

Abstract
Cystic fibrosis (CF) is the most common life-threatening monogenic disease afflicting Caucasian people. It affects the respiratory, gastrointestinal, glandular and reproductive systems. The major cause of morbidity and mortality in CF is the respiratory disorder caused by a vicious cycle of obstruction of the airways, inflammation and infection that leads to epithelial damage, tissue remodeling and end-stage lung disease. Over the past decades, life expectancy of CF patients has increased due to early diagnosis and improved treatments; however, these patients still present limited quality of life. Many attempts have been made to rescue CF transmembrane conductance regulator (CFTR) expression, function and stability, thereby overcoming the molecular basis of CF. Gene and protein variances caused by CFTR mutants lead to different CF phenotypes, which then require different treatments to quell the patients’ debilitating symptoms. In order to seek better approaches to treat CF patients and maximize therapeutic effects, CFTR mutants have been stratified into six groups (although several of these mutations present pleiotropic defects). The research with CFTR modulators (read-through agents, correctors, potentiators, stabilizers and amplifiers) has achieved remarkable progress, and these drugs are translating into pharmaceuticals and personalized treatments for CF patients. This review summarizes the main molecular and clinical features of CF, emphasizes the latest clinical trials using CFTR modulators, sheds light on the molecular mechanisms underlying these new and emerging treatments, and discusses the major breakthroughs and challenges to treating all CF patients.
Keywords: CFTR, cystic fibrosis, protein misfolding, intracellular trafficking, proteostasis network, personalized medicine, ABC transporters
Introduction
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause cystic fibrosis (CF) – the most common life-limiting autosomal recessive inherited disorder in Caucasian people. The mutated gene translates into a defective CFTR protein with loss-of-activity (Kerem et al., 1989; Riordan et al., 1989). CFTR encodes an anion channel expressed in several cell types that: (1) transports chloride across the apical membrane; (2) modulates the activity of other ion channels, thereby regulating fluid and electrolyte balance in the mucosal membranes; and (3) secretes bicarbonate, which is crucial for pH regulation, host defense and protection against noxious stimuli (Gadsby et al., 2006; Gentzsch et al., 2010; Gustafsson et al., 2012). As a protein of the ATP-binding cassette (ABC) family, CFTR is comprised of two transmembrane domains (TMDs), two nucleotide-binding domains (NBDs) and a unique regulatory domain (RD) (Gadsby et al., 2006) (Figure 1). Cyclic AMP (cAMP)-dependent protein kinase A (PKA), protein kinase C (PKC) and ATP control its activity (Winter and Welsh, 1997; Chappe et al., 2003). The most prevalent CFTR mutation was discovered almost 30 years ago; it was the deletion of a phenylalanine at position 508 (ΔF508) (Kerem et al., 1989; Riordan et al., 1989), which affects ∼80% of CF patients worldwide (Bobadilla et al., 2002; Sosnay et al., 2013). The mutation ΔF508 reduces thermal and kinetic stability of NBD1 and precludes interdomain interactions (Serohijos et al., 2008; Mendoza et al., 2012). Hence, the endoplasmic reticulum (ER) retains misfolded ΔF508-CFTR, which forms only a partially glycosylated protein, and the proteasome promptly degrades it (Cheng et al., 1990; Jensen et al., 1995).
FIGURE 1.
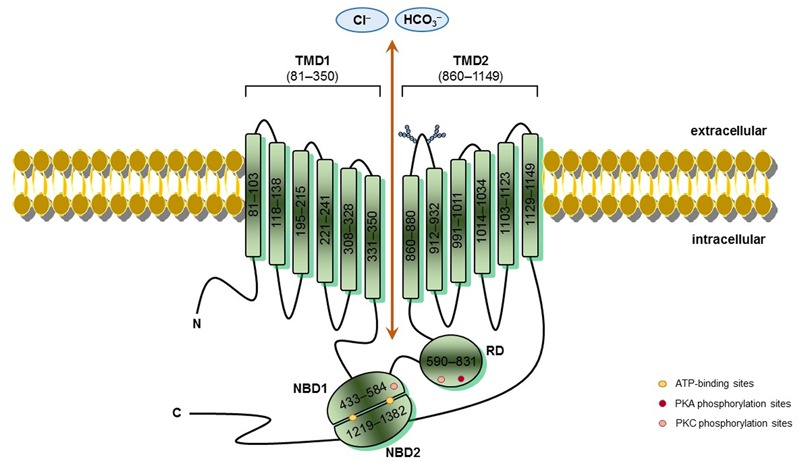
CFTR schematic structure – Cystic fibrosis transmembrane conductance regulator (CFTR) is a 1,480-amino acids protein inserted into the cell surface. CFTR possesses five domains: two transmembrane domains (TMD1/2), containing six hydrophobic alpha-helices, which cross the cell surface lipid bilayer, and are joined by two intracellular loops and three extracellular loops, and with glycosylated residues linked in the extracellular loop 4 (N894, N900); two nucleotide-binding domains (NBD1/2) with highly conserved sequenced for ATP-binding, where occur hydrolysis; and one regulatory domain (RD) with multiple phosphorylation sites. CFTR channel open when protein kinase A (PKA) and protein kinase C (PKC) phosphorylate RD and ATPs bind to side chain charged amino acids in NBDs, thereby activating CFTR function. TMDs form the gate where occurs chloride conductance. The positions denoted into the boxes correspond to the first and last amino acid of each fragment and CFTR sequence was obtained in the Cystic Fibrosis Mutation Database (CFTR1 database; http://www.genet.sickkids.on.ca/Home.html).
CF is a systemic disease that affects ∼85,000 people and presents heterogeneous distribution globally (Bobadilla et al., 2002; Sosnay et al., 2013) (Figure 2, Table 1). According to the latest registry reports, 38,985 CF patients are in Europe (European Cystic Fibrosis Society [ECFS], 2016), 28,676 in the United States (Cystic Fibrosis Foundation [CFF], 2015), 4,128 in Canada (Cystic Fibrosis Canada [CFC], 2016), 3,294 in Australia (Cystic Fibrosis Federation Australia [CFFA], 2016) and 3,511 in Brazil (Brazilian Cystic Fibrosis Study Group [GBEFC], 2016) (Figure 3). The major cause of morbidity and mortality in CF is the respiratory disorder caused by the lack of CFTR at the plasma membrane (PM) (Moskowitz et al., 2008), which decreases the anion permeability in airway cells and leads to a progressive pathophysiological cascade (Figure 4): (1) A faulty conductance of other ions, such as excessive sodium transport mediated by epithelial Na+ channel (ENaC), since it is negatively regulated by CFTR (Kunzelmann et al., 1995; Gentzsch et al., 2010). Controversially, some reports have shown that CFTR loss-of-activity reduces chloride conductance without increasing sodium absorption in CF epithelia (Chen et al., 2010; Itani et al., 2011). (2) The imbalance of ion regulation depletes water content and/or decreases pH in the airway surface liquid (Tarran et al., 2005; Tang et al., 2016). (3) Dehydration and/or acidification of epithelial lining, as well as increased mucin polymer cross-links, raise the amount and viscosity of mucus in gel phase (Tarran et al., 2005; Yuan et al., 2015; Shah et al., 2016; Tang et al., 2016), making the mucus tenacious and difficult to remove by ciliary beating. (4) Accumulation of mucus leads to obstruction of the airways, inflammation and bronchiectasis. (5) Frequently, pathogens colonize the airways and increase the recruitment of inflammatory cells (Lyczak et al., 2002; Bhatt, 2013). Furthermore, oxygen depletion below the sputum-air interface favors biofilm formation (Worlitzsch et al., 2002; Cowley et al., 2015). (6) The destruction of airway and lung parenchyma epithelial cells causes tissue remodeling, reduction of gas exchange area and impairment of lung function. (7) As the disease progresses, the patient succumbs to death due to respiratory failure.
FIGURE 2.
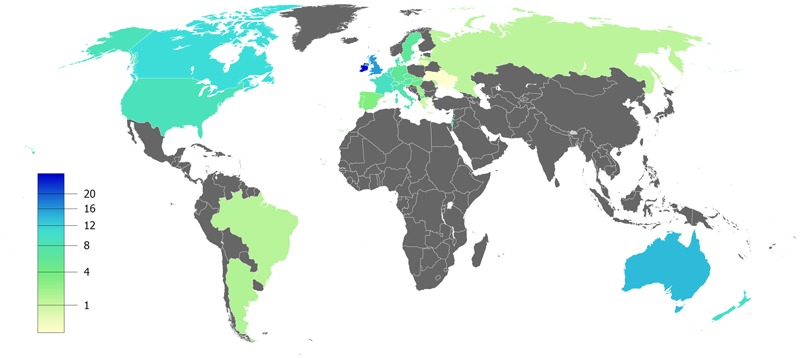
Estimated prevalence of cystic fibrosis per 100,000 habitants – Data compiled from the latest registry reports of Europe (European Cystic Fibrosis Society [ECFS], 2016), United States (Cystic Fibrosis Foundation [CFF], 2015), Canada (Cystic Fibrosis Canada [CFC], 2016), Australia (Cystic Fibrosis Federation Australia [CFFA], 2016) and Brazil (Brazilian Cystic Fibrosis Study Group [GBEFC], 2016).
Table 1.
Top 10 countries with the highest number of CF patients.
| Registered patients | Per 100,000 habitants | |
|---|---|---|
| 1° | United States | Ireland |
| 2° | United Kingdom | United Kingdom |
| 3° | France | Australia |
| 4° | Germany | Canada |
| 5° | Italy | Belgium |
| 6° | Canada | New Zealand |
| 7° | Brazil | France |
| 8° | Australia | United States |
| 9° | Russia | Switzerland |
| 10° | Spain | Denmark |
FIGURE 3.
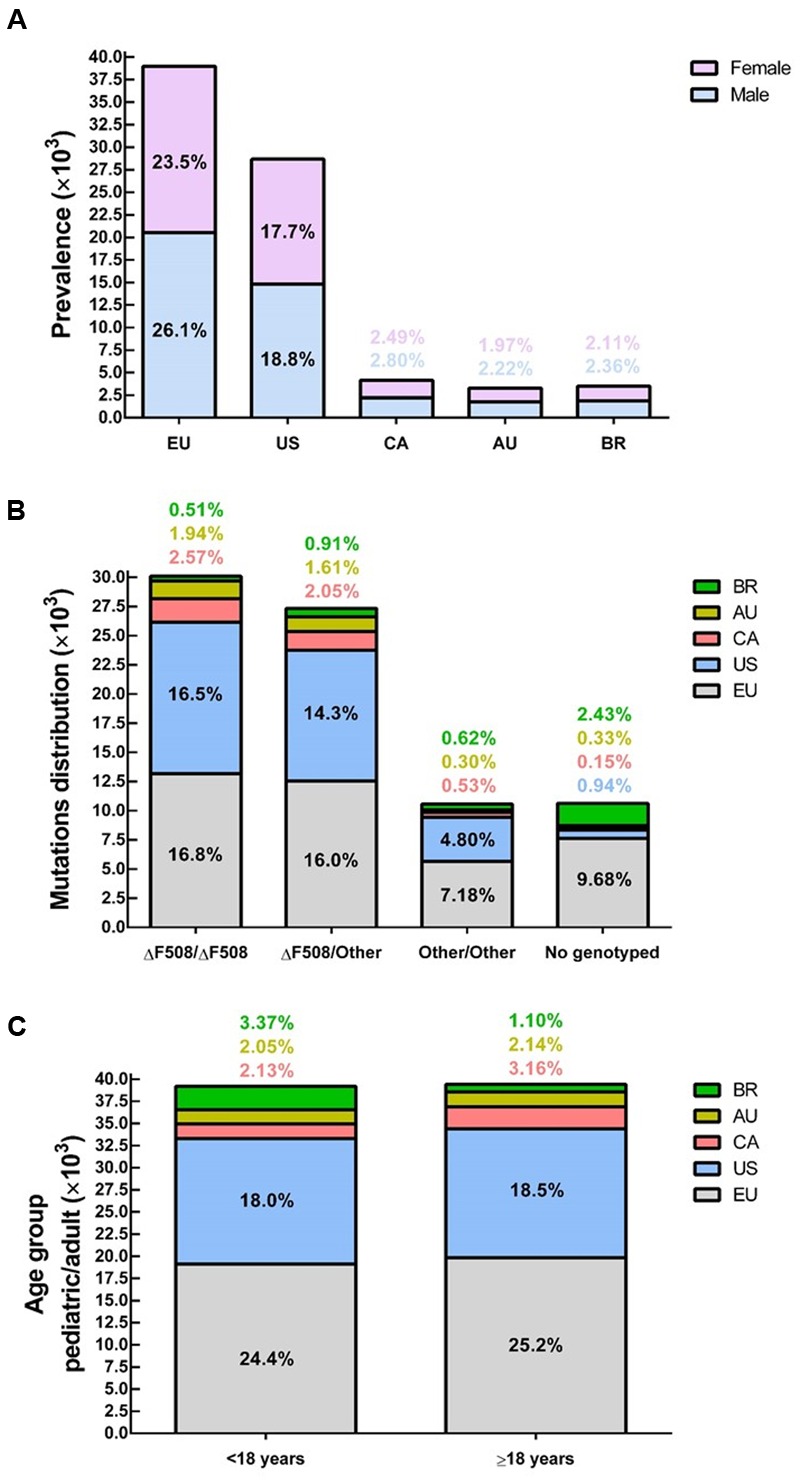
Demography of cystic fibrosis in a sample of 78,627 patients in different countries or demographic regions – (A) Prevalence by gender: average of 52% male and 48% female. (B) Within the sample group, 86% have been genotyped and approximately 38% are ΔF508-homozygous, 35% ΔF508-heterozygous and 13% bearing other CFTR (cystic fibrosis transmembrane conductance regulator) mutants in both alleles. (C) About 50% of patients are under 18 years and 50% are 18 years or older. Data compiled from the latest registry reports of Europe (EU; European Cystic Fibrosis Society [ECFS], 2016), United States (US; Cystic Fibrosis Foundation [CFF], 2015), Canada (CA; Cystic Fibrosis Canada [CFC], 2016), Australia (AU; Cystic Fibrosis Federation Australia [CFFA], 2016) and Brazil (BR; Brazilian Cystic Fibrosis Study Group [GBEFC], 2016).
FIGURE 4.
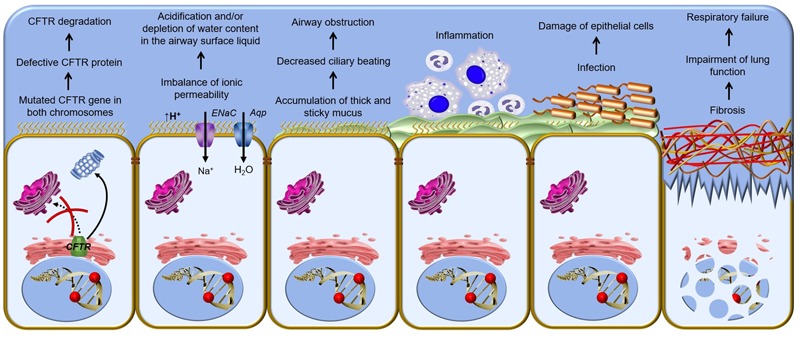
Pathophysiological cascade of respiratory disorder in cystic fibrosis – Cellular mechanism of cystic fibrosis begins with the defective CFTR (cystic fibrosis transmembrane conductance regulator) gene and shortage of CFTR channel at the plasma membrane. A vicious cycle of airways obstruction, inflammation and infection leads to epithelial damage, lung remodeling and end-stage lung disease. ENaC, epithelial Na+ channel; Aqp, aquaporin.
In addition to the substantial impact of CFTR dysfunction in the upper and lower respiratory tract, CF patients also experience clinical manifestations related to the reproductive, gastrointestinal and glandular systems (Table 2). Male infertility is present in 98% of cases, and about 85% of CF patients have pancreatic insufficiency, often associated to malnutrition.
Table 2.
Approximate age of onset of CF clinical manifestations and comorbidities.
| Upper and lower respiratory tract | Gastrointestinal and hepatobiliary systems | Endocrine and reproductive systems | Salt-wasting syndrome and others | |
|---|---|---|---|---|
| Babyhood and childhood | Chronic cough Sputum overproduction Tenacious mucus Airway obstruction Recurrent and persistent pneumonia or lung infections Bronchiectasis Nasal polyps/sinus disease |
Meconium ileus Steatorrhea Deficiency of fat-soluble vitamins Intussusception Recurrent pancreatitis Pancreatic insufficiency |
Absence of the vas deferens Impaired growth | Salty sweat Mucosal dehydration Hypochloremia Hyponatremia Hypokalemia Digital clubbing |
| Adolescence and adulthood | Atelectasis Impaired pulmonary function Hemoptysis Chronic infection with multidrug-resistant pathogens Allergic bronchopulmonary aspergillosis Pneumothorax Respiratory failure |
Biliary fibrosis/cirrhosis Hepatic steatosis Gastroesophageal reflux Distal intestinal obstruction syndrome Digestive tract cancer |
Delayed puberty Oligomenorrhea Reduced fertility in women Obstructive azoospermia in males CF-related diabetes mellitus Reduced bone mineral density/osteoporosis |
Arthritis/vasculitis Nephrolithiasis Chronic kidney disease Anxiety/depression |
Please note that the clinical signs and symptoms in an approximate age of onset do not exclude the possibility of these arose from early or late manner.
Salt loss syndrome also affects CF patients, since chloride reabsorption via CFTR in the sweat glands is compromised, increasing chloride elimination (>60 mmol.L-1), as well as of other ions. In fact, the iontophoretic sweat test is considered the ‘gold standard’ for the diagnosis of CF. It precedes only the confirmation of a diagnosis by genetic testing (Lyczak et al., 2002; Farrell et al., 2008; Castellani et al., 2016). In addition, sweat chloride measurement is a biomarker of CFTR activity and response for new treatments (Accurso et al., 2014; Collaco et al., 2016).
Life Expectancy in Cystic Fibrosis
Multifactorial improvements have increased the survival of CF patients (Figure 5): early diagnosis, genotype–phenotype detection, nutritional support, more efficient and effective pulmonary interventions, multidisciplinary professional monitoring, establishment of CF specialist centers and, most recently, the development of precision medicine (Farrell et al., 2008; Cohen-Cymberknoh et al., 2011; Smyth et al., 2014; Quon and Rowe, 2016).
FIGURE 5.
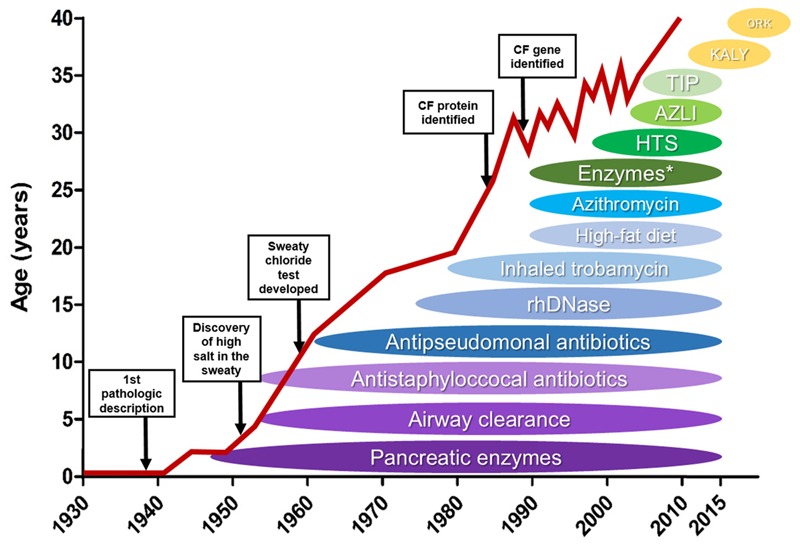
Effect of novel therapies on life expectancy of cystic fibrosis patients – Schematic illustration of how the discovery and introduction of novel cystic fibrosis (CF) treatments have influenced the patients’ survival over the decades. HTS: high throughput screening, AZLI, aztreonam for inhalation solution; TIP, tobramycin inhalation solution; KALY, KalydecoTM; ORK, OrkambiTM. ∗enteric-coated pancreatic enzymes. (Reproduced and adapted with permission of European Respiratory Society©: The European Lung White Book Respiratory Health and Disease in Europe, 2nd Ed. © 2013 European Respiratory Society, Sheffield, UK. Print ISBN: 978-1-84984-042-2, Online ISBN: 978-1-84984-043-9).
In the last decades, many countries have adopted newborn screening programs. Newborns are diagnosed through the measurement of immunoreactive trypsinogen. This early diagnosis and the confirmation of the disease through the sweat test are important for a better prognosis (Farrell et al., 2008; Djik and Fitzgerald, 2012; Castellani et al., 2016). Thereafter, identifying which are the mutations in the CFTR gene is crucial, since different ethnic or regional populations can have a different spectrum of CFTR variants, each leading to different degrees of disease severity (Moskowitz et al., 2008; Pique et al., 2016; Schrijver et al., 2016).
A multidisciplinary team of health professionals should perform periodic monitoring of the disease progression and make any necessary adjustments in treatments in order for patients to achieve the best clinical outcomes (Cohen-Cymberknoh et al., 2011; Smyth et al., 2014). CF patients show weight loss and impaired growth due to pancreatic insufficiency and intestinal malabsorption. Supplementations with pancreatic enzymes and fat-soluble vitamins have improved their nutritional status (Feranchak et al., 1999; Kalnins and Wilschanski, 2012). Lung injuries are the most common clinical characteristic due to mucociliary clearance impairment in the airways. Production of very viscous mucus impairs the ciliary beating, and treatments with mucolytic (e.g., dornase-alpha) (Fuchs et al., 1994) or hydrator (e.g., mannitol, hypertonic saline) (Elkins et al., 2006; Bilton et al., 2011) are helpful in eliminating the retained mucus. Antibiotic therapy is also essential, since the mucus becomes trapped in the respiratory tract, causing recurrent and persistent infections by a group of opportunistic pathogens: Burkholderia cepacia, Haemophilus influenzae, Pseudomonas aeruginosa (hallmark of CF), Staphylococcus aureus and Stenotrophomonas maltophilia (Lyczak et al., 2002; Worlitzsch et al., 2002; Bonestroo et al., 2010; Bhatt, 2013). It is noteworthy that during infection by P. aeruginosa, the bacterial population can segregate and evolve differently according to the lung microenvironment, leading to differences in the bacterial characteristics, such as nutritional requirements, defense and microbial resistance. This may explain, partly, why this pathogen is so prevalent and difficult to eradicate from CF patient lungs (Jorth et al., 2015; LaFayette et al., 2015).
Non-pharmacological treatments, such as aerobic exercise, physiotherapy, feeding and physiological supports, are also important to high-quality care and better outcomes (Cohen-Cymberknoh et al., 2011; Smyth et al., 2014). As the disease progresses, patients require continuous therapy with oxygen, and in the end-stage lung disease, the only alternative is lung transplantation, which still presents a high risk of cellular rejection (Adler et al., 2009; Calabrese et al., 2015).
Over the past decades, all the above-mentioned improvements have lengthened the life expectancy of CF patients. In 1960, CF patients only lived through childhood. Nowaday, they live to see their forties (Figure 5), and those born more recently are expected to make it to their fifties (Cohen-Cymberknoh et al., 2011; MacKenzie et al., 2014; Burgel et al., 2015). However, CF patients still present even more reduced life expectancy – between 20 and 30 years – in some regions of the world, including Brazil and the African continent (Marson et al., 2015; Stewart and Pepper, 2016).
Several new complications associated with increased survival (Table 2), which were rare or not previously observed, pose new challenges for CF scholars. CF-related diabetes, metabolic bone disease and multidrug-resistant pulmonary pathogens are some comorbidities in older CF patients (Plant et al., 2013). Although still uncommon, adult CF patients have a higher risk of gastrointestinal cancer compared to an age-matched non-CF population (Maisonneuve et al., 2013), which may be explained by the fact that CFTR is a tumor suppressor gene (Than et al., 2016). Another severe complication is the allergic bronchopulmonary aspergillosis, which is correlated to accelerated decline of lung function in adult CF patients (Armstead et al., 2014).
Despite the increase in life expectancy, CF patients still present a limited quality of life due to the high costs and the burden of treatments needed. In particular, CF patients living in developing countries tend to have worse clinical outcomes compared to those who live in developed countries, since lower-income countries have scarce financial resources to optimize therapies, including the application of precision medicine (Cohen-Cymberknoh et al., 2016). Precision medicine (or personalized treatment) is a new approach that takes into account the genetic variances of each individual. Therefore, CF patients with different CFTR mutations may require different treatments to quell their debilitating symptoms (Quon and Rowe, 2016). The knowledge obtained about the molecular scenario in which CFTR mutants are degraded has contributed to understanding the different defects caused by different mutants, and thus to developing drugs that rectify the primary defect of specific mutations.
A Proteostasis Network Engaged in CFTR Degradation
CFTR biogenesis is a cellular process that involves several steps: post-transcriptional splicing, protein translation, folding at the ER, glycosylation at the Golgi apparatus, trafficking to the apical membrane, endosomal recycling and retrieval. The cellular and transcellular protein trafficking involves multiple quality control systems to compensate the limited fidelity of each system. Therefore, a protein homeostasis (proteostasis) network carefully checks CFTR maturation pathway (Balch et al., 2011). Among the quality control systems, ubiquitin-proteasome pathway destroys the largest fraction of misfolded CFTR (Cheng et al., 1990; Jensen et al., 1995), and aggresomes degrade by autophagy CFTR molecules that proteasomes cannot degrade (Kawaguchi et al., 2003; Luciani et al., 2010). In addition, lysosomes eliminate non-native CFTR that escapes from ER-associated degradation (ERAD) (Sharma et al., 2004; Glozman et al., 2009; Okiyoneda et al., 2010). The mutant ΔF508 fails to achieve the native conformation and ERAD machinery arrests CFTR, thereby precluding the protein from being delivered to the PM (Cheng et al., 1990; Jensen et al., 1995).
CFTR folding is facilitated by many molecular chaperones and co-chaperones, which form a CFTR interactome; however, unstable and misfolded CFTR remains bound to chaperones, promoting premature degradation of CFTR (Balch et al., 2011; Amaral and Farinha, 2013; Kim et al., 2013; Pankow et al., 2015) (Figure 6). The heat shock protein (Hsp)27 (or HspB1) binds ΔF508 with small ubiquitin-like modifier (SUMO)-2, directing it to be degraded via SUMOylation (Ahner et al., 2013; Gong et al., 2016). Hsp27 also prevents protein aggregation during stress (Ahner et al., 2007). Hsp40 (or DnaJ) sequesters misfolded CFTR for ERAD and it works as a co-chaperone for Hsp70 (Meacham et al., 1999; Farinha et al., 2002; Kakoi et al., 2013). Hsp70 is a core ER chaperone and its prolonged association with CFTR mutant results in CFTR ubiquitination and degradation by the 26S proteasome (Yang et al., 1993; Jensen et al., 1995; Meacham et al., 2001; Sun et al., 2006). Hsp70 connects to heat shock cognate (Hsc)70, forming the first ER checkpoint that retains the mutant ΔF508. Hsc70 couples to its co-chaperone, the carboxyl terminus of Hsc70-interacting protein (CHIP), and leads CFTR mutant to degradation (Meacham et al., 1999, 2001; Farinha and Amaral, 2005). Hsp90 helps wild type (wt)-CFTR to achieve the complete folding, but it also forms a ‘chaperones trap’ with Hsp40/Hsp70 that targets ΔF508 for proteasome degradation (Wang et al., 2006; Ahner et al., 2007; Koulov et al., 2010; Coppinger et al., 2012). The ATPase activity of Hsp90 is regulated by its co-chaperone, the activator of 90kDa Hsp APTase homolog 1 (AHSA1 or Aha1) (Wang et al., 2006; Koulov et al., 2010). Calnexin is an ER transmembrane chaperone that acts as second checkpoint, assessing CFTR folding status and sending the misfolded protein for degradation (Farinha and Amaral, 2005; Okiyoneda et al., 2008).
FIGURE 6.
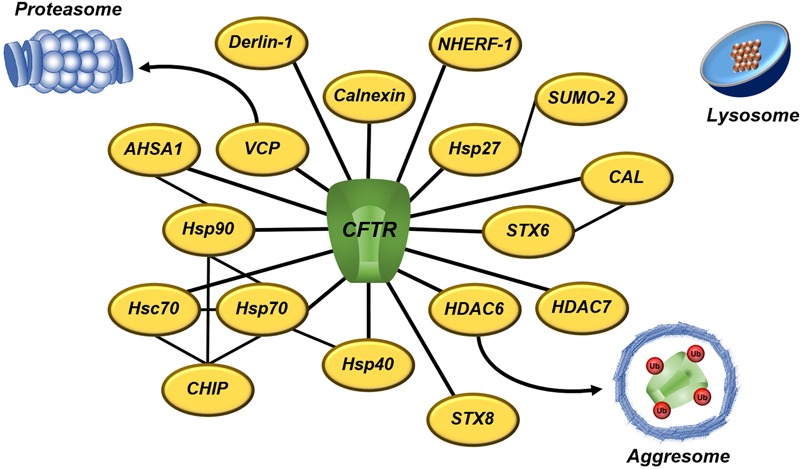
A subset of proteostasis network engaged to CFTR degradation – CFTR interactome involves several quality control proteins that directly or indirectly target CFTR to degradation. Proteasomes and aggresomes eliminate CFTR that fails in acquire the native conformation. Lysosomes degrade CFTR removed from the cell surface during the recycling. The black lines denoted the interaction between CFTR and proteostasis components. AHSA1, activator of 90 kDA Hsp ATPase homolog 1; CAL, CFTR-associated ligand; CHIP, carboxyl terminus of Hsc70-interacting protein; CFTR, cystic fibrosis transmembrane conductance regulator; HDAC, histone deacetylase; Hsc, heat-shock cognate; Hsp, heat-shock protein; NHERF, Na+/H+ exchanger regulatory factor; SUMO, small ubiquitin-like modifier; STX, syntaxin; Ub, ubiquitin; and VCP, vasolin-containing protein.
In addition to the molecular chaperones, inhibition of the histone deacetylase (HDAC) (Hutt et al., 2010; Pankow et al., 2015) or the vasolin-containing protein (VCP or p97) (Vij et al., 2006) rescues the trafficking and gating of ΔF508-CFTR. VCP (Boyault et al., 2006) and HDAC6 (Kawaguchi et al., 2003) translocate misfolded proteins to proteasomes and aggresomes, respectively. VCP is associated to ubiquitinated CFTR, whereas derlin-1 recognizes misfolded, non-ubiquitinated CFTR and initiates its dislocation and degradation in the early course of CFTR biogenesis (Sun et al., 2006). The CFTR-associated ligand (CAL) is a PDZ domain-containing protein, located primarily at Golgi apparatus, which modulates surface expression of CFTR (Cheng et al., 2002). CAL forms a complex with syntaxin (STX)6 that promotes CFTR degradation in lysosomes (Cheng et al., 2010; Cheng and Guggino, 2013). STX8 is an endosomal protein that impairs CFTR trafficking and inhibits its chloride channel activity by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) machinery (Bilan et al., 2004). Vasoactive intestinal peptide (VIP) is a neuropeptide that promotes interaction between CFTR and Na+/H+ exchanger regulatory factor 1 (NHERF-1) (Alshafie et al., 2014). NHERF-1 is a CFTR-binding protein that regulates CFTR distribution and function at the PM (Guerra et al., 2005; Kwon et al., 2007; Favia et al., 2010). Moreover, a system of peripheral proteins quality control (PPQC) removes CFTR from the PM if that is recognized as improperly folded (Okiyoneda et al., 2010).
In healthy conditions, the proteostasis network maintains a healthful proteome by integrating transcription, translation, folding and trafficking systems. However, a decline in the cellular proteostasis capacity occurs with aging, as well as protein misfolding/aggregation disorders cause an imbalance in the expression and/or binding of proteostasis components with client proteins, which accentuates the importance of a healthy quality control system (Balch et al., 2011; Kim et al., 2013; Villella et al., 2013). CFTR protein must fold into well-defined three-dimensional structure to attain stability and functionality (Balch et al., 2011; Kim et al., 2013). The protein lack or its incorrect folding prevents CFTR from performing its normal function and cells may respond to that by maintaining an activated heat shock response and leading to a maladaptive stress response. This effort to neutralize misfolding protein accumulation causes an additional stress to cells that may result in the exacerbation of disease symptoms. These findings were observed in cell lines and primary epithelium from CF and other misfolding protein diseases: alpha-1-antitrypsin, Alzheimer’s disease and Niemann-Pick type C1 disease (Roth et al., 2014). Another important determinant in inherited disorders is that the disease severity depends of the genetic background of each individual and not solely on the defect of a particular gene (Vu et al., 2015). CFTR gene is located at the chr7q31.2 and a genome-wide association analysis identified five loci that display significant relevance in the variable manifestation and progression of lung disease in CF (chr3q29, chr5p15.3, chr6p21.3, chr11p12-p13 and chrXq22-q23) (Corvol et al., 2015; Dang et al., 2016).
Classes of CFTR Mutations and CFTR Modulators
The ΔF508 is the most prevalent CFTR mutation with about 60% of CF patients’ chromosomes worldwide presenting this mutant (Bobadilla et al., 2002; Sosnay et al., 2013) (Figure 3). The remaining 40% present other CFTR mutations and there have been nearby 2,000 mutations reported in the Cystic Fibrosis Mutation Database (CFTR1 database)1. CFTR mutants can reduce protein expression, function, stability or a combination of these, and to help in understanding the nature of such gene and protein variability, CFTR defects have been classified into six classes (Amaral and Farinha, 2013; Quon and Rowe, 2016; Veit et al., 2016a) (Figure 7).
FIGURE 7.
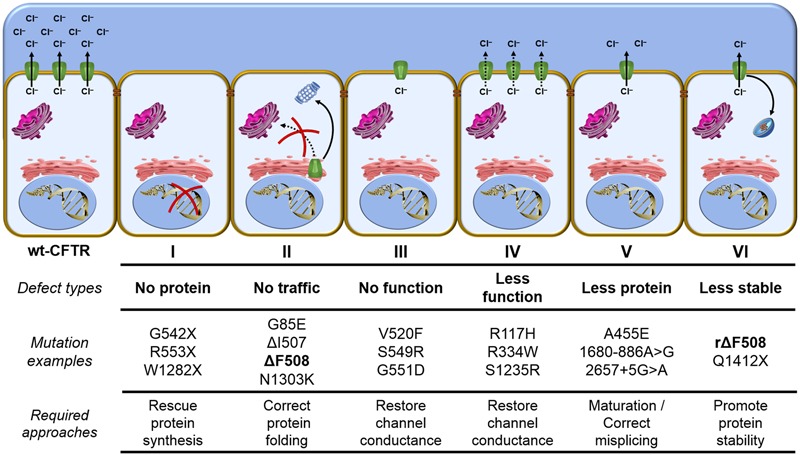
Classes of CFTR mutations – Distribution of CFTR mutations into six functional classes according to the primary molecular defect: Class I mutants are no protein synthesis, since the presence of premature stop codons (class Ia) or frameshifts for deletions or insertions (class Ib) preclude translation of full-length CFTR. Class II mutants are impaired trafficking protein, since CFTR fails to acquire complete folding and ER-associated degradation (ERAD) machinery eliminate the protein. Class III mutants are defective channel gating, since CFTR reach the cell surface, but it does not exhibit channel gating due to diminished ATP binding and hydrolysis. Class IV mutants are less functional proteins, since channel amount that achieve the plasma membrane could be similar to wt-CFTR, but it presents reduced chloride conductance. Class V mutants are less protein maturation caused by amino acid substitution or alternative splicing, since the protein amount that reaches the cell surface is reduced and it also leads to loss of chloride transport due to reduction in the quantity of CFTR channels. Class VI mutants are less stable protein, since CFTR at the plasma membrane is removed during the recycling and it is sent for lysosome degradation. wt, wild type; CFTR, cystic fibrosis transmembrane conductance regulator; rΔF508, rescued ΔF508 by low-temperature incubation; and ER, endoplasmic reticulum.
Distribution of CFTR mutants into classes may contribute to the application of precision medicine, since similar strategies might rescue CFTR from similar defects. However, the classification has a few caveats: (1) Numerous mutations have not been characterized, with respect to which group they should be allocated. The mutations’ characteristics for a subset of known mutations can be found at the Clinical and Functional Translation of CFTR (CFTR2 database)2. (2) At first glance, CFTR mutations in the same group show similar characteristics, but they may respond differently to the same treatment. (3) Several mutations (e.g., ΔF508) present pleiotropic defects, which means they could fit in more than one class. The major characteristic of ΔF508 is the incomplete folding of the protein caused by NBD1 instability (class II) (Cheng et al., 1990; Jensen et al., 1995; Lukacs and Verkman, 2012), but this mutation also affects channel gating (class III) (Dalemans et al., 1991; Serohijos et al., 2008; Mendoza et al., 2012) and cell surface residence time (class VI) (Sharma et al., 2004; Swiatecka-Urban et al., 2005; Okiyoneda et al., 2010). Based on this limitation, new classifications are under debate, including a scheme that would be composed of the traditional classes I, II, III/IV, V, VI and their 26 combinations, totaling 31 CFTR mutations classes (Veit et al., 2016a).
Monotherapies could be efficient in overcoming the molecular defect of some CFTR mutations; however, given the complexity of pleiotropic CFTR variants, combination of treatments may be required to rectify their defects and thus achieve therapeutic levels in the patients (Amaral and Farinha, 2013; Quon and Rowe, 2016; Veit et al., 2016a). Libraries of compounds have been screened by high-throughput screening (HTS) to identify more efficacious and non-cytotoxic drugs for application of precision medicine. Among these new pharmacological treatments, ‘CFTR modulators’ are small molecules that target specific defects caused by mutations in the CFTR gene and they are classified into five main groups: read-through agents, correctors, potentiators, stabilizers and amplifiers.
Rescuing the Protein Synthesis
Read-through agents could benefit CF patients bearing class I mutations, since the presence of a premature stop codon (class Ia) precludes protein synthesis of full-length CFTR (Quon and Rowe, 2016). Class Ia mutations represent about 9% of the mutants causing CF and they are present in more than 50% of Israeli CF patients (Kerem et al., 1997; Bobadilla et al., 2002). In this line, ribosomal ‘over-reading’ of a premature stop codon should permit the continuing translation to the normal end of the transcript. This read-through effect was first observed with aminoglycoside antibiotics, such as gentamicin and tobramycin, which are commonly used for eradication of P. aeruginosa. Both antibiotics promoted expression of full-length CFTR at the PM and restored partially its chloride secretion in cell lines and transgenic mice (Howard et al., 1996; Du et al., 2002; Wilschanski et al., 2003). Despite these preclinical results, CF patients bearing nonsense mutations did not present CFTR activity after nasal application of aminoglycosides (Clancy et al., 2007). Furthermore, high systemic levels or long-term use of gentamycin has potential toxic effects in CF patients (Prayle et al., 2010).
Ataluren (formerly PTC124) is among the new drugs discovered by HTS. This drug suppressed the human G542X nonsense mutant in transgenic mice, restoring CFTR functional expression at the PM (Du et al., 2008). Thereafter, this drug resulted in some improvement in forced expiratory volume in 1 s (FEV1) of CF patients bearing nonsense mutations in phase II trials (Kerem et al., 2008; Sermet-Gaudelus et al., 2010; Wilschanski et al., 2011). However, these findings were inconclusive in the first long-term phase III trial, with only a subgroup of patients who were not receiving chronic inhaled aminoglycosides presenting a slight effect in FEV1 (Kerem et al., 2014). Ataluren has also shown limited premature stop codon suppression in CF rectal organoids (Zomer-van Ommen et al., 2016). Moreover, other read-through agents have shown promising results in preclinical experiments, including synthetic aminoglycosides (Rowe et al., 2011; Xue et al., 2014), ataluren derivatives (Pibiri et al., 2015, 2016), and the US Food and Drug Administration (FDA)-approved herbal agent, escin (Mutyam et al., 2016).
For other class I mutations (class Ib), such as frameshifts caused by small deletions or insertions during protein synthesis, very little has been achieved in treatments yet.
Rescuing the Protein Folding and Trafficking
Correctors are small-molecules that enhance the conformational stability of CFTR, resulting in greater efficacy of protein folding and rescuing the trafficking of the mature CFTR to the PM (Pedemonte et al., 2005; Okiyoneda et al., 2013; Lopes-Pacheco et al., 2015; Quon and Rowe, 2016). CF patients bearing class II mutations, including ΔF508, could benefit from correctors treatment, since these CFTR mutants fail to reach complete folding and the ER machinery targets the protein to be degraded (Van Goor et al., 2006; Atawade et al., 2015; Rapino et al., 2015; Lopes-Pacheco et al., 2016).
New treatments may target the defective CFTR structure directly by binding to the mutated protein (pharmacological chaperone) and/or indirectly by modulating CFTR interactome (proteostasis regulator). Some reports have shown that correctors act either as pharmacological chaperones (Wang et al., 2007a; Sampson et al., 2011; Eckford et al., 2014; Sinha et al., 2015) or as proteostasis regulators (Hegde et al., 2015; Lopes-Pacheco et al., 2015, 2016; Rapino et al., 2015). Based on the possible mechanism of action as pharmacological chaperones, correctors have been classified into three groups: 1) correctors that stabilize the interactions between NBD1 and intracellular loops 1 and 4 (e.g., C3, C18 and VX-809); 2) correctors that restore NBD2 stability and its interfaces with other CFTR domains (e.g., C4); and 3) correctors that directly stabilize NBD1 (Okiyoneda et al., 2013).
Lumacaftor (formerly VX-809) restored ΔF508-CFTR expression and function in human bronchial epithelial (HBE) cells (Van Goor et al., 2011). The higher efficacy of lumacaftor, compared to other correctors (C3 and C4), seems to be due to its effect on the early CFTR synthesis (Farinha et al., 2015). Despite progress in vitro, lumacaftor treatment by itself showed a significant decrease only in sweat chloride levels and no improvements in FEV1 in a phase II trial with ΔF508-homozygous patients (Clancy et al., 2012). Lumacaftor presented the most variable effects in primary HBE cells and rectal organoids bearing ΔF508 in only one allele or other CFTR mutants in both alleles, with some mutants being ‘un-rescuable’ (e.g., N1303K) (Atawade et al., 2015; Dekkers et al., 2016a,b). Furthermore, a synonymous mutation changing ATC to ATT at position 507 (I507) alters mRNA and protein structure (Lazrak et al., 2013), consequently, affecting the efficacy of correctors to rescue ΔF508-CFTR (Bali et al., 2016a,b). Aiming at greater effects, combinations of correctors have been evaluated and have been shown to enhance the rescue of CFTR bearing ΔF508, as well as other class II mutants, compared to monotherapies (Okiyoneda et al., 2013; Phuan et al., 2014; Hegde et al., 2015; Lopes-Pacheco et al., 2015, 2016; Rapino et al., 2015). Some correctors could also increase CFTR maturation of class V mutants caused by amino acid substitution, such as A455E (Dekkers et al., 2016a,b; Lopes-Pacheco et al., 2016). In addition, VIP rescued functional expression of ΔF508-CFTR by stimulating both PKA- and PKC-dependent pathways in nasal and bronchial epithelial cells (Rafferty et al., 2009; Alcolado et al., 2011). Many newly discovered correctors are being investigated, including VX-661 that presented more a favorable pharmacokinetic profile than lumacaftor (Cholon et al., 2014; Veit et al., 2014; Phuan et al., 2015) and is in phase III trials to ΔF508-homozygous and -heterozygous patients (NCT02347657, NCT02392234 and NCT02412111).
Besides the correctors, drugs that modulate proteostasis have been evaluated to restore CFTR functional expression at the PM, since wt-CFTR and ΔF508 present a rather different interactome during their processing and trafficking (Pankow et al., 2015). Cysteamine, a proteostasis regulator approved by the FDA for nephropathic cystinosis, has shown promise for treating CF. Cysteamine (reduced form of cystamine), in association with epigallocatechin gallate, restored beclin 1-dependent autophagy protein levels and depleted sequestrosome 1/p62, thereby correcting autophagy flux, and rescuing CFTR trafficking, function and stability at the PM. These findings were observed in lungs from CftrΔF508 mice, CFBE41o- cell line expressing ΔF508-CFTR, and primary nasal epithelial cells freshly harvested from ΔF508-homozygous patients (Luciani et al., 2012; Villella et al., 2013; De Stefano et al., 2014). Recently, a combination of cysteamine and epigallocatechin gallate decreased sweat chloride levels, as well as inflammatory biomarkers levels in the sputum, and tended to improve FEV1 in ΔF508-homozygous and -heterozygous (with a class II mutant in the second allele) patients in a phase II trial (Tosco et al., 2016). Several other proteostasis regulators have been investigated to rescue CFTR, including sildenafil analogs (Robert et al., 2008), oubain (Zhang et al., 2012), roscovitine (Norez et al., 2014), suberoylanilide hydroxamic acid (Hutt et al., 2010; Pankow et al., 2015) and latonduine analogs (Carlile et al., 2016). Silencing of RPL12, a ribosomal stalk protein, also rescued ΔF508-CFTR and presented a synergistic effect with lumacaftor, restoring the mutant function to about 50% of the wt-CFTR in primary HBE cells bearing ΔF508 in both alleles (Veit et al., 2016b).
Restoring the Channel Conductance
Potentiators are drugs that increase channel open probability, improving CFTR channel activity. Potentiators could benefit CF patients bearing class III and IV mutations, since CFTR is present at the PM, but it exhibits no gating or reduced activity (Van Goor et al., 2006, 2009, 2014; Eckford et al., 2012). Furthermore, patients bearing class I or II mutations for which protein synthesis or trafficking were rescued, but not proper channel activity, may benefit from this additional approach.
Ivacaftor (formerly VX-770) rescued CFTR channel gating in HBE cells bearing G551D mutation (Van Goor et al., 2009) through a nonconventional ATP-independent mechanism (Eckford et al., 2012). Ivacaftor also potentiated channel activity of CFTR bearing other class III or IV mutants in Fisher rat thyroid cells (Yu et al., 2012; Van Goor et al., 2014) and rescued forskolin-induced swelling in rectal organoids bearing CFTR mutants with residual function (Dekkers et al., 2016a). A subsequent series of phase II and III clinical trials showed that ivacaftor reduced sweat chloride levels and improved FEV1 in CF patients bearing G551D (Accurso et al., 2010; Ramsey et al., 2011; Davies et al., 2013a,b; McKone et al., 2014) or one of other eight class III mutants (G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, and G1349D) (De Boeck et al., 2014) in at least one allele. For R117H, a residual function mutant, ivacaftor decreased sweat chloride levels of all patients, but only individuals older than 18 years and with a polythymidine tract variant of 5T presented improvements in FEV1 (Carter et al., 2015; Moss et al., 2015; Ronan et al., 2015). Currently, ivacaftor is available for CF patients bearing the aforementioned mutations.
Monotherapy with ivacaftor (Flume et al., 2012) or lumacaftor (Clancy et al., 2012) was unsuccessful in improving FEV1 of ΔF508-homozygous patients in phase II trials. A closer investigation of the pleiotropic defects caused by ΔF508 in CFTR reveals that combinations of drugs with different mechanism of actions will be required to obtain more efficient rescue of the mutated protein that may ultimately result in therapeutic levels in the patients (Okiyoneda et al., 2013; Phuan et al., 2014; Lopes-Pacheco et al., 2015; Veit et al., 2016a). Toward this goal, phase II and III trials tested the effects of co-administration of lumacaftor/ivacaftor. This approach reduced pulmonary exacerbations, slightly decreased sweat chloride levels and induced significant, but modest, improvements in FEV1 of ΔF508-homozygous patients (Boyle et al., 2014; Wainwright et al., 2015; Elborn et al., 2016). Recently, co-administration of lumacaftor/ivacaftor was licensed for ΔF508-homozygous patients.
Some flavonoids also increase channel open probability of CFTR mutants (Galietta et al., 2001; Pyle et al., 2010; Yu et al., 2011). In particular, genistein and curcumin have shown synergy with lumacaftor to enhance forskolin-induced swelling in rectal organoids bearing CFTR mutants ΔF508, G551D and S1251N (Dekkers et al., 2016c). Ivacaftor has shown clinical benefits when administered by itself, but its effects could be even greater when used with other compounds for CFTR bearing G551D or R117H (Gentzsch et al., 2016; Lin et al., 2016; Yu et al., 2016). In addition, new potentiators are under investigation to restore channel activity of CFTR mutants, including QBW251 (NCT02190604) and GLPG1837 (NCT02690519 and NCT02707562), as well as dual activity compounds that act as both correctors and potentiators, such as aminoarylthiazoles (Pedemonte et al., 2011; Pesce et al., 2015) and rattlesnake phospholipase A2 (Faure et al., 2016).
Stabilizing the Protein at the Cell Surface
Stabilizers are agents that anchor CFTR channel at the PM and decrease protein degradation rate, thereby correcting the instability of class VI mutants. Low-temperature incubation of cells bearing ΔF508 rescue CFTR to the PM (rΔF508) (Sharma et al., 2001); however, the protein still presents reduced half-life due to both increased endocytosis (Swiatecka-Urban et al., 2005) and decreased recycling (Sharma et al., 2004). Lumacaftor also did not confer long-term stability of wt-CFTR for the mutant ΔF508 (He et al., 2013).
New treatments must rectify the intrinsic protein instability to rescue the steady state levels and augment CFTR residence time at the PM. In this line, hepatocyte growth factor (HGF) activated Rac1 signaling and promoted ΔF508 stabilization favoring interaction of CFTR and NHERF-1 (Moniz et al., 2013). Lumacaftor also promoted interaction of CFTR and NHERF-1 (Arora et al., 2014), but co-administration of HGF/lumacaftor further increased the rescue of ΔF508-CFTR and enhanced protein anchoring at the PM, compared to lumacaftor alone (Loureiro et al., 2015). Administration of VIP (Rafferty et al., 2009; Alshafie et al., 2014) or activation of EPAC1, an exchange protein directly activated by cAMP (Lobo et al., 2016), also stabilized CFTR at the PM by promoting its interaction with NHERF-1 and by decreasing its endocytosis rate.
In addition, inhibition of S-nitrosoglutathione reductase enhanced ΔF508 maturation and stability by preventing interaction of CFTR with Hsp70/Hsp90 organizing protein (Marozkina et al., 2010; Zaman et al., 2016). Cavosonstat (formerly N91115) increased S-nitrosoglutathione levels in pre-clinical studies and it is the first CFTR stabilizer in phase II trials being evaluated for ΔF508-homozygous patients using the combination lumacaftor/ivacaftor (NCT02589236) or for those patients bearing gating mutants and receiving ivacaftor (NCT02724527).
Correcting the Splicing
Antisense oligonucleotides-based therapy is an emerging approach to correct class V mutations caused by alternative splicing that generate aberrant mRNA variants. About 11% of mutations-causing CF occur by incorrect splicing and this approach has shown to modulate the splicing and restore normal full-length CFTR transcript, as well as rescue functional CFTR protein (Bonini et al., 2015; Igreja et al., 2016).
A synthetic RNA oligonucleotide, QR-010, is being evaluated in two phase I trials to assess: (1) the effects of inhaled single and multiple doses in ΔF508-homozygous patients (NCT02532764), and (2) the effects of intranasal administration in ΔF508-homozygous and -heterozygous patients (NCT02564354).
Precision Medicine: Breakthroughs and Challenges to Treating all CF Patients
From the first pathological description of the disease until the discovery of the CFTR gene and its correlation with CF, therapeutic interventions were only aimed at reducing clinical symptoms and end-organ complications. Nowadays, based on the accumulated knowledge about CFTR protein (synthesis, folding, trafficking and degradation), many studies have sought more specific treatments to restore expression, function and stability of CFTR mutants, thereby overcoming the primary molecular defect that causes CF. As such, CFTR modulators provided new perspectives and advances in the treatment of patients bearing common and rare CFTR mutations.
Many clinical trials have completed the tests of safety and efficacy of CFTR modulators (Table 3). The first breakthrough in personalizing CF treatments came with the approval of ivacaftor (KalydecoTM from Vertex Pharmaceuticals) for patients bearing G551D-CFTR in at least one allele, initially licensed by the FDA and later by the European Medicines Agency (EMA). Clinical studies showed that ivacaftor treatment improves FEV1 and almost normalizes sweat chloride levels (Accurso et al., 2010; Ramsey et al., 2011; Davies et al., 2013a,b), as well as reduces frequency of infections caused by P. aeruginosa (Rowe et al., 2014), enhances nutritional status (Borowitz et al., 2016), insulin secretion (Bellin et al., 2013) and quality of life (Quittner et al., 2015). Acute administration of ivacaftor also corrects smooth muscle abnormalities, improving airway distensibility and vascular tone (Adam et al., 2016). Clinical benefits obtained by ivacaftor treatment have proven durable effects, with no new safety concerns (Sawicki et al., 2015). Ivacaftor was later licensed for patients bearing one of the other eight gating mutations (De Boeck et al., 2014) and a mutant with residual function in at least one allele (Moss et al., 2015), increasing to 5–7% the number of individuals worldwide who may benefit from this pharmaceutical treatment. Despite these optimistic results, patients using ivacaftor should continue with conventional therapies, which have also been optimized, to prevent pulmonary exacerbations and complications (Smyth et al., 2014).
Table 3.
Completed clinical trials of CFTR modulators in CF patients.
| ClinicalTrials.gov ID (formerly name) | Phase | Subjects | Age (years) | Drug(s) | Follow up | Reference(s) |
|---|---|---|---|---|---|---|
| NCT00237380 | 2 | Nonsense mutationsa,b | ≥18 | Ataluren | 56 days | Kerem et al., 2008 |
| NCT00351078 | 2 | Nonsense mutationsa,b | ≥18 | Ataluren | 112 days | Wilschanski et al., 2011 |
| NCT00457821 | 2 | G551D-homozygous and -heterozygous | ≥18 | Ivacaftor | 28 days | Accurso et al., 2010, 2014 |
| NCT00458341 | 2 | Nonsense mutationsa,c,d | 6–18 | Ataluren | 28 days | Sermet-Gaudelus et al., 2010 |
| NCT00803205 | 3 | Nonsense mutationsa,c | ≥6 | Ataluren | 48 weeks | Kerem et al., 2014 |
| NCT00865904 | 2 | ΔF508-homozygous | ≥18 | Lumacaftor | 28 days | Clancy et al., 2012 |
| NCT00909532 (STRIVE) | 3 | G551D-homozygous and -heterozygous | ≥12 | Ivacaftor | 48 weeks | Ramsey et al., 2011; Quittner et al., 2015 |
| NCT00953706 (DISCOVER) | 2 | ΔF508-homozygous | ≥12 | Ivacaftor | 16 weeks | Flume et al., 2012 |
| NCT01117012 (PERSIST) | 3 | G551D-homozygous and -heterozygous | ≥6 | Ivacaftor | 96 weeks | McKone et al., 2014 |
| NCT01225211 | 2 | ΔF508-homozygous and -heterozygous | ≥18 | Lumacaftor and Ivacaftor | 56 days | Boyle et al., 2014 |
| NCT01262352 (ENVISION) | 2 | G551D-homozygous and -heterozygous | ≥6 | Ivacaftor | 48 weeks | Davies et al., 2013a,b |
| NCT01521338 (GOAL) | 4 | G551D-homozygous and -heterozygous | ≥6 | Ivacaftor | 6 months | Rowe et al., 2014; O’Connor and Seegmiller, 2016 |
| NCT01531673 | 2 | ΔF508-homozygous and -heterozygous | ≥12 | VX-661 and/or Ivacaftor | 28 days | ∗∗∗ |
| NCT01614457 (KONDUCT) | 3 | R117H-homozygous and -heterozygous | ≥6 | Ivacaftor | 24 weeks | Moss et al., 2015 |
| NCT01614470 (KONNECTION) | 3 | Non-G551D gating mutations in at least one allelee | ≥6 | Ivacaftor | 24 weeks | De Boeck et al., 2014 |
| NCT01685801 | 2 | R117H and/or CFTR mutations with residual function in at least one allele b,f, G551D and/or other gating mutations in at least one allelee | ≥12 | Ivacaftor | 24 weeks | ∗∗∗ |
| NCT01705045 (KIWI) | 3 | G551D-homozygous and heterozygous | 2–5 | Ivacaftor | 24 weeks | Davies et al., 2016 |
| NCT01707290 | 3 | Non-G551D gating or residual function mutations in at least one alleleb,e,f |
≥6 | Ivacaftor | 24 weeks | ∗∗∗ |
| NCT01807923 (TRAFFIC) | 3 | ΔF508-homozygous | ≥12 | Lumacaftor and Ivacaftor | 24 weeks | Wainwright et al., 2015; Elborn et al., 2016 |
| NCT01807949 (TRANSPORT) | 3 | ΔF508-homozygous | ≥12 | Lumacaftor and Ivacaftor | 24 weeks | Wainwright et al., 2015; Elborn et al., 2016 |
| NCT01897233 | 3 | ΔF508-homozygous | 6–11 | Lumacaftor and Ivacaftor | 24 weeks | ∗∗∗ |
| NCT01931839 | 3 | ΔF508-homozygous | ≥12 | Lumacaftor and Ivacaftor | 96 weeks | ∗∗∗ |
aG542X, W1282X; b3849 + 10 kbC→T; CR553X, R1162X; dQ492X, E1104X, W846X, W882X, Q1313X; eG178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P, G1349D; fR117H, E56K, P67L, D110E, D110H, R117C, R347H, R352Q, A455E, D579G, S945L, L206W, R1070W, F1074L, D1152H, S1235R, D1270N, 2789 + 5G→A, 3272-26A→G, 711 + 5G→A, 3120G→A, 1811 + 1.6 kbA- > G, 711 + 3A→G, 1898 + 3A→G, 1898 + lG→A, 1717-lG→A, 1717-8G→A, 1342-2A→C, 405 + 3A→C, 1716G/A 1811 + lG→C, 1898 + 5G→T, 3850-3T→G, IVS14b + 5G→A, 1898 + lG→T, 4005 + 2T→C, 621 + 3A→G, 621 + lG→T. ∗∗∗Manuscript not available yet.
Ataluren (TranslarnaTM from PTC Therapeutics) was approved by the EMA for individuals who have Duchenne muscular dystrophy caused by nonsense mutation. After the inconclusive results from the first phase III trial, a second one is ongoing to evaluate the effects of ataluren in CF patients bearing nonsense mutations and not receiving inhaled aminoglycosides (NCT02139306).
Recently, the FDA and the EMA licensed the combination lumacaftor/ivacaftor (OrkambiTM from Vertex Pharmaceuticals) for ΔF508-homozygous patients, adding a new pharmaceutical treatment to 40–45% of CF patients worldwide. Results from a phase II trial evaluating this combination in ΔF508-homozygous patients (Boyle et al., 2014) encouraged the pursuance of two long-term phase III trials (24 weeks) involving more than 1,100 people. Co-administration of lumacaftor/ivacaftor proved clinical effectiveness, despite fairly small improvement in FEV1 (Wainwright et al., 2015; Elborn et al., 2016). A longer-term (96 weeks) phase III trial showed that co-administration of lumacaftor/ivacaftor presents sustained benefit with decreased pulmonary exacerbations, reduced rate of lung function decline and improved nutritional status of ΔF508-homozygous patients (Konstan et al., 2016). Although it is still a glimmer of light at the end of the tunnel, this combination provides proof of concept that pleiotropic CFTR mutants can be rescued at therapeutic levels, representing a new hope to restore a healthy life to patients.
Intriguingly, studies using cell lines bearing ΔF508-CFTR showed that: (1) Chronic ivacaftor exposure (>1 μM) reduced CFTR correction obtained by lumacaftor treatment (Cholon et al., 2014; Veit et al., 2014); however, exposure to a lower concentration (<1 μM) of ivacaftor did not inhibit the functional correction obtained by lumacaftor (Matthes et al., 2016). Therefore, co-administration of lumacaftor/ivacaftor may present a dose-dependent inhibitory effect. Addition of C4 also reversed the negative effects of ivacaftor on lumacaftor-corrected CFTR (Bali et al., 2016b). (2) P. aeruginosa infection reduces CFTR chloride secretion stimulated by lumacaftor and/or ivacaftor treatments (Stanton et al., 2015). (3) In cells previously treated with lumacaftor, the PPQC system still recognizes the CFTR delivered at the PM as improperly folded and removes the protein from the cell surface (Loureiro et al., 2015). Taken together, the experimental results indeed may explain the modest findings in the clinical trials and highlight the importance of better evaluating drug-drug and drug-protein interactions for future drug combinations.
The introduction of KalydecoTM and OrkambiTM to the market add feasible treatment for about 50% of all CF patients worldwide (Table 4). Unfortunately, there is still an unmet need for the other half of the CF population, who are ΔF508-heterozygous and bear a wide range of other variants (so-called orphan mutants). The patient registries and the clinical studies network have optimized the clinical phase of drug development due to heterogeneous distribution of CFTR variants (De Boeck et al., 2011; Rowe et al., 2012). Randomized controlled trials are suitable for common mutations, but many CFTR mutants are rare or even unique, requiring different approaches. As such, modified single-patient (‘N-of-1’) is an alternative trial design in which single patients serve as their own control by measuring outcome parameters after multiple cycles of on-off treatment (Zucker et al., 2010; Duan et al., 2013).
Table 4.
Summary of KalydecoTM and OrkambiTM approval for CF patients’ treatment.
| Pharmaceutical treatment | CFTR mutations | Jurisdiction approved | Age group licensed |
|---|---|---|---|
| KalydecoTM | G551D∗ | United States, Europe and Canada | >2 years |
| Australia and New Zealand | >6 years | ||
| G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, G1349D∗ | United States, Europe and Canada | >2 years | |
| Australia | >6 years | ||
| R117H∗ | United States | >2 years | |
| Europe and Canada | >18 years | ||
| OrkambiTM | ΔF508-homozygous | United States, Europe and Canada | >12 years |
∗Mutation in at least one allele.
Rectal organoids have also shown to be a good model for identifying drug-responsive individuals with rare CFTR genotypes (Dekkers et al., 2016a; Vijftigschild et al., 2016). In addition, both KalydecoTM and OrkambiTM are in ongoing clinical trials to: (1) evaluate the longer-term safety and efficacy of the treatments; (2) increase the panel of mutations that may benefit from these drugs; and (3) evaluate the safety and efficacy in younger patients. Since the progressive damage caused by CF starts during babyhood, earlier treatments are critical to offering the best chances of improving long-term outcomes.
In an attempt to achieve greater clinical outcomes, two new classes of CFTR modulators are under investigation in association with OrkambiTM: (1) PTI-428 is an amplifier that doubled the activity of the combination lumacaftor/ivacaftor in HBE cells bearing ΔF508-CFTR in both alleles (Proteostasis Therapeutics, 2016); and (2) cavosonstat is a stabilizer that improved ΔF508-CFTR stability at the PM after co-administration of lumacaftor/ivacaftor (Nivalis Therapeutics, 2016). Moreover, other correctors and potentiators are advancing in triple combination for treatment of ΔF508-homozygous and -heterozygous patients: GLPG1837/GLPG2222/GLPG2665 (Galapagos, 2015), VX-152/VX-661/ivacaftor and VX-440/VX-661/ivacaftor (Vertex Pharmaceuticals, 2015).
An important limitation that has to be addressed regarding these new pharmaceutical treatments is the high cost (over US$250,000 per patient per year), which renders difficult their acquisition for CF patients living in low-income countries. Investments in the identification of drugs already available and able to overcome the primary molecular defect of CF could be another way, cheaper and fast, to optimize treatments.
Soon after the CFTR gene was discovered and cloned (Kerem et al., 1989; Riordan et al., 1989), gene therapy was proposed to cure lung disease in CF. The idea of introducing a copy of wt-CFTR in airway cells to re-establish the production of functional protein seemed graceful and ‘simple’. Although CF is a ‘simple’ monogenic disorder, most approaches (viral and non-viral) failed to cross the trapped mucus, thereby resulting in inefficient transduction of CFTR gene, and some of them promoting immune response activation (Di Gioia et al., 2015; Quon and Rowe, 2016). Nevertheless, researchers have sought more efficient gene delivery systems. Poly (β-amino esters)-based biodegradable polymers were able to deliver a plasmid that encodes full-length CFTR in murine lungs and CFBE41o- cell lines expressing wt-CFTR or ΔF508 (Suk et al., 2014), as well as to penetrate in freshly expectorated mucus from CF patients (Mastorakos et al., 2015). Furthermore, repeated nebulization of pGM169/GL67A, a gene-liposome complex, showed feasible, safe and reproducible results in murine lungs (Alton et al., 2014), as well as stabilized lung function of CF patients, thereby presenting a significant, albeit modest, benefit in FEV1 in a phase II trial (NCT01621867) (Alton et al., 2015). These systems may also be an interesting approach to delivery of CFTR modulators, bringing some benefits in usefully exploited patients’ treatment: (1) non-invasive administration route by inhalation/nebulization; (2) delivering directly to a specific lung region or defined cell type; (3) reduced systemic side effects and no serum proteins sequestered; and (4) sustained release, which decrease times and dose of therapies to maintain the beneficial effects (Vij et al., 2010; Suk et al., 2014; Alton et al., 2015; Kim et al., 2015).
CFTR Modulators Rescuing Mutants in Other ABC Transporters
Plasma membrane proteins belong to some of the largest families, encoding ion channels, transporters, aquaporins and ATP-powered pumps. Forty-nine proteins constitute the ABC transporters family in the human genome (Loo and Clarke, 2008; Vasiliou et al., 2009) and among these proteins, CFTR is unique in possessing a RD and in functioning as a chloride channel (Winter and Welsh, 1997; Gadsby et al., 2006; Serohijos et al., 2008). Mutations in ABC transporters have been linked to diseases and, since these transporters share a similar domain organization and structure, some CFTR modulators have been evaluated to rescue protein trafficking and function of other ABC transporters. Here are some examples: ABCA4 (Sabirzhanova et al., 2015), ABCB1 (or P-glycoprotein) (Wang et al., 2007b), ABCC6 (or multidrug resistance-associated protein 6) (Zhou et al., 2013), ABCC8 (or SUR1) (Sampson et al., 2013), and ABCG2 (Woodward et al., 2013). These findings show that CFTR modulators could be a promising treatment for several diseases caused by mutations in other ABC transporters.
Conclusion
Since the discovery that CFTR loss-of-activity causes CF, the biological understanding of CFTR processing has advanced steadily and opened new perspectives for more sophisticated treatments, which act directly on the molecular defect that causes the disease. The presence of CFTR modulators in the market has affected positively the clinical outcomes of CF patients, representing a new dawn in their lives. Although there is still a long way to go in completely restoring a healthy life for all CF patients, research from the bench to the clinic is moving forward at an accelerated pace toward precision medicines, which would enable “the highest attainable standard of health,” as enshrined in the constitution of the World Health Organization.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Solon Leite (Pharm.D.), Elga Bandeira (M.Sc, Ph.D) and Fernanda Cruz (M.D., Ph.D.) for the suggestions and comments during the manuscript writing and revision, Rachel Veloso (from Trends Language) and Fernanda Graciolli (from The Editoria) for editing the manuscript, and Nilo Lopes (CF patient) for being a motivation in the writing of this state-of-art.
Abbreviations
- ABC
ATP-binding cassette
- AHSA1
activator of 90 kDa Hsp ATPase homolog 1
- ATP
adenosine triphosphate
- cAMP
cyclic adenosine monophosphate
- CAL
CFTR-associated ligand
- CF
cystic fibrosis
- CFBE
CF bronchial epithelial
- CFTR
CF transmembrane conductance regulator
- CHIP
carboxyl terminus of Hsc70-interacting protein
- EPAC
exchange protein directly activated by cAMP
- ER
endoplasmic reticulum
- EMA
European Medicines Agency
- ENaC
epithelial Na+ channel
- ERAD
ER-associated degradation
- FDA
Food and Drug Administration
- FEV1
forced expiratory volume in 1 s
- HBE
human bronchial epithelial
- HDAC
histone deacetylase
- HGF
hepatocyte growth factor
- Hsc
heat shock cognate
- Hsp
heat shock protein
- HTS
high-throughput screening
- NBD
nucleotide-binding domain
- NHERF-1
Na+/H+ exchanger regulatory factor
- PKA
protein kinase A
- PKC
protein kinase C
- PM
plasma membrane
- PPQC
peripheral protein quality control
- RD
regulatory domain
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SUMO
small ubiquitin-like modifier
- STX
syntaxin
- TMD
transmembrane domain
- VCP
vasolin-containing protein
- VIP
vasoactive intestinal peptide
- wt
wild type
Footnotes
References
- Accurso F. J., Rowe S. M., Clancy J. P., Boyle M. P., Dunitz J. M., Durie P. R., et al. (2010). Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 363 1991–2003. 10.1056/NEJMoa0909825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso F. J., Van Goor F., Zha J., Stone A. J., Dong Q., Ordonez C. L., et al. (2014). Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J. Cyst. Fibros. 13 139–147. 10.1016/j.jcf.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam R. J., Hisert K. B., Dodd J. D., Grogan B., Launspach J. L., Barnes J. K., et al. (2016). Acute administration of ivacaftor to people with cystic fibrosis and a G551D-CFTR mutation reveals smooth muscle abnormalities. JCI Insight 1 e86183. 10.1172/jci.insight.86183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler F. R., Aurora P., Barker D. H., Barr M. L., Blackwell L. S., Bosma O. H., et al. (2009). Lung transplantation for cystic fibrosis. Proc. Am. Thorac. Soc. 6 619–633. 10.1513/pats.2009008-088TL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahner A., Gong X., Schmidt B. Z., Peters K. W., Rabeh W. M., Thibodeau P. H., et al. (2013). Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell 24 74–84. 10.1091/mbc.E12-09-0678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahner A., Nakatsukasa K., Zhang H., Frizzell R. A., Brodsky J. L. (2007). Small heat-shoch proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol. Biol. Cell 18 806–814. 10.1091/mbc.E06-05-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcolado N., Conrad D. J., Rafferty S., Chappe F. G., Chappe V. M. (2011). VIP-dependent increase in F508del-CFTR membrane localization is mediated by PKC𝜀. Am. J. Physiol. Cell Physiol. 301 C53–C65. 10.1152/ajpcell.00568 [DOI] [PubMed] [Google Scholar]
- Alshafie W., Chappe F. G., Li M., Anini Y., Chappe V. M. (2014). VIP regulates CFTR membrane expression and function in Calu-3 cells by increasing its interaction with NHERF1 and P-ERM in a VPAC1- and PKC𝜀-dependent manner. Am. J. Physiol. Cell Physiol. 307 C107–C119. 10.1152/ajpcell.00296.2013 [DOI] [PubMed] [Google Scholar]
- Alton E. W., Armstrong D. K., Asbhy D., Bayfield K. J., Bilton D., Bloomfield E. V., et al. (2015). Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 3 684–691. 10.1016/S2213-2600(15)00245-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alton E. W., Boyd A. C., Cheng S. H., Davies J. C., Davies L. A., Dayan A., et al. (2014). Toxicology study assessing efficacy and safety of repeated administration of lipid/DNA complexes to mouse lung. Genet. Ther. 21 89–95. 10.1038/gt.2013.61 [DOI] [PubMed] [Google Scholar]
- Amaral M. D., Farinha C. M. (2013). Rescuing mutant CFTR: a multi-task approach to a better outcome in treating cystic fibrosis. Curr. Pharm. Des. 19 3497–3508. 10.2174/13816128113199990318 [DOI] [PubMed] [Google Scholar]
- Armstead J., Morris J., Denning D. W. (2014). Multi-country estimate of different manifestations of aspergillosis in cystic fibrosis. PLoS ONE 9:e98502 10.1371/journal.pone.0098502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora K., Moon C., Zhang W., Yarlagadda S., Penmatsa H., Ren A., et al. (2014). Stabilizing rescued surface-localized ΔF508 CFTR by potentiation of its interaction with Na(+)/H(+) exchanger regulatory factor 1. Biochemistry 53 4369–4379. 10.1021/bi401263h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atawade N. T., Uliyakina I., Farinha C. M., Clarke L. A., Mendes K., Solé A., et al. (2015). Measurements of functional responses in human primary lung cells as a basis for personalized therapy for cystic fibrosis. EBioMedicine 2 147–153. 10.1016/j.ebiom.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch W. E., Roth D. M., Hutt D. M. (2011). Emergent properties of proteostasis in managing cystic fibrosis. Cold Spring Harb. Perspect. Biol. 3 a004499 10.1101/cshperspect.a004499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali V., Lazrak A., Guroji P., Fu L., Matalon S., Bebok Z. (2016a). A synonymous códon change alters the drug sensitivity of ΔF508 cystic fibrosis transmembrane conductance regulator. FASEB J. 30 201–213. 10.1096/fj.15-273714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali V., Lazrak A., Guroji P., Matalon S., Bebok Z. (2016b). Mechanistic approaches to improve correction of the most common disease-causing mutation in cystic fibrosis. PLoS ONE 77:e0155882 10.1371/journal.pone.0155882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellin M. D., Laguna T., Leschyshyn J., Regelmann W., Dunitz J., Billings J., et al. (2013). Insulinsecretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediat. Diabetes 14 417–421. 10.1111/pedi.12026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt J. M. (2013). Treatment of pulmonary exacerbations in cystic fibrosis. Eur. Respir. Rev. 22 205–216. 10.1183/09059180.00006512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilan F., Thoreau V., Nacfer M., Dérand R., Norez C., Cantereau A., et al. (2004). Syntaxin 8 impairs trafficking of cystic fibrosis transmembrane conductance regulator (CFTR) and inhibits its channel activity. J. Cell Sci. 117 1923.1935 10.1242/jcs.01070 [DOI] [PubMed] [Google Scholar]
- Bilton D., Robinson P., Cooper P., Gallagher C. G., Kolbe J., Fox H., et al. (2011). Inhaled dry powder mannitol in cystic fibrosis: an efficacy and safety study. Eur. Respir. J. 38 1071–1080. 10.1183/09031936.00187510 [DOI] [PubMed] [Google Scholar]
- Bobadilla J. L., Macek M., Jr., Fine J. P., Farrell P. M. (2002). Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence dataand application to screening. Hum. Mutat. 19 575–606. 10.1002/humu.10041 [DOI] [PubMed] [Google Scholar]
- Bonestroo H. J., de Winter-de Groot K. M., van der Ent C. K., Arets H. G. (2010). Upper and lower airway cultures in children with cystic fibrosis: do not negect the upper airways. J. Cyst. Fibros. 9 130–134. 10.1016/j.jcf.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Bonini J., Varilh J., Raynal C., Thèze C., Beyne E., Audrezet M. P., et al. (2015). Small-scale high-throughput sequencing-based identification of new therapeutic tools in cystic fibrosis. Genet. Med. 17 796–806. 10.1038/gim.2014.194 [DOI] [PubMed] [Google Scholar]
- Borowitz D., Lubarsky B., Wilschanski M., Munck A., Gelfond D., Bodewes F., et al. (2016). Nutritional status improves in cystic fibrosis patients with the G551D mutation after treatment with ivacaftor. Dig. Dis. Sci. 61 198–207. 10.1007/s10620-015-3834-2 [DOI] [PubMed] [Google Scholar]
- Boyault C., Gilquin B., Zhang Y., Rybin V., Garman E., Meyer-Klaucke W., et al. (2006). HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 25 3357–3366. 10.1038/sj.emboj.7601210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle M. P., Bell S. C., Konstan M. W., MocColley S. A., Rowe S. M., Rietschel E., et al. (2014). A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir. Med. 2 527–538. 10.1016/S2213-2600(14)70132-8 [DOI] [PubMed] [Google Scholar]
- Brazilian Cystic Fibrosis Study Group [GBEFC] (2016). Registro Brasileiro de Fibrose Cística 2014. Available at: http://conteudosportal.com.br/GBEFC/wp-content/uploads/2016/03/Registro2014_v09.pdf [accessed July 27, 2016]. [Google Scholar]
- Burgel P. R., Bellis G., Olesen H. V., Viviani L., Zolin A., Blasi F., et al. (2015). Future trends in cystic fibrosis demography in 34 European countries. Eur. Respir. J. 46 133–141. 10.1183/09031936.00196314 [DOI] [PubMed] [Google Scholar]
- Calabrese F., Lunardi F., Nannini N., Balestro E., Loy M., Marulli G., et al. (2015). Higher risk of acute cellular rejection in lung transplant recipients with cystic fibrosis. Ann. Transplant. 20 769–776. 10.12659/AOT.894785 [DOI] [PubMed] [Google Scholar]
- Carlile G. W., Robert R., Matthes E., Yang Q., Solari R., Hatley R., et al. (2016). Latonduine analogues restore F508del-CFTR trafficking through modulation of PARP-3 and PARP-16 activity. Mol. Pharmacol. 90 65–79. 10.1124/mol.115.102418 [DOI] [PubMed] [Google Scholar]
- Carter S., Kelly S., Caples E., Grogan B., Doyle J., Gallagher C. G., et al. (2015). Ivacaftor as salvage therapy in a patients with cystic fibrosis genotype F508del/R117H/IVS8-5T. J. Cyst. Fibros. 14 e4–e5. 10.1016/j.jcb.2015.01.010 [DOI] [PubMed] [Google Scholar]
- Castellani C., Massie J., Sontag M., Southern K. W. (2016). Newborn screening for cystic fibrosis. Lancet Respir. Med. 4 653–661. 10.1016/S2213-2600(16)00053-9 [DOI] [PubMed] [Google Scholar]
- Chappe V., Hinkson D. A., Zhu T., Chang X. B., Riordan J. R., Hanrahan J. W. (2003). Phosphorylation of protein kinase C sites in NBD1 and the R domains control CFTR channel activation by PKA. J. Physiol. 548 39–52. 10.113/jphysiol.2002.035790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. H., Stoltz D. A., Karp P. H., Ernst S. E., Pezzulo A. A., Moninger T. O., et al. (2010). Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 143 911–923. 10.1016/j.cell.2010.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Cebotaru V., Cebotaru L., Guggino W. B. (2010). Syntaxin 6 and CAL mediate the degradation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell 21 1178–1187. 10.1091/mbc.E09-03-0229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Guggino W. (2013). Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS ONE 8:e68001 10.1371/journal.pone.0068001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Moyer B. D., Milewski M., Loffing J., Ikeda M., Mickle J. E., et al. (2002). A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J. Biol. Chem. 277 3520–3529. 10.1074/jbc.M110177200 [DOI] [PubMed] [Google Scholar]
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., et al. (1990). Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63 827–834. 10.1016/0092-8674(90)90148-8 [DOI] [PubMed] [Google Scholar]
- Cholon D. M., Quinney N. L., Fulcher M. L., Esther C. R., Jr., Das J., Dokholyan N. V., et al. (2014). Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci. Transl. Med. 6 246ra96 10.1126/scitranslmed.3008680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J. P., Rowe S. M., Accurso F. J., Aitken M. L., Amin R. S., Ashlock M. A., et al. (2012). Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 67 12–18. 10.1136/thoraxjnl-2011-200393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J. P., Rowe S. M., Bebok Z., Aitken M. L., Gibson R., Zeitlin P. L., et al. (2007). No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations. Am. J. Respir. Cell. Mol. Biol. 37 57–66. 10.1165/rcmb.2006-0173OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cymberknoh M., Shoseyov D., Breuer O., Shamali M., Wilschanski M., Kerem E. (2016). Treatment of cystic fibrosis in low-income countries. Lancet Respir. Med. 4 91–92. 10.1016/S2213-2600(15)00507-X [DOI] [PubMed] [Google Scholar]
- Cohen-Cymberknoh M., Shoseyov D., Kerem E. (2011). Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am. J. Respir. Crit. Care Med. 183 1463–1471. 10.1164/rccm.201009-1478CI [DOI] [PubMed] [Google Scholar]
- Collaco J. M., Blackman S. M., Raraigh K. S., Corvol H., Rommens J. M., Pace R. G., et al. (2016). Sources of variation in sweat chloride measurements in cystic fibrosis. Am. J. Repir. Crit. Care Med. 10.1164/rccm.201603-0459OC [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppinger J. A., Hutt D. M., Razvi A., Koulov A. V., Pankow S., Yates J. R., III, et al. (2012). A chaperone trap contributes to the onset of cystic fibrosis. PLoS ONE 7:e37682 10.1371/journal.pone.0037682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvol H., Blackman S. M., Boëlle P. Y., Gallins P. J., Pace R. G., Stonebraker J. R., et al. (2015). Genome wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 6 8382 10.1038/ncomms9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley E. S., Kopf S. H., LaRiviere A., Ziebis W., Newman D. K. (2015). Pediatric cystic fibrosis sputum can be chemically dynamic, anoxic and extremely reduced due to hydrogen sulfide formation. MBio 6:e00767-15 10.1128/mBio.00767-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cystic Fibrosis Canada [CFC] (2016). The Canadian Cystic Fibrosis Registry: 2014 Annual Report. Available at: https://cysticfibrosis.uberflip.com/i/705240-cystic-fibrosis-canada-registry [accessed July 27, 2016]. [Google Scholar]
- Cystic Fibrosis Federation Australia [CFFA] (2016). Cystic Fibrosis in Australia 2014: 17th Annual Report Australian Cystic Fibrosis Data Registry. https://www.cysticfibrosis.org.au/media/wysiwyg/CF-Australia/medical-documents/CFA_ataRegistryReport_2014_Final.pdf [accessed Aug 05, 2016]. [Google Scholar]
- Cystic Fibrosis Foundation [CFF] (2015). Patient Registry: Annual Data Report 2014. Available at: https://www.cff.org/2014_CFF_Annual_Data_Report_to_the_Center_Directors.pdf/ [accessed May 08, 2016] [Google Scholar]
- Dalemans W., Barbry P., Campigny G., Jallat S., Dott K., Dreyer D., et al. (1991). Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354 526–528. 10.1038/354526a0 [DOI] [PubMed] [Google Scholar]
- Dang H., Gallins P. J., Pace R. G., Guo X. L., Stonebraker J. R., Corvol H., et al. (2016). Novel variation at chr11p13 associated with cystic fibrosis lung disease severity. Hum. Genome Var. 3 16020 10.1038/hgv.2016.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. C., Cunningham S., Harris W. T., Lapey A., Regelmann W. E., Sawicki G. S., et al. (2016). Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir. Med. 4 107–115. 10.1016/S2213-2600(15)00545-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. C., Sheridan H., Bell N., Cunningham S., Davis S. D., Elborn J. S., et al. (2013a). Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D-CFTR mutation and preserved spirometry: a randomised controlled trial. Lancet Respir. Med. 1 630–638. 10.1016/S2213-2600(13)70182-6 [DOI] [PubMed] [Google Scholar]
- Davies J. C., Wainwright C. E., Canny G. J., Chilvers M. A., Howestine M. S., Munck A., et al. (2013b). Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 187 1219–1225. 10.1164/rccm.201301-0153OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck K., Bulteel V., Tiddens H., Wagner T., Fajac I., Conway S., et al. (2011). Guideline on the design and conduct of cystic fibrosis clinical trials: the European Cystic Fibrosis Society-Clinical Trials Network (ECFS-CTN). J. Cyst. Fibros. 10 S67–S74. 10.1016/S1569-1993(11)60010-6 [DOI] [PubMed] [Google Scholar]
- De Boeck K., Munck A., Walker S., Saro A., Hiatt P., Gilmartin G., et al. (2014). Efficacy and safety of ivacaftor with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 13 674–680. 10.1016/j.jcf.2014.09.005 [DOI] [PubMed] [Google Scholar]
- De Stefano D., Villella V. R., Esposito S., Tosco A., Sepe A., De Gregorio F., et al. (2014). Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 10 2053–2074. 10.4161/15548624.2014.973737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers J. F., Bekers G., Kruisselbrink E., Vonk A., de Jonge H. R., Janssens H. M., et al. (2016a). Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 8 344ra84 10.1124/scitranslmed.aad8278 [DOI] [PubMed] [Google Scholar]
- Dekkers J. F., Gogorza Gondra R. A., Kruisselbrink E., Vonk A. M., Janssens H. M., de Winter-de Groot K. M., et al. (2016b). Optimal correction of distinct CFTR folding mutants in rectal cystic fibrosis organoids. Eur. Respir. J. 48 451–458. 10.1183/13993003.01192-2015 [DOI] [PubMed] [Google Scholar]
- Dekkers J. F., Van Mourik P., Vonk A. M., Kruisselbrink E., Berkers G., de Winter-de Groot K. M., et al. (2016c). Potentiator synergy in rectal organoids carrying S1251N, G551D, or F508del CFTR mutation. J. Cystc. Fibros. 10.1016/j.jcf.2016.04.007 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Di Gioia S., Trapani A., Castellani S., Carnoe A., Belgiovine G., Craparo E. F., et al. (2015). Nanocomplexes for gene therapy of respiratory diseases: targeting and overcoming the mucus barrier. Pulm. Pharmacol. Ther. 34 8–24. 10.1016/j.pupt.2015.07.003 [DOI] [PubMed] [Google Scholar]
- Djik F. N., Fitzgerald D. A. (2012). The impact of newborn screening and earlier intervention on the clinical course of cystic fibrosis. Paediatr. Respir. Rev. 13 220–225. 10.1016/j.prrv.2012.05.003 [DOI] [PubMed] [Google Scholar]
- Du M., Jones J. R., Lanier J., Keeling K. M., Lindsey J. R., Tousson A., et al. (2002). Amoniglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J. Mol. Med. 80 595–604. 10.1007/s00109-002-0363-1 [DOI] [PubMed] [Google Scholar]
- Du M., Liu X., Welch E. M., Hirawat S., Peltz S. W., Bedwell D. M. (2008). PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc. Natl. Acad. Sci. U.S.A. 105 2064–2069. 10.1073/pnas.071195105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan N., Kravitz R. L., Schmid C. H. (2013). Single-patient (n-of-1) trials: pragmatic clinical decision methodology for patient-centered comparative effectiveness research. J. Clin. Epidemiol. 66 S21–S28. 10.1016/j.jclinepi.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford P. D., Li C., Ramjeesingh M., Bear C. E. (2012). Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 287 36639–36649. 10.1074/jbc.M112.393637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford P. D., Ramjeesingh M., Molinski S., Pasyk S., Dekkers J. F., Li C., et al. (2014). VX-809 and related corrector compounds exhibit secondary activity stabilizing active F508del-CFTR after its partial rescue to the cell surface. Chem. Biol. 21 666–678. 10.1016/j.chembiol.2014.02.021 [DOI] [PubMed] [Google Scholar]
- Elborn J. S., Ramsey B. W., Boyle M. P., Konstan M. W., Huang X., Marigowda G., et al. (2016). Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir. Med. 4 617–626. 10.1016/S2213-2600(16)30121-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins M. R., Robinson M., Rose B. R., Harbour C., Moriarty C. P., Marks G. B., et al. (2006). A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 354 229–240. 10.1056/NEJMoa043900 [DOI] [PubMed] [Google Scholar]
- European Cystic Fibrosis Society [ECFS] (2016). ECFS Patient Registry: Annual Data Report 2013. Available at: https://www.ecfs.eu/sites/default/files/images/ECFSPR_Report2013_02.2016.pdf [accessed May 08, 2016]. [Google Scholar]
- Farinha C. M., Amaral M. D. (2005). Most F508del-CFTR is targeted to degradation at na early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 25 5242–5252. 10.1128/MCB.25.12.5242-5252.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha C. M., Nogueira P., Mendes F., Penque D., Amaral M. D. (2002). The human DnaJ homologue (Hdj)-1/heat shock protein (Hsp) 40 co-chaperone is required for the in vivo statilization of cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 336 797–806. 10.1042/bj20011717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha C. M., Sousa M., Canato S., Scdmidt A., Uliyakina I., Amaral M. D. (2015). Increased efficacy of VX-809 in different cellular systems results from na early stabilization effect of F508del-CFTR. Pharmacol. Res. Perspect. 3 e00152 10.1002/prp2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell P. M., Rosenstein B. J., White T. B., Accurso F. J., Castellani C., Cutting G. R., et al. (2008). Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis doundation consensus report. J. Pediatr. 153 S4–S14. 10.1016/j.jpeds.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G., Bakouh N., Lourdel S., Odolczyk N., Premchandar A., Servel N., et al. (2016). Rattlesnake phospholipase A2 increases CFTR-chloride channel current and corrects ΔF508CFTR dysfunction: impact in cystic fibrosis. J. Mol. Med. 428 2898–2915. 10.1016/j.jmb.2016.05.016 [DOI] [PubMed] [Google Scholar]
- Favia M., Guerra L., Faneli T., Cardone R. A., Monterisi S., Di Sole F., et al. (2010). Na+/H+ exchanger regulatory fator 1 onverexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol. Biol. Cell 21 73–86. 10.1091/mbc.E09-03-0185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feranchak A. P., Sontag M. K., Wagener J. S., Hammond K. B., Accurso F. J., Sokol R. J. (1999). Prospective, long-term study of fat-soluble status in children with cystic fibrosis identified by newborn screen. J. Pediatr. 135 601–610. 10.1016/S0022-3476(99)70059-4 [DOI] [PubMed] [Google Scholar]
- Flume P. A., Liou T. G., Borowitz D. S., Li H., Yen K., Ordoñez C. L., et al. (2012). Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 142 718–724. 10.1378/chest.11.2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs H. J., Borowitz D. S., Christiansen D. H., Morris E. M., Nash M. L., Ramsey B. W., et al. (1994). Effect of aerolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The pulmozyme study group. N. Engl. J. Med. 331 637–642. 10.1056/NEJM199409083311003 [DOI] [PubMed] [Google Scholar]
- Gadsby D. C., Vergani P., Csanády L. (2006). The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 23 477–483. 10.1038/nature04712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galapagos N. V. (2015). Galapagos Advances Triple Combination Therapy in Cystic Fibrosis. Available at: http://www.glpg.com/docs/view/8829a660-en [accessed May 18, 2016]. [Google Scholar]
- Galietta L. J., Springsteel M. F., Eda M., Niedzinski E. J., By K., Haddadin M. J., et al. (2001). Novel CFTR chloride channel activators identified by screening of combinatorial libraries based on flavone and benzoquinolizinium lead compounds. J. Biol. Chem. 276 19723–19728. 10.1074/jbc.M101892200 [DOI] [PubMed] [Google Scholar]
- Gentzsch M., Dang H., Dang Y., Garcia-Caballero A., Suchindran H., Boucher R. C., et al. (2010). The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J. Biol. Chem. 285 32227–32232. 10.1074/jbc.M110.155259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M., Ren H. Y., Houck S. A., Quinney N. L., Cholon D. M., Sopha P., et al. (2016). Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am. J. Physiol. Lung Cell. Mol. Physiol. 10.1152/ajplung.00186.2016 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozman R., Okiyoneda T., Mulvihill C. M., Rini J. M., Barriere H., Lukacs G. L. (2009). N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J. Cell Biol. 184 847–862. 10.1083/jcb.200808124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X., Ahner A., Roldan A., Lukacs G. L., Thibodeau P. H., Frizzell R. A. (2016). Non-native conformers of cystic fibrosis transmembrane conductance regulator NBDq are recognized by Hsp27 and conjugated to SUMO-2 for degradation. J. Biol. Chem. 291 2004–2017. 10.1074/jbc.M115.685628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra L., Fanelli T., Favia M., Riccardi S. M., Busco G., Cardone R. A., et al. (2005). Na+/H+ exchanger regulatory factor isoform 1 overexpression modulates cystic fibrosis transmembrane conductance regulator (CFTR) expression and activity in human airway 16HBE14o- cells and rescues deltaF508 CFTR functional expression in cystic fibrosis cells. J. Biol. Chem. 280 40925–40933. 10.1074/jbc.M505103200 [DOI] [PubMed] [Google Scholar]
- Gustafsson J. K., Ermund A., Ambort D., Johansson M. E., Nilsson H. E., Thorell K., et al. (2012). Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. J. Exp. Med. 209 1263–1272. 10.1084/jem.20120562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Kota P., Aleksandrov A. A., Cui L., Jensen T., Dokholyan N. V., et al. (2013). Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 27 536–545. 10.1096/fj.12-216119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde R. N., Parashuraman S., Iorio F., Ciciriello F., Capuani F., Carissimo A., et al. (2015). Unravelling druggable signalling networks that control F508del-CFTR proteostasis. Elife 4 e10365 10.7554/eLife.10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M., Frizzell R. A., Bedwell D. M. (1996). Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 2 467–469. 10.1038/nm0496-467 [DOI] [PubMed] [Google Scholar]
- Hutt D. M., Herman D., Rodrigues A. P., Noel S., Pilewski J. M., Marreson J., et al. (2010). Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 6 25–33. 10.1038/nchembio.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igreja S., Clarke L. A., Botelho H. M., Marques L., Amaral M. D. (2016). Correction of a cystic fibrosis splicing mutation by antisense oligonucleotides. Hum. Mutat. 37 209–215. 10.1002/humu.22931 [DOI] [PubMed] [Google Scholar]
- Itani O. A., Chen J. H., Karp P. H., Ernst S., Keshavjee S., Parekh K., et al. (2011). Human cystic fibrosis airway epithelia have reduced Cl- conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. U.S.A. 108 10260–10265. 10.1073/pnas.1106695108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., Riordan J. R. (1995). Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83 129–135. 10.1016/0092-8674(95)90241-4 [DOI] [PubMed] [Google Scholar]
- Jorth P., Staudinger B. J., Wu X., Hisert K. B., Hayden H., Garudathri J., et al. (2015). Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe 18 307–319. 10.1016/j.chom.2015.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakoi S., Yorimitsu T., Sato K. (2013). COPII machinery cooperates with ER-localized Hsp40 to sequester misfolded membrane protein into ER-associated compartments. Mol. Biol. Cell 24 633–642. 10.1091/mbc.E12-08-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalnins D., Wilschanski M. (2012). Maintenance of nutritional status in patients with cystic fibrosis: new and emerging therapies. Drug Des. Devel. Ther. 6 151–161. 10.2147/DDDT.S9258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003). The acetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115 727–738. 10.1016/S0092-8674(03)00939-5 [DOI] [PubMed] [Google Scholar]
- Kerem B., Chiba-Falek O., Kerem E. (1997). Cystic fibrosis in Jews: frequency and mutation distribution. Genet. Test. 1 35–39. 10.1089/gte.1997.1.35 [DOI] [PubMed] [Google Scholar]
- Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., et al. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science 245 1073–1080. 10.1126/science.2570460 [DOI] [PubMed] [Google Scholar]
- Kerem E., Hirawat S., Armoni S., Yaakov Y., Shoseyov D., Cohen M., et al. (2008). Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 372 719–727. 10.1016/S0140-6736(08)61168-X [DOI] [PubMed] [Google Scholar]
- Kerem E., Konstan M. W., De Boeck K., Accurso F. J., Sermet-Gaudelus I., Wilschanski M., et al. (2014). Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2 539–547. 10.1016/S2213-2600(14)70100-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., O’Neill J. D., Dorrello N. V., Baccheatta M., Vunjak-Novakovic G. (2015). Targeted delivery of liquid microvolumes into the lung. Proc. Natl. Acad. Sci. U.S.A. 112 11530–11535. 10.1073/pnas.1512613112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., Hartl F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Ann. Rev. Biochem. 82 323–355. 10.1146/annurev-biochem.060208-092442 [DOI] [PubMed] [Google Scholar]
- Konstan M. W., McKone E., Moss R. B., Marigowda G., Cooke J., Huang X., et al. (2016). “Evidence for reduced rate of lung function decline and sustained benefit with combination lumacaftor and ivacaftor (LUM/IVA) therapy in patients (pts) ≥ 12 years of age with cystic fibrosis (CF) homozygous for the F508del-CFTR mutation,” in Poster at the 8th European Conference on Rare Diseases & Orphan Products (ECRD): Poster 108 Edinburgh. [Google Scholar]
- Koulov A. V., LaPointe P., Lu B., Razvi A., Coppinger J., Dong M. Q., et al. (2010). Biological and structural basis for Aha1 regulation of Hsp90 ATP activity in maintaining proteostasis in human disease cystic fibrosis. Mol. Biol. Cell 21 871–884. 10.1091/mbc.E09-12-1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann K., Kathöfer S., Greger R. (1995). Na+ and Cl- conductances in airway epithelial cells: increased Na+ conductance in cystic fibrosis. Pflugers. Arch. 431 1–9. 10.1007/BF00374371 [DOI] [PubMed] [Google Scholar]
- Kwon S. H., Pollard H., Guggino W. B. (2007). Knockdown of NHERF1 enhances degradation of temperature rescued deltaF508 CFTR from the cell surface of human airway cells. Cell. Physiol. Biochem. 20 763–772. 10.1159/000110436 [DOI] [PubMed] [Google Scholar]
- LaFayette S. L., Houle D., Beaudoin T., Wojewodka G., Radzioch D., Hoffman L. R., et al. (2015). Cystic fibrosis-adapted Pseudomonas aerugiona quorum sensing lasR mutants cause hyperinflammatory responses. Sci. Adv. 1 e1500199 10.1126/sciadv.1500199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazrak A., Fu L., Bali V., Bartoszewski R., Rab A., Havasi V., et al. (2013). The silent codon change I507-ATC- > ATT contributes to the severity of the ΔF508 CFTR channel dysfuntion. FASEB J. 27 4630–4645. 10.1096/fj.13-227330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W. Y., Sohma Y., Hwang T. C. (2016). Synergistic potentiation of CFTR gating by two chemically distinct potentiators, ivacaftor (VX-770) and NPPB. Mol. Pharmacol. 90 275-285 10.1124/mol.116.104570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo M. J., Amaral M. D., Zaccolo M., Farinha C. M. (2016). EPAC1 activation by cAMP stabilizes CFTR at the membrane by promoting its interaction with NHERF1. J. Cell Sci. 129 2599–2612. 10.1242/jcs.185629 [DOI] [PubMed] [Google Scholar]
- Loo T. W., Clarke D. M. (2008). Mutational analysis of ABC proteins. Arch. Biochem. Biophys. 476 51–64. 10.1016/j.abb.2008.02.025 [DOI] [PubMed] [Google Scholar]
- Lopes-Pacheco M., Boinot C., Sabirzhanova I., Morales M. M., Guggino W. B., Cebotaru L. (2015). Combination of correctors rescue ΔF508-CFTR by reducing its association with Hsp40 and Hsp27. J. Biol. Chem. 290 25636–25645. 10.1074/jbc.M115.671925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Pacheco M., Sabirzhanova I., Rapino D., Morales M. M., Guggino W. B., Cebotaru L. (2016). Correctors rescue CFTR mutation in nucleotide-bunding domain 1 (n.d.) by modulating proteostasis. Chembiochem 17 493–505. 10.102/cbic.201500620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loureiro C. A., Matos A. M., Dias-Alves Â, Pereira J. F., Uliykina I., Barros P., et al. (2015). A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci. Signal 8 ra48 10.1126/scisignal.aaa1580 [DOI] [PubMed] [Google Scholar]
- Luciani A., Villella V. R., Esposito S., Brunetti-Pierri N., Medina D., Settembre C., et al. (2010). Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 12 863–875. 10.1038/ncb2090 [DOI] [PubMed] [Google Scholar]
- Luciani A., Villella V. R., Esposito S., Gavina M., Russo I., Silano M., et al. (2012). Targeting autophagy as a novel strategy for faciliting the therapeutic action of potentiatios on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 8 1657–1672. 10.4161/auto.21483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs G. L., Verkman A. S. (2012). CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trend Mol. Med. 18 81–91. 10.1016/j.molmed.2011.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak J. B., Cannon C. L., Pier G. B. (2002). Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 15 194–222. 10.1128/CMR.15.2.194-222.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie T., Gifford A. H., Sabadosa K. A., Quinton H. B., Knapp E. A., Goss C. H., et al. (2014). Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry. Ann. Intern. Med. 161 233–241. 10.7326/M13-0636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve P., Marshall B. C., Knapp E. A., Lowenfels A. B. (2013). Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J. Natl. Cancer. Inst. 105 122–129. 10.1093/jnci/djs481 [DOI] [PubMed] [Google Scholar]
- Marozkina N. V., Yemen S., Borowitz M., Liu L., Plapp M., Sun F., et al. (2010). Hsp 70/Hsp 90 organizing protein as a nitrosylation target in cystic fibrosis therapy. Proc. Natl. Acad. Sci. U.S.A. 107 11393–11398. 10.1073/pnas.0909128107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson F. A., Hortencio T. D., Aguiar K. C., Ribeiro J. D. (2015). Demographic, clinical and laboratory parameters of cystic fibrosis during the last two decades: acomparative analysis. BMC Pulm. Med. 15:3 10.1186/1471-2466-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastorakos P., da Silva A. L., Chisholm J., Song E., Choi W. K., Boyle M. P., et al. (2015). Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene delivery. Proc. Natl. Acad. Sci. U.S.A. 112 8720–8725. 10.1073/pnas.1502281112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes E., Goepp J., Carlile G. W., Luo Y., Dejgaard K., Billet A., et al. (2016). Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor). Br. J. Pharmacol. 173 459–470. 10.1111/bph.13365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKone E. F., Borowitz D., Devinek P., Griese M., Konstan M. W., Wainwright C., et al. (2014). Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir. Med. 2 902–910. 10.1016/S2213-2600(14)70218-8 [DOI] [PubMed] [Google Scholar]
- Meacham G. C., Lu Z., King S., Sorscher E., Tousson A., Cyr D. M. (1999). The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 18 1492–1505. 10.1093/emboj/18.6.1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. (2001). The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3 100–105. 10.1038/35050509 [DOI] [PubMed] [Google Scholar]
- Mendoza J. L., Schmidt A., Li Q., Nuvaga E., Barrett T., Bridges R. J., et al. (2012). Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 148 164–174. 10.1016/j.cell.2011.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz S., Souza M., Moraes B. J., Mendes B. J., Palma M., Barreto C., et al. (2013). HGF stimutation of Rac signaling enhances pharmacological correction of the most prevalente cystic fibrosis mutant F508del-CFTR. ACS Chem. Biol. 8 432–442. 10.1021/cb300484r [DOI] [PubMed] [Google Scholar]
- Moskowitz S. M., Chmiel J. F., Sernen D. L., Cheng E., Gibson R. L., Marshall S. G., et al. (2008). Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genet. Med. 10 851–868. 10.1097/GIM.0b013e31818e55a2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss R. B., Flume P. A., Elborn J. S., Cooke J., Rowe S. M., McColley S. A., et al. (2015). Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His-CFTR mutation: a double-blind randomised controlled trial. Lancet Respir. Med. 3 524–533. 10.1016/S2213-2600(15)00201-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutyam V., Du M., Xue X., Keeling K. M., White E. L., Bostwick J. R., et al. (2016). Discovery of clinically approved agents that promote suppression of CFTR nonsense mutations. Am. J. Respir. Crit. Care Med. 10.1164/rccm.201601-0154OC [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivalis Therapeutics (2016). Nivalis Therapeutics Expands N91115 Clinical Development Program in Cystic Fibrosis. Available at: http://ir.nivalis.com/press-releases/detail/40/nivalis-therapeutics-expands-n91115-clinical-development [accessed May 18, 2016]. [Google Scholar]
- Norez C., Vandebrouck C., Bertrand J., Noel D., Durieu E., Oumata N., et al. (2014). Roscovitine is a proteostasis regulator that corrects the trafficking defect of F508del-CFTR by a CDK-independent mechanism. Br. J. Pharmacol. 171 4831–4849. 10.1111/bph.12859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor M. G., Seegmiller A. (2016). The effects of ivacaftor on CF fatty acid metabolism: an analysis from GOAL study. J. Cyst. Fibros. 10.1016/j.jcf.2016.07.006 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T., Barrière H., Bagdány M., Rabeh W. M., Du K., Höhfeld J., et al. (2010). Peripheral protein quality controle removes unfolded CFTR from de plasma membrane. Science 329 805–810. 10.1126/science.1191542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T., Niibori A., Harada K., Kohno T., Michalak M., Duszyk M., et al. (2008). Role of calnexin in the ER quality control and productive folding of CFTR; differential effect of calnexin knockout on wild-type and deltaF508 CFTR. Biochim. Bhiophys. Acta 1783 1585–1594. 10.1016/j.bbamcr.2008.04.002 [DOI] [PubMed] [Google Scholar]
- Okiyoneda T., Veit G., Dekkers J. F., Bagdany M., Soya N., Xu H., et al. (2013). Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 9 444–454. 10.1038/nchembio.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S., Bamberger C., Calzolari D., Martínez-Bartolomé S., Mavallée-Adam M., Balch W. E., et al. (2015). ΔF508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528 510–516. 10.1038/nature15729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N., Lukacs G. L., Du K., Caci E., Zegarra-Moran O., Galietta L. J., et al. (2005). Small-molecule correctors of defective deltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 115 2564–2571. 10.1172/JCI24898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N., Tomati V., Sondo E., Caci E., Millo E., Armirotti A., et al. (2011). Dual activity of aminoarylthiazoles on the trafficking and gating defects of the cystic fibrosis transmembrane conductance regulator chloride channel caused by cystic fibrosis mutations. J. Biol. Chem. 286 15215–15226. 10.1074/jbc.M110.184267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce E., Belloti M., Liessi N., Guariento S., Damonte G., Cichero E., et al. (2015). Synthesis and structure-activity relationship of aminoarylthiazole derivatives as correctors of the chloride transport defect in cystic fibrosis. Eur. J. Med. Chem. 99 14–35. 10.1016/j.ejmech.2015.05.030 [DOI] [PubMed] [Google Scholar]
- Phuan P. W., Veit G., Tan J. A., Finkbeiner W. E., Lukacs G. L., Verkman A. S. (2015). Potentiators of defective ΔF508-CFTR gating that do not interfere with corrector action. Mol. Pharmacol. 88 791–799. 10.1124/mol.115.099689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuan P. W., Veite G., Tan J., Roldan A., Finkbeiner W. E., Lukacs G. L., et al. (2014). Synergy-based small-molecule screen using a human lung epithelial cell line yields ΔF508-CFTR correctors that augment VX-809 maximal efficacy. Mol. Pharmacol. 86 42–51. 10.1124/mol.114.092478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pibiri I., Lentini L., Melfi R., Galluci R., Pace A., Spinello A., et al. (2015). Enhancement of premature stop códon readthrough in the CFTR gene by ataluren (PTC124) derivatives. Eur. J. Med. Chem. 101 236–244. 10.1016/j.ejmech.2015.06.038 [DOI] [PubMed] [Google Scholar]
- Pibiri I., Lentini L., Tutone M., Melfi R., Pace A., Di Leonardo A. (2016). Exploring the readthrough of nonsense mutations by non-acidic ataluren analogues selected by ligand-based virtual screening. Eur. J. Med. Chem. 122 429–435. 10.1016/j.ejmech.2016.06.048 [DOI] [PubMed] [Google Scholar]
- Pique L., Graham S., Pearl M., Kharrazi M., Schrijver I. (2016). Cystic fibrosis newborn screening programs: implications of the CFTR variant spectrum in nonwhite patients. Genet. Med. 10.1038/gim.2016.48 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Plant B. J., Goss C. H., Plant W. D., Bell S. C. (2013). Management of comorbidities in older patients with cystic fibrosis. Lancet Respir. Med. 1 164–174. 10.1016/S2213-2600(13)70025-0 [DOI] [PubMed] [Google Scholar]
- Prayle A., Watson A., Fortnum H., Smyth A. (2010). Side effects of aminoglycosides on the kidney, ear and balance in cystic fibrosis. Thorax 65 654–658. 10.1136/thx.2009.131532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proteostasis Therapeutics. (2016). Proteostasis Therapeutics, Inc. Presents New Data Demonstrating Potential for Genotype-Agnostic and Combination Therapies for People with Cystic Fibrosis. Available at: http://ir.proteostasis.com/phoenix.zhtml?c=254052&p=irol-newsArticle&ID=2151856 [accessed May 18, 2016]. [Google Scholar]
- Pyle L. C., Fulton J. C., Sloane P. A., Backer K., Mazur M., Prasain J., et al. (2010). Activation of cystic fibrosis transmembrane conductance regulator by the flavonoide quercetin: potential use as a biomarker of ΔF508 cystic fibrosis transmembrane conductance regulator rescue. Am. J. Respir. Cell Mol. Biol. 43 607–616. 10.1165/rcmb.2009-0281OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quittner A., Suthoff E., Rendas-Baum R., Bayliss M. S., Sermet-Gaudelus I., Castiglione B., et al. (2015). Effect of ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation: patient-reported outcomes in the STRIVE randomized, controlled trial. Health Qual. Life Outcomes 13 93 10.1186/s12955-015-0293-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quon B. S., Rowe S. M. (2016). New and emerging targeted therapies for cystic fibrosis. BMJ 352 i859 10.1136/bmj.i859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafferty S., Alcolado N., Norez C., Chappe F., Pelzer S., Becq F., et al. (2009). Rescue of functional F508del cystic fibrosis transmembrane conductance regulator by vasoactive intestinal peptide in the human nasal epithelial cell line JME/CF15. J. Pharmacol. Exp. Ther. 331 2–13. 10.1124/jpet.109.155341 [DOI] [PubMed] [Google Scholar]
- Ramsey B. W., Davies J., McElvaney N. G., Tullis E., Bell S. C., Dřevínek P., et al. (2011). A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 365 1663–1672. 10.1056/NEJMoa1105185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapino D., Sabirzhanova I., Lopes-Pacheco M., Grover R., Guggino W. B., Cebotaru L. (2015). Rescue of NBD2 mutants N1303K and S1235R of CFTR by small-molecule correctors and transcomplementation. PLoS ONE 10:e0119796 10.1371/journal.pone.0119796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., et al. (1989). Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245 1066–1073. 10.1126/science.2475911 [DOI] [PubMed] [Google Scholar]
- Robert R., Carlile G. W., Pavel C., Liu N., Anjos S. M., Liao J., et al. (2008). Structural analog of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Mol. Pharmacol. 73 478–489. 10.1124/mol.107.040725 [DOI] [PubMed] [Google Scholar]
- Ronan N. J., Fleming C., O’Callaghan G., Maher M. M., Murphy D. M., Plant B. J. (2015). The role of ivacaftor in severe cystic fibrosis in a patient with the R117H mutation. Chest 148 e72–e75. 10.1378/chest.14-3215 [DOI] [PubMed] [Google Scholar]
- Roth D. M., Hutt D. M., Tong J., Bouchecareih M., Wang N., Seeley T., et al. (2014). Modulation of the maladaptive stress responde to manage diseases of protein folding. PLoS Biol. 12:e1001998 10.1371/journal.pbio.1001998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. M., Borowitz D. S., Burns J. L., Clancy J. P., Donaldson S. H., Retsch-Bogart G., et al. (2012). Progress in cystic fibrosis and the CF Therapeutics Development Network. Thorax 67 882–890. 10.1136/thoraxjnl-2012.202550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. M., Heltshe S. L., Gonska T., Donaldson S. H., Borowitz D., Gelfond D., et al. (2014). Clinical mechanism of cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 190 175–184. 10.1164/rccm.201404-0703OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. M., Sloane P., Tang L. P., Backer K., Mazur M., Buckley-Lanier J., et al. (2011). Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. 89 1149–1161. 10.1007/s00109-011-0787-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabirzhanova I., Lopes-Pacheco M., Rapino D., Grover R., Handa J. T., Guggino W. B., et al. (2015). Rescuing trafficking mutants of the ATP-binding cassette protein, ABCA4, with small molecule correctors as a treatment for Stargardt eye disease. J. Biol. Chem. 290 19743–19755. 10.1074/jbc.M115.647685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson H. M., Lam H., Chen P. C., Zhang D., Mottillo C., Mirza M., et al. (2013). Compounds that correct F508del-CFTR trafficking can also correct other protein trafficking diseases: an in vitro study using cell lines. Orphanet J. Rare Dis. 8 11 10.1186/1750-1172-8-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson H. M., Robert R., Liao J., Matthes E., Carlile G. W., Hanrahan J. W., et al. (2011). Identification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTR. Chem. Biol. 18 231–242. 10.1016/j.chembiol.2010.11.016 [DOI] [PubMed] [Google Scholar]
- Sawicki G. S., McKone E. F., Pasta D. J., Millar S. J., Wagener J. S., Johnson C. A., et al. (2015). Sustained benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. Am. J. Respir. Crit. Care Med. 192 836–842. 10.1164/rccm.201503-0578OC [DOI] [PubMed] [Google Scholar]
- Schrijver I., Pique L., Graham S., Pearl M., Cherry A., Kharrazi M. (2016). The spectrum of CFTR variants in nonwhite cystic fibrosis patients: implication for molecular diagnostic testing. J. Mol. Diagn. 18 39–50. 10.1016/j.jmoldx.2015.07.005 [DOI] [PubMed] [Google Scholar]
- Sermet-Gaudelus I., Boeck K. D., Casimir G. J., Vermeulen F., Leal T., Mogenet A., et al. (2010). Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am. J. Respir. Crit. Care Med. 182 1262–1272. 10.1164/rccm.201001-0137OC [DOI] [PubMed] [Google Scholar]
- Serohijos A. W., Hegedus T., Aleksandrov A. A., He L., Cui L., Dokhokyan N. V., et al. (2008). Phynyalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105 3256–3261. 10.1073/pnas.0800254105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah V. S., Meyerholz D. K., Tang X. X., Reznikov L., Abou Alaiwa M., Ernst S. E., et al. (2016). Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 351 503–507. 10.1126/science.aadd5589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M., Benharouga M., Hu W., Lukacs G. L. (2001). Conformational and temperature-sensitive satibility defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J. Biol. Chem. 276 8942–8950. 10.1074/jbc.M009172200 [DOI] [PubMed] [Google Scholar]
- Sharma M., Pampinella F., Nemes C., Benharouga M., So J., Du K., et al. (2004). Misfolding diverts CFTR from recycling to degradation: quality controla t early endosomes. J. Cell Biol. 164 923–933. 10.1083/jcb.200312018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha C., Zhang W., Moon C. S., Actis M., Yarlagadda S., Arora K., et al. (2015). Capturing the direct binding of CFTR correctors to CFTR by using click chemistry. Chembiochem 16 2017–2022. 10.1002/cbic.201500123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth A. R., Bell S. C., Bojcin S., Bryon M., Duff A., Flume P., et al. (2014). European cystic fibrosis society standards of care: best practice guidelines. J. Cyst. Fibros. 13 (Suppl. 1) S23–S42. 10.1016/j.jcf.2014.03.010 [DOI] [PubMed] [Google Scholar]
- Sosnay P. R., Siklosi K. R., Van Goor F., Kaniecki K., Yu H., Sharma N., et al. (2013). Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 45 1160–1167. 10.1038/ng.2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton B. A., Coutermarsh B., Barnaby R., Hogan D. (2015). Pseudomonas aeruginosa reduces VX-809 stimulated F508del-CFTR chloride secretion by airway epithelial cells. PLoS ONE 10:e0127742 10.1371/journal.pone.0127742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C., Pepper M. S. (2016). Cystic fibrosis on the African continent. Genet. Med. 18 653–662. 10.1038/gim.2015.157 [DOI] [PubMed] [Google Scholar]
- Suk J. S., Kim A. J., Trehan K., Schneider C. S., Cebotaru L., Woodward O. M., et al. (2014). Lung gene therapy with highly compacted DNA nanoparticles that overcome the mucus barrier. J. Control Release 178 8–17. 10.1016/j.jconrel.2014.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F., Zhang R., Gong X., Geng X., Drain P. F., Frizzell R. A. (2006). Derlin-1 promotes the efficient degradation if the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J. Biol. Chem. 281 36856–36863. 10.1074/jbc.M607085200 [DOI] [PubMed] [Google Scholar]
- Swiatecka-Urban A., Brown A., Moreau-Marquis S., Renuka J., Coutermarsh B., Barnaby R., et al. (2005). The short apical membrane half-life of rescue {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 280 36762–36772. 10.1074/jbc.M508944200 [DOI] [PubMed] [Google Scholar]
- Tang X. X., Ostedgaard L. S., Hoegger M. J., Moninger T. O., Karp P. H., McMenimen J. D., et al. (2016). Acidic pH increases airway surface liquid viscosity in cystic fibrosis. J. Clin. Invest. 126 879–891. 10.1172/JCI83922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R., Button B., Picher M., Paradiso A. M., Ribeiro C. M., Lazarowski E. R., et al. (2005). Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J. Biol. Chem. 280 35751–35759. 10.1074/jbc.M505832200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Than B. L., Linnekamp J. F., Starr T. K., Largaespada D. A., Rod A., Zhang Y., et al. (2016). CFTR is a tumor suppressor gene in murine and human intestinal cancer. Oncogene 35 4179–4187. 10.1038/onc.2015.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosco A., De Gregorio F., Esposito S., De Stefano D., Sana I., Ferrari E., et al. (2016). A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 23 1380–1393. 10.1038/cdd.2016.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Cao D., Neuberger T., et al. (2009). Rescue of CF airway cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 106 18825–18830. 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Stack J. H., Straley K. S., et al. (2011). Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108 18843–18848. 10.1073/pnas.1105787108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goor F., Straley K. S., Cao D., González J., Hadida S., Hazlewood A., et al. (2006). Rescue of deltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung. Cell Mol. Physiol. 290 L1117–L1130. 10.1152/ajplung.00169.2005 [DOI] [PubMed] [Google Scholar]
- Van Goor R., Yu H., Burton B., Hoffman B. J. (2014). Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 13 29–36. 10.1016/j.jcf.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Vasiliou V., Vasiliou K., Nebert D. W. (2009). Human ATP-binding cassette (ABC) transporter family. Hum. Genomics 3 281.290 10.1186/1479-7364-3-3-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G., Avramescu R. G., Chiang A. N., Houch S. A., Cai Z., Peters K. W., et al. (2016a). From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 27 424–433. 10.1091/mbc.E14-04-0935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G., Avramescu R. G., Perdomo D., Phuan P. W., Dagdany M., Apaja P. M., et al. (2014). Some gating potentiators, including VX-770, diminish ΔF508-CFTR functional expression. Sci. Transl. Med. 6 246ra97 10.1126/scitranslmed.3008889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit G., Oliver K., Apaja P. M., Perdomo D., Bidaud-Meynard A., Lin S. T., et al. (2016b). Ribosomal stalk protein silencing partially corrects the ΔF508-CFTR functional expression defect. PLoS Biol. 14:e1002462 10.1371/journal.pbio.1002462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertex Pharmaceuticals (2015). Vertex Announces Significant Progress in Its Development Efforts to Treat the Cause of Cystic Fibrosis in the Vast Majority of People with the Disease. Available at: http://investors.vrtx.com/releasedetail.cfm?ReleaseID=935806 [accessed May 18, 2016]. [Google Scholar]
- Vij N., Fang S., Zeitlin P. L. (2006). Selective inhibition of endoplasmic reticulum-associated degradation rescues deltaF508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: therapeutic implications. J. Biol. Chem. 281 17369–17378. 10.1074/jbc.M600509200 [DOI] [PubMed] [Google Scholar]
- Vij N., Min T., Marasigan R., Belcher C. N., Mazur S., Ding H., et al. (2010). Development of PEGylated PLGA nanoparticle for controlled and sustained drug delivery in cystic fibrosis. J. Nanobiotechnology 8 22 10.1186/1477-3155-8-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijftigschild L. A., Berkers G., Dekkers J. F., Zomer-van Ommen D. D., Matthes E., Kruisselbrink E., et al. (2016). β2-Adrenergic receptor agonists activate CFTR in intestinal organoids and subjects with cystic fibrosis. Eur. Respir. J. 10.1183/13993003.01661-2015 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Villella V. R., Esposito S., Bruscia E. M., Vicinanza M., Cenci S., Guido S., et al. (2013). Disease relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death. Differ. 80 1101–1115. 10.1038/cdd.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu V., Verster A. J., Schertzberg M., Chuluunbaatar T., Spensley M., Pajkic D., et al. (2015). Natural variation in gene expression modulates the severity of mutant phenotypes. Cell 162 391–402. 10.1016/j.cell.2015.06.037 [DOI] [PubMed] [Google Scholar]
- Wainwright C. E., Elborn J. S., Ramsey B. W., Marigowda G., Huang X., Cipolli M., et al. (2015). Lumacaftor-Ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N. Engl. J. Med. 373 220–231. 10.1056/NEJMoa1409547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., et al. (2006). Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127 803–812. 10.1016/j.cell.2006.09.043 [DOI] [PubMed] [Google Scholar]
- Wang Y., Loo T. W., Bartlett M. C., Clarke D. M. (2007a). Correctors promote maturation of cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by binding to the protein. J. Biol. Chem. 282 33247–33251. 10.1074/jbc.C700175200 [DOI] [PubMed] [Google Scholar]
- Wang Y., Loo T. W., Bartlett M. C., Clarke D. M. (2007b). Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol. Pharmacol. 71 751–758. 10.1124/mol.106.029926 [DOI] [PubMed] [Google Scholar]
- Wilschanski M., Miller L. L., Shoseyov D., Blau H., Rivlin J., Aviram M., et al. (2011). Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur. Respir. J. 38 59–69. 10.1183/09031936.00120910 [DOI] [PubMed] [Google Scholar]
- Wilschanski M., Yahav Y., Yaacov Y., Blau H., Bentur L., Rivlin J., et al. (2003). Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N. Engl. J. Med. 349 1433–1441. 10.1056/NEJMoa022170 [DOI] [PubMed] [Google Scholar]
- Winter M. C., Welsh M. J. (1997). Stimulation of CFTR activity by its phosphorylated R domain. Nature 389 294–296. 10.1038/38514 [DOI] [PubMed] [Google Scholar]
- Woodward O. M., Tukayne D. N., Cui J., Greenwell P., Constantoulakis L. M., Parker B. S. (2013). Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. U.S.A. 110 5223–5228. 10.1073/pnas.1214530110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worlitzsch D., Tarran R., Ulrich M., Schwab U., Cekici A., Meyer K. C., et al. (2002). Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109 317–325. 10.1172/JCI13870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Mutyam V., Tang L., Biswas S., Du M., Jackson L. A., et al. (2014). Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell Mol. Biol. 50 805–816. 10.1165/rcmb.2013-0282OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Janich S., Cohn J. A., Wilson J. M. (1993). The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. U.S.A. 90 9480–9484. 10.1073/pnas.30.20.9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Burton B., Huang C. J., Worley J., Cao D., Johnson J. P., Jr., et al. (2012). Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 11 237–245. 10.1016/j.jcf.2011.12.005 [DOI] [PubMed] [Google Scholar]
- Yu Y. C., Miki H., Nakamura Y., Hanyuda A., Matsuzaki Y., Abe Y., et al. (2011). Curcumin and genistein additively potentiate G551D-CFTR. J. Cystc. Fibros. 10 243–252. 10.1016/j.jcf.2011.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. C., Sohma Y., Hwang T. C. (2016). On the mechanism of gating defects caused by the R117H mutation in CFTR. J. Physiol. 594 3227–3244. 10.1113/JP271723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Hollinger M., Lachowicz-Scroggins M. E., Kerr S. C., Dunican E. M., Daniel B. M., et al. (2015). Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sic. Transl. Med. 7 276ra27 10.1126/scitranslmed.3010525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman K., Sawczak V., Zaidi A., Butler M., Bennett D., Getsy P. (2016). Augmentation of CFTR maturation by S-nitroglutathione reductase. Am. J. Physiol. Lung Cell. Mol. Physiol. 310 L263–L270. 10.1152/ajplung.00269.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Ciciriello F., Anjos S. M., Carissimo A., Liao J., Carlile G. W., et al. (2012). Ouabain mimics low temperature rescue of F508del-CFTR in cystic fibrosis epithelial cells. Front. Pharmacol. 3:176 10.3389/fphar.2012.00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Jiang Q., Takahagi S., Shao C., Uitto J. (2013). Premature termination codon read-through in the ABCC6 gene: potential treatment for pseudoxanthoma elasticum. J. Invest. Dermatol. 133 2672–2677. 10.1038/jid.2013.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomer-van Ommen D. D., Vijftigschild L. A., Kruisselbrink E., Vonk A. M., Dekkers J. F., Janssens H. M., et al. (2016). Limited premature termination codon suppression by read-through agents in cystic fibrosis intestinal organoids. J. Cyst. Fibros. 15 158–162. 10.1016/j.jcf.2015.07.007 [DOI] [PubMed] [Google Scholar]
- Zucker D. R., Ruthazer R., Schmid C. H. (2010). Individual (N-of-1) trials can be combined to give population comparative treatment effect es: methodologic considerations. J. Clin. Epidemiol. 63 1312–1323. 10.1016/j.jclinepi.2010.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]