum access to water and rodent chow in the animal facility at The Ohio State University. All procedures were in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee.
2.2 RSD
RSD was performed as described previously (Wohleb et al., 2011). In brief, an intruder male CD-1 mouse was introduced into home cages of male C57BL/6 mice (three per cage) for 2 hours (hrs) on 6 consecutive nights. Behavior was observed to make certain that the intruder was aggressive. If the CD-1 mouse did not initiate an attack within 5–10 minutes or was attacked by resident mice, a new CD-1 mouse was introduced. At the end of the 2 hrs the CD-1 mouse was removed and the resident mice were left undisturbed until the next day when the same paradigm was repeated. During RSD, resident mice display submissive behaviors such as upright posture, fleeing, and crouching (Stark et al., 2001; Hanke et al., 2012). Home cage control (HCC) cohorts were left undisturbed in a separate room.
2.3 Pharmacological treatments and administration procedures
C57BL/6 mice were randomly selected for inclusion into different experimental treatment groups. The groups were: RSD/imipramine, RSD/vehicle, HCC/imipramine, and HCC/vehicle. Mice in the RSD/imipramine received daily imipramine (15 mg/kg) treatment in their drinking water as previously described (Ramirez et al., 2015), starting two days before the initiation of social defeat. HCC/imipramine received daily imipramine treatment in their drinking water at the same dose while RSD/vehicle and HCC/vehicle groups drank untreated water (Figure 1a). Bottles of water were changed daily before and during the experimental protocol. The dose of imipramine was based on a previous study with C57BL/6 mice, in which chronic administration at 15mg/kg in drinking water effectively reversed social avoidance behavior and neuroinflammatory signaling after 24 days of social defeat cessation (Ramirez et al., 2015).
Figure 1.

(A) Experimental schematic is shown. C57BL/6 mice were randomly allocated in different experimental groups. Mice were exposed to 6 cycles of Repeated Social Defeat (RSD) or left undisturbed and treated with imipramine in their drinking water or vehicle (untreated water) starting two days before social defeat. (A) RSD promoted higher levels of corticosterone in plasma, immediately after the last cycle of RSD. Animals subjected to RSD and treated with imipramine had increased levels of corticosterone than HCC, but lower than RSD animals treated with vehicle. The next day after the last cycle of RSD, blood was collected to determine levels of IL-6 in plasma. (B) Mice subjected to RSD and treated with vehicle had increased levels of IL-6 in plasma. Levels of IL-6 in plasma of mice subjected to RSD and treated with imipramine had lower levels of this cytokine, comparable to HCC. Bars represent the mean±SEM. Means with different letters (a,b,c) are significantly different from each other (p<0.05). Control=Home cage controls; RSD=Repeated Social Defeat
The amount of water consumed for each cage was registered daily throughout the experiment (from Day 0 to Day 6). The calculation of the concentration of imipramine in drinking water was based on the evaluated mean volume of daily water consumption, assessed by weighing the bottles daily (from Day 0 to Day 6) from our previous imipramine study (Ramirez et al., 2015). An average of 9.0 ml per day/per cage intake was calculated. Water consumption in the four groups of mice was in fact similar. Based on this average, the desirable dosage of treatment (15 mg/kg/day) was established.
2.4 Plasma corticosterone determination
Blood was collected from the submandibular vein immediately following the last cycle of RSD (i.e. at 1900 EST). Blood samples were immediately placed on ice and plasma was collected after centrifuging samples at 1800 x g for 15 minutes at 4°C. Plasma was stored at −80°C until assayed in duplicate using a Corticosterone ELISA kit from Enzo Life Sciences (Farmingdale, New York) following manufacturer’s instructions (n=6 per group).
2.5 Anxiety-like behavior testing and social avoidance test
Behavior was determined the morning following the last cycle of RSD as previously described (Wohleb et al., 2011). Each behavioral test was assessed in different cohorts of mice. Anxiety-like behavior was measured in the open field test, consisting of a 40 x 40 x 25 cm square plexiglass box with a solid floor and a 6 x 6 grid drawn on the floor separating the open field into 36 identical squares. This test takes advantage of mice’s natural tendencies to explore the environment while avoiding open spaces. For this experiment, the dependent variables were: total time spent in the center of the open field, latency to entering the center and the number of entries into the center of the open field in a 5 min period. The test apparatus was cleaned with water between experimental subjects. Behavior was taped, digitized and coded using the Observer program (Noldus Information Technologies, Netherlands) (n=6 per group).
The social avoidance test was performed as previously described (Ramirez et al., 2015) in a social interaction open-field arena (42 cm wide, 42 cm depth, and 42 cm height). The interaction zone of the test arena consisted of a 14 cm × 24 cm rectangular area that projected 8 cm around the wire-mesh enclosure. The duration of the test was 5 minutes and consisted of two trials (2.5 minutes each trial). In the first trial, an experimental mouse was placed into the arena with an empty wire mesh cage and activity was recorded for 2.5 minutes. In the second trial, the experimental mouse was placed in the arena with an unfamiliar CD-1 mouse in the wire mesh cage and activity was recorded for the same amount of time. Time in the interaction zone and time spent in the corners was video-recorded and analyzed using Noldus EthoVision Software (Leesburg, Virginia). Fourteen hours after the sixth cycle of RSD, mice were euthanized by CO2 asphyxiation after the completion of either anxiety-like behavior testing or the social avoidance test and tissues were collected for processing.
2.6 Plasma IL-6 determination
Blood was collected by cardiac puncture the next day after the last cycle of RSD with EDTA lined 1ml syringes. Whole blood was centrifuged; plasma was collected and stored at −80°C until assayed. IL-6 levels were determined using the BD OptEIA Mouse IL-6 ELISA as previously described (Wohleb et al., 2011) according to the manufacturer’s instructions (BD Biosciences, San Jose, California). Briefly, 96-well enzyme immunoassay plates were coated with anti-mouse IL-6 capture antibody and incubated overnight at 4°C. Samples and IL-6 standards (0–1000 pg/ml) were added and incubated for 2 hrs at room temperature. Plates were washed and incubated with biotinylated anti-mouse IL-6 antibody. Then, plates were washed and incubated with streptavidin-horseradish peroxidase conjugate. After 1 hr incubation at room temperature, plates were washed and incubated with tetramethylbenzidine liquid substrate for 15 min. Reactions were ended and absorbance was read at 450 nm using a Spectramax Plus 384 plate reader (Molecular Devices, Sunnyvale, California). The assay was sensitive to 10 ng/ml IL-6 and the interassay and intra-assay coefficients of variation were <10% (n=5–6 per group).
2.7 Microglia isolation and flow cytometry
Microglia were isolated from whole-brain homogenates as previously described (Henry et al., 2008; 2009; Wynne et al., 2010, Wohleb et al., 2011) the next day after the last cycle of RSD. In brief, brains were homogenized in Hanks’ balanced salt solution (HBSS), with a pH of 7.4, by mashing the brain through a 70 μm nylon mesh cell strainer. The homogenates were then centrifuged at 500 X g for 6 min at 10°C. Supernatants were decanted and the obtained pellets were resuspended in 70% isotonic Percoll (GE Healthcare, Pittsburgh, Pennsylvania) at room temperature. A Percoll density gradient was layered in this manner: 70%, 50%, 35%, and 0% isotonic Percoll. The gradient was centrifuged at 2000 X g for 20 minutes at 10°C and microglia was taken by aspirating the interphase between the 50% and 70% Percoll layers (Frank et al., 2006; Nair et al., 2007, Wohleb et al., 2011). The retrieved cells were washed and resuspended in sterile HBSS and centrifuged at 600 X g for 6 minutes at 10°C. The supernatant was decanted and viable cells were counted using 0.1% trypan blue staining in an automated cell counter (Luna-FL™ dual fluorescence cell counting, Logos Biosystems, Annandale, Virginia). Each brain yielded approximately 6.5 × 10 5 cells. Studies (Henry et al., 2009; Wynne et al., 2010) demonstrate that viable cells isolated from brain homogenates using this protocol yields >90% microglia. For flow cytometry, staining of microglia surface antigens was executed as previously described (Henry et al., 2008; 2009 and Wohleb et al., 2011). In brief, Fc receptors were blocked with anti-CD16/CD32 antibody (eBioscience, San Diego, California). The cells were then incubated with anti-CD11b-APC, anti-CD45-FITC, and anti-MHC-II-PE antibodies (eBioscience, San Diego, California). Expression of these surface receptors was evaluated using a Becton-Dickinson FACSCalibur™ four-color cytometer. Ten thousand events were recorded and microglia were identified by the expression of CD11b+ and CD45low markers and macrophages by the expression of CD11b+ and CD45high (Nair and Bonneau, 2006). For each antibody, gating was determined based on appropriate isotype-stained controls. Flow data were analyzed using FlowJo software (Tree Star, San Carlos, California).
2.8 Microglia RNA isolation and real-time PCR
The next day after the last cycle of RSD microglia was isolated as described in section 2.7. RNA from Percoll-isolated microglia was isolated using the RNeasy plus mini-kit (Qiagen®). RNA concentration was determined by a spectrophotometer (Eppendorf, Hamburg, Germany). RNA was reverse transcribed to get cDNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems®) and quantitative PCR was done using the Applied Biosystems® by Life Technologies Assay-on-Demand Gene Expression protocol as previously described (Wohleb et al., 2011). Briefly, amplification of cDNA was performed by real-time PCR. A target cDNA (IL-1 β, IL-6, TNF-α, TLR4, CCL2, and CX3CR1) and a reference cDNA (glyceraldehyde-3-phosphate dehydrogenase) were amplified at the same time using an oligonucleotide probe with fluorescent reporter dye (FAM). An ABI PRISM® 7300-sequence detection system (Applied Biosystems® by Life Technologies, Grand Island, New York) was used to assess fluorescence. Data were analyzed by the comparative threshold cycle and the results are given as the fold difference detected (n=5–6 per group).
2.9 Total splenocytes RNA isolation and real-time PCR
Spleens were collected, weighed, and flash frozen in liquid nitrogen. Samples were stored at −80°C until assayed. Samples were homogenized using a tissue tearor and then centrifuged at 4°C and 12,000g for 10 minutes. Supernatant was collected and each sample received 200ul of chloroform, followed by vortexing and centrifugation at 12,000x g for 15 minutes. The upper aqueous phase was decanted into a fresh tube, to which isopropanol was added to precipitate the RNA. This solution was centrifuged at 12,000 x g for 10 minutes, supernatant was removed, and the resulting pellet was washed with 1ml 75% EtOH to remove residual protein. The pellet was resuspended in 20ul nuclease free water. RNA concentration was measured by spectrophotometry (Implen, Westlake Village, CA) and RNA was reverse transcribed to cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using a Taqman Gene Expression Assay to observe for potential changes in expression of pro-inflammatory cytokines (i.e. IL-1β, IL-6, TNF-α). Expression was analyzed using an ABI Prism 7000 Sequence Detection System (Applied Biosystems; Foster City, CA) using the 2-ΔΔ Ct method with normalization glyceraldehyde-3 phosphate dehydrogenase (GAPDH) (n=5–6 per group).
2.10 Flow cytometry of bone marrow, blood, and spleen
Blood was collected after cardiac puncture with an EDTA filled syringe and bone marrow was removed, placed in ice-cold HBSS and processed. Bone marrow was flushed from bone into a 70μm strainer with 1xPBS into a 50 ml conical tube, until it reached a volume of 7.5 ml. Samples were centrifuged at 1800 rpm for 8 minutes. Supernatant was poured off and the pellet of cells was brought up in 1ml FACS buffer and vortexed. Fifty μl of each sample was added into a flow tube. Fc receptors of bone marrow cells and blood leukocytes were blocked with anti-CD16/CD32 antibody (eBioscience, San Diego, California) as previously described (Wohleb et al., 2014). Cells were washed and then incubated with the appropriate antibodies (CD45, CD11b, CD34; eBioscience, San Diego, California), and Ly6C (BD Biosciences, San Jose, California) for 1 h at 4°C. Cells were washed and re-suspended in FACS buffer for analysis. Non-specific binding was assessed by using isotype-matched antibodies. Antigen expression was determined using a Becton-Dickinson FACSCalibur four-color cytometer (BD Biosciences, San Jose, California).
For flow cytometry assessment of splenocytes, spleens were collected, weighed, placed in ice-cold HBSS and disrupted mechanically to obtain single cell suspensions as previously described (Hanke et al., 2012). Lysis of red blood was performed by adding 2 ml of room temperature lysis buffer (0.16 M NH4Cl, 10 mM KHCO3, and 0.13 mM EDTA) for 2 min, and then by a single wash with HBSS/10% heat inactivated fetal bovine serum (FBS). Each cell pellet was resuspended in HBSS, filtered and washed a final time in HBSS. Cells were counted and samples were resuspended (2.5 × 106 cells/ml) in supplemented RPMI medium (10% heat-inactivated FBS, 0.075% sodium bicarbonate, 10 mM HEPES buffer, 100 U/ml penicillin G, 100 mg/ml streptomycin sulfate, 1.5 mM L-glutamine, and 0.0035% 2-mercaptoethanol). Single cell suspensions derived from spleen tissue (1 x106 cells per sample) were incubated with 1 μg each of fluorescently labeled monoclonal antibodies (or the appropriate isotype controls). Antibody labeling was performed at 4° C for 45 min. The cells were then washed twice in PBS containing 1% FBS and 0.09% NaN3. All antibodies were obtained from BD PharMingen (San Jose, California), APC-labeled anti-CD11b, PerCP-labeled anti-Gr-1/Ly-6C. Leukocytes were gated based on forward versus side scatter and a total of 10,000–100,000 events were analyzed on a FACSCalibur flow cytometer using Cell Quest and Cell Quest Pro analysis software (Becton-Dickenson; San Jose, California) (n=6 per group).
3. Statistical Analysis
To ensure a normal distribution, gathered data were subjected to Shapiro-Wilk tests using SPSS Statistics version 21 (IBM, Armonk, New York). Data are expressed as means ± SEM. To determine significant main effects and interactions between variables being evaluated, data were subjected to one-way (trial, stress, imipramine), two-way (trial x stress); (trial x imipramine); (stress x imipramine) or three-way (trial x stress x imipramine) analysis of variance (ANOVAs). When appropriate, differences between treatment group means were analyzed by an F-protected t test using the least-significant difference method. In all cases, the level of significance was set at p ≤ .05.
4. Results
4.1 Imipramine attenuated RSD-induced corticosterone in plasma and blocked IL-6 response in plasma
Previous studies showed that mice had enhanced levels of serum corticosterone immediately after the last cycle of RSD compared to HCC mice (Hanke et al., 2012). Additionally, RSD consistently increased plasma levels of IL-6 (Stark et al., 2001; 2002). Hence, corticosterone and IL-6 responses were used as biomarkers to gauge the robustness of the response to social defeat. Therefore, our first objective was to determine the effect of imipramine on stress-induced increased levels of plasma corticosterone, and the second objective the effect of this drug on IL-6 level in plasma. Figure 1B shows that immediately after the last cycle of RSD, increased levels of corticosterone were found in plasma of mice subjected to RSD (main effect of stress; F (1,23) = 32.15, p < 0.01; main effect of imipramine; F (1,23) = 6.76, p < 0.05; stress x imipramine interaction; F (1,23) = 1.30, p = 0.26). Post-hoc analyses revealed imipramine attenuated plasma corticosterone in socially defeated animals (p<0.05) compared to socially defeated mice treated with vehicle. However, plasma corticosterone was high in RSD mice treated with imipramine compared to HCC (p<0.05).
Blood was collected the day after the last cycle of RSD and plasma IL-6 levels were determined. As reported previously, social defeat increased IL-6 levels in plasma (Figure 1C), and this increase was blocked by imipramine treatment (main effect of stress; F (1,21) = 25.95, p<0.01; stress x imipramine interaction; F(1,21)= 4.46, p <0.05). Together, these data indicate that imipramine was effective in attenuating stress-related increased corticosterone levels in plasma and blocked levels of circulating IL-6 in plasma of C57BL/6 mice subjected to social defeat.
4.2 Effect of Imipramine on stress-driven alteration of bone marrow hematopoiesis
RSD enhanced myelopoiesis and promoted the development, priming, and egress of a MPC population from the BM (Engler et al., 2004). Thus, the aim of this experiment was to determine the effects of imipramine on stress-induced alterations in hematopoiesis in the BM (Figures 2A and 2B). RSD mice treated with vehicle and imipramine, had a decreased percentage of lymphocyte progenitors in the BM (main effect of stress; F (1,23)= 33.73, p<0.01) compared to HCC. Imipramine prevented stress-induced decrease in erythrocyte progenitors (main effect of stress; F (1,23) = 6.15, p<0.05; stress x imipramine interaction; F(1,23)=9.61, p<0.01) in the BM. Moreover, imipramine blocked stress-induced increase percentage of granulocytes (main effect of stress; F (1,23)= 19.28, p<0.01; stress x imipramine interaction; F (1,23) =11.32, p<0.01) and blocked stress-induced increase of monocyte progenitors in the BM (main effect of stress; F (1,23)=10.76, p<0.01; stress x imipramine interaction; F (1,23) =9.77, p<0.01). In sum, imipramine blocked RSD-induced enhanced production of myeloid cells (granulocytes and monocytes).
Figure 2.

The morning after the last cycle of RSD, bone marrow (BM) was collected from the different experimental groups, and CD31 and Ly-6C+ expression levels were determined using flow cytometry. A is a representative bivariate dot plot of CD31/Ly-6c staining. Average numbers of monocytic (M) (CD31+/Ly-6chigh), granulocytic (G) (CD31+/Ly-6c+), lymphoid (L) (CD31+/Ly-6c-), erythroid (E) (CD31−/Ly-6c-) lineage-committed progenitor cells are illustrated. (A and B) Imipramine reversed stress-induced decrease of erythrocyte progenitors in the bone marrow but had no effect on stress-induced decrease of lymphocytic progenitors in the bone marrow. Stress-induced increase of granulocyte and monocyte progenitors in the BM was reduced with imipramine treatment. Bars represent the mean±SEM. Means with different letters (a,b) are significantly different from each other (p<0.05). VCON=Vehicle Controls; ICON=Imipramine Controls; VRSD=Vehicle Repeated Social Defeat; ICON=Imipramine Controls.
4.3 Effect of imipramine in stress-induced increased percentage of monocytes and granulocytes in circulation and in the spleen
Previous findings reported an increased number of monocytes and granulocytes in socially defeated mice (Hanke 2012; Powell et al., 2013; Wohleb et al., 2013). Figure 3A and 3B, shows mice exposed to RSD had increased numbers of monocytes (main effect of stress; F (1,23) = 4.53, p=0.05) and granulocytes in circulation (main effect of stress; F (1,23) = 114.73, p<0.01). Post-hoc analyses revealed imipramine decreased stress-induced increase in monocytes (p<0.05) in circulation. Additionally, imipramine also ameliorated granulocyte trafficking in circulation (stress x imipramine interaction; F (1,23) =100.89, p<0.01).
Figure 3.

Blood and spleens were collected, the morning after the last cycle of RSD and Ly-6C and CD11b expression was assessed by flow cytometry. A is representative flow bivariate dot plots of Ly-6C/CD11b labeling showing that RSD mice treated with vehicle had elevated percentage of blood monocytes (M) (CD11b+/Ly6Chigh) and blood granulocytes (G) (CD11b+/Ly6Cintermediate) in circulation. Imipramine decreased stress-induced increase in circulating (B) monocytes and (C) granulocytes in mice subjected to RSD. D shows a bivariate dot plot of Ly-6C/CD11b labeling in the spleen demonstrating that RSD mice treated with vehicle had elevated percentage of monocytes (M) (CD11b+/Ly6Chigh) and (G) granulocytes (CD11b+/Ly6Cintermediate) in this organ. (E) Imipramine blocked stress-induced increase of monocytes in the spleens of mice subjected to RSD. However, mice subjected to RSD and treated with (F) imipramine still displayed a high percentage of granulocytes when compared to HCC. (G) Moreover, mice subjected to RSD and treated with vehicle had increased spleen weight compared to HCC. Imipramine did not decrease splenomegaly in socially defeated animals. Bars represent the mean±SEM. Means with different letters (a,b) are significantly different from each other (p<0.05). Control=Home cage control; RSD=Repeated Social Defeat
RSD caused an increase in the number and percentage of monocytes and granulocytes in the spleen, which was associated with the development of splenomegaly (Avitsur et al., 2003). Thus, total splenocytes were analyzed using flow cytometry to determine if imipramine would affect the percentage of these cells in the spleens of socially defeated mice (Figures 3D, 3E and 3F). Flow cytometry analysis indicated that stress-driven increase in monocytes (main effect of stress; F (1,23) =4.77, p<0.05) was completely blocked by imipramine treatment (stress x imipramine interaction; F (1,23) = 4.38, p<0.05). However, this drug did not reverse the stress driven increase in splenic granulocytes (main effect of stress; F (1,23) = 4.74, p <0.05; stress x imipramine interaction, F (1,23) = 1.24, p>0.05). Mice subjected to RSD and treated with imipramine had a greater percentage of these cells in the spleen compared to HCC (p<0.05).
Mice subjected to RSD showed increased spleen size and weight (i.e., splenomegaly) that is associated with an increased number of monocytes/macrophages (Avitsur et al., 2003). Therefore, we next aimed to determine if treatment with imipramine would attenuate splenomegaly in socially defeated mice. Imipramine did not attenuate splenomegaly (main effect of stress; F (1,47) = 8.04, p<0.01; stress x imipramine interaction; F (1,47) = 1.78, p>0.05). Post-hoc analysis indicate spleen weight in RSD mice treated with this drug was still high compared to HCC (p<0.05) as shown in Figure 3G.
These data suggest that imipramine prevented the release of monocytes and granulocytes from the bone marrow into circulation, in socially defeated mice. Imipramine decreased the pool of monocytes in the spleens of socially defeated animals, but was not effective in decreasing the pool of granulocytes in the spleen or splenomegaly associated with RSD.
4.4 Imipramine did not decrease stress-induced production of pro-inflammatory cytokines in splenocytes
We sought to determine if imipramine would decrease RSD increased pro-inflammatory cytokine production by splenocytes, specifically, IL-1β, IL-6, and TNF-α (Table 1A). Imipramine did not block mRNA expression of IL-1β (main effect of stress; F(1,20)= 5.12, p <0.05; stress x imipramine interaction; F(1,20)= 3.09, p=0.09), IL-6 (main effect of stress; F(1,21)=7.76, p=0.01; stress x imipramine interaction; F(1,21)=3.54, p=0.07), and TNF-α in total splenocytes (main effect of stress; F(1,21) = 7.36, p=0.01; stress x imipramine interaction; F(1,21) = 0.69, p>0.05). In sum, the production of pro-inflammatory cytokines in total splenocytes was enhanced after six cycles of RSD and imipramine treatment was not effective in preventing increased mRNA levels of IL1-β, IL-6, and TNF-α in socially defeated mice.
Table 1.
Total splenocytes of socially defeated animals had increased mRNA expression of IL1-β, IL-6, and TNF-α. (A) Imipramine was not effective in decreasing mRNA expression of these pro-inflammatory cytokines in mice subjected to RSD.
|
A. Total Splenocytes mean relative gene expression
| ||||
|---|---|---|---|---|
| (6 Days After RSD) | ||||
| Vehicle
|
Imipramine
|
|||
| Gene | HCC | RSD | HCC | RSD |
| IL-1β | 1.12±0.20a | 2.42±0.43b | 1.28±0.12a | 1.46±0.49a,b |
| IL-6 | 1.01±0.41a | 5.63±1.41b | 1.59±0.77a | 2.49±1.11a,b |
| TNF-α | 1.28±0.40a | 9.27±3.23b | 1.10±0.59a | 5.35±2.97a,b |
Means with different letters (a,b) are significantly different from each other (p<0.05). HCC= Home cage controls; RSD= Repeated Social Defeat.
4.5 Imipramine prevented stress-induced trafficking of bone marrow-derived CD11b+ myeloid cells to the brain
RSD altered the phenotype of myeloid-derived CD11b+ cells in the periphery (Bailey et al., 2009) and increased their propensity to traffic from the bone marrow to the brain (Wohleb et al., 2011). In line with this idea, we next sought to determine the effect of imipramine on the trafficking of circulating monocytes to the CNS in socially defeated mice. Figure 4A shows representative bivariate dot plots of CD11b and CD45 staining for macrophages (CD11b+/CD45high) (Nair et al., 2007) in the CNS for the various experimental groups. The percentage of macrophages after RSD was increased, however, this was ablated by imipramine (main effect of stress; F (1,23) = 10.13, p<0.01; stress x imipramine interaction; F (1,23) = 6.70, p=0.01) as seen in Figure 4B. These data suggest that imipramine was effective in preventing RSD-induced accumulation of macrophages in the brain.
Figure 4.

Brains were collected the day after the last cycle of RSD, CD11b+ cells were enriched by Percoll gradient separation, CD45 and CD11b expression levels were determined using flow cytometry. (A) Representative bivariate dot plots of the different experimental groups with CD11b/CD45 staining, showing that (B) imipramine blocked stress-induced increased percentage of macrophages in the brain. Bars represent the mean±SEM. Means with different letters (a,b) are significantly different from each other (p<0.05). Control=Home cage controls; RSD=Repeated Social Defeat
4.6 Imipramine ameliorated neuroinflammatory signaling
To determine if RSD-driven neuroinflammatory signaling was attenuated with imipramine treatment, gene expression of pro-inflammatory cytokines and activation markers was examined in microglia (Table 2). RSD mice treated with vehicle had increased relative gene expression of IL1-β (main effect stress; F(1,21)= 31.15, p< 0.01), IL-6 (main effect stress; F(1,21)= 9.57, p< 0.01), TNF-α (main effect stress; F(1,21)= 13.54, p< 0.01), and activation markers TLR-4, CCL2, and CX3CR1 (main effect stress; F(1,20)= 7.76, p=0.01; main effect stress; F(1,21)= 11.68, p< 0.01; main effect stress; F(1,20)= 9.99, p< 0.01; respectively) and this effect was prevented with imipramine (stress x imipramine interaction; F(1,21) = 8.89, p<0.01;stress x imipramine interaction; F(1,21) = 6.28, p<0.05; stress x imipramine interaction; F(1,21) = 5.22, p<0.05; stress x imipramine interaction; F(1,20) = 4.05, p=0.06; stress x imipramine interaction; F(1,21) = 4.97, p<0.05; stress x imipramine interaction; F(1,20) = 4.17, p=0.05; respectively ). Post-hoc analyses indicated there was no difference in mRNA expression of these pro-inflammatory molecules evaluated between RSD mice treated with imipramine and HCC (p<0.05 for all molecules assessed). These data suggest that imipramine abated RSD-induced neuroinflammatory signaling by decreasing gene expression of pro-inflammatory molecules in microglia.
Table 2.
Microglia was collected by Percoll gradient separation. (A) Increased neuroinflammatory signaling was observed in RSD mice treated with vehicle and ameliorated in RSD mice treated with imipramine. RSD markedly increased relative gene expression of pro-inflammatory cytokines IL-1β, IL-6, TNF-α, and activation markers TLR-4, CCL2 and CX3CR1.
|
A. Microglia mean relative gene expression
| ||||
|---|---|---|---|---|
| (6 Days After RSD) | ||||
| Vehicle
|
Imipramine
|
|||
| Gene | HCC | RSD | HCC | RSD |
| IL-1β | 1.02±0.43a | 4.47±0.71b | 0.40±0.08a | 2.22±0.14a |
| IL-6 | 0.99±0.18a | 6.24±1.94b | 1.64±0.13a | 2.28±0.74a |
| TNF-α | 1.22±0.47a | 3.75±0.48b | 0.25±0.12a | 0.89±0.12a |
| TLR-4 | 1.01±0.19a | 4.31±1.16b | 1.08±0.15a | 1.61±0.56a |
| CCL2 | 0.99±0.36a | 6.44±1.59b | 1.49±0.18a | 2.51±1.42a |
| CX3CR1 | 0.99±0.23a | 6.75±1.61b | 2.63±0.95a | 1.11±0.07a |
Means with different letters (a,b) are significantly different from each other (p<0.05). HCC= Home cage controls; RSD= Repeated Social Defeat.
4.7 Imipramine prevented stress-induced anxiety-like behavior
RSD promoted anxiety-like behavior in the open field test (Wohleb et al., 2011). Therefore, the aim of this experiment was to evaluate if imipramine treatment of socially defeated mice could reverse anxiety-like behavior. The day after the last cycle of RSD, individual mice were placed in the corner of an open field box and allowed to freely explore the apparatus. Figure 5A show mice subjected to RSD had a decreased number of center entries compared to HCC mice, and this was prevented with imipramine treatment (main effect of stress; F (1,23) = 4.27, p=0.05; stress x imipramine interaction; F (1,23) =4.28, p<0.05). Figure 5B shows that mice subjected to RSD and treated with vehicle took more time to first enter the center of the open field compared to HCC treated with vehicle and this was reversed with imipramine (main effect of stress; F (1,23) = 4.54, p<0.05; stress x imipramine interaction, F (1,23) =3.85, p=0.06). Post-hoc analyses revealed mice subjected to RSD and treated with imipramine took less time to enter the center compared to RSD mice treated with vehicle (p<0.05), the time was no significantly different from HCC mice (p>0.05) Likewise, Figure 5C shows that imipramine was effective in increasing center duration in mice treated with imipramine and subjected to RSD (main effect of stress; F (1,23) =4.38, p<0.05; stress x imipramine interaction; F (1,23) = 0.86, p>0.05). Post-hoc analyses indicated this increase in center duration was significantly different from RSD mice treated with vehicle (p<0.05), but not from HCC mice (p>0.05). These data indicate imipramine acted as an anxiolytic in socially defeated mice.
Figure 5.

Anxiety-like behavior was evaluated the next day after the last cycle of RSD in the open field test. (A) Mice subjected to RSD and treated with vehicle, had significantly less number of center entries than HCC and RSD mice treated with imipramine. (B) Mice treated with vehicle and subjected to RSD took more time to enter the center of the open field, and this was reversed with imipramine treatment. (C) A decreased time spent in the open field was assessed in RSD mice treated with vehicle and this was prevented with imipramine treatment. Bars represent the mean±SEM. Means with asterisk (*) are significantly different from each other (p<0.05). Control=HCC; RSD=Repeated Social Defeat; #= number; (s)=seconds
4.8 Imipramine reversed social avoidance behavior
Recent findings indicate that RSD promotes long-lasting depressive-like behavior that can be measured as social avoidance (Wohleb et al., 2013) and imipramine was able to reverse this type of behavior 24 days after stress cessation (Ramirez et al., 2015). Therefore, in this experiment the aim was to determine if eight-day imipramine treatment during RSD could prevent stress-induced social avoidance behavior. The day after the 6th cycle of RSD, social avoidance was determined using a two-trial interaction paradigm with an empty social target trial (Trial 1) followed by a social target trial (Trial 2). Figure 6 show that mice subjected to RSD spent less time in the interaction zone when an intruder was introduced in (main effect stress; F (1,20) =7.38, p=0.01). Imipramine was effective in reversing stress-induced social avoidance behavior in mice subjected to RSD (main effect trial; F (1,20) = 12.86; trial x stress interaction; F (1,20) = 31.17, p<0.01; trial x imipramine; F (1,20) = 17.25, p<0.01; trial x stress x imipramine interaction; F (1,20) = 29.28, p<0.01). Together, these data indicate that eight days of imipramine treatment during RSD effectively reversed stress-induced depressive-like behavior in the test of social avoidance.
Figure 6.
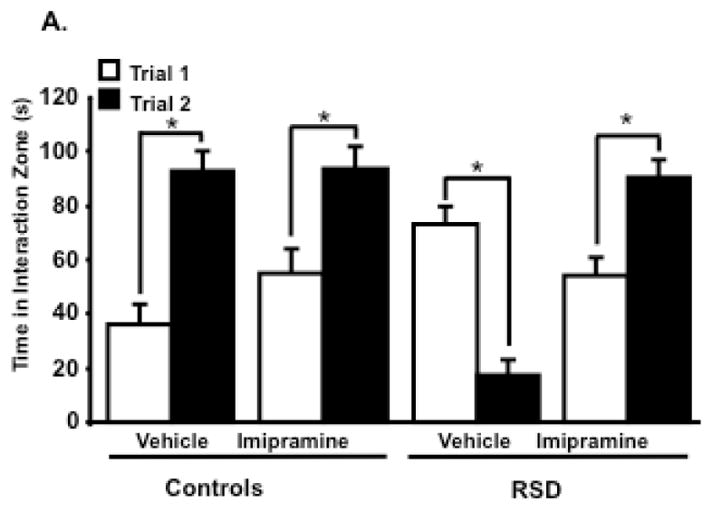
Depressive-like behavior was evaluated using the social avoidance test. Social withdrawal was exacerbated when an aggressor was introduced in the social target trial (Trial 2) in RSD mice treated with vehicle, with a decreased time spent in the interaction zone, and this was reversed with (A) imipramine treatment. Bars represent the mean ± SEM. Means with asterisk (*) are significantly different from each other (p<0.05). Trial 1: time without intruder; Trial 2= time with intruder; RSD=Repeated Social Defeat; (s)=seconds
5. Discussion
The present study confirms that imipramine treatment in the drinking water (15mg/kg) prevented stress-induced neuroimmune dysregulation and associated behavioral alterations, after six cycles of social defeat. At this time point, imipramine attenuated the corticosterone response in plasma, a product of HPA activation. Also, imipramine prevented stress-induced increase in plasma IL-6, and the development and trafficking of MPCs from the BM to circulation and brain. Finally, treatment with imipramine, during six days of social defeat, decreased neuroinflammatory signaling, paralleled by a blockade of co-morbid anxiety- and depressive-like behaviors in mice exposed to RSD.
It is possible that monoaminergic drugs may correct neuroendocrine dysregulation through normalization of stress-induced increase of cortisol/corticosterone secretion. There is evidence that mirtazapine, a tricyclic antidepressant like imipramine, decreased cortisol levels in depressed patients (Antonioli et al., 2012). In clinical studies, the reduction of cortisol in depressed patients was associated with remission (Antonioli et al., 2012). In the present study, imipramine attenuated RSD-induced increase of corticosterone.
In the present study, the antidepressant imipramine blocked IL-6 in plasma. Over the last few years, evidence from in vivo studies show a clear trend to increased IL-6 plasma levels in both acute and chronic depression, and a tendency to normalize with antidepressants (Sluzewska et al., 1995, Frommberger et al., 1997; Basterzi et al., 2005). Interestingly, immune cells from animals and patients that have undergone psychological stressors do not show enhancement of cytokine production in vitro, even though they did show increased circulating levels of IL-6 (Zhou et al., 1993; Jankord et al., 2010), suggesting that the production of IL-6 might be from another source other than peripheral immune cells. In rodents, exposure to physical and psychological stressors elevated plasma levels of IL-6 and this elevation was correlated with the intensity/severity of the stressor (Zhou et al., 1993). In this same study, adrenalectomy attenuated the elevation of this cytokine in plasma due to stress, suggesting that this cytokine may be under the regulation of the endocrine and neural system (Zhou et al., 1993). For example, there is evidence that psychosocial stress triggers activation of IL-6 neurons in the hypothalamo-neurohyphophyseal system, and this activation is paralleled by an increase in plasma IL-6 (Jankord et al., 2010). Studies indicate magnocellular neurons produce IL-6, and then it is transported to the neurohypophysis, where it is released into the peripheral circulation (Ghorbel et al., 2003; Jankord et al., 2007). The data presented here confirms once more, IL-6 is an important modulator of RSD-induced social avoidance behavior. After 6 cycles of RSD, imipramine blocked increases of IL-6 peripherally and centrally and restored social interaction. In the same manner, 24 days after stress cessation, social avoidant behavior was prevented with imipramine treatment, paralleled by a blockade of mRNA expression of IL-6 in microglia (Ramirez et al., 2015).
It has been suggested that imipramine attenuates noradrenergic receptor function (Gandolfi et al., 1984). Alpha-adrenergic receptor antagonists have been found to block increased peripheral blood IL-6 related to stress in humans (Miller et al., 2013). Central injections that block adrenergic signaling attenuated stress-induced increase of circulating IL-6 (Jankord et al., 2010). Previous work has demonstrated that RSD triggered anxiety-like behavior and enhanced the inflammatory state in the periphery and in the CNS in a β-adrenergic dependent manner (Hanke et al., 2012; Wohleb et al., 2011). Pre-treatment of RSD mice with propranolol blocked RSD-induced microglia/macrophage activation and social defeat-induced anxiety, as well as expression of c-Fos in brain regions associated with fear and threat appraisal (Wohleb et al., 2011). It is plausible that imipramine in the RSD stress model, antagonized directly the activation of these adrenergic receptors and decreased pro-inflammatory cytokine production, thus preventing associated stress-related anxiety-like behavior.
A proposed mechanism for the antidepressant effect on cytokine function is that in vitro, they inhibit pro-inflammatory cytokine activity through cAMP levels, (Xia et al., 1996; Brustolim et al., 2006; Kast, 2003). For example, increased adenylate cyclase, leads to intracellular cyclic adenosine monophosphate (cAMP) levels to activate the cAMP-dependent protein kinase (PKA) pathway. An increase of intracellular cAMP levels in different cell types such as monocytes, macrophages, and microglia, leads to a decrease in pro-inflammatory cytokine levels (Hashioka et al., 2007; Maes, 2001; Xia et al., 1996).
Another proposed mechanism for the antidepressant effect of imipramine on cytokine function is that immune cells, such as monocytes, store serotonin, and transport of serotonin into these cells is promoted by pro-inflammatory cytokines (Morikawa et al., 1998; Mossner et al., 1998; Ramamoorthy et al., 1995). Pro-inflammatory cytokines are responsible for the up-regulation of serotonin transport, thus increasing its depletion extracellularly (Morikawa et al., 1998; Mossner et al., 1998; Ramamoorthy et al., 1995). In vitro, serotonergic antidepressants increase release of serotonin and inhibit its reuptake, leading to low intracellular and increased extracellular levels of serotonin. Low intracellular serotonin levels appear to decrease cytokine levels through the inhibition of mRNA expression, while increased extracellular serotonin appears to exert and immunosuppressive effect (Idova and Cheido 1987; Nordlind et al., 1992; Bondesson et al., 1993; Smejkal-Jagar and Boranic 1994; Young and Matthews 1995; Kubera et al., 2000).
In the experiments described in this manuscript, we provided evidence that by treating animals exposed to stress with an antidepressant, neuroinflammatory-signaling by resident microglia was ablated, and BM-derived monocytes did not accumulate in the CNS. This confirms that BM-derived monocytes produced in response to social defeat are an important in the underlying stress-induced anxiety. However, neuronal activation plays a major part in microglia activation. Therefore, it is hypothesized one of the major mechanisms of actions of these drugs is inhibiting neuronal activation. It has been suggested that imipramine blocks reuptake of norepinephrine into presynaptic neurons (Michelson et al., 1997). The locus coeruleus, contains inhibitory-2 auto receptors, thus, accumulation of norepinephrine at the synapse would decrease the firing of noradrenergic neurons. Since norepinephrine stimulates CRH release, imipramine may contribute by decreasing noradrenergic activation in the brainstem. Ablation of noradrenergic locus coeruleus projections is accompanied by a reduction in stress-induced IL-1β production (Johnson et al., 2005). In other words, central noradrenergic responses exert a major role in stress-induced microglia activation. This suggests these drugs act on noradrenergic function and may influence HPA axis and SNS activity, thus rescuing RSD-induced changes in hematopoiesis. Repeated activation of the SNS due to stress increases norepinephrine in the BM promoting the production of myeloid cells, including monocytes and granulocytes (Hanke et al., 2012). It is plausible that imipramine may also be inhibiting the activation of the SNS in mice subjected to RSD.
In agreement with other studies, imipramine decreased the percentage of circulating monocytes and granulocytes enhanced by stress exposure (Seidel et al., 1996). It has been shown antidepressants normalized increased counts of monocytes, leukocytes and neutrophils in depressed patients (Bondesson et al., 1993; Seidel et al., 1996). This makes sense since imipramine as well, blocked the development of monocytes and granulocytes in the BM. This supports the notion that these drugs impede the release of pro-inflammatory cells from the BM. The mechanism leading to this reduction in circulating monocytes and granulocytes in mice subjected to RSD and treated with imipramine needs to be further elucidated.
In a previous study, we had demonstrated that imipramine partially blocked the production of pro-inflammatory cytokines in total splenocytes at 24 days after stress termination (Ramirez et al., 2015). Once more, in the present study, imipramine was not effective in recovering stress-induced increased mRNA expression of IL-1β, IL-6, and TNF-α in spleen cells of socially defeated animals, while it was effective in attenuating the production of these pro-inflammatory cytokines and other factors in microglia. Interestingly, there was a significant decrease of monocytes in the spleen after stress exposure in mice that were treated with the antidepressant, but the percentage of granulocytes was still high in this experimental group. The increased percentage of granulocytes was paralleled by splenomegaly in stressed mice treated with imipramine.
Antidepressant pharmacotherapy is the most often used treatment for depression, thus the exact mechanism of action is a matter of research. Many hypotheses have been taken into consideration for antidepressant activity. It has been thought that depressed patients have impairment in neuroimmune endocrine functions. Thus, high levels of circulating corticosteroids and a hyperactivation of the neuroimmune system, causes high levels of pro-inflammatory cytokines in the periphery and CNS as a result of psychosocial stress. We had previously suggested that imipramine prevented microglia activation and could have neuroprotective properties besides exerting an antidepressant effect. Here, we go beyond that idea and describe the impact of imipramine on the reduction of stress-induced neuroendocrine effects in both the periphery and CNS, along with the amelioration of symptoms. Imipramine treatment in the RSD model seems to correct, or at least interfered with these alterations.
Highlights.
Imipramine ameliorated stress-induced inflammation in central nervous system
Imipramine prevented stress- related anxiety- and depressive-like behaviors
Imipramine attenuated stress-induced corticosterone and IL-6 responses in plasma
Imipramine decreased the percentage of myeloid progenitor cells in bone marrow, circulation, and brain
Imipramine blocked neuroinflammatory signaling
Acknowledgments
This research was supported by NIH/NIMH R01 MH097243-03 and R01 MH093473-04.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Karol Ramirez, Email: karol.ramirez@ucr.ac.cr.
John F. Sheridan, Email: sheridan.1@osu.edu.
References
- Antonioli M, Rybka J, Carvalho LA. Neuroimmune endocrine effects of antidepressants. Neuropsychiatric disease and treatment. 2012;8:65–83. doi: 10.2147/NDT.S16409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitsur R, Padgett DA, Dhabhar FS, Stark JL, Kramer KA, Engler H, Sheridan JF. Expression of glucocorticoid resistance following social stress requires a second signal. Journal of Leukocyte Biology. 2003;74(4):507–13. doi: 10.1189/jlb.0303090. [DOI] [PubMed] [Google Scholar]
- Bailey MT, Kierstein S, Sharma S, Spaits M, Kinsey SG, Tliba O, Amrani Y, Sheridan JF, Panettieri RA, Haczku A. Social stress enhances allergen-induced airway inflammation in mice and inhibits corticosteroid responsiveness of cytokine production. Journal of Immunology. 2009;182(12):7888–96. doi: 10.4049/jimmunol.0800891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basterzi AD, Aydemir C, Kisa C, et al. IL-6 levels decrease with SSRI treatment in patients with major depression. Human Psychopharmacology. 2005;20:473–476. doi: 10.1002/hup.717. [DOI] [PubMed] [Google Scholar]
- Bondesson L, Nordlind K, Liden S, Sundstrom E. Inhibiting effects of serotonin and serotonin antagonists on the migration of mononuclear leucocytes. Immunopharmacology and Immunotoxicology. 1993;15:243–250. doi: 10.3109/08923979309025997. [DOI] [PubMed] [Google Scholar]
- Brustolim D, Ribeiro-dos-Santos R, Kast RE, Altschuler EL, Soares MB. A new chapter opens in anti-inflammatory treatments: the antidepressant bupropion lowers production of tumor necrosis factor-alpha and interferon-gamma in mice. International Immunopharmacology. 2006;6:903–907. doi: 10.1016/j.intimp.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Castanon N, Leonard B, Neveu P, Yirmiya R. Effects of antidepressants on cytokine production and actions. Brain Behavior and Immunity. 2002;16(5):569–574. doi: 10.1016/s0889-1591(02)00008-9. [DOI] [PubMed] [Google Scholar]
- Engler H, Bailey MT, Engler A, Sheridan JF. Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. Journal of Neuroimmunology. 2004;148(1–2):1–2. doi: 10.1016/j.jneuroim.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Eshleman AJ, Carmolli M, Cumbay M, Martens CR, Neve KA, Janowsky A. Characteristics of drug interactions with recombinant biogenic amine transporters expressed in the same cell type. Journal of Pharmacology and Experimental Therapy. 1999;289(2):877–85. [PubMed] [Google Scholar]
- Engler H, Engler A, Bailey MT, Sheridan JF. Tissue-specific alterations in the glucocorticoid sensitivity of immune cells following repeated social defeat in mice. Journal of Neuroimmunoly. 2005;163(1–2):1–2. doi: 10.1016/j.jneuroim.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Frank, Matthew G, Wieseler-Frank Julie L, Watkins Linda R, Maier Steven F. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: Immunophenotypic and functional characteristics. Journal of Neuroscience Methods. 2006;151(2):121–130. doi: 10.1016/j.jneumeth.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Frommberger UH, Bauer J, Haselbauer P, Fraulin A, Riemann D, Berger M. Interleukin-6-(IL-6) plasma levels in depression and schizophrenia: comparison between the acute state and after remission. European Archives of Psychiatry and Clinical Neuroscience. 1997;247:228–233. doi: 10.1007/BF02900219. [DOI] [PubMed] [Google Scholar]
- Gandolfi O, Barbaccia ML, Costa E. Comparison of iprindole, imipramine and mianserin action on brain serotonergic and beta adrenergic receptors. Journal of Pharmacogical and Experimental Therapeutics. 1984 Jun;229(3):782–6. [PubMed] [Google Scholar]
- Ghorbel MT, Sharman G, Leroux M, Barrett T, Donovan DM, Becker KG, Murphy D. Microarray analysis reveals interleukin-6 as a novel secretory product of the hypothalamo-neurohypophyseal system. The Journal of Biological Chemistry. 2003;278(21):19280–5. doi: 10.1074/jbc.M209902200. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Axelrod J. Inhibition of uptake of tritiated-noradrenaline in the intact rat brain by imipramine and structurally related compounds. Nature. 1964;204:1318–1319. doi: 10.1038/2041318a0. [DOI] [PubMed] [Google Scholar]
- Goshen I, Yirmiya R. Interleukin-1 (IL-1): A central regulator of stress responses. Frontiers in Neuroendocrinology. 2009;30(1):30–45. doi: 10.1016/j.yfrne.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFN gamma signaling-does it mean JAK-STAT? Cytokine & Growth Factor Reviews. 2008;19:383–394. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF. Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behavior and Immunity. 2012;26(7):1150–1159. doi: 10.1016/j.bbi.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashioka S, Klegeris A, Monji A, Kato T, Sawada M, McGeer PL, Kanba S. Antidepressants inhibit interferon-@c-induced microglial production of IL-6 and nitric oxide. Experimental Neurology. 2007;206(1):33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. Journal of Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behavior and Immunity. 2009;23(3):309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Zheng LT, Ock J, Lee MG, Kim SH, Lee HW, Lee WH, Park HC, Suk K. Inhibition of glial inflammatory activation and neurotoxicity by tricyclic antidepressants. Neuropharmacology. 2008;55(5):826–34. doi: 10.1016/j.neuropharm.2008.06.045. [DOI] [PubMed] [Google Scholar]
- Idova GV, Cheido MA. Stimulation of the immune response during blockade of serotonin receptors by cyproheptadine. Biull Eksp Biol Med. 1987;103:440–442. [PubMed] [Google Scholar]
- Iversen L. Neurotransmitter transporters and their impact on the development of psychopharmacology. British Journal of Pharmacology. 2006;147(Suppl 1):S82–8. doi: 10.1038/sj.bjp.0706428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankord R, Turk JR, Schadt JC, Casati J, Ganjam VK, Price EM, Keisler DH, Laughlin MH. Sex difference in link between interleukin-6 and stress. Endocrinology. 2007;148(8):3758–64. doi: 10.1210/en.2006-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankord R, Zhang R, Flak JN, Solomon MB, Albertz J, Herman JP. Stress activation of IL-6 neurons in the hypothalamus. American Journal of Physiology Regulatory, integrative and comparative physiology. 2010;299(1):343–51. doi: 10.1152/ajpregu.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005;135(4):1295–1307. doi: 10.1016/j.neuroscience.2005.06.090. [DOI] [PubMed] [Google Scholar]
- Kast RE. Anti- and pro-inflammatory considerations in antidepressant use during medical illness: bupropion lowers and mirtazapine increases circulating tumor necrosis factor-alpha levels. General Hospital Psychiatry. 2003;25:495–496. doi: 10.1016/s0163-8343(03)00093-8. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behavior and Immunity. 2007;21(4):458–466. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimek V, Stockmeier C, Overholser J, Meltzer HY, Kalka S, Dilley G, Ordway GA. Reduced levels of norepinephrine transporters in the locus coeruleus in major depression. Journal of Neuroscience. 1997 Nov 1;17(21):8451–8. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubera M, Holan V, Mathison R, Maes M. The effect of repeated amitriptyline and desipramine administration on cytokine release in C57BL/6 mice. Psychoneuroendocrinology. 2000;25(8):785–797. doi: 10.1016/s0306-4530(00)00026-3. [DOI] [PubMed] [Google Scholar]
- Lim CM, Kim SW, Park JY, Kim C, Yoon SH, Lee JK. Fluoxetine affords robust neuroprotection in the post-ischemic brain via its anti-inflammatory effect. Journal of Neuroscience Research. 2009;87(4):1037–45. doi: 10.1002/jnr.21899. [DOI] [PubMed] [Google Scholar]
- Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853–8. doi: 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- Maes M. The immunoregulatory effects of antidepressants. Human Psychopharmacology: Clinical and Experimental. 2001;16(1):95–103. doi: 10.1002/hup.191. [DOI] [PubMed] [Google Scholar]
- Michelson D, Galliven E, Hill L, Demitrack M, Chrousos G, Gold P. Chronic imipramine is associated with diminished hypothalamic-pituitary-adrenal axis responsivity in healthy humans. Journal of Clinical Endocrine Metabolism. 1997;82(8):2601–6. doi: 10.1210/jcem.82.8.4172. [DOI] [PubMed] [Google Scholar]
- Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depression and Anxiety. 2013;30(4):297–306. doi: 10.1002/da.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa O, Sakai N, Obara H, Saito N. Effects of interferon-alpha, interferon-gamma and cAMP on the transcriptional regulation of the serotonin transporter. European Journal of Pharmacology. 1998;349:317–324. doi: 10.1016/s0014-2999(98)00187-3. [DOI] [PubMed] [Google Scholar]
- Mossner R, Heils A, Stober G, Okladnova O, Daniel S, Lesch KP. Enhancement of serotonin transporter function by tumor necrosis factor alpha but not by interleukin-6. Neurochemistry International. 1998;33:251–254. doi: 10.1016/s0197-0186(98)00026-6. [DOI] [PubMed] [Google Scholar]
- Nair A, Hunzeker J, Bonneau RH. Modulation of microglia and CD8^+ T cell activation during the development of stress-induced herpes simplex virus type-1 encephalitis. Brain Behavior and Immunity. 2007;21(6):791–806. doi: 10.1016/j.bbi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Nordlind K, Sundstrom E, Bondesson L. Inhibiting effects of serotonin antagonists on the proliferation of mercuric chloride stimulated human peripheral blood T lymphocytes. International Archives of Allery and Immunology. 1992;97:105–108. doi: 10.1159/000236104. [DOI] [PubMed] [Google Scholar]
- Powell ND, Tarr AJ, Sheridan JF. Psychosocial stress and inflammation in cancer. Brain Behavior Immunity. 2013;30:41–7. doi: 10.1016/j.bbi.2012.06.015. [DOI] [PubMed] [Google Scholar]
- Ramirez KG, Shea DT, McKim DB, Reader BF, Sheridan JF. Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behavior and Immunity. 2015;46:212–20. doi: 10.1016/j.bbi.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy S, Ramamoorthy JD, Prasad PD, Bhat GK, Mahesh VB, Leibach FH, Ganapathy V. Regulation of the human serotonin transporter by interleukin-1 beta. Biochemical and Biophysical Research Communications. 1995;216:560–567. doi: 10.1006/bbrc.1995.2659. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nature reviews Neuroscience. 2013;14(9):609–25. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel A, Arolt V, Hunstiger M, Rink L, Behnisch A, Kirchner H. Major depressive disorder is associated with elevated monocyte counts. Acta Psychiatrica Scandinavica. 1996;94(3):198–204. doi: 10.1111/j.1600-0447.1996.tb09849.x. [DOI] [PubMed] [Google Scholar]
- Sluzewska A, Rybakowski J, Laciak M, Mackiewicz A, Sobieska M, Wiktorowicv K. Interleukin-6 Serum Levels in Depressed Patients before and after Treatment with Fluoxetine. Annals of the New York Academy of Sciences. 1995;762(1):474–476. doi: 10.1111/j.1749-6632.1995.tb32372.x. [DOI] [PubMed] [Google Scholar]
- Smejkal-Jagar L, Boranic M. Serotonin, serotonergic agents and their antagonists suppress humoral immune reaction in vitro. Research in Experimental Medicine. 1994;194:297–304. doi: 10.1007/BF02576391. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Hunzeker J, Padgett DA, Sheridan JF. Interleukin-6 and the development of social disruption-induced glucocorticoid resistance. Journal of Neuroimmunology. 2002;124(1–2):1–2. doi: 10.1016/s0165-5728(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. American Journal of Physiology: Regulatory, Integrative & Comparative Physiology. 2001;49(6) doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- Strekalova T, Anthony DC, Dolgov O, Anokhin K, Kubatiev A, Steinbusch HM, Schroeter C. The differential effects of chronic imipramine or citalopram administration on physiological and behavioral outcomes in naïve mice. Behavioral Brain Research. 2013;245:101–6. doi: 10.1016/j.bbr.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. Journal of Neuroscience. 2011;31(17):6277–88. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Frontiers in neuroscience. 2014;8:447. doi: 10.3389/fnins.2014.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. Journal of Neuroscience. 2013;33(34):13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX 3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behavior and Immunity. 2010;24(7):1190–1201. doi: 10.1016/j.bbi.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, DePierre JW, Nassberger L. Tricyclic antidepressants inhibit IL-6, IL-1b and TNF-a release in human blood monocytes and IL-2 and interferon-gamma in T cells. Immunopharmacology. 1996;34(2–3):27. doi: 10.1016/0162-3109(96)00111-7. [DOI] [PubMed] [Google Scholar]
- Yirmiya R. Endotoxin produces a depressive-like episode in rats. Brain Research. 1996;711(1):163–174. doi: 10.1016/0006-8993(95)01415-2. [DOI] [PubMed] [Google Scholar]
- Young MR, Matthews JP. Serotonin regulation of T cell subpopulations and of macrophage accessory function. Immunology. 1995;84:148–152. [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS. Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology. 1993;133(6):2523–30. doi: 10.1210/endo.133.6.8243274. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007 Sep 7;317(5843):1390–3. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]