

Abstract
The functions of many proteins are regulated through allostery, whereby effector binding at a distal site changes the functional activity (e.g., substrate binding affinity or catalytic efficiency) at the active site. Most allosteric studies have focused on thermodynamic properties, in particular, substrate binding affinity. Changes in substrate binding affinity by allosteric effectors have generally been thought to be mediated by conformational transitions of the proteins, or alternatively, by changes in the broadness of the free energy basin of the protein conformational state without shifting the basin minimum position. When effector binding changes the free energy landscape of a protein in conformational space, the change not only affects thermodynamic properties but also dynamic properties, including the amplitudes of motions on different timescales and rates of conformational transitions. Here we assess the roles of conformational dynamics in allosteric regulation. Two cases are highlighted where NMR spectroscopy and molecular dynamics simulation have been used as complementary approaches to identify residues possibly involved in allosteric communication. Perspectives on contentious issues, e.g., the relation between picosecond-nanosecond local and microsecond-millisecond conformational exchange dynamics, are presented.
Graphical Abstract
1. Introduction
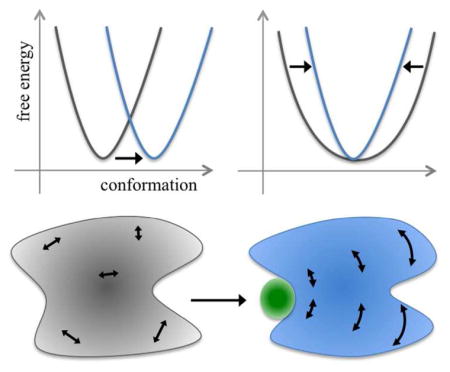
The functions of many proteins are regulated through allostery, whereby effector binding at a distal site changes the functional activity (e.g., substrate binding affinity or catalytic efficiency) at the active site (Figure 1a). Since the introduction of the word “allosteric” by Monod and Jacob1 in 1961, allosteric studies have mainly focused on thermodynamic properties, in particular, substrate binding affinity. Changes in substrate binding affinity by allosteric effectors have generally been thought to be mediated by conformational transitions of the proteins (Figure 1b, Left), as illustrated by Monod, Wyman, and Changeux (MWC) using the T to R quaternary conformational transition of hemoglobin.2 An alternative idea, considered by Wyman and Allen3 as early as 1951 and popularized by Cooper and Dryden,4 is that an allosteric effector may simply change the broadness of the free energy basin of the protein conformational state, rather than shifting the basin to a distinctly different region in conformational space (Figure 1b, Right). This type of allostery, known as entropically driven, has gained considerable attention, as many allosteric proteins show only subtle structural differences before and after binding effectors (see ref 5 for a reappraisal of such structural differences). Recently a change in the broadness (as opposed to the minimum position) of the free energy basin of the ligand-binding domain was proposed to underlie the partial agonism of a ligand-gated ion channel.6–7 When effector binding changes the free energy landscape of a protein in conformational space, the change not only affects thermodynamic properties but also dynamic properties, including the amplitudes of motions on different timescales (Figure 1c) and rates of conformational transitions.
Figure 1.

Conformational and dynamic effects of allosteric binding. (a) Binding of an effector at the allosteric site primes the binding of the substrate at the active site, and consequently the thermodynamic or kinetic properties of the latter binding are different from those in the absence of the effector. (b) Allosteric binding may result in a change in (Left) the conformational state, as signified by the movement of the corresponding free energy basin from one region to another region in conformational space, or (Right) the broadness of the free energy basin. (c) Conformation dynamics may be changed as well, e.g., from uncorrelated, fast (e.g., sub-nanosecond) motions in the apo form to correlated, slow (e.g., > microsecond) motions in the effector-bound form. Internal motions are represented by double-headed arrows.
Whereas the functional consequences (e.g., an increase in substrate binding affinity) of allosteric binding can be directly measured, the mechanisms of action are concluded with the help of inference and simplification. Allosteric mechanisms have been delineated using two types of pathways. The first type, referred to as transition pathway here, connects the end states of an allosteric transition, e.g., a conformational change upon allosteric binding (Figure 2). In the induced-fit pathway,8 the allosteric transition proceeds with the effector first binding loosely to the protein in the “apo” conformation and the protein then switching to the “bound” conformation. Alternatively, in the conformational-selection (also known as population-shift) pathway,9–10 the apo protein first switches to the bound conformation and the effector then binds. There has been much debate about transition pathways in defining allosteric mechanism.11–12 However, the historical debate between the MWC model2 and the model of Koshland, Nemethy, and Filmer (KNF),13 though sometimes presented as representing conformational selection and induced fit, respectively, is not really about transition pathways. Rather, this debate is about the origin of binding cooperativity in hemoglobin (and other oligomeric proteins). MWC attributed the free energy of cooperation to a concerted change in quaternary structure, but KNF to sequential changes in tertiary structures that affect inter-subunit physical interactions. Another point of contention is whether the detection of a minor population of conformations characteristic of the effector-bound form when the protein is free of the effector can be taken as proof of the conformational-selection pathway. As has been argued,11,14 every conformation has an equilibrium probability according to the Boltzmann distribution; whether “bound” conformations can be detected depends on the sensitivity of the experimental probe and therefore should not dictate the mechanism of allosteric transition.
Figure 2.

Transition pathways connecting the end states of allosteric binding and communication pathways from the allosteric site to the active site. The allosteric transition illustrated here is a conformational change, with “apo” and “bound” conformations represented by gray and blue shading, respectively. Transition pathways, indicated by brown arrows, are mainly concerned with kinetic intermediates, with induced fit passing through the intermediate in which the effector is loosely bound with the protein in the apo conformation, whereas conformational selection passing through the intermediate in which the apo protein adopts the bound conformation. Communication pathways, indicated by green arrows, are concerned with intermediate residues through which the allosteric site is coupled with the active site. The two types of pathways thus differ in their emphases, but are not orthogonal to each other. In the induced-fit transition pathway, the conformational change initiated by loose effector binding propagates to the active site while the effector consolidates its binding. In the conformational-selection pathway, the stabilization of the bound conformation starts at the allosteric site and propagates to the active site.
In any event, strictly speaking transition pathways are irrelevant when considering thermodynamic properties of allostery. That is because, fundamentally, thermodynamic properties depend only on end states, not on transition pathways connecting them. By contrast, kinetic properties do depend on transition pathways.15 In particular, the relative contributions of the induced-fit and the conformational-selection pathways to the rate of protein-effector binding can be measured.14,16 The relative contributions of the two pathways depend on both intrinsic factors, in particular the rates of conformational transition,14,17 and extrinsic factors, in particular effector concentration.14,16 With increasing rates of conformational transition or effector concentration, the induced-fit pathway becomes dominating.
The second type of pathway concerns the communication between the allosteric site and the active site (Figure 2). Perutz18 presented an early model of communication pathway for hemoglobin, based on structure comparison and structure-function correlation. In this model, oxygen binding to the T conformation triggers movement of the iron into the heme plane, realignment of the neighboring helices, and breakage of inter-subunit salt bridges, thereby shifting the quaternary equilibrium toward the R conformation (in line with the MWC model; see refs 19–20 for subsequent development of allosteric models for hemoglobin). Many workers (though with notable exceptions21) presume that networks of residues that exhibit spatial correlations in conformations or temporal correlations in motions mediate the communication between allosteric and active sites. One may identify these communication pathways by comparing residue-specific differences in conformations and dynamics between the apo form, effector-bound form, and ternary complex with both effector and substrate bound. NMR spectroscopy has now been established as a powerful tool for characterizing allosteric communication, due to its ability to provide atomic-level information on chemical environment and on picosecond-nanosecond local (backbone or side chain) and microsecond-millisecond conformational exchange dynamics.22–28
In principle long molecular dynamics simulations can provide all the details regarding the communication between allosteric and active sites, and simulations longer than a microsecond are beginning to shed light on communication mechanisms.29–31 Computational studies are still mostly based on sub-microsecond simulations, or based on contact analysis or elastic network modeling of static crystal structures.32 Notably, allosteric communication has been assumed to involve evolutionarily conserved33 or positionally correlated34 networks of residues. The complementarity of computational and experimental approaches, NMR spectroscopy in particular, in studying allostery can be easily appreciated.
In recent years there has been intense interest in the potential roles of conformational dynamics in allosteric regulation. In referring to entropically driven allostery, a number of workers have used the word “dynamic” (e.g., ref 4), but what was really meant is just that proteins are not static but, under equilibrium fluctuation, can sample an ensemble of conformations.20,35 The extent of conformational sampling in a (meta)stable state is measured by conformational entropy, which, like all thermodynamic properties, is microscopically determined by the shape of the free energy basin in conformational space. Beyond thermodynamic properties, dynamic properties characterizing time-dependent processes are also observables, including the timescales, amplitudes, and spatial-temporal correlations of internal motions as well as rates of conformational transitions. Microscopically, dynamic properties are governed by the equations of motion, and dictated by not only free energy basins but also barriers, along with dynamic parameters (e.g., effective diffusion coefficients).
Given the latter strict sense of the word “dynamic,” there may be three views on conformational dynamics as it relates to allostery. The first view is that allosteric regulation should be limited to thermodynamic properties only; any effect on conformational dynamics is a mere byproduct.36 For example, an allosteric effector may increase the substrate binding affinity by reducing side chain flexibility, leading to a lower conformational entropic cost or preorganization of the active site.25,37 As a consequence the amplitudes of the picosecond-nanosecond orientational dynamics of the side chains may also be reduced.
The second view is that, even if conformational dynamics does not dictate the final thermodynamic outcome, characterizing it can lead to a fuller understanding of allosteric regulation.38 As already noted, the rates of conformational transitions are a key determinant of transition pathways. Conformational dynamics may also be involved in mediating allosteric communication.24–25,39 Absent such involvement, conformational dynamics may still be helpful in identifying communication pathways, as illustrated by the residues with suppressed sub-nanosecond side chain dynamics in the last example.
The third view is that allosteric regulation also extends to kinetic properties and surely conformational dynamics is a determinant of these properties. In particular, enzyme kinetics is characterized by two parameters, the turnover number kcat and the Michaelis constant KM. These parameters in turn are determined by the rate constants of the three basic steps of the enzyme-catalyzed reaction: substrate binding, chemical transformation of the substrate into the product, and product release. Allosteric effectors have been suggested to affect millisecond timescale dynamics implicated in these basic steps and thereby modulate the corresponding rate constants.40–43
In this review we aim to assess the roles of conformational dynamics in allosteric regulation. We highlight two proteins for which NMR spectroscopy and molecular dynamics simulation have been used as complementary approaches to identify residues possibly involved in allosteric communication, e.g., by following effector-induced changes in dynamic properties or positional correlations. We also present our perspectives on several contentious issues, e.g., the relation between picosecond-nanosecond local and microsecond-millisecond conformational exchange dynamics, and on future developments, including designing molecular dynamics simulations to test ideas about allosteric communication.
2. Approaches for Characterizing Allosteric Communication
Effector-induced changes in conformational dynamics and positional correlations, probed by NMR spectroscopy and molecular dynamics simulation, respectively, are often used to characterize allosteric regulation. Below we give some basic ideas behind these experimental and computational approaches, hopefully to allow the reader better appreciation of the results to be covered in Section 3.
2.1. NMR Relaxation and Conformational Dynamics
Different types of NMR relaxation experiments can report conformational dynamics on different timescales.44 The magnetization arising from nuclear spins can be decomposed into a longitudinal component (i.e., parallel to the static magnetic field B0) and a transverse component. In two classical experiments, one observes the time-dependent recovery of the equilibrium value of the longitudinal component and the time-dependent decoherence of the transverse component. The main mechanisms that contribute to the longitudinal and transverse relaxation for a nuclear spin S (e.g., backbone 15N) involve the magnetic dipole-dipole interaction (e.g., with the backbone amide 1H, to be referred to as spin I) and the chemical shift anisotropy (CSA) of the S spin.45 These act as time-dependent (due to stochastic molecular motions) perturbations to the Hamiltonian of the S spin. The dipolar contribution depends on the magnitude (rIS) and direction of the internuclear vector, whereas the CSA contribution depends on the magnitude (ΔσS) of the CSA and orientation of the CSA tensor. For a backbone amide 15N-1H spin pair, the 15N CSA tensor is usually nearly axially symmetric and the symmetry axis is nearly collinear with internuclear vector. Here we assume axial symmetry and collinearity, along a unit vector n.
Because of stochastic molecular motions, n is a randomly fluctuating variable, but the correlation, <n(t) · n(0)>, between n at a given moment and n a time interval t later, upon averaging over the time course of the molecule and also over an ensemble of molecules, is a smooth, generally decaying function of t. Both the dipolar and CSA contributions to NMR relaxation can be expressed in terms of the spectral density function, which is a Fourier transform of another time-correlation function of the vector n
| (1) |
where P2(x) = (3x2 − 1)/2. The resulting longitudinal and transverse relaxation rates are
| (2) |
| (3) |
where and , with μ0 denoting the vacuum permeability, ħ denoting the reduced Planck’s constant, and γX and ωX (= γXB0) denoting the gyromagnetic ratio and Larmor frequency, respectively, of spin X.
Backbone 15N R1 and R2 measurements thus mainly probe orientation dynamics of the NH bond vector and the peptide plane, which typically occur on the picosecond-nanosecond timescale. In a hypothetical situation where the protein is a rigid body, orientation dynamics of the unit vector n is solely due to the overall rotational tumbling of the protein (Figure 3a). The time-correlation function is then
Figure 3.

Different types of conformational dynamics. (a) Overall rotational diffusion. The unit vector n is assumed to be rigidly attached to the protein represented by a sphere with blue shading. (b) Local orientational dynamics of n, illustrated here as diffusion in a cone, in the body-fixed frame. (c) Conformational exchange between a major state (shaded blue) and a minor state (shaded green). The magnetization (green arrow) precesses at different frequencies in the two states, resulting in dephasing. (d) Conformational sampling on different timescales. Sub-microsecond molecular dynamics simulations sample local fluctuations within a single conformational state (or substate therein). Transitions between conformational states require non-local correlated motions and cross high free energy barriers, typically occurring on the microsecond-millisecond timescale.
| (4) |
where τc = 6D is the rotational correlation time, typically of the order of 10 nanoseconds, and D is the overall rotational diffusion coefficient (assumed to be isotropic). Generally the unit vector also experiences local orientational dynamics (Figure 3b), which often is much faster than and consequently can be decoupled from the overall rotation, resulting in a time-correlation function46
| (5) |
where τe is the effective correlation time for the local orientational dynamics, and 𝒮2, known as the order parameter, reflects the fact that, in the body-fixed reference frame, the local orientational dynamics may not completely randomize the direction of n. Upon removing the overall rotation, the time-correlation function decays not to zero but to
| (6a) |
| (6b) |
where Y2m(θ, ϕ) are spherical harmonics, and θ, ϕ, and nα are the polar and azimuthal angles and the Cartesian components, respectively, of the unit vector n in the body-fixed frame. By using eq 5 to fit R1 and R2 data, one obtains information on the timescale (i.e., te) and the amplitude (i.e., 1 − 𝒮2) of the fast local dynamics. The order parameter can also be directly calculated from molecular dynamics simulations according to eq 6b.47–48
As illustration (Figure 3b), consider the local orientation dynamics that is modeled as diffusion in cone spanning a polar angle θ0. Evaluation of eq 6a leads to46
| (7) |
The conformational entropy associated with motion in the cone is
| (8) |
By eliminating cos θ0, one can directly relate the order parameter to conformational entropy49–50
| (9) |
where kB is Boltzmann’s constant. Summing the entropies derived from order parameters at different sites in the protein provides a measure of the total conformational entropy. If these calculations are done for the apo and effector-bound form, the difference can be interpreted as the effector-induced change in protein conformational entropy.51
If the S spin can stochastically exchange between two chemical environments (labeled 1 and 2), as occurs when the protein switches between two conformations, the exchange will contribute to the dephasing of the transverse magnetization and hence enhance R2, because spins in the two environments have different precession frequencies (Figure 3c). Relative to the difference Δν between the resonance frequencies ν1 and ν2 in the two environments, the exchange rate kex (sum of the forward and backward transition rates k1 and k2) falls into three regimes. In the fast exchange regime (i.e., kex ≫ 2πΔν ≡ Δω), the NMR spectrum of spin S has a single peak, positioned at the population average of ν1 and ν2. With the enhancement by the chemical exchange, the transverse relaxation rate becomes
| (10) |
where is the population average of the transverse relaxation rates in the two environments when exchange is absent, and p1 and p2 are the equilibrium population fractions in the two environments. In the slow exchange regime (i.e., kex ≪ Δω), the NMR spectrum of spin S has two peaks, positioned at ν1 and ν2, and the transverse relaxation rates in the two environments are increased by k1 and k2, respectively. In the intermediate exchange regime, the two peaks coalescence, with a broadened lineshape. This exchange broadening can often lead to the loss of NMR signal. Hence, just like R2 values that are higher than can be accounted for by overall rotational tumbling and local orientational dynamics, broadened lineshapes and unobserved resonances are often interpreted as indicating microsecond-millisecond conformational exchange. However, spin relaxation experiments are generally not suited for quantitative characterization of conformational exchange kinetics (i.e., determination of the transition rates).
Although the different precession frequencies at the two environments lead to dephasing of the transverse magnetization (Figure 3c), a 180° radio-frequency pulse applied in a transverse direction has the effect of rephasing, and thereby suppressing exchange-mediated transverse relaxation. This is the essence of a Carr–Purcell Meiboom–Gill relaxation dispersion (CPMG RD) experiment, where one employs a pulse sequence in which 180° pulses are separated by periods (of duration 2τcp) during which relaxation occurs.52–53 By varying τcp, one observes a range of effective transverse relaxation rates (R2,eff), and only when τcp → ∞ is the effect of chemical exchange fully exhibited. That is, R2,eff is “dispersed” or spread out as a function of τcp (or its inverse). In particular, in the fast exchange regime, R2,eff approaches the value given by eq 10 when τcp → ∞, but approaches when τcp → 0. A formula that bridges these two limits of τcp in the fast exchange regime is54
| (11) |
Fitting the dependence of observed R2,eff on τcp to eq 11 yields kex and p1p2Δω2.
The conformational exchange that is of practical interest is one between a major state and a minor state (i.e., p1 ≫ p2). A general formula, valid in all the three exchange regimes, has been derived for R2,eff(τcp) of the major state.55–56 Fitting experimental data can yield the transition rates k1 and k2 (and hence the population fraction of the minor state) as well as Δω. The latter is equivalent to the difference in chemical shift between the major and minor states, and thus contains structural information about the minor state (chemical shifts in the major state are directly observable). In favorable cases, this information can be used to validate structural models of the minor state.42 However, the nature of the conformational changes from the major state to the minor state is often poorly defined by CPMG RD experiments, although the millisecond timescale of kex suggests that the conformational changes must be more than local (Figure 3d).
2.2. Path and Community Analysis
To model protein stability and function in general and allosteric communication in particular, terminologies, ideas, and methods from graph theory57 have been extensively borrowed and expanded. Typically the protein (composed of N residues) is mapped to a weighted graph, in which each node represents a residue (Figure 4). Two graph properties of interest are the shortest path between two nodes and the partitioning of the graph into communities.
Figure 4.

A protein represented as a weighted graph. (a) Shortest path between two nodes 1 and 7, composed of three connected edges shown in blue. Each edge (indicated by a line between two nodes) is assigned a path length (e.g., eq 16 or 17). The path lengths of the three blue edges are shown. Summing over the individual path lengths, the total length of the shortest path is 3.0. (b) The nodes are partitioned into two communities, one with green nodes inside a lime oval and one with red nodes inside a pink oval. An edge between nodes 5 and 6 links the two communities; these nodes are known as critical nodes.
Earlier graph models used the static crystal structure of the protein to specify the weights between nodes. In the simplest version,58 two nodes were assumed to form an “edge” and assigned a weight 1 if the distance between two representative atoms of the corresponding residues was within a cutoff; otherwise the weight was 0. (For later reference, the edges in a protein comprise the contact map.) In graph theory,57 these inter-node weights define the adjacency matrix:
| (12) |
The sum of the ith row (or column) elements of 𝒜 is the degree, i.e., the number of edges formed by node i. Denoting the diagonal matrix of degrees as 𝒟, the difference
| (13) |
is known as the Laplacian matrix, which, upon multiplying by a spring constant, happens to coincide with the Hessian matrix of the Gaussian network model59 (a form of elastic network model). Assuming here a value of 1 for the path length of each edge, then the total length of a path along connected edges is just the number of edges involved. Any two nodes can be connected by multiple paths; the shortest paths between residues in the allosteric site and residues in the active site may be especially important for allosteric communication. Atilgan et al.58 calculated the average of shortest path lengths of a given residue to other residues in a protein, and found that average shortest path lengths are highly correlated with the amplitudes of residue position thermal fluctuation predicted by the Gaussian network model. Intuitively, “hub” residues (i.e., those with high degrees) should have both small average shortest path lengths and low amplitudes of thermal fluctuation.
Instead of directly using a cutoff in inter-residue distance for specifying edges, Brinda and Vishveshwara60 used the inter-residue interaction strength
| (14) |
where nij is the number of atom-atom contacts between residues i and j within a distance cutoff, and ni and nj are normalization factors. An edge is formed if ℐij exceeds a threshold ℐmin. (If a molecular dynamics simulation is done, one can further stipulate that an edge is formed only if this condition is satisfied over a specified fraction of the simulation time.61) At very high ℐmin, nodes are disconnected from each other; at very low ℐmin, all nodes become interconnected; at intermediate ℐmin, nodes segregate into disconnected communities. A recommended ℐmin is one that results in the largest community containing approximately half of the residues in the protein.
Chennubhotla and Bahar62 used eq 14 as the definition of the adjacency matrix. Following graph theory,57 they assigned the normalized element
| (15) |
as the probability that a random walker through the graph makes a jump from node i to node j. The resulting stationary probabilities of the random walker on the nodes were used to find the probabilities for partitioning each node into different communities. The latter probabilities were further used to define the entropy for the partitioning of the node. Nodes with high entropies are shared with high probabilities among multiple communities. These “messenger” nodes may be critical for inter-community communication in allosteric regulation. For the chaperonin GroEL-GroES complex, these nodes also had small amplitudes in the lowest-frequency mode (i.e., global mode) of the Gaussian network model; the latter occur in hinge regions for inter-domain global motions. It remains to be determined whether messenger nodes coincide with another group of residues, also proposed for allosteric communication; the latter residues are identified by a large response in a normal mode of interest when the spring constants for the edges from these residues are perturbed.63 For path length, rather than a constant value 1 for each edge, Chennubhotla and Bahar introduced the definition
| (16) |
such that high transition probability corresponds to short path length and vice versa.
Sethi et al.34 took a major step by using information from molecular dynamics simulations to define the contact map and the path lengths of edges. An edge was formed between two residues, i and j, when the distances between their heavy atoms were below a cutoff for a specified fraction of the simulation time. More importantly, the path lengths of the edges were defined using the (normalized) co-variance matrix of the residue positions:
| (17) |
in analogy to eq 16. The co-variance matrix itself is given by
| (18) |
where xi = ri−<ri> are displacements from mean residue positions. When residues i and j are highly correlated in displacement from their mean positions, |𝒞ij| → 1 and, according to eq 17, the path length lij → 0. Conversely, when the two residues have totally uncorrelated displacements, |𝒞ij| → 0 and lij → ∞. The total length of a path along connected edges is the sum of the path lengths of the individual edges (Figure 4a); the shortest path between nodes i and j is the path that has the minimum total length among all possible paths connecting i and j. The shortest path is specified by the series of constituent nodes, starting at i and ending at j.64
Sethi et al. used the Girvan-Newman algorithm65 to partition the graph into communities, within which edges are dense but between which edges are sparse (Figure 4b). The Girvan-Newman algorithm uses edge betweenness, defined as the number of shortest paths that cross a particular edge, for partitioning. The edge betweenness of an inter-community edge is high because all the inter-community shortest paths must cross it (or any small number of other such edges). In contrast, the edge betweenness of an intra-community edge is low because many other neighboring edges can provide alternatives for shortest paths. The Girvan-Newman algorithm is an iterative procedure, in which the edge with the highest betweenness is cut and the betweennesses of the remaining edges are recalculated, until every node becomes isolated and hence a community of its own. The partitioning finally chosen is the one with optimal modularity, where the fraction of intra-community edges maximally exceeds the expected value if the edges are randomly placed between nodes in the graph. Nodes linked by inter-community edges are termed critical, since all the inter-community shortest paths cross these edges (Figure 4b). Critical nodes serve a similar role as the messenger nodes of Chennubhotla and Bahar.62
Rivalta et al.66 took Sethi et al.’s approach but redefined the edge path lengths in terms of a generalized correlation matrix introduced by Lange et al.,67
| (19) |
where ℐij are the mutual information. The latter,
| (20) |
measures the deviation of the joint probability density p(xi, xj) of the displacements xi and xj from the product pi(xi)pj(xj) of the two marginal probabilities, which is the expected probability density if xi and xj are totally uncorrelated. reduces to 𝒞ij if p(xi, xj) is Gaussian. When xi and xj are perpendicular, 𝒞ij = 0 even if the displacements are highly correlated. This correlation is captured by .
We remark that the co-variance matrix and its generalization measure residue-residue positional correlation during equilibrium fluctuation. Mutual information, expressed in terms of a probability density in conformational space, can be easily recognized as an equilibrium property. The co-variance matrix is similarly an equilibrium property, not a dynamic property, even though sometimes it is referred to as the dynamic cross-correlation matrix. Calculation of 𝒞ij from molecular dynamics trajectories, by averaging over snapshots, may give it a “dynamic” appearance, but the time sequence of the snapshots have no effect – the result is the same if the snapshots are scrambled.
Likewise, even though path and community analysis based on residue-residue positional correlation is referred to as dynamical network analysis,32,34,66,68 strictly speaking no dynamical information is involved. Given that molecular dynamics simulations used to prepare for path and community analysis typically are sub-microsecond long, and therefore cannot sample microsecond-millisecond conformational exchanges, the information supplied is only quasi-equilibrium. That is, the average is limited to a single conformational state (or a substate therein; Figure 3d).
It is of interest to note that exchange between conformational states have recently been investigated by building Markov state models from molecular dynamics simulations.30–31,69 In these models, microstates are obtained by clustering snapshots according to structural similarity, and transition probabilities between microstates are estimated from molecular dynamics trajectories.
3. Case Studies of Dynamic Effects in Allosteric Regulation
As noted in the preceding section, allosteric communication is generally thought to be mediated by groups of residues that exhibit effector-induced changes in conformational dynamics or positional correlations. The two types of residues can be identified by NMR spectroscopy and molecular dynamics simulation, respectively. Information from the two approaches can be combined to develop allosteric mechanisms, as illustrated below.
3.1 Pin1
Pin1 is a peptidyl-prolyl cis/trans isomerase (PPIase) that acts on phosphoSer/Thr-Pro (p(S/T)P) motifs present in mitotic phosphoproteins,70 thereby controlling their fates.71 The full-length Pin1 consists of an N-terminal WW domain (residues 1-39) and the C-terminal PPIase domain (residues 50-163) (Figure 5a). Both domains can selectively bind p(S/T)P containing substrate motifs, but only the PPIase domain can isomerize the peptidyl-prolyl bond,72–73 at a catalytic site lined by three loops (labeled as catalytic loop, β5-α4, and β6-β7 in Figure 5a). In addition to other roles, several lines of evidence suggest that substrate-WW binding allosterically regulates the PPIase activity. First, the substrate affinity and catalytic activity of the isolated PPIase domain are different from those of the full-length protein.26,72–73 Second, crystal structures of Pin1 show that the two domains are tightly packed against each other, although the linker between them is disordered (Figure 5a).74–76 Third, NMR studies showed that binding of both substrates and a nonpeptidic ligand (polyethylene glycol) to the WW domain resulted in tighter coupling between the two domains.77–78 The allosteric communication in Pin1 has been investigated in recent studies based on NMR spectroscopy26,79–81 and molecular dynamics simulations.37,82
Figure 5.

Structure and allosteric communication of Pin1. (a) Structure of Pin1 with FFpSPR bound at the WW site. The catalytic site of the PPIase domain is lined by the three loops labeled as catalytic loop, β5-α4, and β6-β7. (b) Two clusters of paths connecting the WW domain to the catalytic-site loops. The cluster shown as light blue arrows preexists in apo Pin1, but the paths in the second cluster, shown as pink arrows, are broken in the apo form and are completed only in the FFpSPR-bound form. (c, d) Community analysis results for apo and FFpSPR-bound Pin1. The communities are shown in different colors as cartoon structures (Left) or as ovals (Middle). Intercommunity connections are shown as lines, with width proportional to the cumulative betweenness of inter-community edges (Middle). Shown in (Right) is a dynamic model for allostery. A spring depicts a representative internal coordinate from each of communities 1, 2, and 3 that is modeled as undergoing diffusive motion in a harmonic potential. The internal coordinates are weakly coupled in apo Pin1 and become strongly coupled in the FFpSPR-bound form.
Namanja et al.26,79 obtained the order parameters ( ) for methyl symmetry axes from measurements of methyl 2D relaxation rates. They found that binding of two substrates, one with sequence FFpSPR and the other a pTP containing peptide from the mitotic phosphatase Cdc25C, both resulted in increases in in three regions (Figure 5b): the WW-PPIase interface, the interface of the α1 helix with the PPIase core, and the catalytic site. Namanja et al. proposed that these residues, with suppressed sub-nanosecond methyl orientational dynamics, formed a hydrophobic conduit for interdomain allosteric regulation. Other methyls also exhibited compensatory decreases in upon peptide binding, with unknown functional implications; more such methyls were found in Cdc25C-bound Pin1 than in FFpSPR-bound Pin1. It should be noted that less than a third of Pin1 residues contain methyls.
To gain a sense of the effect of peptide binding on microsecond-millisecond dynamics, Namanja et al.26,79 measured methyl 13C longitudinal and transverse relaxation rates and looked for residues with high values for their product, R1;13CR2;13C, which may indicate contributions from conformational exchange. Upon FFpSPR binding, the number of residues exhibiting R1;13CR2;13C increases was more than that for decreases,83 implicating enhancement of exchange dynamics. Residues with increased R1;13CR2;13C map to the WW-PPIase interface and to the interface of the α1 and α2 helices with the PPIase core. However, the nature of the minor state with which the major state exchanges remains elusive, if the elevated R1;13CR2;13C indeed reflects conformational exchange. For Cdc25C, the trend in R1;13CR2;13C changes was apparently reversed, with more decreases than increases. Recent CPMD RD data provided validation of reduced exchange dynamics upon binding Cdc25C and were interpreted as reflecting weakened interdomain contact.81
To gain detailed insight into the allosteric communication in Pin1, we carried out molecular dynamics simulations of Pin1 in apo form as well as bound with several peptides, either at the WW site or the catalytic site or both sites.37,82 In the 100-ns simulations, FFpSPR binding at the WW site resulted in decreases in root-mean-square-fluctuation (RMSF), indicating suppression of sub-nanosecond local dynamics, in the three loops lining the catalytic site, to the level exhibited when a trans-locked alkene isostere of FFpSPR was bound to both the WW site and the catalytic site. The FFpSPR-induced reduction in RMSF for the catalytic-site loops is a clear indication of allosteric communication between the two binding sites. The communication evidently was unidirectional, as a cis-locked alkene isostere bound only at the catalytic site did not produce loss in flexibility in the WW domain.
We used the graph-partitioning method of Brinda and Vishveshwara,60 which is based on residue-residue physical proximity, to identify allosteric pathways. The partitioning produced two clusters of paths for FFpSPR-bound Pin1. The first emanates from the WW backside and propagates through the interdomain interface and the PPIase domain core to the β5-α4 and β6-β7 loops; the second emanates from the WW front pocket and propagates through the bound peptide, the α1 helix, and the latter’s interface with the PPIase core, to the catalytic loop. The first cluster of paths preexists in apo Pin1, but the second cluster is broken as the gap between the α1 helix and the WW front pocket is too wide without the bound peptide (Figure 5b). In essence, the bound peptide serves as a bridge to complete the second cluster of paths from the WW site to the catalytic site.
We also applied the community analysis method of Sethi et al.,34 which is based on both physical proximity and positional correlation. The overall conclusion was the same, but new insight emerged. The WW front pocket, the β5-α4 loop, and the β6-β7 and catalytic loops are located in three different communities (numbered 1, 2, and 3, respectively, in Figure 5c, d, Left and Middle). In apo Pin1, community 1 is not directly linked to either community 2 or 3. In FFpSPR-bound Pin1, community 1 is enlarged to include the whole WW domain and the N-terminal region of the peptide. It is now directly linked to community 3, with the N-terminal residues of the peptide providing critical nodes for the linkage. Moreover, the enlarged community 1 becomes directly and strongly linked to community 2. The strengthened coupling between these communities thus explains the allosteric communication.
The forgoing analyses suggested that FFpSPR elicits allosteric effects by serving as a bridge to connect the α1 helix and the WW domain. To test this idea, we carried out simulations in which subsets of residues in different regions of apo Pin1 were artificially restrained to limit conformational fluctuations and monitored the RMSF of the catalytic-site loops. The RMSF was hardly changed with a restraint of the WW domain or within the PPIase core, and only partially reduced with a restraint of the PPIase core and α1 helix interface, but fully reduced when four residues from the WW domain were added into the restraint. Importantly, the reference conformation in these restraints was taken from the simulation of apo Pin1. So these restrained simulations directly demonstrated the idea4 that merely restricting the fluctuations around a mean structure can generate allosteric effects, while also showing that restrictions in different regions of a protein are not equally effective.
Our follow-up study82 showed that, compared to FFpSPR, Cdc25C more tightly interacted with the WW domain but less so with the α1 helix and the PPIase core. As a result, community 1 lost direct linkage to communities 2 and 3 (as in apo Pin1), and the suppression of sub-nanosecond local dynamics (as indicated by RMSF) was not as effective, in line with the NMR studies.26,79 NMR80 and molecular dynamics simulation37 studies of the I28A mutant, within the WW-PPIase interface, provided additional evidence that weakened inter-community coupling leads to reduced suppression of sub-nanosecond local dynamics. Note that, when the cis-locked alkene isostere is bound at the PPIase catalytic site, the bridge between the α1 helix and the WW domain is missing, and hence communication from the PPIase domain to the WW domain is ineffective. This provides a simple explanation for the apparent unidirectionality of the allosteric communication between the WW and catalytic sites.
Why should the amplitude of sub-nanosecond local dynamics be related to inter-community coupling, which is based on physical proximity and positional correlation? To illustrate this point, let us consider the local dynamics of the unit vector n shown in Figure 3a, b, in two extremes. In one extreme, local dynamics is fully quenched, with the unit vector rigidly attached to the protein so that the order parameter 𝒮2 is 1. Of course this unit vector would be fully correlated with any other such unit vector since they both follow the same overall tumbling of the protein. The correlation time of n is that of overall tumbling, i.e., τc, which is of the order of 10 nanoseconds. In the other extreme, the local dynamics completely randomizes the orientation of n, resulting in 𝒮2 = 0. Obviously n would be totally uncorrelated with any other such unit vector. Now the correlation time of n (see eq 5 with 𝒮2 = 0) is τcτe/(τc + τe) ≈ τe since τc ≫ τe, which is sub-nanosecond. So in this example, increased inter-community coupling is correlated with quenching of fast, sub-nanosecond dynamics and emergence of slower, 10-nanosecond dynamics.
To further explore the relation between inter-community coupling and dynamics on fast and slow timescales, we introduced a dynamic model of allostery (Figure 5c, d, Right). This model was inspired by the molecular dynamics simulation and community analysis results for Pin1 summarized above (Figure 5c, d, Left and Middle), but highly simplified. We modeled a representative internal coordinate from each of communities 1, 2, and 3 as undergoing diffusive motion in a harmonic potential. The strength of coupling between the internal coordinates was tunable, from very weak for apo Pin1 to very strong for the FFpSPR-bound form. This model predicts that, with weak coupling, the internal coordinates have a single-exponential time-correlation function, with a short correlation time (e.g., sub-nanosecond). However, with strong coupling, the time-correlation function is a double exponential; one exponential has a short correlation time and a small amplitude, while the second exponential has a long correlation time (e.g., microsecond-millisecond) and a large amplitude. That is, with increasing inter-community coupling, the fast motion is suppressed, and replaced by slow motion. The internal coordinates from the different communities also become highly correlated. The slow motion may represent conformational exchange, insofar as the latter requires inter-community correlated motions (Figure 3d).
Our model suggests that, with strengthened inter-community coupling, suppression of fast local dynamics may be expected along with initiation of slow conformational exchange dynamics. The NMR results summarized above provide some support. With FFpSPR, data indicated suppression of local dynamics whereas R1;13CR2;13C data indicated enhanced conformational exchange. With Cdc25C, both effects were moderated. It appears that allosteric activators like FFpSPR, by strengthening inter-community coupling, in particular through providing critical nodes, may elicit disparate dynamic responses on fast and slow timescales.
3.2 Imidazole Glycerol Phosphate Synthase
Imidazole glycerol phosphate synthase (IGPS) is a bifunctional enzyme with active sites located in two separate domains named HisH and HisF (which are associated noncovalently in bacteria but are covalently linked in eukaryotes; Figure 6). HisH catalyzes the hydrolysis of glutamine into glutamate and ammonia, using a conserved catalytic triad, consisting of Cys84, His178, and Glu180 (Thermotoga maritima IGPS numbering), located ~10 Å away from the HisH-HisF interface. This reaction starts with thioester bond formation between glutamine and Cys84, with the resulting oxyanion tetrahedral intermediate stabilized by the backbone amide of Val51, part of a conserved PGVG motif. The ammonia product then travels down a 20 Å hydrophobic tunnel formed by the β strands of the HisF (β/α)8 barrel to the second active site at the bottom, where it combines with a metabolite N′-[(5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (PRFAR) to yield two products that enter the histidine and purine biosynthetic pathways. Coordination of the two reactions is achieved through PRFAR serving as an allosteric effector. Binding of PRFAR to the HisF active site enhances the kcat of HisH by ~5000-fold.84 The long PRFAR molecule, with the two ends near residues Val100 and Leu222, respectively, bisects the bottom of the HisF (β/α)8 barrel, with β1/α1, β2/α2, and β3/α3 on one side and β5/α5, β6/α6, and β7/α7 on the other side (referred to as sideR and sideL, respectively66). The allosteric effects of PRFAR binding were studied by both NMR spectroscopy and molecular dynamics simulations.28,40,66,68
Figure 6.

Structure of IGPS. SideL and sideR are in light orange and magenta, respectively, for of HisF, and in gray and green, respectively for HisH. HisF sideL consists of residues 101-220, and HisH sideR consists of β1-β4 strands, α1, α2, α2′, and α4 helices, and Ω-loop. The bound PRFAR in HisF is shown in cyan spheres, and the catalytic triad and PGVG motif are labeled, as are some secondary structure elements (α1-α3 and loop1 in HisF and α1, α2, α2′, and Ω-loop in HisH).
From CPMG RD experiments on backbone 15N and alanine methyl 13C, Lipchock et al.40 found no evidence of HisF exchange dynamics in apo IGPS. Methyl 13C RD data on isoleucines, leucines, and valines (ILV) showed signs of conformational exchange for 17 of 116 assigned resonances from HisF, mostly on sideR. Upon PRFAR binding, 63 backbone amide resonances from HisF were broadened beyond detection, indicative of the intermediate exchange regime, mostly on sideR. Backbone 15N and ILV methyl 13C RD data together identified 68 HisF residues as undergoing millisecond conformational exchange, and the active ILV residues spread to the entire central beta barrel and the β/α interface. On the other hand, 1H-1H dipolar cross-correlated relaxation rates of HisF ILV residues did not indicate a significant overall change in sub-nanosecond dynamics upon PRFAR binding. It was unknown whether PRFAR had any effect on fast dynamics of other side chains or the protein backbone.
In the crystal structure of apo IGPS, the peptide plane between Gly50 and Val51 of HisH is in an orientation opposite to what would provide stabilization to the oxyanion tetrahedral intermediate, with the Gly50 carbonyl oxygen instead of the Val51 amide proton pointing to the negatively charged oxygen atom of the would-be tetrahedral intermediate.85 The Val51 amide instead hydrogen bonds to the carbonyl of Pro10. Lipchock et al.’s 1H, 15N NMR spectrum for HisH in apo IGPS showed a sharp cross peak for Gly50, which was broadened beyond detection upon titration with PRFAR. This observation showed that PRFAR-stimulated millisecond exchange dynamics extended across the HisF-HisH interface and into the active site of HisH. Lipchock et al. proposed that this exchange dynamics allowed the Gly50-Val51 peptide plane to flip and thus present the stabilizing amide proton of Val51.
Rivalta et al.66 reported 100-ns molecular dynamics simulations of apo and PRFAR-bound IGPS. PRFAR binding resulted in an overall decrease in RMSF in HisF, especially its loop1 (residues 16-30), but not in HisH. The decrease in RMSF means that the amplitudes of local sub-nanosecond dynamics were suppressed. The PRFAR-induced changes in positional correlation, as captured by the generalized correlation matrix calculated from mutual information, were heterogeneous. Positional correlations were reduced in HisF sideL (residues 101-220) and in the HisH catalytic triad and the surrounding residues, including the PGVG motif. However, correlations were enhanced within and between HisF sideR (including loop1, α2 and α3 helices, and β3 strand) and HisH elements on the same side (including β1-β3 strands, α1 helix, and the intervening Ω-loop) (Figure 6).
Using the generalized correlation matrix to define edge pass lengths, Rivalta et al. carried out community analysis. One of the most notable changes induced by upon PRFAR binding was the strengthened coupling between a community in HisF and a community in HisH, both on sideR (Figure 6). The HisF community included β2-β3 strands and α2-α3 helices; the HisH community included β1-β4 strands, α1, α2, α2′, and α4 helices, and Ω-loop. The two communities were linked by edges between residues in HisF α2-α3 helices and in HisH α1 helix and Ω-loop. The strengthened inter-community coupling was in line with the increased positional correlations between these elements.
In these molecular dynamics simulations, the HisH Val51-Pro10 hydrogen bond was stable in apo IGPS but unstable in the PRFAR-bound form. Concomitantly, the Gly50-Val51 peptide plane partially rotated, which the authors suggested as an early sign of an anticipated full flip of the peptide plane in the millisecond timescale.
The foregoing NMR and computational results appear to be qualitatively consistent with the predictions of our simple dynamic model of allostery (Figure 5c, d, Right). That is, upon PRFAR binding, the coupling between two communities across the interdomain interface was strengthened, while the amplitudes of sub-nanosecond local dynamics were suppressed (as indicated by reduced RMSF) and millisecond exchange dynamics was stimulated (as revealed by NMR spectroscopy). However, instead of the disparate responses of fast and slow dynamics that we emphasize, Rivalta et al.28,66 appear to suggest that fast and slow dynamics go hand in hand. According to them, disruptions of residue-residue interactions on the nanosecond timescale “may represent the initial loosening of the protein core that precedes the wholesale enhancement of ms motions observed by solution NMR.” Clearly, the nature of millisecond exchange dynamics, although beyond the scope of sub-microsecond molecular dynamics simulations, deserves further studies.
Vanwart et al.68 also carried out molecular dynamics simulations and community analysis for apo and PRFAR-bound IGPS. Their focus was how different representatives of residues positions, e.g., Cα atom versus residue center of mass, affected community partitioning. The latter representative was recommended.
4. Perspectives
We have assessed the potential roles of conformational dynamics in the allosteric regulation of two proteins. From NMR spectroscopy, a hydrophobic conduit was proposed for interdomain communication in Pin1, based on suppressed side chain sub-nanosecond dynamics upon effector binding;26,79 effector-stimulated millisecond conformational exchange dynamics was proposed to allow for a peptide plane flip anticipated for IGPS activation.40 From molecular dynamics simulation and community analysis, effector binding was found to strengthen the coupling between communities across the interdomain interfaces in both Pin137,82 and IGPS.66 It is clear that characterizing the conformational dynamics in these proteins has led to better understanding of their allosteric regulation. It is also clear that NMR spectroscopy and molecular dynamics simulation are highly complementary in developing allosteric mechanisms.
Much remains to be learned. One contentious issue is the relation between fast local dynamics and slow conformational exchange dynamics. One view is that stimulation of the slow dynamics may be accompanied by suppression of the fast dynamics, both of which are related to strengthening of inter-community coupling.82 An opposite view is that disruptions of residue-residue interactions, mediated by strong fast dynamics, may precedes enhancement of slow exchange dynamics. One can fault the first view for the simplicity of the theoretical model used to predict it, and fault the second view for making inference across six orders of magnitude in time. It might be possible to settle this issue through theoretical models that have more molecular ingredients.
Many workers have invoked the term allosteric signal. If there is such a signal, can one experimentally measure its speed of propagation? A recent experiment on a ligand-gated ion channel has the appearance of such a measurement, in which a brief application of an agonist was followed by single-channel recordings of currents through the transmembrane channel.86 The delayed response in currents might be construed as indicating the time needed for propagating an allosteric signal from the ligand-binding domain to the transmembrane channel. However, following the common practice in single-channel electrophysiology, the current response was fit to a kinetic model, with rate constants for agonist binding and unbinding and for conformational transitions of the multi-domain channel protein (including opening and closing of the transmembrane channel). It certainly is more insightful to interpret the delay in current response as due to events including agonist binding and channel closed-to-open transition than as due to the propagation of some allosteric signal. “Pump-probe” molecular dynamics simulations in which selected atoms are pumped by oscillating forces87 and time-resolved femtosecond crystallography enabled by X-ray free electron lasers88–89 might be able to shed light on issues surrounding allosteric signal.
From the apo form to the ternary complex, the same free energy change is accumulated whether effector binding is followed by substrate binding or vice versa. Accordingly, the change in free energy for substrate binding by the pre-binding of an effector is the same as the change in free energy of effector binding by the pre-binding of the substrate (both equal to the free energy of cooperation). This thermodynamic reciprocity implies that allosteric communication is always bidirectional. Our molecular dynamics simulations evidently showed unidirectional allosteric communication in Pin1: while substrate binding to the WW domain resulted in suppression of local dynamics in the PPIase catalytic-site loops, substrate binding to the PPIase did not produce suppression of the local dynamics in the WW domain. Note that this unidirectionality does not violate thermodynamic reciprocity because the former only concerns the events of binding a single ligand (at either the WW site or the catalytic site), whereas the latter involves simultaneous occupation of both sites. This unidirectionality in Pin1 comes about because the WW-bound substrate serves as a bridge to strengthen interdomain communication, whereas this bridge is missing when the substrate is bound to the PPIase. Unidirectional allosteric communication was also seen in molecular dynamics simulations of sortase A, where allosteric activation appears to be mediated by the disorder-to-order transition of a long loop.90 Yet another example was revealed by NMR spectroscopy, for the communication between the two cAMP binding sites (named A and B) in the RIα subunit of protein kinase A.91–92 Upon site A binding, spectral changes spread to the whole protein, but upon site B binding, spectral changes were confined to the B domain only. Examples like these can serve as important test cases for validating ideas about allosteric communication.
Are allosteric proteins endowed with special structural, energetic, or dynamic properties, or can any protein potentially be allosteric? The latter view, argued by Gunasekaran et al.,93 seems to have support. For example, dynamic and conformational changes in response to Val to Ala mutations in a small protein eglin c, previously not known to be allosteric, were detected at sites as far as 16 Å away.94 Moreover, screening with compound libraries identified secondary binding sites on many proteins, and binding of small molecules to these newly identified sites modulated protein functions.95 On the other hand, not all the dynamic and conformational changes in an allosteric protein are equal in mediating allosteric communication. It is important to design control systems to validate proposed mechanisms of allosteric communication.
Such validation can be very effectively done by designed molecular dynamics simulations. Our simulations with artificial restraints on Pin1 offered a glimpse into the potential of this approach. To make our idea about the critical importance of the α1-WW bridge experimentally testable, we introduced an α1-WW crosslink and our molecular dynamics simulations of this construct indeed showed suppression of fast dynamics for the catalytic-site loops, similar to the effect induced by substrate-WW binding.82 This predicted effect on fast dynamics awaits experimental test. On another front, a method called accelerated molecular dynamics designed to artificially reduce free energy barriers has already met with success in exploring the roles of microsecond-millisecond timescale dynamics in allosteric communication.43
While this review has focused on structured proteins, allostery in intrinsically disordered proteins and proteins with intrinsically disordered regions is emerging as an exciting frontier.90,96–97 Conformational dynamics in these proteins can be anticipated to play even greater roles in allosteric communication, although they are only starting to be appreciated.90
Acknowledgments
This work was supported by Grant GM0585187 from the National Institutes of Health.
Biographies
Jingjing Guo received her PhD in medicinal chemistry from Lanzhou University (China) in 2015 under the supervision of Prof. Huanxiang Liu. From 2013 to 2015, she studied as a visiting student with Prof. Huan-Xiang Zhou at Florida State University, conducting research on protein allostery. She is now a lecturer at Henan Normal University (China).
Huan-Xiang Zhou received his PhD from Drexel University in 1988. He did postdoctoral work at the National Institutes of Health with Attila Szabo. After faculty appointments at Hong Kong University of Science and Technology and Drexel, he moved in 2002 to Florida State University, where he is now Distinguished Research Professor. He has served on a number of grant review panels and journal editorial boards. His group currently does theoretical, computational, and experimental research on protein association, on crowding and emergent properties of cellular environments, on structures and functional mechanisms of ion channels and other membrane proteins, and on self-assembly of peptides.
References and Notes
- 1.Monod J, Jacob F. General Conclusions: Telenomic Mechanisms in Cellular Metabolism, Growth, and Differentiation. Cold Spring Harbor Symp Quant Biol. 1961;26:389–401. doi: 10.1101/sqb.1961.026.01.048. [DOI] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Wyman J, Allen DW. The Problem of the Heme Interactions in Hemoglobin and the Basis of the Bohr Effect. J Polym Sci. 1951;7:499–518. [Google Scholar]
- 4.Cooper A, Dryden DT. Allostery without Conformational Change. A Plausible Model. Eur Biophys J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 5.Nussinov R, Tsai CJ. Allostery without a Conformational Change? Revisiting the Paradigm. Curr Opin Struct Biol. 2015;30:17–24. doi: 10.1016/j.sbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Dai J, Zhou HX. Reduced Curvature of Ligand-Binding Domain Free-Energy Surface Underlies Partial Agonism at NMDA Receptors. Structure. 2015;23:228–236. doi: 10.1016/j.str.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai J, Wollmuth LP, Zhou HX. Mechanism-Based Mathematical Model for Gating of Ionotropic Glutamate Receptors. J Phys Chem B. 2015;119:10934–10940. doi: 10.1021/acs.jpcb.5b00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgen AS. Conformational Changes and Drug Action. Fed Proc. 1981;40:2723–2728. [PubMed] [Google Scholar]
- 10.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding Funnels and Binding Mechanisms. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 11.Formaneck MS, Ma L, Cui Q. Reconciling the “Old” and “New” Views of Protein Allostery: A Molecular Simulation Study of Chemotaxis Y Protein (Chey) Proteins. 2006;63:846–867. doi: 10.1002/prot.20893. [DOI] [PubMed] [Google Scholar]
- 12.Boehr DD, Nussinov R, Wright PE. The Role of Dynamic Conformational Ensembles in Biomolecular Recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 14.Greives N, Zhou HX. Both Protein Dynamics and Ligand Concentration Can Shift the Binding Mechanism between Conformational Selection and Induced Fit. Proc Natl Acad Sci USA. 2014;111:10197–10202. doi: 10.1073/pnas.1407545111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammes GG, Wu CW. Kinetics of Allosteric Enzymes. Annu Rev Biophys Bioeng. 1974;3:1–33. doi: 10.1146/annurev.bb.03.060174.000245. [DOI] [PubMed] [Google Scholar]
- 16.Hammes GG, Chang YC, Oas TG. Conformational Selection or Induced Fit: A Flux Description of Reaction Mechanism. Proc Natl Acad Sci USA. 2009;106:13737–13741. doi: 10.1073/pnas.0907195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou HX. From Induced Fit to Conformational Selection: A Continuum of Binding Mechanism Controlled by the Timescale of Conformational Transitions. Biophys J. 2010;98:L15–L17. doi: 10.1016/j.bpj.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perutz MF. Stereochemistry of Cooperative Effects in Haemoglobin. Nature. 1970;228:726–739. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 19.Eaton WA, Henry ER, Hofrichter J, Bettati S, Viappiani C, Mozzarelli A. Evolution of Allosteric Models for Hemoglobin. IUBMB Life. 2007;59:586–599. doi: 10.1080/15216540701272380. [DOI] [PubMed] [Google Scholar]
- 20.Cui Q, Karplus M. Allostery and Cooperativity Revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wrabl JO, Gu J, Liu T, Schrank TP, Whitten ST, Hilser VJ. The Role of Protein Conformational Fluctuations in Allostery, Function, and Evolution. Biophys Chem. 2011;159:129–141. doi: 10.1016/j.bpc.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-State Allosteric Behavior in a Single-Domain Signaling Protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 23.Yan J, Liu Y, Lukasik SM, Speck NA, Bushweller JH. CBFbeta Allosterically Regulates the Runx1 Runt Domain Via a Dynamic Conformational Equilibrium. Nat Struct Mol Biol. 2004;11:901–906. doi: 10.1038/nsmb819. [DOI] [PubMed] [Google Scholar]
- 24.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically Driven Protein Allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden Dynamic Allostery in a PDZ Domain. Proc Natl Acad Sci USA. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Namanja AT, Wang XJ, Xu B, Mercedes-Camacho AY, Wilson KA, Etzkorn FA, Peng JW. Stereospecific Gating of Functional Motions in Pin1. Proc Natl Acad Sci USA. 2011;108:12289–12294. doi: 10.1073/pnas.1019382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruschak AM, Kay LE. Proteasome Allostery as a Population Shift Between Interchanging Conformers. Proc Natl Acad Sci USA. 2012;109:E3454–3462. doi: 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manley G, Rivalta I, Loria JP. Solution NMR and Computational Methods for Understanding Protein Allostery. J Phys Chem B. 2013;117:3063–3073. doi: 10.1021/jp312576v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, et al. Structural Basis for Modulation of a G-Protein-Coupled Receptor by Allosteric Drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 30.Malmstrom RD, Kornev AP, Taylor SS, Amaro RE. Allostery through the Computational Microscope: cAMP Activation of a Canonical Signalling Domain. Nat Commun. 2015;6 doi: 10.1038/ncomms8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pontiggia F, Pachov DV, Clarkson MW, Villali J, Hagan MF, Pande VS, Kern D. Free Energy Landscape of Activation in a Signalling Protein at Atomic Resolution. Nat Commun. 2015;6:7284. doi: 10.1038/ncomms8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feher VA, Durrant JD, Van Wart AT, Amaro RE. Computational Approaches to Mapping Allosteric Pathways. Curr Opin Struct Biol. 2014;25:98–103. doi: 10.1016/j.sbi.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily Conserved Networks of Residues Mediate Allosteric Communication in Proteins. Nat Struct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 34.Sethi A, Eargle J, Black AA, Luthey-Schulten Z. Dynamical Networks in tRNA:Protein Complexes. Proc Natl Acad Sci USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The Ensemble Nature of Allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wand AJ. On the Dynamic Origins of Allosteric Activation. Science. 2001;293:1395. doi: 10.1126/science.293.5534.1395a. [DOI] [PubMed] [Google Scholar]
- 37.Guo J, Pang X, Zhou HX. Two Pathways Mediate Interdomain Allosteric Regulation in Pin1. Structure. 2015;23:237–247. doi: 10.1016/j.str.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tzeng SR, Kalodimos CG. Protein Dynamics and Allostery: An NMR View. Curr Opin Struct Biol. 2011;21:62–67. doi: 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 39.Zhuravleva A, Gierasch LM. Substrate-Binding Domain Conformational Dynamics Mediate Hsp70 Allostery. Proc Natl Acad Sci USA. 2015;112:E2865–2873. doi: 10.1073/pnas.1506692112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lipchock JM, Loria JP. Nanometer Propagation of Millisecond Motions in V-Type Allostery. Structure. 2010;18:1596–1607. doi: 10.1016/j.str.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Srivastava AK, McDonald LR, Cembran A, Kim J, Masterson LR, McClendon CL, Taylor SS, Veglia G. Synchronous Opening and Closing Motions Are Essential for cAMP-Dependent Protein Kinase a Signaling. Structure. 2014;22:1735–1743. doi: 10.1016/j.str.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyen D, Fenwick RB, Stanfield RL, Dyson HJ, Wright PE. Cofactor-Mediated Conformational Dynamics Promote Product Release from Escherichia Coli Dihydrofolate Reductase Via an Allosteric Pathway. J Am Chem Soc. 2015;137:9459–9468. doi: 10.1021/jacs.5b05707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gasper PM, Fuglestad B, Komives EA, Markwick PR, McCammon JA. Allosteric Networks in Thrombin Distinguish Procoagulant vs. Anticoagulant Activities. Proc Natl Acad Sci USA. 2012;109:21216–21222. doi: 10.1073/pnas.1218414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer AG., 3rd NMR Characterization of the Dynamics of Biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 45.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 Relaxation by Mutual Cancellation of Dipole-Dipole Coupling and Chemical Shift Anisotropy Indicates an Avenue to NMR Structures of Very Large Biological Macromolecules in Solution. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lipari G, Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 47.Showalter SA, Bruschweiler R. Validation of Molecular Dynamics Simulations of Biomolecules Using NMR Spin Relaxation as Benchmarks: Application to the Amber99SB Force Field. J Chem Theory Comput. 2007;3:961–975. doi: 10.1021/ct7000045. [DOI] [PubMed] [Google Scholar]
- 48.Maragakis P, Lindorff-Larsen K, Eastwood MP, Dror RO, Klepeis JL, Arkin IT, Jensen MO, Xu HF, Trbovic N, Friesner RA, et al. Microsecond Molecular Dynamics Simulation Shows Effect of Slow Loop Dynamics on Backbone Amide Order Parameters of Proteins. J Phys Chem B. 2008;112:6155–6158. doi: 10.1021/jp077018h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang D, Kay LE. Contributions to Conformational Entropy Arising from Bond Vector Fluctuations Measured from NMR-Derived Order Parameters: Application to Protein Folding. J Mol Biol. 1996;263:369–382. doi: 10.1006/jmbi.1996.0581. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Raychaudhuri S, Wand AJ. Insights into the Local Residual Entropy of Proteins Provided by NMR Relaxation. Protein Sci. 1996;5:2647–2650. doi: 10.1002/pro.5560051228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wand AJ. The Dark Energy of Proteins Comes to Light: Conformational Entropy and Its Role in Protein Function Revealed by NMR Relaxation. Curr Opin Struct Biol. 2013;23:75–81. doi: 10.1016/j.sbi.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carr HY, Purcell EM. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys Rev. 1954;94:630–638. [Google Scholar]
- 53.Meiboom S, Gill D. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev Sci Instrum. 1958;29:688–691. [Google Scholar]
- 54.Luz Z, Meiboom S. Nuclear Magnetic Resonance Study of Protolysis of Trimethylammonium Ion in Aqueous Solution - Order of Reaction with Respect to Solvent. J Chem Phys. 1963;39:366–370. [Google Scholar]
- 55.Carver JP, Richards RE. General Two-Site Solution for Chemical Exchange Produced Dependence of T2 Upon Carr-Purcell Pulse Separation. J Magn Reson. 1972;6:89–105. [Google Scholar]
- 56.Baldwin AJ. An Exact Solution for R2,Eff in CPMG Experiments in the Case of Two Site Chemical Exchange. J Magn Reson. 2014;244:114–124. doi: 10.1016/j.jmr.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung F. Spectral Graph Theory. AMS Publications; 1997. [Google Scholar]
- 58.Atilgan AR, Akan P, Baysal C. Small-World Communication of Residues and Significance for Protein Dynamics. Biophys J. 2004;86:85–91. doi: 10.1016/S0006-3495(04)74086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bahar I, Atilgan AR, Erman B. Direct Evaluation of Thermal Fluctuations in Proteins Using a Single-Parameter Harmonic Potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 60.Brinda KV, Vishveshwara S. A Network Representation of Protein Structures: Implications for Protein Stability. Biophys J. 2005;89:4159–4170. doi: 10.1529/biophysj.105.064485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seeber M, Felline A, Raimondi F, Muff S, Friedman R, Rao F, Caflisch A, Fanelli F. Wordom: A User-Friendly Program for the Analysis of Molecular Structures, Trajectories, and Free Energy Surfaces. J Comput Chem. 2011;32:1183–1194. doi: 10.1002/jcc.21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chennubhotla C, Bahar I. Markov Propagation of Allosteric Effects in Biomolecular Systems: Application to Groel-Groes. Mol Syst Biol. 2006;2:36. doi: 10.1038/msb4100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng W, Brooks BR, Thirumalai D. Allosteric Transitions in the Chaperonin GroEL Are Captured by a Dominant Normal Mode That Is Most Robust to Sequence Variations. Biophys J. 2007;93:2289–2299. doi: 10.1529/biophysj.107.105270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Floyd RW. Algorithm-97 - Shortest Path. Commun ACM. 1962;5:345–345. [Google Scholar]
- 65.Girvan M, Newman ME. Community Structure in Social and Biological Networks. Proc Natl Acad Sci USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rivalta I, Sultan MM, Lee NS, Manley GA, Loria JP, Batista VS. Allosteric Pathways in Imidazole Glycerol Phosphate Synthase. Proc Natl Acad Sci USA. 2012;109:E1428–1436. doi: 10.1073/pnas.1120536109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lange OF, Grubmuller H. Generalized Correlation for Biomolecular Dynamics. Proteins. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 68.Vanwart AT, Eargle J, Luthey-Schulten Z, Amaro RE. Exploring Residue Component Contributions to Dynamical Network Models of Allostery. J Chem Theory Comput. 2012;8:2949–2961. doi: 10.1021/ct300377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vanatta DK, Shukla D, Lawrenz M, Pande VS. A Network of Molecular Switches Controls the Activation of the Two-Component Response Regulator NtrC. Nat Commun. 2015;6:7283. doi: 10.1038/ncomms8283. [DOI] [PubMed] [Google Scholar]
- 70.Lu KP, Hanes SD, Hunter T. A Human Peptidyl–Prolyl Isomerase Essential for Regulation of Mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 71.Liou YC, Zhou XZ, Lu KP. Prolyl Isomerase Pin1 as a Molecular Switch to Determine the Fate of Phosphoproteins. Trends Biochem Sci. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu PJ, Zhou XZ, Shen M, Lu KP. Function of Ww Domains as Phosphoserine- or Phosphothreonine-Binding Modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 73.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Küllertz G, Stark M, Fischer G, Lu KP. Pin1-Dependent Prolyl Isomerization Regulates Dephosphorylation of Cdc25c and Tau Proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 74.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and Functional Analysis of the Mitotic Rotamase Pin1 Suggests Substrate Recognition Is Phosphorylation Dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 75.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural Basis for Phosphoserine-Proline Recognition by Group IV WW Domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 76.Zhang M, Wang XJ, Chen X, Bowman ME, Luo Y, Noel JP, Ellington AD, Etzkorn FA, Zhang Y. Structural and Kinetic Analysis of Prolyl-Isomerization/Phosphorylation Cross-Talk in the Ctd Code. ACS Chem Biol. 2012;7:1462–1470. doi: 10.1021/cb3000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jacobs DM, Saxena K, Vogtherr M, Bernado P, Pons M, Fiebig KM. Peptide Binding Induces Large Scale Changes in Inter-Domain Mobility in Human Pin1. J Biol Chem. 2003;278:26174–26182. doi: 10.1074/jbc.M300796200. [DOI] [PubMed] [Google Scholar]
- 78.Matena A, Sinnen C, van den Boom J, Wilms C, Dybowski JN, Maltaner R, Mueller JW, Link NM, Hoffmann D, Bayer P. Transient Domain Interactions Enhance the Affinity of the Mitotic Regulator Pin1 toward Phosphorylated Peptide Ligands. Structure. 2013;21:1769–1777. doi: 10.1016/j.str.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 79.Namanja AT, Peng T, Zintsmaster JS, Elson AC, Shakour MG, Peng JW. Substrate Recognition Reduces Side-Chain Flexibility for Conserved Hydrophobic Residues in Human Pin1. Structure. 2007;15:313–327. doi: 10.1016/j.str.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 80.Wilson KA, Bouchard JJ, Peng JW. Interdomain Interactions Support Interdomain Communication in Human Pin1. Biochemistry. 2013;52:6968–6981. doi: 10.1021/bi401057x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, Mahoney BJ, Zhang M, Zintsmaster JS, Peng JW. Negative Regulation of Peptidyl-Prolyl Isomerase Activity by Interdomain Contact in Human Pin1. Structure. 2015;23:2224–2233. doi: 10.1016/j.str.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo J, Zhou HX. Dynamically Driven Protein Allostery Exhibits Disparate Responses for Fast and Slow Motions. Biophys J. 2015;108:2771–2774. doi: 10.1016/j.bpj.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Namanja AT. PhD Dissertation. University of Notre Dame; 2009. Molecular Basis for Signal Transduction in the Bimodular Cell-Cycle Enzyme Pin1. [Google Scholar]
- 84.Myers RS, Jensen JR, Deras IL, Smith JL, Davisson VJ. Substrate-Induced Changes in the Ammonia Channel for Imidazole Glycerol Phosphate Synthase. Biochemistry. 2003;42:7013–7022. doi: 10.1021/bi034314l. [DOI] [PubMed] [Google Scholar]
- 85.Douangamath A, Walker M, Beismann-Driemeyer S, Vega-Fernandez MC, Sterner R, Wilmanns M. Structural Evidence for Ammonia Tunneling across the (βα)8 Barrel of the Imidazole Glycerol Phosphate Synthase Bienzyme Complex. Structure. 2002;10:185–193. doi: 10.1016/s0969-2126(02)00702-5. [DOI] [PubMed] [Google Scholar]
- 86.Kazi R, Dai J, Sweeney C, Zhou HX, Wollmuth LP. Mechanical Coupling Maintains the Fidelity of NMDA Receptor-Mediated Currents. Nat Neurosci. 2014;17:914–922. doi: 10.1038/nn.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharp K, Skinner JJ. Pump-Probe Molecular Dynamics as a Tool for Studying Protein Motion and Long Range Coupling. Proteins. 2006;65:347–361. doi: 10.1002/prot.21146. [DOI] [PubMed] [Google Scholar]
- 88.Tenboer J, Basu S, Zatsepin N, Pande K, Milathianaki D, Frank M, Hunter M, Boutet S, Williams GJ, Koglin JE, et al. Time-Resolved Serial Crystallography Captures High-Resolution Intermediates of Photoactive Yellow Protein. Science. 2014;346:1242–1246. doi: 10.1126/science.1259357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barends TR, Foucar L, Ardevol A, Nass K, Aquila A, Botha S, Doak RB, Falahati K, Hartmann E, Hilpert M, et al. Direct Observation of Ultrafast Collective Motions in CO Myoglobin Upon Ligand Dissociation. Science. 2015;350:445–450. doi: 10.1126/science.aac5492. [DOI] [PubMed] [Google Scholar]
- 90.Pang X, Zhou HX. Disorder-to-Order Transition of an Active-Site Loop Mediates the Allosteric Activation of Sortase A. Biophys J. 2015;109:1706–1715. doi: 10.1016/j.bpj.2015.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Byeon IJ, Dao KK, Jung J, Keen J, Leiros I, Doskeland SO, Martinez A, Gronenborn AM. Allosteric Communication between cAMP Binding Sites in the RI Subunit of Protein Kinase a Revealed by NMR. J Biol Chem. 2010;285:14062–14070. doi: 10.1074/jbc.M110.106666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McNicholl ET, Das R, SilDas S, Taylor SS, Melacini G. Communication between Tandem cAMP Binding Domains in the Regulatory Subunit of Protein Kinase A-Iα as Revealed by Domain-Silencing Mutations. J Biol Chem. 2010;285:15523–15537. doi: 10.1074/jbc.M110.105783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gunasekaran K, Ma BY, Nussinov R. Is Allostery an Intrinsic Property of All Dynamic Proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 94.Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Dynamic Coupling and Allosteric Behavior in a Nonallosteric Protein. Biochemistry. 2006;45:7693–7699. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H. Detection of Secondary Binding Sites in Proteins Using Fragment Screening. Proc Natl Acad Sci USA. 2015;112:15910–15915. doi: 10.1073/pnas.1518946112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hilser VJ, Thompson EB. Intrinsic Disorder as a Mechanism to Optimize Allosteric Coupling in Proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of Allostery by Protein Intrinsic Disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]