

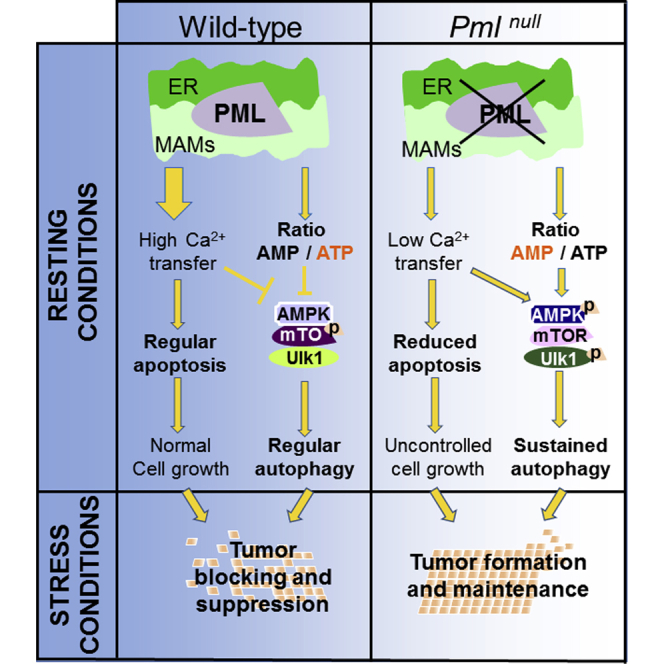
Summary
The precise molecular mechanisms that coordinate apoptosis and autophagy in cancer remain to be determined. Here, we provide evidence that the tumor suppressor promyelocytic leukemia protein (PML) controls autophagosome formation at mitochondria-associated membranes (MAMs) and, thus, autophagy induction. Our in vitro and in vivo results demonstrate how PML functions as a repressor of autophagy. PML loss promotes tumor development, providing a growth advantage to tumor cells that use autophagy as a cell survival strategy during stress conditions. These findings demonstrate that autophagy inhibition could be paired with a chemotherapeutic agent to develop anticancer strategies for tumors that present PML downregulation.
Graphical Abstract
Highlights
-
•
PML regulates autophagic processes from ER/MAM domains in a Ca2+-dependent manner
-
•
Localization of PML away from the MAMs is dependent on p53
-
•
Activation of autophagy by PML depletion promotes survival under stress conditions
-
•
Block of autophagy restores the activity of chemotherapy in PML-downregulated tumors
Missiroli et al. demonstrate that the tumor suppressor promyelocytic leukemia protein (PML) works as a repressor of autophagy by controlling autophagosome formation at mitochondria-associated membranes (MAMs) in a p53-dependent manner. Together, their studies generate alternative anticancer strategies for tumors that present PML downregulation.
Introduction
To become cancerous, cells must overcome the foolproof mechanism of cell death, thereby reducing their propensity to activate self-destructive catabolic pathways in response to hostile environmental clues (Hanahan and Weinberg, 2011). The induction of apoptosis is the major route of cell death that is targeted by many chemotherapeutic drugs, yet it is antagonized by multiple cellular processes, including autophagy.
Autophagy plays dual roles in cancer; it can function as either a tumor suppressor by preventing the accumulation of damaged proteins and organelles or a survival pathway by suppressing apoptosis and promoting the growth of established tumors (Booth et al., 2014, Eisenberg-Lerner et al., 2009, Maiuri et al., 2007, Mariño et al., 2014, Su et al., 2015).
Recent studies have indicated that autophagy represents an important mechanism underlying chemotherapy resistance in leukemia (Sehgal et al., 2015) and in solid cancers (Eisenberg-Lerner et al., 2009, Sehgal et al., 2015), although the exact molecular mechanism underlying the effects of autophagy on tumorigenesis must be further elucidated.
Here, we propose a role for promyelocytic leukemia protein (PML) in the negative regulation of autophagy. PML is a tumor suppressor that was initially identified because of its dysregulation during the pathogenesis of acute promyelocytic leukemia (APL) (Piazza et al., 2001). The fusion oncoprotein PML/RARα can activate constitutive autophagy in APL cells, thereby contributing to the anti-apoptotic function of PML/RARα. However, the precise mechanisms by which PML regulates autophagy remain unknown.
Because PML dysregulation is associated with an extensive range of malignancies, including solid tumors (Gurrieri et al., 2004), we reasoned that completely understanding all of the molecular pathways that require PML for the control of cell death and, consequently, of cancer progression is fundamental.
Results
PML Represses the Autophagic Process
To determine the possible involvement of Pml in the autophagic process, we monitored autophagosome levels in wild-type (Pml+/+) and Pml−/− primary mouse embryonic fibroblasts (MEFs) either under normal conditions (fed) or after serum deprivation (starved). Autophagosomes were detected in live-imaging experiments as fluorescent cytoplasmic dots that concentrated microtubule-associated proteins 1 light chain 3A (MAP1LC3A, best known as LC3) fused to GFP. Such GFP-LC3-positive dots were more frequent in Pml−/− MEFs than in wild-type (WT) MEFs under basal conditions (Figures 1A and 1B). The redistribution of LC3 to autophagosomes is usually accompanied by its lipidation, causing an increase in its electrophoretic mobility (and hence a shift from LC3-I to LC3-II) (Klionsky et al., 2012). Accordingly, detection of the conversion of LC3-I to LC3-II via immunoblotting confirmed that Pml−/− MEFs contained higher levels of LC-3-II than WT MEFs under basal conditions (Figure 1C). Following nutrient deprivation, autophagosome formation was induced in WT MEFs at levels similar to those found in Pml−/− cells under fed conditions; conversely, autophagosome formation did not significantly change after starvation and other pro-autophagic stimuli in Pml−/− MEFs (Figures 1A–1C and S1A). Transmission electron microscopy (TEM) confirmed the increase in baseline autophagosomes in Pml−/− MEFs (Figure 1D).
Figure 1.

PML Represses Autophagic Processes
(A) Percentages of GFP-LC3 puncta-positive cells in Pml WT and KO MEFs transfected with the GFP-LC3 plasmid under basal conditions (fed) and after starvation (starved). Bars, SEM. ∗∗p < 0.01, n = 4.
(B) Representative images of GFP-LC3 puncta in MEFs. Scale bar, 10 μm.
(C) Conversion of endogenous LC3-I to LC3-II, monitored via immunoblotting in Pml+/+ and Pml−/− MEFs cultured in regular medium (fed) or without nutrients (starved).
(D) Ultrastructural evidence of higher autophagy levels in MEFs in the absence of PML compared with WT conditions basal conditions (fed) or after serum deprivation (starved). Scale bar, 500 nm. Bars, SEM. ∗p < 0.05, n = 3.
(E) Immunoblotting of endogenous LC3 in the liver and skeletal muscle of fed or starved (24 hr) Pml+/+ and Pml−/− mice.
(F) Immunoblot of subcellular fractions isolated from Pml+/+ and Pml−/− MEFs, where IP3R3 was used as an ER marker, Sigma 1-R as a MAM marker, Cyt c as a mitochondria marker and ATG14/STX17 as autophagosome formation markers. C, cytosol; ER, endoplasmic reticulum; MAMs, mitochondria-associated membranes; Mp, pure mitochondria.
Increased LC3-II abundance was also detected in the liver and skeletal muscle of adult Pml−/− mice (Figure 1E) compared with WT animals. Interestingly, as observed above in vitro, LC3-II could be induced via starvation (food deprivation for 24 hr) only in WT mice (but not in Pml−/− mice), in which LC3-II reached the same level as that observed in Pml−/− mice under fed conditions. Thus, we analyzed whether Pml might affect the formation of autophagosomes. We found that two autophagosome markers, ATG14 and STX17 (Hamasaki et al., 2013), were shifted to the MAM compartments in Pml−/− MEFs, suggesting increased autophagosome biogenesis in the absence of Pml (Figures 1F and S1B).
Control experiments revealed that short hairpin RNA (shRNA)-mediated knockdown of Pml also increased the number of GFP-LC3 puncta in WT MEFs, while reintroduction of PML into Pml−/− MEFs reduced GFP-LC3 puncta (Figures S1C and S1D). Pharmacological blockade of autophagy using 3-methyladenine (3-MA), an inhibitor of the Beclin-1 (Becl1)-dependent class III phosphoinositide 3-kinase (PI3K), blocked the excessive formation of autophagosomes in Pml−/− MEFs, indicated by the similar amounts of LC3-GFP puncta contained in WT and Pml−/− MEFs (Figure S1E). Accordingly, knockdown of Becl1 with two distinct shRNAs (Boya et al., 2005) reduced the levels of LC3-II in Pml−/− MEFs (Figure S1F). Conversely, in vivo blockade of the last step of autophagy (which depends on lysosomal proteases) by means of leupeptin (Esteban-Martínez and Boya, 2015) failed to reduce the difference in LC3-II formation observed between WT and Pml−/− mouse livers (Figure S1G). Similarly, bafilomycin A1 or NH4Cl, which both abolish the acidification of lysosomes, caused an increased abundance of LC3-II in both WT and Pml−/− MEFs, yet it did not abolish the difference in LC3-II formation between MEFs with the two genotypes, suggesting that the absence of Pml truly induces an increase in autophagic flux (Figures S1H and S1I).
PML Localization at Endoplasmic Reticulum/MAM Contact Sites Is Necessary for Modulating Autophagy
The localization of PML at ER/MAM contact sites (Figures S2A–S2C) is fundamental for its pro-apoptotic activity via a calcium (Ca2+)-mediated pathway (Giorgi et al., 2010). Driven by this consideration, we determined whether PML also plays a role in the control of autophagy in these specialized domains. To address this question, we took advantage of an erPML chimeric protein, which contains the entire PML protein targeted to the outer surface of the ER (Figure S2D) and can rescue sensitivity to apoptosis in Pml−/− MEFs (Giorgi et al., 2010). ErPML (but not a control construct targeted to nuclei, nuPML) localized at MAMs and suppressed the elevated levels of autophagy when re-introduced into Pml−/− MEFs (Figures 2A–2D) comparable to those measured in WT MEFs (Figure 1). Pml−/− MEFs re-transfected with erPML (but not with nuPML) acquired the ability to increase autophagy in response to starvation (Figures 2A–2C). Hence, the presence of PML at MAMs may be important for the repression of autophagy. Accordingly, nutrient deprivation-dependent autophagy induction in WT cells was accompanied by the delocalization of Pml from ER-mitochondria contact sites (Figure S2E).
Figure 2.

PML Localization at ER/MAM Domains Controls the Levels of Autophagy
(A) Percentages of GFP-LC3 puncta-positive cells induced by the transfection of erPML and nuPML chimeras into Pml−/− MEFs under resting conditions (fed) and after serum deprivation (starved). Bars, SEM. ∗∗p < 0.01, n = 4.
(B) Representative images of GFP-LC3 puncta in Pml−/− MEFs before and after re-introduction of the two chimeras, erPML and nuPML. Scale bar, 10 μm.
(C) Immunoblotting to detect LC3 in PML KO MEFs after the re-introduction of erPML and nuPML chimeras both under basal conditions (fed) and after serum deprivation (starved).
(D) Images of autophagic ultrastructures in Pml−/− MEFs following the transfection of erPML and nuPML chimeras. Scale bar, 500 nm. Bars, SEM. ∗∗∗p < 0.005, n = 3.
p53 Drives PML Localization at MAMs
Previously, the role of p53 in the regulation of autophagy was proposed to depend on its localization (Tasdemir et al., 2008). Cytoplasmic p53, particularly when associated with the ER membranes, represses basal pro-survival autophagy, whereas nuclear p53 fails to do so (Tasdemir et al., 2008). However, the precise molecular mechanism by which p53 suppresses autophagy remains elusive.
Because of the tight interaction between p53 and PML (Ablain et al., 2014, Guo et al., 2000, Papa et al., 2012, Pearson and Pelicci, 2001), we sought to investigate the possible effect of PML on autophagy in relation to p53. Therefore, we evaluated p53 and PML localization at points of close contact between the ER and mitochondria in Pml−/− and p53−/− backgrounds, respectively. Subcellular fractionation of MEFs with distinct genotypes revealed that p53 protein was associated with MAMs irrespective of the absence or presence of Pml (Figure S3A), suggesting that Pml does not control the subcellular localization of p53. In stark contrast, in the absence of p53, Pml delocalized from MAMs without an overall change in its expression (Figures S3B and S3C), as shown by comparative subcellular fractionation followed by immunoblots, immunogold detection by electron microscopy (EM) and immunofluorescence (IF) analyses of WT and p53−/− MEFs (Figures 3A–3C and S3D). These results suggest that PML might function as a master regulator of autophagy downstream of p53.
Figure 3.

Delocalization of PML from the MAMs Is p53 Dependent
(A) Immunoblot detection of PML in p53−/− MEF fractions. C, cytosol; ER, endoplasmic reticulum; MAMs, mitochondria-associated membranes; Mp, pure mitochondria.
(B) Pre-embedding immunogold labeling of PML in p53+/+ and p53−/− MEFs. In WT cells, PML localizes mainly at discrete sites in the nucleus (PML nuclear bodies) and on the ER, outer mitochondria membrane (OMM) and MAMs (arrows indicate PML-positive MAMs). In p53−/− MEFs, while PML still localizes at PML nuclear bodies and on the ER membranes, the association with OMM and in particular the presence at MAMs is reduced (arrows indicate PML negative MAMs). Scale bar, left panel, 1 μm; upper-right panel, 500 nm; lower-right panel, 250 nm.
(C and D) The co-localization of PML (red) and Sigma 1-R-EGFP (used as a MAM marker, green) in (C) p53−/− MEFs and in (D) p53−/− MEFs following re-introduction of the erPML chimera was analyzed based on immunofluorescence using confocal images. The lower-left panels display the merged image of the two stains. The lower-right panels display the PML signal overlaid with MAMs (MAM boundaries are highlighted in yellow) in a rainbow lookup table (LUT) (MAMsPML: Manders coefficient for PML staining was calculated according to Manders coefficient method as the proportion of PML signal overlapping with the Sigma 1-R marker). Scale bar, 10 μm.
(E–G) Reduced levels of autophagy were observed in p53−/− MEFs following erPML transfection as determined based on the percentage of LC3-GFP puncta (E and F) and on immunoblotting to detect LC3-I conversion into LC3-II (G). Representative images are shown. Bars, SEM. ∗∗p < 0.01, n = 4. Scale bar, 10 μm.
(H) PML and p53 localization following the re-introduction of p53wt as analyzed via immunoblotting in H1299 p53−/− cell fractions, where IP3R3 was used as an ER marker, Sigma 1-R as a MAM marker, Tubulin as a cytosolic marker and Cyt c as a mitochondria marker. C, cytosol; ER, endoplasmic reticulum; MAMs, mitochondria-associated membranes; Mp, pure mitochondria.
(I and J) Immunofluorescence of Pml (red) and p53 (blue) in H1299 cells after the re-introduction of p53wt (I) and mutant p53K382R (J). The lower-left panels display the merged image of the two stains. The lower-right panels display the Pml signal overlaid with MAMs (MAM boundaries are highlighted in yellow) in a rainbow LUT. Scale bar, 10 μm.
This concept was confirmed by the introduction of the erPML chimera into p53−/− MEFs. Indeed, the forced localization of Pml at MAMs in p53−/− cells (Figure 3D) reduced autophagosome formation (Figures 3E–3G). Similarly, the re-introduction of p53wt, which also localizes at the ER and MAMs (Giorgi et al., 2015b), engages PML at those contact sites (Figures 3H, 3I, and S3E) and suppresses signs of enhanced autophagy in p53−/− cells (Figures S3F and S3G). Furthermore, a p53 mutant (p53K382R) that is unable to inhibit autophagy when re-introduced into p53−/− cells (Morselli et al., 2011) (Figures S3F and S3G) failed to localize at the ER/MAMs in p53−/− cells and also failed to rescue Pml localization to MAMs (Figures 3H, 3J, and S3H). Intriguingly, we observed, by means of proximity ligation assay (PLA), that PML interacts with p53wt in large extent in the nucleus, but also in regions enriched of MAMs marker (Figure S3I). Accordingly, this ability is lost in cells expressing the p53K382R as this mutant is also reported to have reduced ability to bind PML (Pearson et al., 2000). Overall, these results strongly suggest that p53 maintains proper Pml localization at MAM domains that is fundamental for the repression of autophagy.
Pml Controls Autophagy through the AMPK/mTOR/Ulk1 Pathway
Autophagy is activated as a pro-survival mechanism in response to various cellular stresses associated with an insufficient energy supply (Amaravadi et al., 2011, Galluzzi et al., 2014). The autophagy-signaling network is controlled by a variety of kinases. In particular, a complex that includes AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR), and unc-51-like kinase 1 (Ulk1) has emerged as important for the regulation of autophagy (Kim et al., 2011, Nazio et al., 2013). Thus, we examined the relationship between PML-dependent autophagy and this multiprotein complex.
AMPK is a conserved energy-sensing kinase that is activated by phosphorylation in response to low cellular energy levels (Mihaylova and Shaw, 2011). Previously, we reported that, in the absence of PML, cells produced less ATP after agonist stimulation (Giorgi et al., 2010). As the ratio of AMP to ATP represents an important means of regulating the activity of AMPK, we examined the role of AMPK by quantifying its phosphorylation. As expected, Pml−/− MEFs exhibited increased levels of phosphorylated AMPK (Figure 4A), which correlated with enhanced autophagy, compared with that of WT MEFs. Accordingly, the downstream AMPK substrate acetyl CoA carboxylase was hyper-phosphorylated (Figure 4A). Higher levels of AMPK phosphorylation, correlating with increased autophagy, were also observed in the liver and skeletal muscle of adult Pml−/− mice compared to WT (Figure 4B).
Figure 4.

PML Modulates Autophagy through the AMPK/mTOR/Ulk1 Pathway in a Ca2+-Dependent Manner
(A) Immunoblot detection of the phosphorylation status of AMPK, ACCα, p70S6K, mTOR, and Ulk1 in Pml+/+ and Pml−/− MEFs.
(B) Detection of autophagy and AMPK-mTOR-Ulk1 phosphorylation levels in the liver and skeletal muscle of Pml+/+ and Pml−/− mice.
(C) Representative traces of increased mitochondrial Ca2+ levels in Pml−/− MEFs after MCU overexpression. Pml−/−: [Ca2+]m peak 33.7 ± 2.55; Pml−/− + MCU: [Ca2+]m peak 59.2 ± 6.22) SEM. ∗∗p < 0.01, n = 3.
(D–F) Quantification of autophagy in Pml−/− MEFs following MCU overexpression via (D and E) analysis of GFP-LC3 puncta or (F) immunoblotting. Representative images are shown. Bars, SEM. ∗∗p < 0.01, n = 3. Scale bar, 10 μm.
(G) Schematic model of autophagy regulation by PML. In the absence of Pml, the release of Ca2+ from the ER into the mitochondria and the production of ATP are reduced. This low-energy status induces AMPK activation, mTOR inhibition, and Ulk1 phosphorylation, leading to increased autophagy.
In contrast to AMPK, mTOR activity reflects a higher energy status with the consequent inhibition of autophagy (Kim et al., 2011). AMPK and mTOR have recently been proposed to regulate autophagy through the coordinated phosphorylation of Ulk1 (Egan et al., 2011, Kim et al., 2011). To determine whether AMPK activation in the absence of Pml promotes autophagy by activating Ulk1, we examined the phosphorylation levels of both Ulk1 and mTOR. In agreement with the findings of Kim et al. (2011), increased levels of LC3 lipidation in either Pml−/− cells or Pml KO mice tissues were associated with Ulk1 hyper-phosphorylation (which reflects Ulk1 activation), together with reduced phosphorylation of mTOR and its substrate p70S6K (which reflects mTOR inhibition) (Figures 4A and 4B). These results are consistent with the possibility that PML controls autophagy through the AMPK/mTOR/Ulk1 pathway.
The critical role of Ca2+ in the formation of autophagosomes has recently been emphasized (Cárdenas et al., 2010). In addition to a high concentration of AMP, AMPK activity is upregulated through the inhibition of mitochondrial Ca2+ uptake due to reduced Ca2+ release via the inositol triphosphate receptor (IP3R) (Cárdenas et al., 2010). Pml−/− cells display a reduced Ca2+ transfer from the ER to mitochondria via IP3R, which protects cells from apoptosis (Giorgi et al., 2010). To verify whether downregulated ER-mitochondrial Ca2+ transfer is important for the induction of autophagy in Pml−/− cells, we increased mitochondrial Ca2+ uptake in Pml−/− cells by overexpressing the mitochondrial Ca2+ uniporter (MCU) (Baughman et al., 2011, De Stefani et al., 2011). MCU overexpression in Pml−/− MEFs increased the ability of mitochondria to accumulate Ca2+ (Figure 4C) and was sufficient to repress autophagy (Figures 4D–4F) by reducing the amount of activated AMPK (Figure 4F), suggesting that PML controls autophagy at ER-mitochondria contact sites by its effects on Ca2+ homeostasis (Figure 4G).
Loss of PML Enhances Resistance to Metabolic Stress
As a homeostatic process, autophagy plays a crucial role during metabolic stress in an attempt to maintain/restore cellular homeostasis (Degenhardt et al., 2006, Rabinowitz and White, 2010). Upon starvation, WT cells exhibited a rapid decrease in ATP production, as demonstrated previously (Tasdemir et al., 2008), whereas cells lacking PML maintained high ATP levels during stress (Figure 5A). Accordingly, Pml−/− cells maintained a higher mitochondrial membrane potential (Ψm) under stress conditions compared to WT controls (Figure 5B). This autophagy-dependent energized status of Pml KO cells conferred enhanced resistance to cell death (Figure 5C), favoring cell growth under unfavorable conditions (Figure 5D), such as those present in tumors.
Figure 5.

PML Deletion Favors Cell Survival under Stress Conditions Due to Autophagy Activation
(A) Cytosolic ATP levels in Pml+/+ and Pml−/− MEFs as measured by luciferase expression under starvation conditions (glucose deprivation for 15 min). ∗∗p < 0.01, compared to WT conditions, n = 3.
(B) Analysis of mitochondrial membrane potential (Ψm) as measured by TMRM intensity in Pml+/+ and Pml−/− MEFs. Where indicated, cells were deprived of glucose or exposed to 1 μM carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP). On the bottom-right side, representative images of the TMRM signal in the presence or absence of glucose are shown. Normalized TMRM intensity is displayed as a rainbow LUT (statistical analysis cross, average; line, median; box, 25 and 75 percentile; bars, maximum and minimum values; ∗p < 0.05, n = 3). Scale bar, 10 μm.
(C and D) Pml−/− cells are more resistant to metabolic stress-induced cell death than Pml+/+ cells as confirmed by (C) immunoblot for apoptotic markers and by (D) cell viability assay. Metabolic stress is induced by glucose deprivation (3 hr for western blot and the indicated hours for cell viability). Bars, SEM. ∗p < 0.05, ∗∗∗p < 0.005, n = 3.
(E) Basal-dependent (i) and ATP synthase-dependent (ii, + Oligomycin) mitochondrial O2 consumption rates (OCR) in Pml−/− and Pml+/+ MEFs under starvation conditions (glucose, pyruvate, and glutamine deprivation for 1 hr). Bars, SEM. ∗∗p < 0.01, ∗∗∗p < 0.005, n = 4.
We confirmed this energized status of Pml KO cells by analyzing mitochondrial function in response to metabolic stress. The oxygen consumption rate (OCR) was used as readout of mitochondrial basal respiration and ATP-linked respiration in Pml+/+ and Pml−/− MEFs. As shown in Figure 5E, the loss of PML under basal conditions induced only a minor reduction in basal and ATP-dependent OCRs. Interestingly, when MEFs were deprived from glucose, WT cells displayed a dramatic reduction in both basal and ATP-dependent OCRs, while the OCR was only slightly affected in Pml−/− cells.
Accordingly, either genetic inhibition of autophagy (Becn1 shRNA) or re-introduction of the erPML chimera into Pml−/− MEFs prevented the protection of such cells from death under glucose-depleting conditions (Figures S4A–S4D). These results indicate that enhanced autophagy is responsible for the resistance of PML-deficient cells to nutrient stress.
Targeting Autophagy in PML-Null Cells Rescues Chemotherapy-Induced Cell Death
Accumulating evidence has indicated that malignant cells in established tumors utilize autophagy to resist chemotherapy-induced cell death. Indeed, solid tumors in particular develop regions of hypoxia and limited nutrient availability in their centers, creating a microenvironment of metabolic stress. Under these hostile conditions, autophagy activation promotes cell survival by providing missing nutrients (Maes et al., 2013, Xu et al., 2013). Accordingly, the inhibition of autophagy augments the response of tumor cells to anticancer drugs (Chittaranjan et al., 2014, Sehgal et al., 2015, Selvakumaran et al., 2013).
To confirm the role of PML in adaptation to metabolic stress, we directly analyzed autophagy levels, mitochondrial respiration, and PML localization in human NB4 promyelocytic leukemia cells expressing the PML-RARα oncogenic fusion protein, which causes neoplastic transformation by disrupting the function of PML.
Treatment with arsenic trioxide (ATO), typically used in APL therapy to degrade the PML-RARα (Lallemand-Breitenbach et al., 2001), modulated autophagic machinery in NB4 cells (Figures 6A–6C). Interestingly, we found that short-term treatment of ATO, promoting the selective proteasome-dependent degradation of PML-RARα fusion protein but not of PML, led to an increase in both p62 and LC3-II levels, indicating a block in the autophagic process (Figure 6C). Accordingly, treatment with an inhibitor of autophagic flux, reported in Figure 6D, suggested that ATO interferes with autophagosome clearance (Ren et al., 2011) and thus reverts the APL condition through reduced autophagy efficacy.
Figure 6.

Selective Degradation of the PML-RARα Oncogenic Fusion Protein in APL Human Cancer Restores a Correct PML Localization and the Sensitivity to Metabolic Stress
(A) Quantification of LC3 dots in NB4 cells transfected with the GFP-LC3 plasmid under basal conditions (0 hr) and after ATO (1 μM) treatment. Bars, SEM. ∗∗p < 0.01, n = 4.
(B) Representative images of GFP-LC3 dots showing the effects of ATO on autophagic process in NB4 cells.
(C) PML-RARα degradation and LC3-II and p62 accumulation after ATO treatment (1 μM) in NB4 cells. PML-RARα was detected monitoring the band at 110 kDa either with a PML antibody or with a RARα antibody.
(D) Analysis of autophagic flux in NB4 cells after ATO treatment (1 μM).
(E) Quantification of basal (i) and Oligomycin ATP synthase-dependent (ii) mitochondrial O2 consumption rates (OCR) either in vehicle or ATO treated NB4 cells under starvation conditions (glucose, pyruvate, and glutamine deprivation for 1 hr). Bars, SEM. ∗p < 0.05, n = 7.
(F) Immunoblot of subcellular fractions isolated from NB4 cells and PML protein quantification at MAM regions and ER normalized to the amount of Sigma 1-R and IP3R3. Where indicated, cells were exposed to ATO treatment (1 μM, 12 hr). IP3R3, Sigma 1-R and Cyt c was used as ER, MAMs, and mitochondria marker, respectively.
Moreover, NB4 cells displayed a higher resistance to metabolic stress (by means of measurements of oxygen consumption during substrate deprivation) that reverted after ATO treatment (Figure 6E), which also restored proper PML localization at ER/MAM contact sites already in the leukemia background (Figure 6F).
To further analyze the role of PML in cancer development, we compared the sensitivity of Pml+/+ and Pml−/− MEFs to the chemotherapeutic agent 5-fluorouracil (5-FU) (Li et al., 2010). As expected, Pml−/− cells were protected from apoptosis (Giorgi et al., 2010) compared to WT cells (Figures 7A and 7B). Pharmacological inhibition of autophagy by either 3-methyladenine (3-MA) or the antimalarial drug chloroquine (CQ) (Figure S5A) alone did not alter cell viability, yet it markedly increased apoptosis when combined with 5-FU, and this effect was particularly remarkable for Pml−/− MEFs (Figures 7A and 7B).
Figure 7.

Inhibition of Autophagy Augments the Cytotoxicity of Chemotherapy Treatment in Pml KO Tumors
(A and B) Pml+/+ MEFs are sensitive to 5-fluoracil-induced cell death (5-FU, 25 μM for 16 hr), whereas Pml−/− cells are resistant. Inhibition of autophagy by treatment with 3-MA (2.5 mM for 16 hr) or CQ (5 μM for 16 hr) increases apoptosis induced via chemotherapy treatment with 5-FU (25 μM for 16 hr) in Pml−/− MEFs.
(C) Tumor growth of Pml−/− and Pml+/+ transformed MEF xenografts.
(D) Representative images of mouse fibrosarcoma xenografts.
(E) Increased levels of autophagy in tumors derived from Pml−/−-transformed MEFs as analyzed by immunoblotting. Tumors were excised on day 19 after inoculation.
(F) Analysis of apoptosis based on the intensity of fluorescence (SR-FLIVO) emitted in tumor tissue sections, accompanied by statistical analysis (cross, average; line, median; box, 25 and 75 percentile; bars, maximum and minimum values; ∗∗∗p < 0.005, n = 3). Scale bar, 50 μm.
(G) Increased cytotoxicity-chemotherapy effects on Pml−/− tumors after autophagy inhibition.
(H) Representative histological sections of human colon cancer immunostained for PML and LC3 accompanied by statistical analysis expressed as the percentage of staining intensity. Bars, SEM. ∗p < 0.05, ∗∗∗p < 0.005, n = 13. Magnification 20×.
Next, we explored whether the synergistic effect of CQ and 5-FU was also applicable in vivo. Transformed Pml+/+ and Pml−/− MEFs that displayed levels of autophagy comparable to those of primary cells (Figure S5B) were injected subcutaneously either into immunodeficient nu/nu mice or in syngeneic mice, and the resultant tumor volumes were monitored. Pml−/− cells exhibited a particularly high tumorigenic potential both in vitro (Figures S5C–S5E) and in vivo (Figures 7C and S5F), developing tumors that were 10-fold larger than those generated from WT cells (Figures 7C, 7D, and S5G) and with elevated signs of autophagy (Figures 7E and S5H).
Compared with the control conditions (PBS), 5-FU treatment slightly suppressed tumor growth, whereas 5-FU combined with CQ greatly reduced the sizes of Pml−/− tumors (Figures 7C and 7D). Treatment with CQ alone had no significant effects on tumor growth (Figures S5I and S5J). Genetic inhibition of autophagy (by means of Becn1 shRNA) in Pml−/− cells reduced their tumorigenic potential either in vitro (Figures S6A and S6B) or in vivo (Figures S6C and S6D) and restored their sensitivity to chemotherapy (Figure S6E).
Intravenous injection of a fluorescent probe (SR-FLIVO) was used to detect apoptosis-associated caspase activation within the tumors (Giorgi et al., 2015a). Pml−/− tumors were completely insensitive to 5-FU-induced apoptosis (Figure 7F), correlating with higher levels of autophagy (Figure 7E), yet they responded to the combined treatment with 5-FU and CQ by vigorous apoptosis induction (Figure 7F) and immunoblot-detectable caspase-3 maturation (Figure 7G). Knockdown of Beclin-1 in Pml−/− cells led to the generation of tumors that contained areas undergoing apoptosis (Figures S6F and S6G) and that were less abundant in Pml−/− tumors transfected with a scrambled control shRNA. Hence, elevated autophagy may be responsible for the growth advantage of Pml−/− cancers. Indeed, in human colonic adenocarcinoma samples with reduced PML protein expression, levels of LC3 detectable by immunohistochemistry were relatively higher compared to those found in cancer tissues with PML protein abundance (Figure 7H).
Discussion
Loss of PML Confers a Metabolic Advantage to Cells under Nutrient Deprivation
Autophagy is an important homeostatic mechanism with a paradoxical dual role: it can function as a cell survival pathway, suppressing apoptosis, or it can lead to death. As a result, the connection between autophagy and apoptosis is complex and affects the treatment of diseases associated with cell death. Alteration of the autophagic machinery is associated with various human disorders, establishing autophagy as a therapeutic target (i.e., a target for drug development). The connection of autophagy with the development of cancer has been well established, although the exact roles played by this process during various stages of tumor progression are not yet clear and, in some cases, are contradictory.
Here, we provide molecular insight into the acquisition of tumor-promoting behavior due to the loss of the PML protein. Pml is a tumor suppressor gene that was originally related to the pathogenesis of acute promyelocytic leukemia (APL). However, the loss of PML was later found to be a critical event in various human cancers (Gurrieri et al., 2004). We recently identified an extra-nuclear transcription-independent function of PML in regulating cell survival through changes in Ca2+ signaling at ER/mitochondria contact sites (Giorgi et al., 2010). Accumulating evidence has suggested that ER and MAM domains play a prominent role in autophagy (Hamasaki et al., 2013, Heath-Engel et al., 2008) and emphasized the important role of IP3R and Ca2+ in autophagosome formation (Cárdenas et al., 2010, Criollo et al., 2007); therefore, we sought to investigate the role of PML in these processes.
We clearly demonstrated that the localization of PML at the ER-mitochondria contact sites is fundamental not only for apoptosis control, but also for autophagy regulation. At these sites, PML represses autophagosome formation and, thus, autophagy induction. We found that the mislocalization of PML away from MAMs is necessary to activate autophagy in response to stress and that the same mechanism occurs in the absence of p53. Our data demonstrate that p53, which also localizes at ER/MAMs (Giorgi et al., 2015b), operates as a bridge to maintain the correct localization of PML. p53K382R mutants, which display reduced interactions with PML (Pearson et al., 2000) and are unable to localize at MAMs, cannot preserve PML localization, making them incapable of suppressing autophagy.
PML loss reduced IP3R-mediated Ca2+ transfer from the ER to mitochondria. Mitochondrial Ca2+ stimulates tricarboxylic acid (TCA) cycle dehydrogenases and subsequent mitochondrial respiration and ATP production. A reduction in [Ca2+]m inhibits this mechanism that activates AMPK (Cheng et al., 2013). AMPK activates pro-survival autophagy (Cárdenas et al., 2010) by a mechanism involving mTOR (Tasdemir et al., 2008) and Ulk-1 pathways (Kim et al., 2011, Nazio et al., 2013). In our scenario, PML-deprived cells use enhanced baseline levels of autophagy to obtain an advantage during metabolic stress conditions. Indeed, restoration of the Ca2+ signal in the context of a Pml−/− or p53−/− background (by overexpression of MCU) restores basal levels of autophagy. Cells lacking PML due to the inhibition of Ca2+ release activity have slightly diminished bioenergetics in terms of mitochondrial respiration and ATP production that are sensed by AMPK, which activates autophagy. Because oxygen and glucose supplies are often low in the tumor microenvironment, autophagy, as catabolic process, is necessary to obtain alternative carbons source to preserve normal cellular bioenergetics (Cárdenas et al., 2010) and to confer resistance to cell death to Pml KO cells. Lipid or glutamine catabolism is considered the major alternative pathway to provide carbons for mitochondria (Boroughs and DeBerardinis, 2015). PML promotes lipid utilization by fatty acid oxidation (Carracedo et al., 2013, Ito et al., 2012), excluding this pathway as a possible stimulated carbon source when PML is lost. Furthermore, autophagy can sustain mitochondrial respiration in a glutamine metabolism-dependent manner (Lin et al., 2012, Strohecker et al., 2013). Thus, we could speculate that in the absence of PML, stimulated glutamine catabolism by autophagy is used as an alternative pathway to sustain mitochondrial activity and, thus, cell survival during stress conditions.
These data, together with data presented in our previous publication, indicate that PML from MAMs acts on the transfer of Ca2+ from the ER to the mitochondria, thus regulating both apoptosis and autophagy. While the effect on apoptosis is manifested in response to Ca2+-dependent apoptotic stimuli, the stimulation of autophagy is a direct consequence of reduced mitochondrial respiration. Thus, the loss of PML from MAMs confers contemporary resistance to apoptotic stimuli and metabolic stress, promoting cell survival in the tumor environment.
Recent studies have suggested that autophagy represents an important mechanism of resistance to cancer treatments. Therefore, we further investigated the cross-talk between autophagy and PML-related cell death using in vivo models. The prototypical PML-RARα-related promyelocytic leukemia model, NB4, displays low levels of WT PML at MAMs. Exposure to ATO induces degradation of PML-RARα and increased PML levels at MAMs. As a result, ATO-treated cells display blocked autophagy and reduced resistance to metabolic stress. Furthermore, we observed that the absence of PML promotes tumor development associated with resistance to anticancer drugs due to increased autophagy levels in the tumor. Treatment with the autophagy inhibitors 3-MA and CQ restored chemotherapy-related apoptosis. Therefore, our findings hold therapeutic implications for the treatment of solid tumors associated with PML downregulation.
Experimental Procedures
Cell Culture and Transfection
Primary Pml+/+ and Pml−/− mouse embryonic fibroblasts (MEFs) and H1299 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) (Life Technologies), 1% penicillin-streptomycin-glutamine (100×) liquid (EuroClone).
Human APL NB4 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% FBS (Gibco) and 1% penicillin-streptomycin (100×) liquid (Gibco).
MEFs were transformed with SV40 large T antigen. MEFs were transfected with the following plasmids: PML, erPML, nuPML (Giorgi et al., 2010), p53WT, p53K382R (Morselli et al., 2011), MCU (De Stefani et al., 2011), and GFP-LC3 using a MicroPorator (Digital Bio). For PML depletion in WT MEFs, a specific shRNA silencing lentiviral vector (Sigma) was used. H1299 cells were transfected using a standard calcium phosphate procedure with p53WT and p53K382R constructs, while NB4 cells were transfected with a GFP-LC3 plasmid by electroporation.
Procedures involving animals and their care were in conformity with institutional guidelines, and all experimental protocols were approved by the animal ethics committee. Pml+/+ and Pml−/− mice were bred and maintained according to both the Federation for Laboratory Animal Science Associations and the Animal Experimental Ethics Committee guidelines.
Autophagy Induction and Inhibition
The autophagic process was triggered in vitro as follows: serum deprivation (EBSS, 1 hr), glucose deprivation (1 hr), rapamycin (100 nM, 24 hr in DMEM with 10% FBS), and lithium treatment (10 mM, 24 hr in DMEM with 10% FBS). Pharmacological inhibition of autophagy was performed by treating cells with 2.5 mM 3-MA (16 hr) or 5 μM CQ (16 hr) in DMEM with 10% FBS (Klionsky et al., 2016, Mizushima et al., 2010). Genetically inhibition of autophagy was performed by using shRNA targeting Beclin1 (psirna42-mbeclin) (Invitrogen). ATO (arsenic trioxide, 1 μM) has been used on NB4 cells for 1 or 12 hr in RPMI with 10% FBS). For in vivo studies on the effects of starvation, mice were deprived of food for 24 hr.
Analysis of Autophagic Flux
Primary Pml+/+ and Pml−/− MEFs were treated either with the autophagy inhibitor NH4Cl (20 mM) or bafilomycin A1 (BafA1, Sigma) (100 nM) in DMEM with 10% FBS (Klionsky et al., 2012, Mizushima et al., 2010). NB4 cells were treated with the autophagy inhibitor NH4Cl (20 mM) in DMEM with 10% FBS. For in vivo determination of autophagic flux, lysosomal activity was blocked by leupeptin (Boya et al., 2005).
TEM
Primary Pml+/+ and Pml−/− MEFs and Pml−/− MEFs expressing nuPML and erPML were cultured in DMEM with 10% FBS and allowed to grow to 70% confluence. Where indicated, the cells were subjected to serum starvation conditions (EBSS, 30 min). After the cells received the indicated treatments, they were fixed in formaldehyde and glutaraldehyde. Fixed samples were post-fixed with osmium tetroxide, dehydrated, and embedded in Araldite Durcupan ACM (Fluka).
Pre-embedding Immunogold Electron Microscopy
Cells were fixed with 4% formaldehyde in 0.1 M phosphate buffer (pH 7.4) for 1 hr at room temperature. Subsequently, cells were permeabilized, blocked, and incubated sequentially with PML primary antibodies (Millipore MAB3738 or Abcam ab72137) and nanogold-conjugated secondary antibodies (Nanoprobes) diluted in blocking buffer. After washes, cells were re-fixed in 1% glutaraldehyde, and nanogold was enlarged with gold enhancement solution (Nanoprobes). Cells were post-fixed with osmium tetroxide, embedded in epon, and processed into ultrathin slices. After contrasting with uranyl acetate and lead citrate, sections were analyzed with a Zeiss LEO 512 electron microscope.
Aequorin Measurements
All aequorin measurements were performed transfecting cells with the appropriate aequorin chimera targeted to the mitochondria (mtAEQmut), as previously described (Bonora et al., 2013).
Luciferase Measurements
Primary Pml+/+ and Pml−/− MEFs were transfected with cytosolic (untargeted) firefly luciferase, and luminescence was measured as described (Jouaville et al., 1999).
Subcellular Fractionation
Fractionations were performed as described previously (Vance, 1990, Wieckowski et al., 2009). IP3R3, Sigma 1-R, tubulin, and cytochrome c (Cyt c) were used as markers for the ER, MAMs, cytosol, and pure mitochondria, respectively.
Immunofluorescence Assay
Cells were fixed in 3.7% formaldehyde, washed, permeabilized with 0.1% Triton X-100, and incubated overnight at 37°C in a wet chamber with indicated antibodies. Images were acquired using an LSM 510 laser scanning confocal microscope (Zeiss).
Immunohistochemistry
Four-micrometer-thick sections were cut from formalin-fixed paraffin-embedded blocks. One section for each block was routinely stained with H&E for histological examination. Immunodetection of PML and LC3B was performed using the Multimeric Detection Kit (Universal DAB Detection Kit Ultraview, Roche Tissue Diagnostics) on a BenchMark XT automated immunostainer (Gurrieri et al., 2004, Rosenfeldt et al., 2012).
Measurements of Mitochondrial Membrane Potential
Mitochondrial membrane potential (Ψm) was assessed by loading cells with 10 nM tetramethyl rhodamine methyl ester (TMRM; Life Technologies, T-668) for 35 min at 37°C in Krebs-ringer bicarbonate (KRB) supplemented with 1 mM CaCl2. Images were acquired using an inverted confocal microscope.
XF Bioenergetic Analysis
OCRs in Pml+/+ and Pml−/− MEFs and NB4 cells were measured using a Seahorse XF96 instrument (Seahorse Biosciences) according to the manufacturer’s protocols.
Cell Proliferation and Viability Assay
Cells seeded in 6-well plates in triplicate and allowed to grow to 80% confluence were exposed to glucose deprivation for different time points (0, 1, 3, and 5 hr). Next, cells were centrifuged, washed with PBS, and counted using a Tali image-based cytometer (Life Technologies).
Cell Survival Assay
Cells seeded in 12-well plates in triplicate and allowed to grow to 80% confluence were treated with vehicle (positive control), 25 μM 5-FU, 5 μM CQ or 25 μM 5-FU, and 5 μM CQ. After 48 hr, the cells were washed with PBS, fixed in 4% paraformaldehyde, and stained with 0.1% crystal violet. Crystal violet was dissolved with 1 mol/l acetic acid, and absorbance at 595 nm was measured.
Wound-Healing Assay
Pml+/+ and Pml−/− MEFs seeded in 6-well plates and grown to 90% confluence were treated with vehicle (positive control), 25 μM 5-FU, 5 μM CQ or 25 μM 5-FU, and 5 μM CQ 1 hr before wounding. The cell monolayer was wounded with a P200 pipette tip. Cells were monitored and captured by phase-contrast microscopy.
Migration Assay
In vitro cell migration assays were performed using Costar Transwell permeable polycarbonate supports (8.0-mm pores) in 24-well plates (Corning). Before seeding, the lower compartment was incubated with DMEM plus 10% FBS supplemented with vehicle (positive control), 25 μM 5-FU, 5 μM CQ or 25 μM 5-FU, and 5 μM CQ. Non-migrated cells were removed using a cotton swab, and migrated cells were fixed and stained with crystal violet.
Mouse Treatment Studies
Tumor xenografts or syngeneic tumors were obtained by subcutaneously implanting 5 × 106 transformed Pml+/+ and Pml−/− MEFs in 50% Matrigel (BD Biosciences) in female nude mice or sv129 mice, respectively. Tumor growth was monitored daily, and tumor volumes were measured every other day.
Detection of Cell Death In Vivo
After an intravenous (i.v.) injection of 100 μl SR-FLIVO (Immunochemistry Technologies) via the lateral tail vein, the FLIVO reagent was allowed to circulate in the mouse for 30 min before sacrifice. The tumors were excised, frozen, sectioned, and stained for nuclei using DRAQ5 according to the manufacturer’s protocol (Cell Signaling Technology). Images were acquired using an inverted confocal microscope.
Statistical Analysis
Unless otherwise indicated, all assays were performed independently and in triplicate, yielding comparable results. The data, which are presented as the means ± SEM, were analyzed using Microsoft Excel (Microsoft), and significance was determined by Student’s t tests. ∗∗∗p < 0.005, ∗∗p < 0.01, and ∗p < 0.05 were considered significant.
Author Contributions
C.G. conceived and designed the experiments. C.G., G.K., and P.P. wrote the manuscript. S.M., M.B., S.P., and M.P. conducted the experiments. F.P. helped with mouse experiments. The immunohistochemistry (IHC) was performed by G.L., R.G., and E.M. TEM and EM analyses were performed by C.T. and A.R. The data were analyzed by S.M., M.B., S.P., C.T., G.L., P.P.P., G.K., P.P., and C.G.
Acknowledgments
This study was supported mainly by the Italian Association for Cancer Research (AIRC) to C.G. (MFAG-13521). C.G. is supported also by the Italian Ministry of Health and local funds from the University of Ferrara. P.P. is grateful to Camilla degli Scrovegni for continuous support and his lab is supported by Telethon (GGP15219/B), AIRC (IG-14442), the Italian Cystic Fibrosis Research Foundation (19/2014), the Italian Ministry of Education, University and Research (COFIN no. 20129JLHSY_002, FIRB no. RBAP11FXBC_002, and Futuro in Ricerca no. RBFR10EGVP_001), local funds from the University of Ferrara and the Italian Ministry of Health. S.P. was supported by a research fellowship FISM—Fondazione Italiana Sclerosi Multipla—cod. 2014/B/3.
Published: August 18, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.082.
Supplemental Information
References
- Ablain J., Rice K., Soilihi H., de Reynies A., Minucci S., de Thé H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat. Med. 2014;20:167–174. doi: 10.1038/nm.3441. [DOI] [PubMed] [Google Scholar]
- Amaravadi R.K., Lippincott-Schwartz J., Yin X.M., Weiss W.A., Takebe N., Timmer W., DiPaola R.S., Lotze M.T., White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011;17:654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman J.M., Perocchi F., Girgis H.S., Plovanich M., Belcher-Timme C.A., Sancak Y., Bao X.R., Strittmatter L., Goldberger O., Bogorad R.L. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M., Giorgi C., Bononi A., Marchi S., Patergnani S., Rimessi A., Rizzuto R., Pinton P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat. Protoc. 2013;8:2105–2118. doi: 10.1038/nprot.2013.127. [DOI] [PubMed] [Google Scholar]
- Booth L.A., Tavallai S., Hamed H.A., Cruickshanks N., Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2014;26:549–555. doi: 10.1016/j.cellsig.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs L.K., DeBerardinis R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P., González-Polo R.A., Casares N., Perfettini J.L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas C., Miller R.A., Smith I., Bui T., Molgó J., Müller M., Vais H., Cheung K.H., Yang J., Parker I. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A., Cantley L.C., Pandolfi P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Guo S., Liu Y., Chu H., Hakimi P., Berger N.A., Hanson R.W., Kao H.Y. Ablation of promyelocytic leukemia protein (PML) re-patterns energy balance and protects mice from obesity induced by a Western diet. J. Biol. Chem. 2013;288:29746–29759. doi: 10.1074/jbc.M113.487595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittaranjan S., Bortnik S., Dragowska W.H., Xu J., Abeysundara N., Leung A., Go N.E., DeVorkin L., Weppler S.A., Gelmon K. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014;20:3159–3173. doi: 10.1158/1078-0432.CCR-13-2060. [DOI] [PubMed] [Google Scholar]
- Criollo A., Maiuri M.C., Tasdemir E., Vitale I., Fiebig A.A., Andrews D., Molgó J., Díaz J., Lavandero S., Harper F. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007;14:1029–1039. doi: 10.1038/sj.cdd.4402099. [DOI] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K., Mathew R., Beaudoin B., Bray K., Anderson D., Chen G., Mukherjee C., Shi Y., Gélinas C., Fan Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Lerner A., Bialik S., Simon H.U., Kimchi A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- Esteban-Martínez L., Boya P. Autophagic flux determination in vivo and ex vivo. Methods. 2015;75:79–86. doi: 10.1016/j.ymeth.2015.01.008. [DOI] [PubMed] [Google Scholar]
- Galluzzi L., Pietrocola F., Levine B., Kroemer G. Metabolic control of autophagy. Cell. 2014;159:1263–1276. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C., Ito K., Lin H.K., Santangelo C., Wieckowski M.R., Lebiedzinska M., Bononi A., Bonora M., Duszynski J., Bernardi R. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330:1247–1251. doi: 10.1126/science.1189157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C., Bonora M., Missiroli S., Poletti F., Ramirez F.G., Morciano G., Morganti C., Pandolfi P.P., Mammano F., Pinton P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget. 2015;6:1435–1445. doi: 10.18632/oncotarget.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C., Bonora M., Sorrentino G., Missiroli S., Poletti F., Suski J.M., Galindo Ramirez F., Rizzuto R., Di Virgilio F., Zito E. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA. 2015;112:1779–1784. doi: 10.1073/pnas.1410723112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A., Salomoni P., Luo J., Shih A., Zhong S., Gu W., Pandolfi P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- Gurrieri C., Capodieci P., Bernardi R., Scaglioni P.P., Nafa K., Rush L.J., Verbel D.A., Cordon-Cardo C., Pandolfi P.P. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 2004;96:269–279. doi: 10.1093/jnci/djh043. [DOI] [PubMed] [Google Scholar]
- Hamasaki M., Furuta N., Matsuda A., Nezu A., Yamamoto A., Fujita N., Oomori H., Noda T., Haraguchi T., Hiraoka Y. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heath-Engel H.M., Chang N.C., Shore G.C. The endoplasmic reticulum in apoptosis and autophagy: Role of the BCL-2 protein family. Oncogene. 2008;27:6419–6433. doi: 10.1038/onc.2008.309. [DOI] [PubMed] [Google Scholar]
- Ito K., Carracedo A., Weiss D., Arai F., Ala U., Avigan D.E., Schafer Z.T., Evans R.M., Suda T., Lee C.H., Pandolfi P.P. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012;18:1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville L.S., Pinton P., Bastianutto C., Rutter G.A., Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V., Zhu J., Puvion F., Koken M., Honoré N., Doubeikovsky A., Duprez E., Pandolfi P.P., Puvion E., Freemont P., de Thé H. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 2001;193:1361–1371. doi: 10.1084/jem.193.12.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Hou N., Faried A., Tsutsumi S., Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer. 2010;46:1900–1909. doi: 10.1016/j.ejca.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Lin T.C., Chen Y.R., Kensicki E., Li A.Y., Kong M., Li Y., Mohney R.P., Shen H.M., Stiles B., Mizushima N. Autophagy: Resetting glutamine-dependent metabolism and oxygen consumption. Autophagy. 2012;8:1477–1493. doi: 10.4161/auto.21228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H., Rubio N., Garg A.D., Agostinis P. Autophagy: Shaping the tumor microenvironment and therapeutic response. Trends Mol. Med. 2013;19:428–446. doi: 10.1016/j.molmed.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Maiuri M.C., Zalckvar E., Kimchi A., Kroemer G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Mariño G., Niso-Santano M., Baehrecke E.H., Kroemer G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova M.M., Shaw R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yoshimori T., Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli E., Shen S., Ruckenstuhl C., Bauer M.A., Mariño G., Galluzzi L., Criollo A., Michaud M., Maiuri M.C., Chano T. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle. 2011;10:2763–2769. doi: 10.4161/cc.10.16.16868. [DOI] [PubMed] [Google Scholar]
- Nazio F., Strappazzon F., Antonioli M., Bielli P., Cianfanelli V., Bordi M., Gretzmeier C., Dengjel J., Piacentini M., Fimia G.M., Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013;15:406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- Papa A., Cordon-Cardo C., Bernardi R., Pandolfi P.P. Compound in vivo inactivation of Pml and p53 uncovers a functional interaction in angiosarcoma suppression. Genes Cancer. 2012;3:599–603. doi: 10.1177/1947601912473604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M., Pelicci P.G. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene. 2001;20:7250–7256. doi: 10.1038/sj.onc.1204856. [DOI] [PubMed] [Google Scholar]
- Pearson M., Carbone R., Sebastiani C., Cioce M., Fagioli M., Saito S., Higashimoto Y., Appella E., Minucci S., Pandolfi P.P., Pelicci P.G. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- Piazza F., Gurrieri C., Pandolfi P.P. The theory of APL. Oncogene. 2001;20:7216–7222. doi: 10.1038/sj.onc.1204855. [DOI] [PubMed] [Google Scholar]
- Rabinowitz J.D., White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y., Xie Y., Chai L., Wang S., Cheng M. Autophagy modification augmented the treatment effects initiated by arsenic trioxide in NB4 cells. Med. Oncol. 2011;28:231–236. doi: 10.1007/s12032-010-9430-6. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt M.T., Nixon C., Liu E., Mah L.Y., Ryan K.M. Analysis of macroautophagy by immunohistochemistry. Autophagy. 2012;8:963–969. doi: 10.4161/auto.20186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal A.R., Konig H., Johnson D.E., Tang D., Amaravadi R.K., Boyiadzis M., Lotze M.T. You eat what you are: Autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015;29:517–525. doi: 10.1038/leu.2014.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumaran M., Amaravadi R.K., Vasilevskaya I.A., O’Dwyer P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013;19:2995–3007. doi: 10.1158/1078-0432.CCR-12-1542. [DOI] [PubMed] [Google Scholar]
- Strohecker A.M., Guo J.Y., Karsli-Uzunbas G., Price S.M., Chen G.J., Mathew R., McMahon M., White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–1285. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z., Yang Z., Xu Y., Chen Y., Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer. 2015;14:48. doi: 10.1186/s12943-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E., Maiuri M.C., Galluzzi L., Vitale I., Djavaheri-Mergny M., D’Amelio M., Criollo A., Morselli E., Zhu C., Harper F. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Wieckowski M.R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- Xu Y., Xia X., Pan H. Active autophagy in the tumor microenvironment: A novel mechanism for cancer metastasis. Oncol. Lett. 2013;5:411–416. doi: 10.3892/ol.2012.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.