

Abstract
Background
Recent studies have shown that low density lipoproteins reconstituted with the natural omega 3 fatty acid docosahexaenoic acid (LDL-DHA) is selectively cytotoxic to liver cancer cells over normal hepatocytes. To date, little is known about the subcellular events which transpire following LDL-DHA treatment.
Methods
Herein, murine noncancer and cancer liver cells, TIB-73 and TIB-75 respectively, were investigated utilizing confocal microscopy, flow cytometry and viability assays to demonstrate differential actions of LDL-DHA nanoparticles in normal versus malignant cells.
Results
Our studies first showed that basal levels of oxidative stress are significantly higher in the malignant TIB-75 cells compared to the normal TIB-73 cells. As such, upon entry of LDL-DHA into the malignant TIB-75 cells, DHA is rapidly oxidized precipitating global and lysosomal lipid peroxidation along with increased lysosomal permeability. This leakage of lysosomal contents and lipid peroxidation products trigger subsequent mitochondrial dysfunction and nuclear injury. The cascade of LDL-DHA mediated lipid peroxidation and organelle damage was partially reversed by the administration of the antioxidant, N-acetylcysteine, or the iron-chelator, deferoxamine. LDL-DHA treatment in the normal TIB-73 cells was well tolerated and did not elicit any cell or organelle injury.
Conclusion
These studies have shown that LDL-DHA is selectively cytotoxic to liver cancer cells and that increased levels of ROS and iron catalyzed reactions promote the peroxidation of DHA which lead to organelle dysfunction and ultimately the demise of the cancer cell.
General significance
LDL-DHA selectively disrupts lysosomal, mitochondrial and nuclear function in cancer cells as a novel pathway for eliminating cancer cells.
Keywords: Liver cancer, Omega-3 fatty acid, Lipid peroxidation, Lysosome membrane permeability, Mitochondrial membrane potential
1. Introduction
Therapeutic selectivity is a highly desired property for anticancer medicines. The ideal cancer therapy should be toxic to malignant cells while invoking minimal harm to normal cells. Traditional chemotherapy indiscriminately damages both cancer and healthy cells resulting in significant adverse effects that compromise their therapeutic benefits. More recently, molecular–targeted drugs, such as Gleevec and trastuzumab, which exploit genetic differences between cancer and normal cells have shown promising therapeutic activity, while producing few toxic side effects [1,2]. However, as a result of their targeting specificity, these agents are only indicated for a select group of cancers [3]. In addition, incurred mutations and genomic instability of the target molecules often lead to drug resistance and limited therapeutic utility of these target specific drugs [3]. Hence, continued efforts for the discovery and development of novel drugs that exploit the unique biology of cancer cells are ongoing.
Natural products have and continue to be an invaluable source for anticancer drug discovery [4]. The natural omega-3 polyunsaturated fatty acid, docosahexaenoic acid (DHA), best known for its cardioprotective and health promoting benefits, has in recent years also been shown to possess promising anticancer properties [5–8]. Epidemiological studies have implicated the intake of this omega-3 fatty acid with reduced rates of cancer development [9–11]. These findings have led many to consider DHA as a cancer preventative agent. Although in vitro cell culture experiments have demonstrated that unesterified DHA at concentrations in excess of 50 μM can inhibit malignant cell growth and induce cancer cell death [12–15], plasma concentrations of unesterified DHA typically saturate at approximately 13 μM when consuming a DHA-enriched diet [16]. Thus, at these levels dietary DHA is likely to only antagonize the carcinogenesis process, and not serve as a viable treatment against established tumors. Alternate delivery strategies that directly transport DHA to tumors cells would greatly enhance the anticancer potency of this natural lipid. Previous studies from our lab have shown that the low-density lipoprotein (LDL) nanoparticle can serve as an ideal transport vehicle for DHA [17]. The unesterified DHA readily incorporates into the LDL nanoplatform at high concentrations and with good stability [17]. The LDL nanoparticle is then able to ferry DHA directly to cells enabling their intracellular transport via LDL receptor mediated endocytosis. LDL particles reconstituted with DHA (hereon referred to as LDL-DHA) are avidly taken up by normal and malignant liver cells [17]. These experiments went on to show that LDL-DHA nanoparticles are selectively toxic to malignant liver cells while normal cells under the same conditions do not experience harm [17]. The hallmark feature of LDL-DHA cytotoxicity is the selective deregulation of reactive oxygen species (ROS) within malignant cells. The intracellular uptake of LDL-DHA induces lethal increases of oxidative stress and lipid peroxidation within cancer cells [17]. Although these global redox disturbances ultimately usher in the demise of the cancer cell, little is known about the subcellular events or organelle dysfunctions that lead to this redox crisis.
The goal of the present study is to elucidate the cytotoxic sequelae that occur within cancer cells following LDL-DHA nanoparticle treatment. These experiments will be performed using the paired Balb/C derived noncancer and cancer liver cell lines, TIB-73 and TIB-75, to demonstrate the differential and contrasting actions of LDL-DHA nanoparticles in normal versus malignant cells.
2. Materials and methods
2.1. Low density lipoprotein isolation and reconstitution
LDL was isolated from the plasma of a hypercholesteremic patients using ultracentrifugation according to the method described by Lund-Katz et al. [18]. Incorporation of DHA (Nu-Chek Prep, Inc.; Elysian, MN) into LDL was performed by the reconstitution (core-loading) method as described previously [17,19]. Control LDL Nanoparticles: Native LDL, LDL reconstituted with oleic acid (LDL-OA). Physicochemical characterization of the LDL nanoparticles (which include composition analyses, mean particle size and surface charge) were performed as previously described [17] and is presented in Supplemental Table 1.
2.2. Preparation of HSA-DHA
Human serum albumin (HSA; 5% w/v) was dissolved in 1 mL of 75 mM KCl solution (pH = 7.4). DHA in ethanol (0.125% w/v; final concentration) was added to the HSA solution, vortexed briefly and incubated at 37 °C for 1 h. HSA bound DHA (HSA-DHA) was then filtered through 0.2 μm syringe filter and stored under N2 atmosphere at 2–8 °C until further use.
2.3. Cell culture
The normal mouse hepatocyte cell line TIB-73 (BNL CL.2) and its malignant counterpart TIB-75 (BNL 1ME A.7R.1) were obtained from ATCC. TIB-73 and TIB-75 cells were cultured in DMEM (Sigma Aldrich, Cat #D6429; St. Louis, MO) supplemented with 10% FBS (Invitrogen, Carlsbad, CA). All cells were grown at 37 °C in a 5% CO2 atmosphere in a humidified incubator.
2.4. Primary hepatocyte isolation and culture from mice
Hepatocytes were isolated from Balb/C mice by non-recirculating collagenase perfusion through the inferior vena cava using liver perfusion reagents from Life Technologies (Carlsbad, CA). Refer to Supplementary Information for details on liver perfusion, isolation, and culture conditions.
2.5. MTS cell viability assay
Cells were plated in 96-well plates and treated for each experiment. After treatment, the media was removed and replaced with 100 μL media mixed with CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega; Madison, WI) for ≈4 h at 37 °C according to the manufacturer’s instructions. After incubation, the absorbance of the well plates was read at 450 nm on a Molecular Devices ThermoMax M5 microplate reader. The relative cell viability was expressed as a percentage of the untreated controls.
2.6. Protein measurement by western blot
Cell proteins were separated on a 10% sodium dodecyl sulfate polyacrylamide gel and transferred to PVDF. Membranes were then probed with primary antibodies from Santa Cruz Biotechnology (Dallas, TX) against superoxide dismutase 1(SOD-1 ((FL-154), sc-11407), catalase ((N-17), sc-34280), glutathione peroxidase 4 (GPx-4 (H-90), sc-50497), and β-actin ((C4) sc-47778). Secondary antibodies conjugated to horseradish peroxidase (HRP) against goat IgG (sc-2350), mouse IgG (sc-2954), and rabbit IgG (sc-2313) from Santa Cruz Biotechnology were used to detect primary antibodies and developed using enhanced chemiluminescence.
2.7. GSH/GSSG assay
Total soluble reduced glutathione (GSH) and glutathione disulfide (GSSG) were measured in cell lysates using the enzymatic recycling method [20]. Briefly, about 3 × 106 cells were homogenized in 10 volumes of cold sulfosalicylic acid (0.6%) plus 0.1% Triton-X. Protein was precipitated and the supernatant was used to determine GSH and GSSG. Results were expressed in mmol per mg of cell protein as determined by Bradford assay.
2.8. Thiobarbituric acid reactive substances assay
The total amount of lipid peroxidation products formed in the cells was determined using the thiobarbituric acid reactive substances (TBARS) method [21]. Refer to Supplementary Information for details.
2.9. Confocal microscopy experiments
For all confocal microscopy experiments, TIB-73 and TIB-75 cells were seeded into 35-mm2 glass bottom dishes coated with 10 μg/mL fibronectin until 80–90% confluent and then serum starved for 6 to 18 h prior to treatment and staining. Cells were imaged at 63× magnification on a Leica SP5 confocal microscope. Images were deconvoluted using AutoQuant ×3 software and analyzed using Imaris imaging software. Refer to Supplementary Information for details on treatment, staining, and imaging analysis.
2.10. Flow cytometry experiments
For all flow cytometry experiments, TIB-73 and TIB-75 cells were grown to 80–90% confluency in 6-well plates, serum starved for 6 to 18 h, and then treated and stained as described in the Supplementary Information. Cells were removed from the plates using 500 μL 0.25% trypsin-EDTA, quenched with 1 mL cold DMEM + 10% FBS, and washed twice with 750 μL PBS and stored on ice before analyzing on a BD FACSCalibur. The flow cytometry data was analyzed on FlowJo software. Data was gated to remove cellular debris and clumps (FSC versus SSC) and then the single cell populations were plotted as described in the Supplementary Information.
2.11. γ-H2AX immunofluorescence for DNA damage
TIB-73 and TIB-75 cells were seeded to 80–90% confluency on an 8-chamber glass slide coated with 10 μg/mL fibronectin and serum starved prior to treatment with serum free DMEM, 40 μM LDL-DHA, 60 μM LDL-DHA, or 60 μM LDL-OA for 6 or 18 h. Following treatment, cells were immunofluorescently stained with Alexafluor 488-conjugated antibody targeted to γ-H2AX (Millipore; Billerica, MA) and coverslipped with ProLong® Gold Antifade Mountant with DAPI (Life Technologies). Refer to Supplemental Information for immunofluorescence staining details. Slides were imaged at 63× magnification on a Leica SP5 confocal microscope. ImageJ software was used to count both the total number of cells and the number of cells with ≥3 γ-H2AX foci per field of view.
2.12. Silver sulfide autometallography
The silver sulfide autometallography staining was adapted from the protocol by Danscher [22]. Refer to Supplemental Information for details on staining and imaging.
2.13. Iron measurement by ICP-MS
Measurement of iron by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) was adapted from the protocol by Au-Yeung et al. [23]. Samples were analyzed on an Agilent 7700× ICP-MS with auto-sampler using argon carrier gas. Iron signal was detected and compared to a 10 ppm standard (Inorganic Ventures, MSFE-10PPM; Christianburg, VA).
2.14. Iron measurement by ferrozine assay
Ferrozine assay for total iron was measured in TIB-73 and TIB-75 cell pellets according to the protocol by Riemer et al. [24]. Iron absorbance values were compared to a FeCl3 standard curve and normalized to cell protein measured by Bradford assay.
2.15. Statistical analysis
The results were expressed as mean ± standard error. Analysis of variance (ANOVA) with Tukey’s multiple comparison post hoc testing was used for evaluation of differences between groups. Differences with a p value < 0.05 were deemed significant.
3. Results
3.1. Selective cytotoxicity of LDL-DHA
Primary Balb/C hepatocytes and the Balb/C derived non-cancerous and cancer liver cells, TIB-73, and TIB-75, respectively, were treated for 72 h with increasing doses of LDL-DHA (Fig. 1A). Cell viability assessed by the MTS assay indicated that LDL-DHA treatment was selectively cytotoxic to the malignant TIB-75 cells (IC50 = 27.7 μM DHA) compared to nonmalignant TIB-73 (IC50 = 91.4 μM DHA) and primary hepatocytes (IC50 = 148.8 μM DHA). Previously published studies showed that control nanoparticles of LDL-OA and LDL-TO were not cytotoxic to either cell line [17]. These control LDL nanoparticles were also non-toxic to the primary hepatocytes at doses exceeding 200 μM. Given that albumin is the principle transporter of unesterified fatty acids in plasma, the sensitivity of TIB-75 cells to HSA-mediated delivery of DHA was evaluated in direct comparison to LDL-DHA. Fig. 2 shows that at 40 μM LDL-DHA TIB-75 cells experience pronounced toxicity, at 80 and 160 μM LDL-DHA near-complete and complete kill of the TIB-75 cells are respectively seen. Meanwhile, at each of these concentrations of HSA-DHA the TIB-75 cells remained unharmed and looked similar to the untreated cells. Collectively, these findings indicate that the LDL nanoplatform is superior to HSA at mediating the cancer killing effects of DHA. The cell death that ensues following LDL-DHA treatment has previously been described to involve both apoptosis and necrosis processes [17]. To clarify the mechanism by which LDL-DHA induces TIB-75 cell death, TIB-75 cells were pretreated with inhibitors of apoptosis, programmed cell necrosis, or autophagy, but inhibition of these individual pathways were not sufficient to prevent the LDL-DHA cytotoxicity (Supplemental Fig. 1).
Fig. 1.
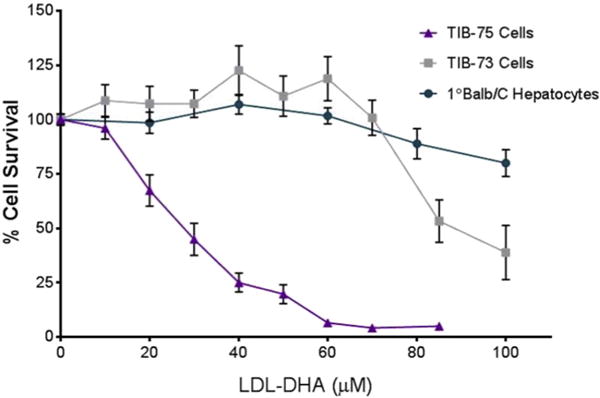
Cytotoxicity of LDL-DHA in TIB-73, TIB-75, and 1° Balb/C hepatocytes TIB-73, TIB-75, and primary Balb/C hepatocytes were treated for 72 h with increasing doses of LDL-DHA (0–200 μM DHA) after serum starvation. TIB-75 cells were more sensitive to LDL-DHA treatment (IC50 = 27.7 μM LDL-DHA) than TIB-73 cells (IC50 = 91.4 μM LDL-DHA) and Balb/C primary hepatocytes (148.8 μM LDL-DHA). The error bars represent the standard error of the mean (n ≥ 5). Viability was significantly lower from untreated cells for TIB-75 cells starting at 20 μM LDL-DHA (p-value < 0.0001), TIB-73 cells starting at 85 μM LDL-DHA (p-value < 0.0001), and primary Balb/C hepatocytes at 80 μM LDL-DHA (p-value = 0.0025) and continuing from 100 μM LDL-DHA (p-value < 0.0001).
Fig. 2.
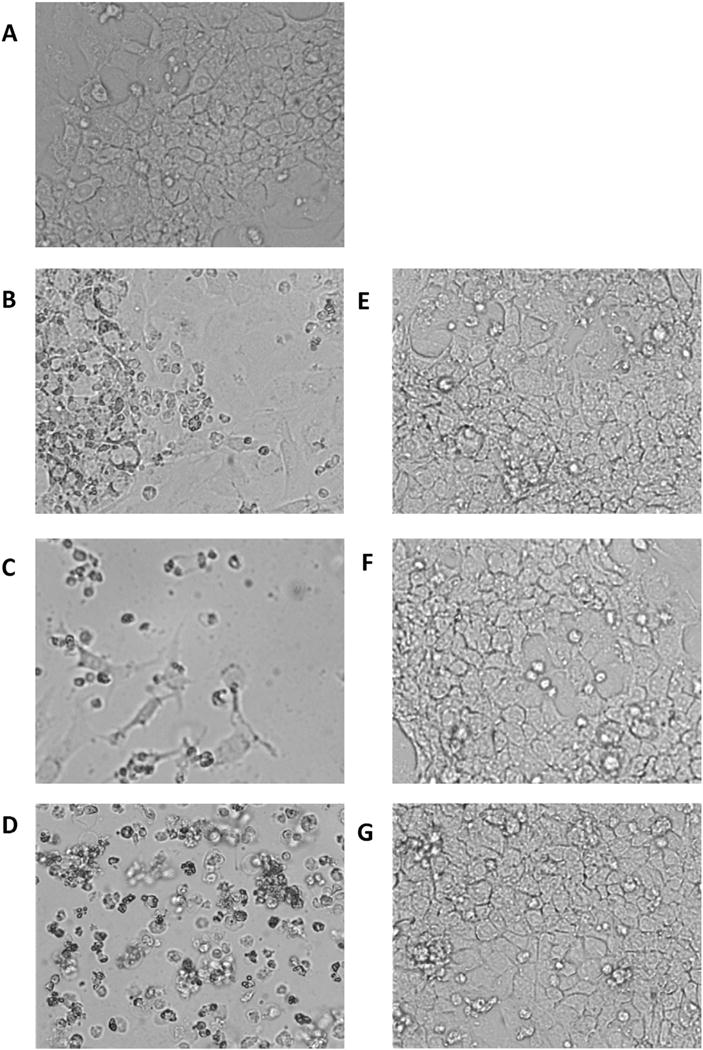
Cytotoxicity of LDL-DHA versus HSA-DHA in TIB-75 cells Micrographs (20× magnification) of TIB-75 cells after 72 h exposure to various concentrations of LDL-DHA or HSA-DHA. (A) Untreated cells; (B) 40 μM LDL-DHA; (C) 80 μM LDL-DHA; (D) 160 μM LDL-DHA; (E) 40 μM HSA-DHA; (F) 80 μM HSA-DHA; (G) 160 μM HSA-DHA.
3.2. Baseline ROS and antioxidant capacity of TIB-73 and TIB-75 cells
Basal levels of oxidative stress play an important role in sensitizing cells to exogenous pro-oxidant agents like DHA. Several assays were used in the present study to assess the baseline levels of oxidative stress in TIB-73 and TIB-75 cells. From the fluorescent ROS reporters DHE and DCF, spectrofluorometry indicated that DHE levels were equivalent between the two cells (Fig. 3A), but DCF fluorescence was significantly higher in the TIB-75 cells (Fig. 3B). Co-localization experiments (Fig. 3C) with the lysosomal probe, LysoTracker, went on show that greater co-registration of DCF fluorescence was evident with LysoTracker in TIB-75 cells than TIB-73 cells, suggesting that lysosomes in TIB-75 contained significantly higher levels of ROS compared to TIB-73 (the Manders coefficient for the co-localization of DCF with LysoTracker was 0.14 ± 0.02 and 0.27 ± 0.04 (p-value = 0.007) for TIB-73 and TIB-75 respectively). Measurements of the glutathione ratio (reduced glutathione (GSH):oxidized glutathione (GSSG)), also indicated that the TIB-75 cells maintained a more highly oxidized state (GSH:GSSG = 40:1) than TIB-73 cells (GSH:GSSG = 115:1) (Fig. 4A). Finally, Western blot analysis of the antioxidant enzyme system revealed that TIB-75 cells had significantly lower GPx-4 and catalase expression, while SOD-1 levels did not differ compared to TIB-73 cells (Fig. 4B). Collectively, these results indicate that TIB-75 cells are more oxidized and are less capable of safely removing ROS than TIB-73 cells, and this difference likely contributed to rendering the TIB-75 cells more susceptible to LDL-DHA-induced cytotoxicity.
Fig. 3. Baseline oxidative stress.
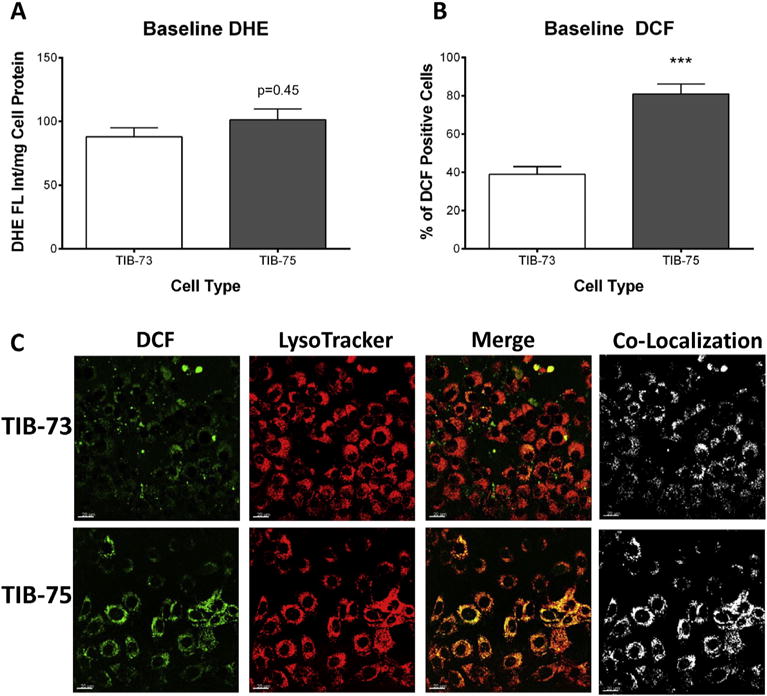
(A) Spectrofluorometry of TIB-73 and TIB-75 cells dyed with 10 μM DHE normalized to cell protein. There was not a significantly higher amount of superoxide levels in TIB-75 cells compared to TIB-73 cells at baseline (p-value = 0.45, n = 3). (B) Flow cytometry of TIB-73 and TIB-75 cells dyed with 15 μM DCF-DA to determine ROS generation. TIB-75 cells had higher DCF fluorescence than TIB-73 cells at baseline (p-value = 0.0008, n = 3). (C) Confocal imaging (40× magnification) of TIB-73 and TIB-75 cells incubated for 30 min with 50 nM LysoTracker Deep Red and 10 μM DCF. Images were deconvoluted prior to analysis to determine signal co-localization. The Manders coefficient for the co-localization of DCF with LysoTracker is 0.14 ± 0.02 and 0.27 ± 0.04 (p-value = 0.007) for TIB-73 and TIB-75, respectively.
Fig. 4. Baseline glutathione levels and antioxidants defenses.
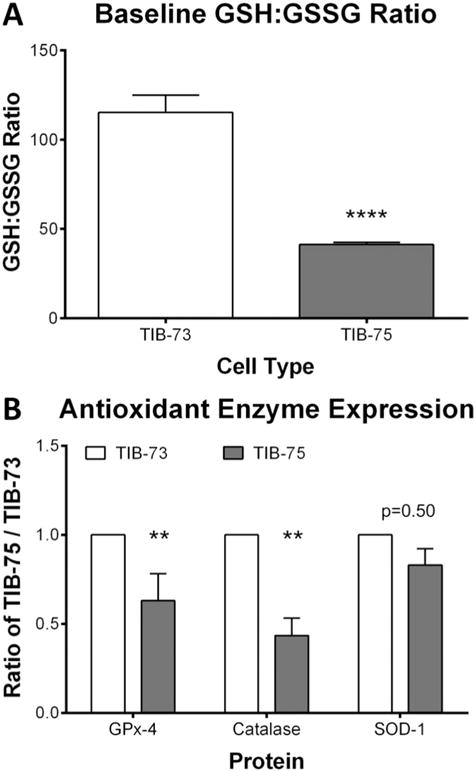
(A) GSH and GSSG levels of TIB-73 and TIB-75 cell pellets were determined using an enzymatic recycling method. TIB-75 cells had a significantly lower GSH:GSSG ratio compared to TIB-73 cells (p-value < 0.0001, n = 5). (B) Antioxidant protein levels from TIB-73 and TIB-75 cell lysates were determined by Western Blot and normalized to β-actin protein. SOD-1 protein expression was not significantly different (p-value = 0.50, n = 8), but TIB-75 cells had significantly lower GPx-4 (p-value = 0.004, n = 10) and catalase (p-value =0.009, n = 5) protein expression compared to TIB-73 cells. * = p-value ≤ 0.05, ** = p-value ≤ 0.01, *** = p-value ≤ 0.001, **** = p-value ≤ 0.0001. The error bars represent the standard error of the mean.
3.3. Effect of LDL-DHA on ROS and lipid peroxidation
Next, we examined the effect of LDL-DHA treatment on the temporal generation of ROS in the cells as measured by DCF fluorescence. Over time LDL-DHA significantly increased the number of DCF staining TIB-75 cells starting as early as 30 min and this continued to increase up to 4 h (Fig. 5A). Elevated populations of DCF staining TIB-75 cells continued over the next 20 h. Conversely, TIB-73 cells did not experience any meaningful increase in the number of cells with elevated DCF staining, which is consistent with previous measurements from our lab using DCF spectrofluorometry [17]. If effective quenching of ROS serves to protect TIB-73 cells from LDL-DHA cytotoxicity it stands to reason that excessive generation of ROS exceeding their antioxidative defense, should increase the sensitivity of TIB-73 cells to LDL-DHA treatment. Increasing doses of exogenous hydrogen peroxide was unable to achieve this effect (Supplemental Fig. 2A); however, pretreatment of TIB-73 cells with menadione, an ROS generating hydroquinone, was able to sensitize the TIB-73 cells to LDL-DHA treatments after 24 h (Supplemental Fig. 2B). A 25 μM dose of menadione was able to reduce the IC50 of TIB-73 cells from 131.8 μM down to 20.3 μM LDL-DHA (Supplemental Fig. 2C). These findings support the important role for ROS generation in sensitizing cells to LDL-DHA cytotoxicity.
Fig. 5. LDL-DHA selectively increases reactive oxygen species in TIB-75 cells.
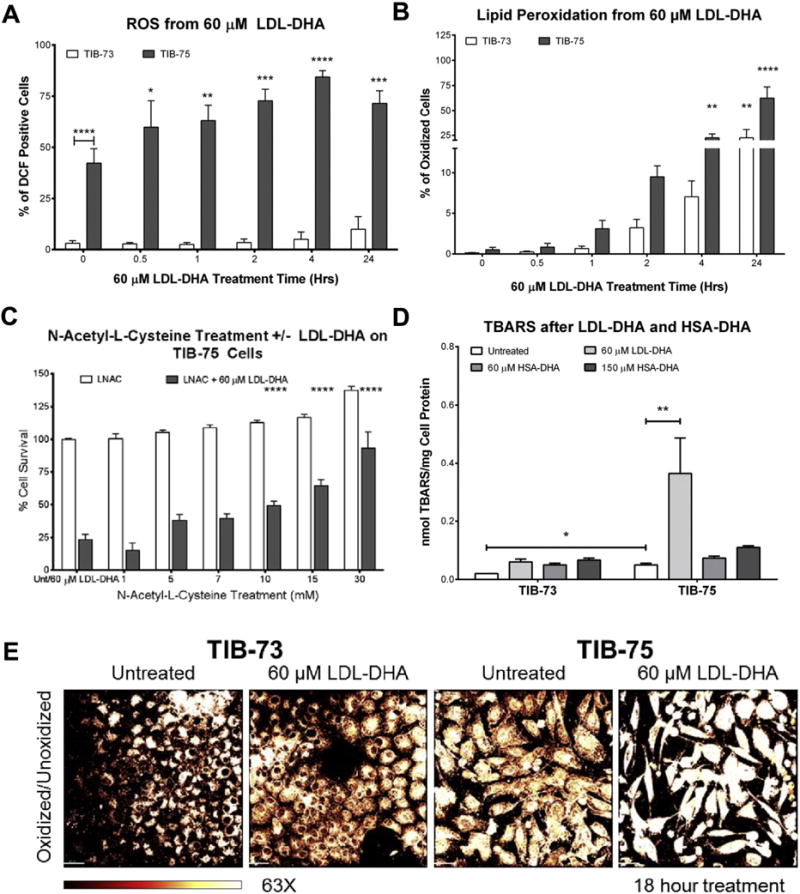
(A) Flow cytometry of TIB-73 and TIB-75 cells after treatment for different time periods with 60 μM LDL-DHA followed by staining with 15 μM DCF-DA to determine ROS generation. TIB-75 cells exhibited increased baseline ROS levels and ROS levels increased over time with LDL-DHA treatment compared to TIB-73 cells (n = 4). (B) Flow cytometry of TIB-73 and TIB-75 cells after staining with 1 μM BODIPY C11 581/591 dye followed by treatment for different time periods with 60 μM LDL-DHA to measure oxidized (green fluorescence) and unoxidized (red fluorescence) lipid species. TIB-75 cells had a higher percentage of cells with oxidized lipids following LDL-DHA treatment and that lipid oxidation increased with time (n = 6). (C) TBARS of TIB-73 and TIB-75 cells treated with serum free DMEM, 60 μM LDL-TO (not shown), LDL-DHA or HSA-DHA or 150 μM HSA-DHA for 24 h. LDL-DHA treatment significantly increased lipid peroxidation in TIB-75 cells, but HSA-DHA and LDL-TO treatment did not have an effect (n = 3). (D) TIB-75 cell viability after a 1 h pretreatment with increasing doses of N-acetyl-L-cysteine (0–30 mM LNAC) followed by a 24 h treatment with 60 μM LDL-DHA. Pretreatment with LNAC to remove lipid peroxide species did significantly rescue TIB-75 cell cytotoxicity to 60 μM LDL-DHA compared to untreated cells (n = 5). (E) Confocal imaging (63× magnification) of TIB-73 and TIB-75 cells after overnight treatment with 60 μM LDL-DHA followed by staining with 2 μM BODIPY C11 581/591 dye for 25 min at 37 °C. A MatLab plug-in of the Imaris imaging software was used to create the heat map images by dividing the fluorescence intensity per pixel of the green channel by the red channel. TIB-75 cells have a higher baseline oxidized state compared to TIB-73 cells and LDL-DHA treatment increases the lipid oxidation of the TIB-75 cells. * = p-value ≤ 0.05, ** = p-value ≤ 0.01, *** = p-value ≤ 0.001, **** = p-value ≤ 0.0001. The error bars represent the standard error of the mean.
Lipid peroxidation is another hallmark feature of LDL-DHA cytotoxicity since DHA is prone to undergo peroxidation in environments with elevated ROS [13,25–27]. Temporal FACS measurement of the number of cells undergoing lipid peroxidation as detected by BODIPY C11 581/591 staining showed that TIB-75 cells treated with LDL-DHA had significantly more cells undergoing lipid peroxidation starting at 4 h and continuing to 24 h where ≈75% of cells experienced enhanced lipid peroxidation (Fig. 5B). TIB-73 cells had a slower and less pronounced increase in the number of cells with lipid peroxidation (≈25%) after a twenty-four hour LDL-DHA treatment. When TIB-75 cells were pretreated with antioxidant and free radical scavenger N-Acetyl-L-Cysteine (LNAC) prior to LDL-DHA treatment, LDL-DHA cytotoxicity was effectively blocked and the TIB-75 cells retained good viability (Fig. 5C).
Supplementary TBAR measurements were also used to assess the LDL-DHA induced lipid peroxidation; however, in these studies albumin transported DHA was also assessed to investigate the capability of HSA-DHA particles to induce cytotoxic lipid peroxides (Fig. 5D). From this experiment it can first be seen that TIB-75 cells have higher baseline TBARS than TIB-73 cells. Secondly, LDL mediated delivery of DHA was able to further induce pronounced increases in lipid peroxidation selectively in the TIB-75 cells. In contrast, HSA-DHA was unable to elicit a significant increase in lipid peroxidation within the TIB-75 cells. The changes in lipid peroxidation were also observed using confocal microscopy imaging BODIPY C11 581/591 and heat maps of the data were displayed as ratios of oxidized to unoxidized lipid dye (Fig. 5E). This ratiometric image analysis also confirmed that TIB-75 cells had higher baseline LPO compared to TIB-73 cells and that LDL-DHA exacerbated lipid peroxidation activity in the TIB-75 cells.
3.4. Effect of LDL-DHA on lysosome lipid peroxidation and membrane permeability
LDL receptor endocytosis deposits LDL-DHA nanoparticles in the lysosome, where DHA is liberated. The resulting high concentrations of DHA in the lysosome can disrupt lysosomal membrane function and integrity [28,29]. Confocal microscopy using BODIPY C11 581/591 dye and colocalization image analysis showed that lipid peroxidation colocalizes within the lysosome in both TIB-73 and TIB-75 cells and that LDL-DHA treatment further increases that colocalization of lipid peroxidation in the lysosomes of TIB-75 cells (Fig. 6A–B). Further confocal microscopy studies using acridine orange (AO) were performed to examine the integrity of the lysosome membranes. Early in the post LDL-DHA treatment period (4 h) TIB-75 cells exhibit marked loss of intracellular red punctate fluorescence which is indicative of increased lysosomal permeability (Fig. 6C and Supplemental Fig. 3). These findings were later confirmed using a second lysosome permeability probe called Magic Red Cathepsin B (Supplemental Fig. 4). Experiments with HSA-DHA and AO were also performed at this time, but even at high doses HSA-DHA was unable to alter the permeability of lysosomes within TIB-75 cells (Supplemental Fig. 5). Temporal FACS analysis was also employed and experiments using AO showed that LDL-DHA increases the number of cells with lysosome membrane permeability (LMP) starting at 8 h, as opposed to 4 h seen with confocal microscopy, this disruption in lysosomal integrity continued through the 24 h post treatment period in the TIB-75 cells (Fig. 6D). TIB-73 cells had a much smaller number of cells with LMP and the kinetics for this increase was slower. Overall, these results indicated that basal levels of lipid peroxidation occurs in the lysosomes in TIB-73 and TIB-75 cells and that LDL-DHA treatment further enhances lysosomal lipid peroxidation and selectively induces large amounts of LMP in the TIB-75 cells.
Fig. 6. LDL-DHA selectively causes lipid peroxidation and lysosome leaking in TIB-75 cells.
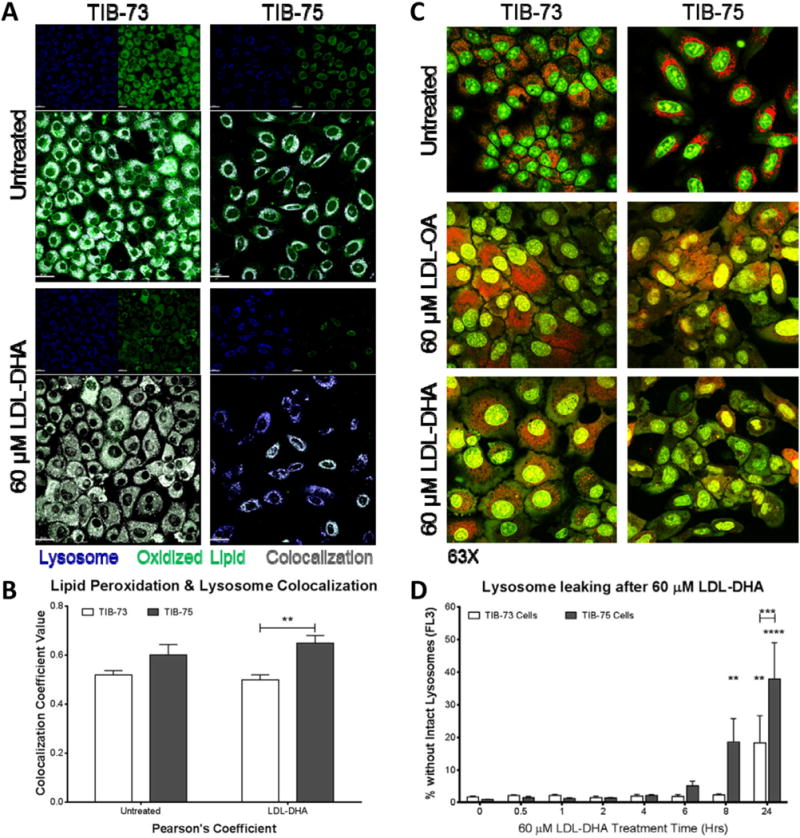
(A) Confocal images (63× magnification) of TIB-73 and TIB-75 cells after staining with 2 μM BODIPY C11 581/591 dye for 25 min at 37 °C, treatment with serum free media or 60 μM LDL-DHA for 4 h, then stained with 75 nM LysoTracker Deep Red for 30 min prior to imaging. (B) Imaris imaging software was used to calculate LysoTracker Deep Red colocalization with green oxidized BODIPY C11 581/591 dye in the cells (n = 3). (C) Confocal images (63× magnification) of TIB-73 and TIB-75 cells dyed with 5 μg/mL acridine orange for 20 min, treated with serum free media or 60 μM of LDL-OA or LDL-DHA for 5 h. Red punctate fluorescence (640 nm emission) indicates intact lysosomes and green diffuse fluorescence (540 nm emission) indicates leaking lysosomes. TIB-75 cells treated with LDL-DHA demonstrated loss of lysosome membrane integrity. (D) Flow cytometry of TIB-73 and TIB-75 cells treated for different time periods with 60 μM LDL-DHA followed by staining with 0.5 μg/mL acridine orange for 15 min at 37 °C. Decreased red acridine orange fluorescence (FL2) determined the number of cells with loss of intact lysosomes. TIB-75 cells exhibited increased loss of intact lysosomes over time with LDL-DHA treatment compared to TIB-73 cells (n = 7). * = p-value ≤ 0.05, ** = p-value ≤ 0.01, *** = p-value ≤ 0.001, **** = p-value ≤ 0.0001. The error bars represent the standard error of the mean.
3.5. Role of iron in LDL-DHA cytotoxicity
Iron plays a critical role in the generation of ROS and lipid peroxide species. As such, the differences in cellular iron between the TIB-73 and TIB-75 cells were assessed. The ferrozine assay showed that TIB-75 cells had higher total iron levels (Fig. 7A), while measurements by ICP-MS were approaching a significant difference between TIB-73 and TIB-75 (p-value = 0.072) (Supplemental Fig. 6). In addition, silver sulfide autometallography for labile iron showed that TIB-75 cells had darker staining than TIB-73 cells (Fig. 7B). Labile iron was also examined by confocal microscopy using the labile iron detecting fluorophore, IP1; this showed that labile iron colocalized with the lysosome in both cell types, but differences in the total amount of labile iron between cells was not observed with IP1 (Supplemental Fig. 7). Pretreatment of TIB-75 cells with an iron chelator, deferoxamine (DFO), was able to partially block LDL-DHA cytotoxicity and retain the viability of the TIB-75 cells to approximately 75% of its untreated controls (Fig. 7C). DFO pretreatment was also able to partially rescue lipid peroxidation and LMP that was induced by LDL-DHA (Fig. 7D–F). Interestingly, DFO pretreatments were unable to prevent other LDL-DHA induced subcellular perturbations involving mitochondrial membrane permeability (MMP) and ROS generation in TIB-75 cells (Supplemental Fig. 8). Additional studies were performed to sensitize TIB-73 cells with exogenous iron, however, the addition of excess FeCl3 was unable to increase the cytotoxicity of LDL-DHA to TIB-73 cells (Supplemental Fig. 9). Overall, these results suggest that labile iron in the lysosome may be catalyzing lipid peroxidation and LMP after LDL-DHA treatment in the TIB-75 cells as DFO was able to partially reverse these effects in TIB-75 cells.
Fig. 7. Iron determination in TIB-73 and TIB-75 cells.
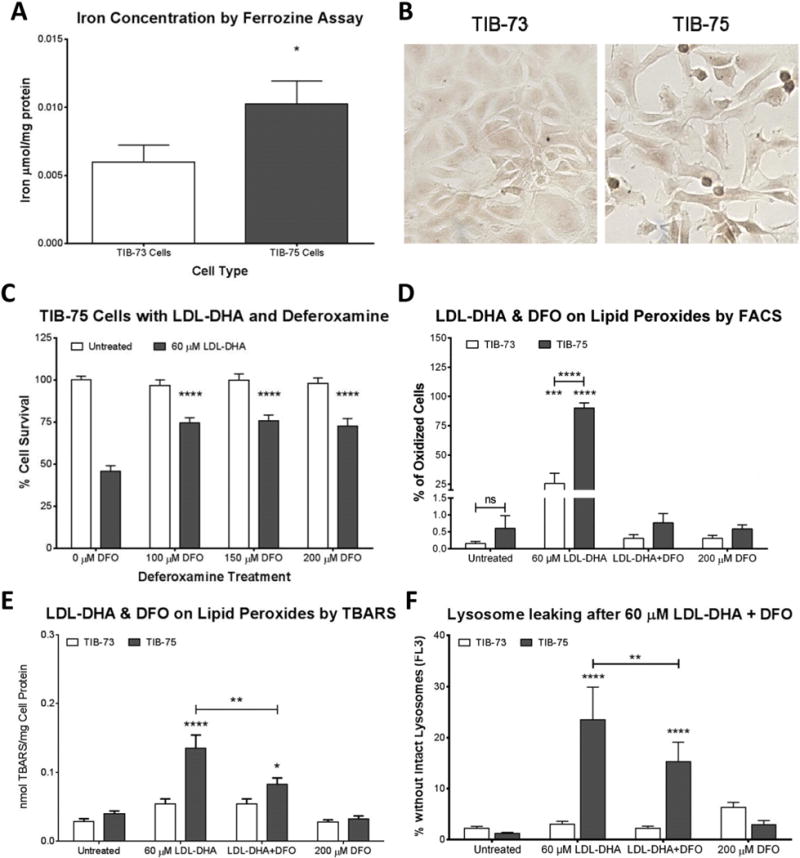
(A) Iron determination in cell lysates by the colorimetric ferrozine assay. Iron concentration of each sample was determined by comparison to a FeCl3 standard curved and normalized to protein (n = 3). (B) Labile iron was visualized in TIB-73 and TIB-75 cells by silver sulfide autometallography (the images above were developed for 10 min). The slides were imaged using a brightfield microscope at 20× magnification. TIB-75 cells displayed a higher amount of silver sulfide staining compared to TIB-73 cells. In the following figures (C–F), cells were serum-starved, pretreated for 3 h with the iron chelator deferoxamine, and then 60 μM LDL-DHA was added for 24 h. (C) Cell viability of TIB-75 cells. Pretreatment with deferoxamine to remove labile iron partially rescued LDL-DHA cytotoxicity in TIB-75 cells (n = 7). (D) Flow cytometry of TIB-73 and TIB-75 cells stained with 1 μM BODIPY C11 581/591 prior to treatment. TIB-75 cells had a higher percentage of cells with oxidized lipids following LDL-DHA treatment and that lipid oxidation was removed with deferoxamine pretreatment (n = 5). (E) TBARS of TIB-73 and TIB-75 cells after treatment. Deferoxamine pretreatment to remove labile iron significantly reduced lipid peroxidation caused by LDL-DHA in TIB-75 cells, but not back to baseline levels (n = 9). (F) Flow cytometry of TIB-73 and TIB-75 cells after treatment and staining with 0.5 μg/mL acridine orange for 15 min. Decreased red acridine orange fluorescence (FL2) of the cells was measured by flow cytometry to determine the number of cells with loss of intact lysosomes. Deferoxamine pretreatment was able to significantly increase the number of TIB-75 cells with intact lysosomes following LDL-DHA treatment (n = 9). * = p-value ≤ 0.05, ** = p-value ≤ 0.01, *** = p-value ≤ 0.001, **** = p-value ≤ 0.0001. The error bars represent the standard error of the mean.
3.6. Effect of LDL-DHA on mitochondria membrane potential and ROS production
Subcellular mechanistic experiments continued with investigations into the effects of LDL-DHA treatment on mitochondrial function. In these studies, TIB-73 and TIB-75 cells were stained with Tetramethylrhodamine Methyl Ester (TMRM) after LDL-DHA treatment and MMP was observed by confocal microscopy. LDL-DHA treatment caused a selective loss of MMP in TIB-75 cells indicated by their significant loss of TMRM fluorescence (Fig. 8A, Supplemental Fig. 10). Flow cytometry using TMRM showed that the number of TMRM negative TIB-75 cells increases significantly at 6 h, peaks at 8 h, and remains elevated at 24 h while TIB-73 cells remained unaffected by this treatment (Fig. 8B). When mitochondrial ROS production was examined using MitoSox dye, it was observed that only the TIB-75 cells treated with LDL-DHA had a significant increase in the number of MitoSox positive cells (Fig. 8C). Given these findings, we attempted to render TIB-73 cells sensitive to LDL-DHA induced cytotoxicity by pretreating them with increasing doses of carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a mitochondria oxidative phosphorylation inhibitor, prior to LDL-DHA treatment (Fig. 8D). TIB-73 cells treated with CCCP or LDL-DHA alone did not experience significant cytotoxicity, but the combination of CCCP and LDL-DHA resulted in increased sensitivity to LDL-DHA dependent on the CCCP dose administered. MitoSox flow cytometry showed that CCCP treatment alone with 50 μM (2.2 ± 0.7%) and 75 μM (2.9 ± 0.7%) gave the TIB-73 cells a similar population of MitoSox positive cells compared to untreated TIB-75 cells (2.6 ± 0.5%) (Fig. 8E). Additionally, TIB-73 cells treated with both CCCP and LDL-DHA had a significant increase in the number of MitoSox positive cells compared to untreated cells indicating that mitochondrial ROS production can sensitize cells to LDL-DHA cytotoxicity.
Fig. 8. LDL-DHA causes selective mitochondrial dysfunction in TIB-75 cells.
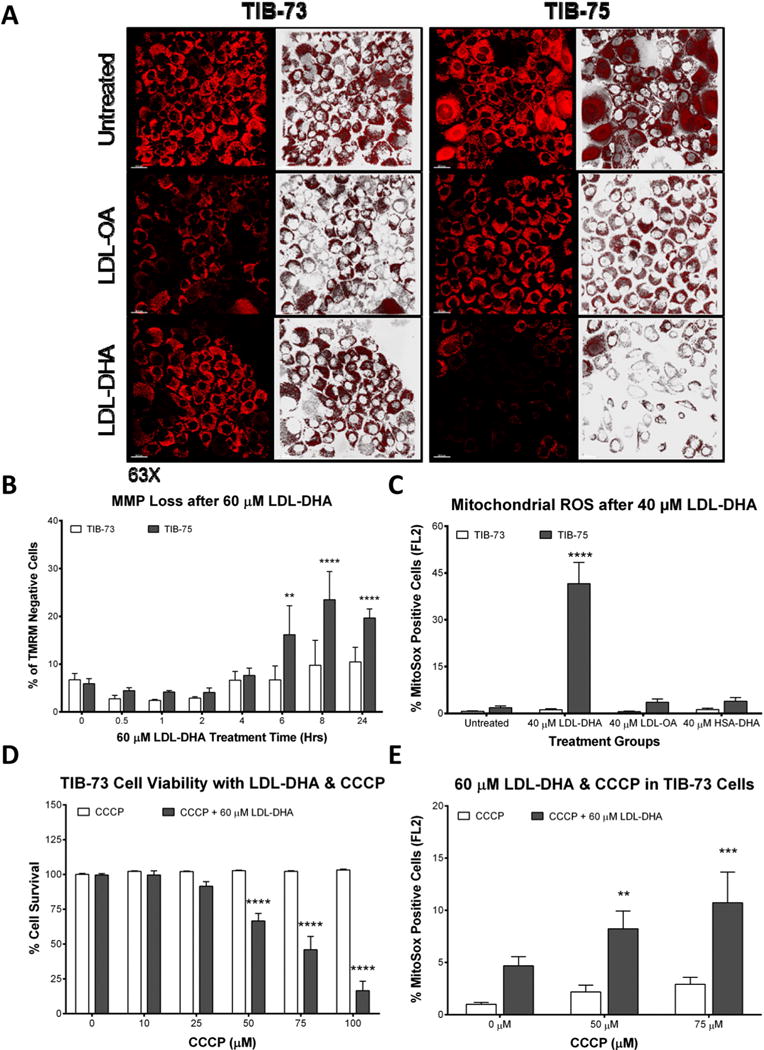
(A) Confocal images (63× magnification) of TIB-73 and TIB-75 cells after treatment with serum free media or 60 μM of LDL-OA or LDL-DHA overnight, 200 nM TMRM for 25 min, and then 50 nM TMRM for imaging. Bright punctate fluorescence indicates normal/hyperpolarized mitochondrial membrane potential and dim fluorescence indicates abnormal/depolarized mitochondrial membrane potential. (B) Flow cytometry of TIB-73 and TIB-75 cells treated for different time periods with 60 μM LDL-DHA and stained with 100 nM TMRM for 15 min. TIB-75 cells had a higher percentage of TMRM negative cells following LDL-DHA treatment that increased with time compared to TIB-73 cells (n = 7). (C) Flow cytometry of TIB-73 and TIB-75 cells treated with serum free media, 40 μM LDL-OA, LDL-DHA or HSA-DHA overnight and then 5 μM MitoSox and 0.2 μM MitoTracker Green for 15 min. TIB-75 cells had a higher percentage of MitoSox positive cells following LDL-DHA treatment compared to TIB-73 cells. LDL-OA and HSA-DHA did not significantly increase MitoSox expression in either cell line (n = 3). For Figs. D and E, TIB-73 cells were pretreated for 1 h with CCCP (0–100 μM) followed by 60 μM LDL-DHA for 24 h (D) or overnight (E). (D) Cell viability of TIB-73 cells after CCCP and LDL-DHA treatment. Inhibition of mitochondrial oxidative phosphorylation by CCCP sensitized TIB-73 to a sub-lethal dose of LDL-DHA in a dose-dependent manner (n = 9). (E) Flow cytometry of TIB-73 cells stained with 5 μM MitoSox and 0.2 μM MitoTracker Green for 15 min after CCCP and LDL-DHA treatment. Pretreatment with CCCP increased the number of MitoSox positive cells following treatment with LDL-DHA compared to the untreated control group (n = 5). * = p-value ≤ 0.05, ** = p-value ≤ 0.01, *** = p-value ≤ 0.001, **** = p-value ≤ 0.0001. The error bars represent the standard error of the mean.
3.7. Effect of LDL-DHA on DNA damage
Lastly, LDL-DHA treatments were evaluated for nuclear or DNA damaging effects. γ-H2AX foci formation were counted in TIB-73 and TIB-75 cells after LDL-DHA treatment (40 or 60 μM). TIB-75 cells treated with 60 μM LDL-DHA for 6 h (Fig. 9A) exhibited an increased number of cells with γ-H2AX foci formation. Similarly, overnight treatment with both 40 μM and 60 μM LDL-DHA (Fig. 9B) also significantly increased γ-H2AX foci formation in TIB-75 cells, but not in TIB-73 cells. The DNA damage along with the lysosome and mitochondria changes indicate that LDL-DHA treatment causes a multitude of subcellular changes selectively in the malignant TIB-75 cells.
Fig. 9. γ-H2AX foci formation after LDL-DHA treatment.
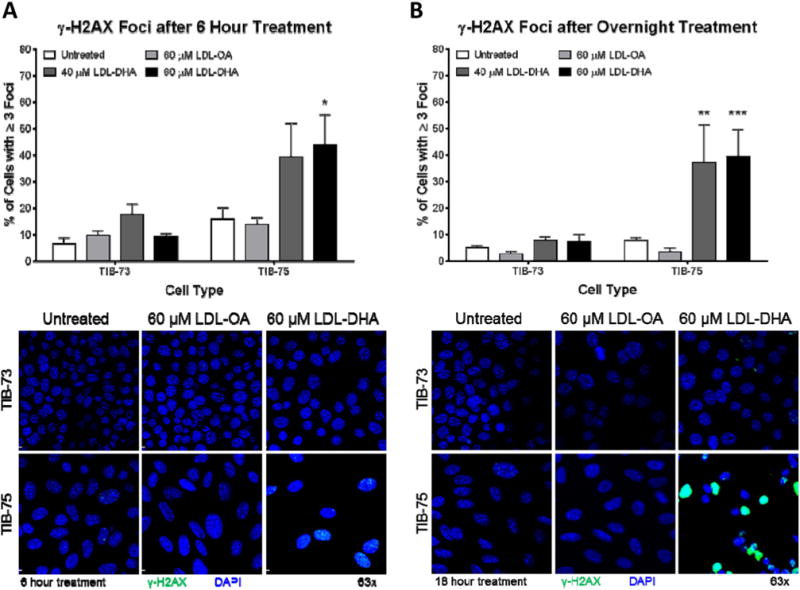
Confocal images (63× magnification) of γ-H2AX stained TIB-73 and TIB-75 cells after treatment for (A) 6 h or (B) overnight with serum free media, 40 μM LDL-DHA, 60 μM LDL-DHA or 60 μM LDL-OA. Images were analyzed on Imaris imaging software and the total number of cells and cells containing ≥3 γ-H2AX foci were counted using ImageJ software (n = 5). * = p-value ≤ 0.05, ** = p-value ≤ 0.01, *** = p-value ≤ 0.001, **** = p-value ≤ 0.0001. The error bars represent the standard error of the mean.
4. Discussion
Conventional chemotherapy and radiation therapy continue to play a central role in cancer treatment. Both therapies induce cancer cell killing through the cytotoxic actions of chemicals or radiation exposure respectively. Unfortunately these therapies also indiscriminately destroy healthy cells, particularly those that divide quickly. These actions can lead to severe side effects such as bone marrow suppression and organ damage which ultimately compromise efficacy of the treatment [30,31]. Modern research efforts are now avidly pursuing the discovery and development of drugs, like Gleevec and trastuzumab, which demonstrate more targeted and selective cytotoxicity towards cancer cells [1,2]. The present and previous papers from our group have shown that LDL-DHA nanoparticles can selectively kill HCC cells and tumors without injury to their nonmalignant counterparts [17,32]. Earlier work by Begin and others reported that unesterified polyunsaturated fatty acids could inhibit the growth and kill various cancer cell lines [33,34]. Normal fibroblasts, however, did not experience cytotoxic effects from these treatments. This differential action of polyunsaturated fatty acids on normal and malignant cells has intrigued many to consider utilizing these lipids in cancer treatment [35,36]. We postulate that the selective cancer killing activity of the LDL-DHA is not the result of increased nanoparticle uptake by the malignant TIB-75 cells (our previous study showed that both TIB-73 and TIB-75 cells avidly bind and internalize LDL nanoparticles to a similar degree) [17], but rather is triggered by the increased basal levels of oxidative stress present in cancer cells. Emerging evidence now suggests that, compared with normal cells, tumor cells maintain increased intracellular levels of reactive oxygen species (ROS) [37,38]. The elevated concentrations of ROS play a critical role in the development and growth of the cancer by promoting genetic instability and uncontrolled proliferation [39,40]. Dysregulated redox homeostasis was evident in the malignant TIB-75 cells. Significantly increased levels of DCF fluorescence and a diminished ratio of the glutathione redox couple, GSH:GSSG, indicated that the TIB-75 cells experienced greater oxidative stress than TIB-73 cells. In addition, TIB-75 cells, also expressed lower levels of the antioxidant enzymes, GPx4 and catalase. Lower levels of the enzyme antioxidants have previously been reported in HCC [41,42], this deficiency is believed to diminish the ROS scavenging capacity in the cell allowing ROS to accumulate to promote oxidative stress. These increased levels of ROS, however, renders the cancer cell more vulnerable to damage from further ROS insults induced by exogenous agents [43]. While moderate amounts of ROS can promote cell differentiation and proliferation, excessive amounts of ROS will cause oxidative damage to lipids, protein and DNA and if not corrected can lead to cell death [44]. When LDL-DHA is internalized into cancer cells the many unsaturated bisallylic protons in DHA are readily attacked by the high intracellular concentrations of ROS initiating the lipid peroxidation cascade. Given the high payload of DHA from each LDL nanoparticle, the self-propagating process of lipid peroxidation can quickly overwhelm and ravage the entire cell. Rapid increases in ROS generation and lipid peroxidation were detected early in the treatment period for TIB-75 cells, and by 24 h of LDL-DHA treatment pronounced levels of ROS and lipid peroxidation were present throughout the population of TIB-75 cells (Fig. 5C and E). Our studies went on to show that oxidative stress and lipid peroxidation play a central role in LDL-DHA mediated cancer cell killing as N-acetylcysteine, a free radical scavenger and precursor of the ROS scavenger glutathione, was able to rescue the TIB-75 cells. For non-malignant cells, like TIB-73, ROS levels are low and the concentration of scavenging antioxidants are high, thus DHA that is taken up by these cells likely undergoes esterification or enzyme-mediated oxidation rather than damaging auto-oxidation. TIB-73 cells did experience small increases in ROS and lipid peroxides with LDL-DHA treatment, however these disturbances were not sufficient to compromise their viability. On the contrary, when TIB-73 cells were pretreated with menadione, a redox cycling oxidant [45], the sensitivity of the cells to LDL-DHA drastically increased such that their IC50 (20.32 μM) was even lower than that of TIB-75. Collectively, these findings support the hypothesis that basal levels of oxidative stress determine the sensitivity of cells/tissues to LDL-DHA treatment. Given the increased levels of ROS in malignant cells, it can be generally expected that LDL-DHA treatments would be selectively damaging to cancer cells over normal cells.
Given that albumin is the principal fatty acid carrier in the plasma [46], we also explored its capacity to transport DHA to cancer cells. DHA loading onto HSA is achieved as this protein possesses approximately seven binding sites for fatty acids with high to moderate affinity [46]. Unlike LDL mediated delivery of DHA, HSA transport of DHA was unable to elicit significant cytotoxicity to the malignant TIB-75 cells. Previous papers from our group and others have reported similar observations for various cancer cells [47,48]. The study by Kanno et al. suggests that albumin is able to modulate the cytotoxic actions of DHA by acting as an absorbent, to stabilize the unsaturation of DHA, and as an antioxidant, directly scavenging ROS and metals from free radical reactions [48]. Indeed, HSA is known to have considerable antioxidant capacity as previous work has shown that >70% of the free radical-trapping activity of serum arises from HSA [49]. This reasoning likely explains why even greatly increased concentrations of HSA-DHA were unable to induce lipid peroxidation or cell kill among the TIB-75 cells.
The endo-lysosome compartment is the first subcellular organelle the endocytosed LDL-DHA nanoparticle will engage. Within the acidic and enzyme-rich milieu of the lysosome the LDL-DHA nanoparticle is hydrolyzed to amino acids, glycerolipids and free fatty acids. The liberated DHA is then free to interact in the lysosome (incorporate into lysosomal membranes) or traverse out of the lysosome to other locations in the cell. Conditions within the lysosomal microenvironment can differ considerably between normal and malignant cells, these differences in turn determine the fate and ultimately the effect of DHA on the cell. Within the cancer cell, the high global levels of ROS may permeate into the lysosomal compartment further increasing the oxidative potential of the lysosomes [50]. Our findings confirm that not only do the lysosomes in the malignant TIB-75 cells experience significantly higher basal levels of oxidative stress (Fig. 3C), but these cells also contain higher concentrations of iron than TIB-73 cells. Thus, within the confines of lysosomes from cancer cells DHA will be exposed to increasing concentrations of ROS and labile iron [51]. Under these conditions the Fenton-like reactions are favored, and the generated hydroxyl radical will readily attack the lysosomal pools of non-esterified and esterified DHA [52]. The ensuing lipid peroxidation proceeds to ravage the lysosomal membrane and compromise its integrity [53]. These events were played out in the TIB-75 cells as lysosomal membrane permeabilization, as measured by Acridine Orange and Magic Red fluorogenic probes, progressed with global and intralysosomal lipid peroxidation. Interestingly, HSA-DHA was unable to evoke LMP in the TIB-75 cells even at increased concentrations of DHA. Since HSA mediated delivery of DHA occurs via diffusion/facilitative transporters, and not through endocytic pathways, LMP is not expected to occur. This differing transport pathway likely also contributes to the reduced cytotoxicity of HSA-DHA in cancer cells.
Cancer cells are known to accumulate higher levels of intracellular iron to promote cell signaling, proliferation and growth [51,54]. However, iron is also a key catalyst in free radical formation and initiation and promotion of lipid peroxidation. The role of iron in mediating LDL-DHA cytotoxicity was evaluated with the iron chelator deferoxamine (DFO). DFO is a hexadentate hydrophilic chelator weighing >600 Da. It is not expected to penetrate cellular membranes, but rather has been proposed to be taken up by fluid-phase endocytosis [55,56]. As such, DFO is cited to act mainly by chelating lysosomal/endosomal iron. Within this environment DFO is able to chelate redox-active iron and inhibit Fenton-type reactions that propagate the lipid peroxidation process. Our experiments showed that DFO proved to be effective in preserving cellular viability, reducing lipid peroxidation and LMP in TIB-75 cells after LDL-DHA treatment. It should be noted that DFO treatment did not completely restore the cellular perturbations brought about by LDL-DHA, this finding suggests while iron does participate in the cytotoxicity induced by LDL-DHA, additional iron-independent processes are also at play.
Numerous cytopathic sequelae follow LMP as a result of many acidic hydrolases that leak into the cytosol of the cell. Although these enzymes have optimal activity at acidic pH, they retain some activity at the neutral pH of the cytosol and are capable damaging other organelles and inducing cell death [57,58]. Moderate lysosomal rupture can trigger apoptosis, while pronounced lysosomal leakage will result in necrosis [59]. Given the heterogeneous uptake of LDL-DHA across the population of exposed cells and heterogeneous distribution of iron within lysosomes, the extent of lysosomal leakage is expected to vary across the cells [60]. This likely accounts for the mixed picture of cell death experienced by the LDL-DHA treated TIB-75 cells. Accompanying the release of acid hydrolases there will also be considerable leakage of DHA and its reactive peroxide and aldehyde end-products from the destabilized lysosomes. The mitochondria and other cellular organelles lie vulnerable in the path of these damaging molecules [61]. Significant disruptions in mitochondrial function and integrity were detected in the TIB-75 cells after LDL-DHA treatment in our study. MMP, which is known to accompany LMP [62], was seen in TIB-75 cells approximately 6 h after treatment (corresponding or slightly trailing LMP). The treated TIB-75 cells also experienced additional mitochondrial dysfunction in the form of increased mitochondrial ROS production. This dysfunction was likely triggered by the MMP which is known to stimulate ROS generation, which in turn can elicit more lysosomal damage [62]. This vicious cycle is likely to continue in the TIB-75 cells given their reduced ROS scavenging capacity. The LDL-DHA treatment caused a smaller and later loss of MMP in the TIB-73 cells. This minor change in MMP, however, did not affect the viability of the TIB-73 cells. Mitochondrial ROS production remained unaltered for the TIB-73 cells over the course of experiments. If mitochondrial ROS production was stimulated with an electron transport chain uncoupler, as was seen with CCCP, the sensitivity of TIB-73 cells to LDL-DHA treatment significantly increased. These findings show that disruptions in mitochondrial function can play a central role in sensitizing cells to LDL-DHA cytotoxicity.
The nucleus proved to be another organelle selectively damaged by LDL-DHA in the TIB-75 cells. The incurred DNA damage in the TIB-75 cells may have resulted from the propagated lipid peroxidation, circulating acid hydrolases, and/or increase in ROS [63,64]. The highly reactive α, β-unsaturated aldehyde end-products of lipid peroxidation are known to readily attack DNA bases to form exocyclic DNA adducts [64]. The DNA damage resulting from these processes can be mutagenic in small quantities promoting tumorigenesis, but in large quantities they are genotoxic and can result in cell death [65,66]. Disruptions in other organelle systems (e.g. endoplasmic reticulum, peroxisomes, etc.) are also expected to follow LDL-DHA treatment in malignant TIB-75 cells. Dysregulation of each of these additional organelle systems contribute to the trauma sustained by the cancer cells. In addition, alterations in mitogenic signaling (suppression of β-catenin [67], HIF1α [68], NFκB [69], etc.) have also been attributed to DHA; collectively, all these disturbances act to ultimately usher in the demise of the cancer cell. While the current study provides valuable insight into the mechanisms governing LDL-DHA selective anticancer activity, further investigation are required to advance this nanotechnology towards clinical translation. Issues related to: the need for plasma-derived components; the finite shelf life of LDL; the need for facile formulation for large-scale production, must all be addressed in order for this nanomedicine to reach its full therapeutic potential.
In conclusion, we have demonstrated that basal levels and balance of ROS can determine the susceptibility of cells to LDL-DHA toxicity. Cancer cells, which are known to upregulate their generation of ROS, inadvertently, render themselves prone to LDL-DHA cytotoxicity. The selective LDL-DHA induced toxicity that is seen in tumor tissue and cells is also experienced at the subcellular level. LDL receptor mediated endocytosis first deposits the LDL-DHA nanoparticle in the lysosome, which in the case of cancer cells is a highly oxidizing environment that triggers the destructive peroxidation of DHA and destabilization of the lysosomal membrane. The subsequent intracellular events which follow arise from the release of lysosomal hydrolases, DHA and oxidized DHA intermediate and end-products into the cell cytosol. Mitochondrial and nuclear damage incurred in these events, promote further organelle dysfunction and trauma which eventually prove fatal to the cancer cell. Collectively, the findings of the present study indicate that the selectivity of LDL-DHA cytotoxicity is mediated via the altered redox and iron biology of cancer cells. These dysregulated processes which favor tumorigenesis and proliferation can also serve to trigger the catastrophic cytotoxic cascade induced by the intralysosomal peroxidation of LDL-DHA.
Supplementary Material
Acknowledgments
We are grateful to Eric Gonzalez and Preston Christensen for their assistance with colorimetric and mass spectrometry measurements of iron. In addition, we would thank Drs. Christopher J. Chang and Mark Vander Wal from the University of California, Berkeley for kindly providing the iron sensing fluorophore, IP1. Finally, we would like to acknowledge the support of the American Gastroenterological Association Research Foundation Scholar Award (IR Corbin), the University of Texas Southwestern Medical Center President’s Research Council Award (IR Corbin), and the UTSW Core Flow Cytometry Facility and Cancer Center Support Grant (5P30 CA 142543-05).
Financial support
This work was supported in part by the American Gastroenterological Association Research Foundation Scholar Award (IR Corbin), the University of Texas Southwestern Medical Center President’s Research Council Award (IR Corbin), and the Cancer Center Support Grant (5P30 CA 14254304).
Abbreviations
- HCC
hepatocellular carcinoma
- DHA
docosahexaenoic acid
- PUFA
polyunsaturated fatty acid
- LPO
lipid peroxidation
- ROS
reactive oxygen species
- COX
cyclooxygenase
- LOX
lipoxygenase
- LDL
low density lipoprotein
- LDL-DHA
low density lipoprotein reconstituted with docosahexaenoic acid
- LDL-OA
low density lipoprotein reconstituted with oleic acid
- LDL-TO
LDL reconstituted with oleic acid triglyceride (triolein)
- HSA
human serum albumin
- HSA-DHA
human serum albumin conjugated to docosahexaenoic acid
- MTS
CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay
- SOD-1
superoxide dismutase-1
- GPx-4
glutathione peroxidase-4
- HRP
horseradish peroxidase
- GSH
reduced glutathione
- GSSG
glutathione disulfide
- TBARS
thiobarbituric acid reactive substances
- ICP-MS
inductively couple plasma mass spectrometry
- DCF
dichlorofluorescein
- LNAC
N-acetyl-L-cysteine
- LMP
lysosome membrane permeability
- AO
acridine orange
- DFO
deferoxamine
- MMP
mitochondrial membrane permeability
- TMRM
tetramethylrhodamine methyl ester
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbagen.2016.07.004.
Conflict of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Author’s contributions
Lacy R. Moss acquisition and analysis of data; contribution to design; critical revision of content; draft article
Rohit S. Mulik acquisition and analysis of data; critical revision of content
Tim Van Treuren acquisition and analysis of data
Soo Young Kim acquisition and analysis of data
Ian R. Corbin conceived study and design; analysis and interpretation of data; draft article and critical revision; Final approval for publication; accountable for all aspects of work.
Transparency document
The Transparency document associated with this article can be found, in online version.
References
- 1.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, Cervantes F, Hochhaus A, Powell BL, Gabrilove JL, Rousselot P, Reiffers J, Cornelissen JJ, Hughes T, Agis H, Fischer T, Verhoef G, Shepherd J, Saglio G, Gratwohl A, Nielsen JL, Radich JP, Simonsson B, Taylor K, Baccarani M, So C, Letvak L, Larson RA. Five-year followup of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 2.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 3.Couzin J. Cancer drugs. Smart weapons prove tough to design. Science. 2002;298:522–525. doi: 10.1126/science.298.5593.522. [DOI] [PubMed] [Google Scholar]
- 4.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 5.Berquin IM, Min Y, Wu R, Wu J, Perry D, Cline JM, Thomas MJ, Thornburg T, Kulik G, Smith A, Edwards IJ, D’Agostino R, Zhang H, Wu H, Kang JX, Chen YQ. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J Clin Invest. 2007;117:1866–1875. doi: 10.1172/JCI31494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwamoto S, Senzaki H, Kiyozuka Y, Ogura E, Takada H, Hioki K, Tsubura A. Effects of fatty acids on liver metastasis of ACL-15 rat colon cancer cells. Nutr Cancer. 1998;31:143–150. doi: 10.1080/01635589809514694. [DOI] [PubMed] [Google Scholar]
- 7.Braden LM, Carroll KK. Dietary polyunsaturated fat in relation to mammary carcinogenesis in rats. Lipids. 1986;21:285–288. doi: 10.1007/BF02536414. [DOI] [PubMed] [Google Scholar]
- 8.Rose DP, Hatala MA, Connolly JM, Rayburn J. Effect of diets containing different levels of linoleic acid on human breast cancer growth and lung metastasis in nude mice. Cancer Res. 1993;53:4686–4690. [PubMed] [Google Scholar]
- 9.Hursting SD, Thornquist M, Henderson MM. Types of dietary fat and the incidence of cancer at five sites. Prev Med. 1990;19:242–253. doi: 10.1016/0091-7435(90)90025-f. [DOI] [PubMed] [Google Scholar]
- 10.Caygill CP, Charlett A, Hill MJ. Fat, fish, fish oil and cancer. Br J Cancer. 1996;74:159–164. doi: 10.1038/bjc.1996.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki S, Horacsek M, Kesteloot H. An ecological study of the relationship between dietary fat intake and breast cancer mortality. Prev Med. 1993;22:187–202. doi: 10.1006/pmed.1993.1016. [DOI] [PubMed] [Google Scholar]
- 12.Lee SE, Lim JW, Kim H. Activator protein-1 mediates docosahexaenoic acid-induced apoptosis of human gastric cancer cells. Ann N Y Acad Sci. 2009;1171:163–169. doi: 10.1111/j.1749-6632.2009.04716.x. [DOI] [PubMed] [Google Scholar]
- 13.Merendino N, Loppi B, D’Aquino M, Molinari R, Pessina G, Romano C, Velotti F. Docosahexaenoic acid induces apoptosis in the human PaCa-44 pancreatic cancer cell line by active reduced glutathione extrusion and lipid peroxidation. Nutr Cancer. 2005;52:225–233. doi: 10.1207/s15327914nc5202_12. [DOI] [PubMed] [Google Scholar]
- 14.Blanckaert V, Ulmann L, Mimouni V, Antol J, Brancquart L, Chenais B. Docosahexaenoic acid intake decreases proliferation, increases apoptosis and decreases the invasive potential of the human breast carcinoma cell line MDA-MB-231. Int J Oncol. 2010;36:737–742. doi: 10.3892/ijo_00000549. [DOI] [PubMed] [Google Scholar]
- 15.Tsujita-Kyutoku M, Yuri T, Danbara N, Senzaki H, Kiyozuka Y, Uehara N, Takada H, Hada T, Miyazawa T, Ogawa Y, Tsubura A. Conjugated docosahexaenoic acid suppresses KPL-1 human breast cancer cell growth in vitro and in vivo: potential mechanisms of action. Breast Cancer Res. 2004;6:R291–R299. doi: 10.1186/bcr789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conquer JA, Holub BJ. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J Lipid Res. 1998;39:286–292. [PubMed] [Google Scholar]
- 17.Reynolds L, Mulik RS, Wen X, Dilip A, Corbin IR. Low-density lipoprotein-mediated delivery of docosahexaenoic acid selectively kills murine liver cancer cells. Nanomedicine (London) 2014;9:2123–2141. doi: 10.2217/nnm.13.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lund-Katz S, Laplaud PM, Phillips MC, Chapman MJ. Apolipoprotein B-100 conformation and particle surface charge in human LDL subspecies: implication for LDL receptor interaction. Biochemistry. 1998;37:12867–12874. doi: 10.1021/bi980828m. [DOI] [PubMed] [Google Scholar]
- 19.Krieger M, McPhaul MJ, Goldstein JL, Brown MS. Replacement of neutral lipids of low density lipoprotein with esters of long chain unsaturated fatty acids. J Biol Chem. 1979;254:3845–3853. [PubMed] [Google Scholar]
- 20.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 21.Erdahl WL, Krebsbach RJ, Pfeiffer DR. A comparison of phospholipid degradation by oxidation and hydrolysis during the mitochondrial permeability transition, A. rch Biochem Biophys. 1991;285:252–260. doi: 10.1016/0003-9861(91)90357-o. [DOI] [PubMed] [Google Scholar]
- 22.Danscher G. Histochemical demonstration of heavy metals. A revised version of the sulphide silver method suitable for both light and electronmicroscopy. Histochemistry. 1981;71:1–16. doi: 10.1007/BF00592566. [DOI] [PubMed] [Google Scholar]
- 23.Au-Yeung HY, Chan J, Chantarojsiri T, Chang CJ. Molecular imaging of labile iron(II) pools in living cells with a turn-on fluorescent probe. J Am Chem Soc. 2013;135:15165–15173. doi: 10.1021/ja4072964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 25.Hardman WE, Munoz J, Jr, Cameron IL. Role of lipid peroxidation and antioxidant enzymes in omega 3 fatty acids induced suppression of breast cancer xenograft growth in mice. Cancer Cell Int. 2002;2:10. doi: 10.1186/1475-2867-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siddiqui RA, Harvey K, Stillwell W. Anticancer properties of oxidation products of docosahexaenoic acid. Chem Phys Lipids. 2008;153:47–56. doi: 10.1016/j.chemphyslip.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Stoll BA. N-3 fatty acids and lipid peroxidation in breast cancer inhibition. Br J Nutr. 2002;87:193–198. doi: 10.1079/BJNBJN2001512. [DOI] [PubMed] [Google Scholar]
- 28.Stillwell W, Ehringer W, Jenski LJ. Docosahexaenoic acid increases permeability of lipid vesicles and tumor cells. Lipids. 1993;28:103–108. doi: 10.1007/BF02535772. [DOI] [PubMed] [Google Scholar]
- 29.Johansson AC, Appelqvist H, Nilsson C, Kagedal K, Roberg K, Ollinger K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010;15:527–540. doi: 10.1007/s10495-009-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Probin V, Zhou D. Cancer therapy-induced residual bone marrow injury-mechanisms of induction and implication for therapy. Curr Cancer Ther Rev. 2006;2:271–279. doi: 10.2174/157339406777934717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Pathobiology of Cancer Regime-Related Toxicities. Springer-Verlag New York; New York: 2013. [Google Scholar]
- 32.Wen X, Reynolds L, Mulik RS, Kim SY, Van Treuren T, Nguyen LH, Zhu H, Corbin IR. Hepatic arterial infusion of low-density lipoprotein docosahexaenoic acid nanoparticles selectively disrupts redox balance in hepatoma cells and reduces growth of orthotopic liver tumors in rats. Gastroenterology. 2016;150:488–498. doi: 10.1053/j.gastro.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Begin ME, Das UN, Ells G, Horrobin DF. Selective killing of human cancer cells by polyunsaturated fatty acids. Prostaglandins Leukot Med. 1985;19:177–186. doi: 10.1016/0262-1746(85)90084-8. [DOI] [PubMed] [Google Scholar]
- 34.Begin ME, Ells G, Das UN, Horrobin DF. Differential killing of human carcinoma cells supplemented with n-3 and n-6 polyunsaturated fatty acids. J Natl Cancer Inst. 1986;77:1053–1062. [PubMed] [Google Scholar]
- 35.Vaughan VC, Hassing MR, Lewandowski PA. Marine polyunsaturated fatty acids and cancer therapy. Br J Cancer. 2013;108:486–492. doi: 10.1038/bjc.2012.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berquin IM, Edwards IJ, Chen YQ. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008;269:363–377. doi: 10.1016/j.canlet.2008.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 38.Toyokuni S, Okamoto K, Yodoi J, Hiai H. Persistent oxidative stress in cancer. FEBS Lett. 1995;358:1–3. doi: 10.1016/0014-5793(94)01368-b. [DOI] [PubMed] [Google Scholar]
- 39.Boonstra J, Post JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene. 2004;337:1–13. doi: 10.1016/j.gene.2004.04.032. [DOI] [PubMed] [Google Scholar]
- 40.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casaril M, Corso F, Bassi A, Capra F, Gabrielli GB, Stanzial AM, Nicoli N, Corrocher R. Decreased activity of scavenger enzymes in human hepatocellular carcinoma, but not in liver metastases. Int J Clin Lab Res. 1994;24:94–97. doi: 10.1007/BF02593907. [DOI] [PubMed] [Google Scholar]
- 42.Yang LY, Chen WL, Lin JW, Lee SF, Lee CC, Hung TI, Wei YH, Shih CM. Differential expression of antioxidant enzymes in various hepatocellular carcinoma cell lines. J Cell Biochem. 2005;96:622–631. doi: 10.1002/jcb.20541. [DOI] [PubMed] [Google Scholar]
- 43.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 44.Perry G, Raina AK, Nunomura A, Wataya T, Sayre LM, Smith MA. How important is oxidative damage? Lessons from Alzheimer’s disease. Free Radic Biol Med. 2000;28:831–834. doi: 10.1016/s0891-5849(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 45.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Petersen OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 46.van der Vusse GJ. Albumin as fatty acid transporter. Drug Metab Pharmacokinet. 2009;24:300–307. doi: 10.2133/dmpk.24.300. [DOI] [PubMed] [Google Scholar]
- 47.Edwards IJ, Berquin IM, Sun H, O’Flaherty T, Daniel JLW, Thomas MJ, Rudel LL, Wykle RL, Chen YQ. Differential effects of delivery of omega-3 fatty acids to human cancer cells by low-density lipoproteins versus albumin. Clin Cancer Res. 2004;10:8275–8283. doi: 10.1158/1078-0432.CCR-04-1357. [DOI] [PubMed] [Google Scholar]
- 48.Kanno S, Kurauchi K, Tomizawa A, Yomogida S, Ishikawa M. Albumin modulates docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma cell lines. Toxicol Lett. 2011;200:154–161. doi: 10.1016/j.toxlet.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Bourdon E, Blache D. The importance of proteins in defense against oxidation. Antioxid Redox Signal. 2001;3:293–311. doi: 10.1089/152308601300185241. [DOI] [PubMed] [Google Scholar]
- 50.Walton JH, Lewis HA. The partition coefficients of hydrogen peroxide between water and certain organic solvents. J Am Chem Soc. 1916;38:633–638. [Google Scholar]
- 51.Kwok JC, Richardson DR. The iron metabolism of neoplastic cells: alterations that facilitate proliferation? Crit Rev Oncol Hematol. 2002;42:65–78. doi: 10.1016/s1040-8428(01)00213-x. [DOI] [PubMed] [Google Scholar]
- 52.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res Fundam Mol Mech Mutagen. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Zdolsek J, Zhang H, Roberg K, Brunk U. H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic Res Commun. 1993;18:71–85. doi: 10.3109/10715769309147344. [DOI] [PubMed] [Google Scholar]
- 54.Le NTV, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta Rev Cancer. 2002;1603:31–46. doi: 10.1016/s0304-419x(02)00068-9. [DOI] [PubMed] [Google Scholar]
- 55.Cable H, Lloyd JB. Cellular uptake and release of two contrasting iron chelators. J Pharm Pharmacol. 1999;51:131–134. doi: 10.1211/0022357991772231. [DOI] [PubMed] [Google Scholar]
- 56.Lloyd JB, Cable H, Rice-Evans C. Evidence that desferrioxamine cannot enter cells by passive diffusion. Biochem Pharmacol. 1991;41:1361–1363. doi: 10.1016/0006-2952(91)90109-i. [DOI] [PubMed] [Google Scholar]
- 57.Kirkegaard T, Jaattela M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta. 2009;1793:746–754. doi: 10.1016/j.bbamcr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 58.Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresic K, Turk V. Apoptotic pathways: involvement of lysosomal proteases. Biol Chem. 2002;383:1035–1044. doi: 10.1515/BC.2002.112. [DOI] [PubMed] [Google Scholar]
- 59.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 60.Nilsson E, Ghassemifar R, Brunk UT. Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem J. 1997;29:857–865. doi: 10.1023/a:1026441907803. [DOI] [PubMed] [Google Scholar]
- 61.Repnik U, Turk B. Lysosomal-mitochondrial cross-talk during cell death. Mitochondrion. 2010;10:662–669. doi: 10.1016/j.mito.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Zhao M, Antunes F, Eaton JW, Brunk UT. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome crelease and apoptosis. Eur J Biochem/FEBS. 2003;270:3778–3786. doi: 10.1046/j.1432-1033.2003.03765.x. [DOI] [PubMed] [Google Scholar]
- 63.Lin Y, Epstein DL, Liton PB. Intralysosomal iron induces lysosomal membrane permeabilization and cathepsin D–mediated cell death in trabecular meshwork cells exposed to oxidative stress. Invest Ophthalmol Vis Sci. 2010;51:6483–6495. doi: 10.1167/iovs.10-5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181–182:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 65.Cao EH, Liu XQ, Wang LG, Wang JJ, Xu NF. Evidence that lipid peroxidation products bind to DNA in liver cells. Biochim Biophys Acta. 1995;1259:187–191. doi: 10.1016/0005-2760(95)00162-6. [DOI] [PubMed] [Google Scholar]
- 66.Burcham PC. Genotoxic lipid peroxidation products: their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis. 1998;13:287–305. doi: 10.1093/mutage/13.3.287. [DOI] [PubMed] [Google Scholar]
- 67.Lim K, Han C, Dai Y, Shen M, Wu T. Omega-3 polyunsaturated fatty acids inhibit hepatocellular carcinoma cell growth through blocking beta-catenin and cyclooxygenase-2. Mol Cancer Ther. 2009;8:3046–3055. doi: 10.1158/1535-7163.MCT-09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mouradian M, Kikawa KD, Dranka BP, Komas SM, Kalyanaraman B, Pardini RS. Docosahexaenoic acid attenuates breast cancer cell metabolism and the Warburg phenotype by targeting bioenergetic function. Mol Carcinog. 2015;54:810–820. doi: 10.1002/mc.22151. [DOI] [PubMed] [Google Scholar]
- 69.Cavazos DA, Price RS, Apte SS, de Graffenried LA. Docosahexaenoic acid selectively induces human prostate cancer cell sensitivity to oxidative stress through modulation of NF-κB. Prostate. 2011;71:1420–1428. doi: 10.1002/pros.21359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.