

Abstract
Pancreatic adenocarcinoma (PAC) is one of the most deadly malignant neoplasms, and the efficacy of conventional cytotoxic chemotherapy is far from satisfactory. Recent research studies have revealed that immunosuppression and inflammation are associated with oncogenesis, as well as tumor development, invasion, and metastasis in PAC. Thus, immunosuppression-related signaling, especially that involving immune checkpoint and inflammation, has emerged as novel treatment targets for PAC. However, PAC is an immune-resistant tumor, and it is still unclear whether immune checkpoint or anti-inflammation therapies would be an ideal strategy. In this article, we will review immune checkpoint and inflammation as potential targets, as well as clinical trials and the prospects for immunotherapy in PAC.
Keywords: Immune checkpoint, Therapeutic anticancer target, Inflammation, Randomized clinical trial, Pancreatic adenocarcinoma
Core tip: Pancreatic adenocarcinoma is recognized as one of the most malignant neoplasms, and more efficacious treatment is desired earnestly. Recent research studies have revealed that the development and progression of pancreatic adenocarcinoma are highly influenced by immune responses, and inflammation is a critical promoter of the disease. In this article, we highlighted the emergence of immunosuppression-related signaling associated with immune checkpoint and inflammation, as a novel treatment target for cancer. Furthermore, the review demonstrated that the current focus on therapeutic strategies involving combination chemotherapy, immunotherapy, and anti-inflammation therapy might provide considerably more clinical benefits to patients than current therapies.
INTRODUCTION
Currently, pancreatic adenocarcinoma (PAC) is recognized as one of the most malignant neoplasms. Most patients with PAC are diagnosed at an advanced, incurable stage because of the absence of routine screening and virtually no specific subjective symptoms before tumor progression. Although advances in a variety treatment approaches have improved the therapeutic management, the 5-year survival rate for patients with metastatic and recurrent PAC remains lower than 5%[1].
For over a decade, gemcitabine monotherapy has been a standard regimen for advanced PAC[2]. Recently, two pivotal studies demonstrated that FOLFIRINOX and nab-paclitaxel plus gemcitabine are more effective than gemcitabine monotherapy. However, these combination regimens prolonged the survival by only several months compared to gemcitabine with significantly increased toxicities[3].
Recent research studies have revealed that the development and progression of PCA are highly influenced by immune responses, and inflammation is a critical promoter of the disease. Therefore, targeting immunosuppressive and inflammatory signaling pathways may be a promising strategy. Here, we will review immunotherapy, especially focusing on the immune checkpoint and inflammatory signaling as potential therapeutic targets in PAC.
IMMUNE CHECKPOINT THERAPY IN PAC TO DATE
It is widely known that tumor cells escape from the host immune surveillance, and some tumors are resistant to the host immune responses. This immune tolerance is closely associated with immune checkpoints, which are inhibitory immune-related pathways that are initiated by ligand-receptor interactions between T cells and antigen-presenting cells (APCs) or tumor cells. T cells recognize tumor-associated antigens (TAAs) presented by the major histocompatibility complex class II from APCs or tumor cells through the T-cell receptor. Simultaneously, costimulatory signals are needed to determine whether T cells activate or inhibit immune responses. When the costimulatory signal is an inhibitory pathway, T cells suppress the immune response, and then tumor cells are subsequently shielded from immune surveillance. Immune checkpoint therapy blocks this inhibitory signal and overcomes the immune tolerance[4].
Among the numerous costimulatory molecules, cytotoxic T-lymphocyte antigen (CLTA)-4 and programmed cell death (PD)-1/PD-ligand (PD-L) 1 have been established as treatment targets. Monoclonal antibodies including ipilimumab, nivolumab, and pembrolizumab blockade these molecules. These agents inhibit costimulatory signals, which results in activation of cytotoxic T lymphocytes (CTLs) against tumor cells. These immune checkpoint blockade demonstrated substantial efficacy in melanoma[5], non-small cell lung cancer (NSCLC)[6], and renal cell cancer[7]. Vigorous research studies are currently ongoing in various tumors.
However, immune checkpoint therapy may not be effective in PAC, at least as a monotherapy. A phase 2 trial evaluated the efficacy of ipilimumab in advanced PAC[8], but no response was observed per Response Evaluation Criteria In Solid Tumors at a dose of 3.0 mg/kg. Furthermore, an anti-PD-L1 antibody (BMS-936559) achieved no response (0/14) in a phase 1 trial of patients with PAC[9]. Although several clinical trials are currently ongoing, single immune checkpoint blockade does not a promising option at this time.
The reason why immune checkpoint therapy is not effective in PAC is unclear, but several hypotheses have been proposed.
One possible reason is associated with PD-L1 expression. Several studies suggested that PD-L1 expression in tumor cells, detected by immunohistochemistry, is associated with responses to PD-1/PD-L1 blockade therapy[10]. However, PD-L1 is expressed in approximately 40% of pancreatic cancer cells[11]. In addition, PD-L1 expression can be induced by oncogenic signaling independent of inflammatory signaling[12]. Therefore, the level of PD-L1 expression in tumor cells alone cannot explain their resistance to immune checkpoint therapy in PAC.
The second possible reason is associated with the immune cell population in the tumor microenvironment (TME), which consists of a highly complicated interaction between tumor cells, immune cells, and stromal cells. With regard to immune cells, from the early tumorigenesis phase, myeloid-driven suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs) occur in the TME. These cells deactivate immune response by various mechanisms. For example DMSCs have potent immunosuppressive properties through the production of reactive oxygen species[13], expression of arginase-1[14], depletion of cysteine[15], and suppression of CTLs. TAMs suppress the function of T cells, NKT cells and NK cells by expression of the ligand receptors for PD-1 and CTLA-4, and induce apoptosis by expression of FASL and TRAIL[16]. MDSCs and TAMs induce Tregs in TME. Tregs suppress APCs via CTLA-4, secretion, secrete inhibitory cytokines (IL-10, TGF-β and IL-35), express granzyme/perforin against effector T cells, and inhibit differentiation of effector T cells by IL-2 consumption by CD25[17]. All of these functions provide immune suppressive environment. On the other hand, few effector T cells, which play essential roles in immune checkpoint therapy infiltrate into the tumor tissue[18]. This imbalanced immune cells population tends to enhance immunosuppression and interrupt immune checkpoint therapy.
The third possible reason is associated with stromal cells in the TME. An abundance of desmoplastic stroma is a distinctive feature of PAC. Desmoplastic stroma consists of cancer-associated fibroblasts (CAFs) and extracellular matrix (ECM) and immune cells. It is well known that CAFs promote tumor progression through the Hedgehog, Wnt/β-catenin, Notch, K-ras signaling and the production of growth factors[19]. In addition, CAFs secrete chemokine ligand 12 (CXCL12) and interleukin 17 (IL-17). These mediators suppress T cells via chemokine receptor 4 (CXCR4)[19]. The CXCR4-CXCL12 axis may be related to resistance to immune checkpoint therapy because the blockade of this signal has a synergistic effect on anti-PD-1 therapy[20].
As mentioned above, PAC induces a highly immunosuppressive environment regulated by immune cells, stromal cells, and mediators. This condition may contribute to its resistance to immune checkpoint therapy.
OVERCOMING RESISTANCE TO IMMUNE CHECKPOINT THERAPY
Comprehensive research studies have revealed strategies to overcoming the resistance to immune checkpoint therapy. One approach involves the establishment of a positive predictive biomarker for immune checkpoint therapy. Expression of PD-L1 may be one candidate predictive marker. In NCSLC, several trials have reported that the objective response rate is associated with PD-L1 expression, although conflicting evidence exists[21]. However, as mentioned above, it is unclear whether the expression of PD-L1 could serve as a useful biomarker in patients with PAC. In addition, the standard immunohistochemical tests for the determination of PD-L1, and the cut off for PD-L1 positive status have not been established yet.
Another candidate predictive marker is the mismatch repair (MMR) status of DNA. MMR deficiency leads to a high number of somatic mutations in tumors. Theoretically, accumulation of these somatic mutations can be recognized by the patient’s immune system. Le et al[22] hypothesized that tumors with MMR deficiency are sensitive to immune checkpoint therapy, and initiated a phase 2 trial in which pembrolizumab was administered to 41 patients with or without MMR deficiency. In that trial, MMR status was assessed by microsatellite instability (MSI) analysis. Microsatellite is region of repetitive DNA, where hundreds to thousands of somatic mutations are occurred in tumor with MMR deficiency. Condition of accumulation of somatic mutations in microsatellite is referred to as MSI, and MSI reflects MMR deficiency. Of the 41 patients, 32 had colorectal cancers, which were regarded as immune resistant tumors. Eleven patients had MMR-deficient colorectal cancer and 21 had MMS-proficient forms. The remaining nine patients had MMR-deficient noncolorectal cancer. PAC was not included in the trial. They reported that the immune-related objective response was 40% in the patients with MMR deficiency, while patients without MMR deficiency did not achieved any response. Therefore, MMR status can be a useful predictive marker for pembrolizumab therapy. That is why PAC with MSI expects to be sensitive to PD-1 therapy. However, PAC with MSI is extremely rare[23] and, therefore, this enrichment strategy based on MSI examination may not be realistic.
Another approach to overcome the resistance to immune checkpoint therapy is establishment of more potent treatment. Combination immunotherapy is currently emerging as a promising treatment. However determining the most effective combinations is a challenge. Candidates for combination therapy with immune checkpoint inhibitors are (1) cytotoxic agents; (2) other immune checkpoint inhibitors; (3) direct cytotoxic T cell stimulators; (4) cancer vaccines; and (5) radiation (Tables 1 and 2). Rationales and problems of each combination therapy are discussed here.
Table 1.
Rationales of each combination therapy
| Treatments | Rationales | Concerns |
| Checkpoint inhibitor plus cytotoxic agents | Enhance cellular immunity | Efficacy may be influenced by timing when cytotoxic agents add |
| Augment dendritic cell maturation | Severe myelosupression may interrupt immune checkpoint therapy | |
| Reduce MDSC and Tregs | ||
| Decreases CAF | ||
| Combination with checkpoint inhibitors | Activate tumor immunity by different mannar | ir AE will increase |
| Provide synergy efficacy even in immune resistant tumor | ||
| Checkpoint inhibitor plus T cells stimulate agents | Activate tumor immunity by different mannar | Severe AE including cytokine storm may occur |
| Deactive Tregs | ||
| Checkpoint inhibitor plus cancer vaccine | Increase the presentation of taas | |
| Enhance PD-L1 expression | ||
| Radiotherapy | Enhance cross priming of ctls | Optimal schedule and dose are not established |
| Enhanse abscopal effect |
MDSC: Myeloid-driven suppressor cell; CAF: Cancer-associated fibroblast; Tregs: Regulatory T cells.
Table 2.
Problems of each combination therapy
| Treatment | Disease | Phase | Clinical trial number | Status |
| Checkpoint inhibitor plus cytotoxic agents | ||||
| Ipilimumab (anti-CDLA-4) | PC | 1 | NCT01473940 | Ongoing |
| Gemcitabine | ||||
| Nivolumab (anti-PD-1) | PC | 1 | NCT02309177 | Ongoing |
| Nab-PTX ± gemcitabine | ||||
| Combination with checkpoint inhibitors | ||||
| Nivolumab (anti-PD-1) | TNBC, GC, PC, | 1, 2 | NCT01928394 | Ongoing |
| Ipilimumab (anti-CTLA-4) | SLCL. BC, OC | |||
| MEDI4736 (anti-PD-1) | Solid tumor | 1 | NCT02261220 | Ongoing |
| Tremelimumab (anti-CTLA-4) | ||||
| Nivolumab (anti-PD-1) | Cervical cancer, BC, CRC, HN, GC, HCC, melanoma, NSCLC | 1, 2 | NCT01968109 | Ongoing |
| BMS-986016 (anti-LAG-3) | ||||
| PDR001 (anti-PD-1) | Solid tumors | 1, 2 | NCT02608268 | Ongoing |
| MBG453 (anti-TIM-3) | ||||
| Ipilimumab (anti-CDLA-4) | B7-H3 expressing tumors (melanoma, HN, NSCLC) | 1, 2 | NCT02381314 | Ongoing |
| MGA271 (anti-B7-H3) | ||||
| Pembrolizumab (anti-PD-1) | B7-H3 expressing tumors | 1, 2 | NCT02475213 | Ongoing |
| MGA271 (anti-B7-H3) | (melanoma, HN, NSCLC) | |||
| Checkpoint inhibitor plus T cells stimulate agents | ||||
| Nivolumab (anti-PD-1) | Solid tumors, B-cell NHL | 1, 2 | NCT02253992 | Ongoing |
| Urelumab (anti-4-1 BB) | ||||
| Pembrolizumab (anti-PD-1) | Solid tumors | 1 | NCT02179918 | Ongoing |
| Urelumab (anti-4-1 BB) | ||||
| Tremelimumab (anti-CTLA-4) | Solid tumors | 1, 2 | NCT02205333 | Ongoing |
| MEDI6469 (anti-OX-40 | ||||
| MEDI4736 (anti-PD-L1) | Solid tumors | 1, 2 | NCT02205333 | Ongoing |
| MEDI6469 (anti-OX-40) | ||||
| Tremelimumab (anti-CTLA-4) | Melanoma | 1 | NCT01103635 | Ongoing |
| CP-870,893 (anti-CD40) | ||||
| Checkpoint inhibitor plus cancer vaccine | ||||
| Ipilimumab (anti-CTLA-4) + GVAX | PC | 2 | Ref 58 | Terminated |
| FOLFIRINOX followed by | PC | 2 | NCT01896869 | Ongoing |
| Ipilimumab (anti-CTLA-4) + GVAX | ||||
| Nivolumeb (anti-PD-1) + GVAX | PC | 2 | NCT02243371 | Ongoing |
| Checkpoint inhibitor plus raditaion | ||||
| Ipilimumab + radiation | Melanoma | 1 | NCT01557114 | Terminated |
| Melanoma | 2 | NCT016899747 | Terminated | |
| NSCLC | 2 | NCT0221739 | Ongoing | |
| Melanoma | 2 | NCT01970527 | Ongoing |
BC: Bladder cancer; CRC: Colorectal cancer; HN: Head and neck cancer; NSCLC: Non-small cell lung cancer; OC: Ovarian cancer; PC: Pancreatic cancer; SCLC: Small cell lung cancer.
Combination therapy with cytotoxic regimens
Originally, it was thought that cytotoxic agents impaired immunity due to myelosuppression, and combination of cytotoxic agents and immunotherapy was not desirable. Accumulating evidences suggest that cytotoxic agents affect the immune system differentially; sometimes they act on the activate tumor immunity and other times on the inactivate[24]. Gemcitabine, oxaliplatin, irinotecan, 5-FU, and paclitaxel (nab-paclitaxel) are key drugs in the treatment of PAC. Preclinical data showed that these agents have immune effects such as enhancement of cellular immunity, augmentation of dendritic cell maturation, and reduction of MDSC and Tregs (reviewed by Duffy et al[25]). These findings suggest cytotoxic agents may be good partners of immune checkpoint therapy. In fact, a synergistic effect was observed with the combination of ipilimumab and cytotoxic agents in a preclinical tumor model[26]. In addition, randomized phase 2 trial showed that combination ipilimumab and CP (paclitaxel and carboplatin) improved immune-related progression-free survival (irPFS) compared to CP alone in NSCLC[27].
The results of the phase 1 trial of the combination of ipilimumab and gemcitabine (NCT01473940) and combination therapy with tremelimumab (CTLA-4 blocking IgG2 antibody) and gemcitabine for advanced PAC were reported that both regimens were well tolerated. However, tremelimumab and gemcitabine achieved responses in only 10.5%, which appeared comparable to the effects of gemcitabine monotherapy[28]. Nab-paclitaxel, which decreases CAFs in preclinical PAC model[29], is also a promising combination candidate. A phase 1 study of combination therapy with nivolumab and nab-paclitaxel with or without gemecitabine is currently ongoing (NCT02309177). Phase 2 trial of FOLFIRINOX followed by ipilimumab with tumor vaccine for metastatic PAC is also ongoing (NCT01896869).
To develop this combination therapy, there are two challenges that need to be overcome; determining what cytotoxic agents to combine and when to use the cytotoxic agent combination. Several studies reported that the absolute lymphocyte count had a positive relationship with improved OS in patients with melanoma who were treated with ipilimumab[30]. Thus, cytotoxic agents with high myelotoxicity may not be desirable. In addition, the timing of the combination may affect efficacy. Phased combination improved the irPFS, but the concurrent combination did not, in NSCLC phase 2 trial[27].
Combination therapy with immune checkpoint signaling blockers
Although both PD-1 pathway and CTLA-4 pathway are related to the regulation of T cell function, the mechanisms of action of each are different[31]. Das et al[32] reported that CTLA-4 blockade leads to a proliferative signature predominantly in a subset of transitional memory T cells, whereas PD-1 blockade leads to changes in genes implicated in cytolysis and NK cell function in vivo. Thus, dual blockade therapy is a reasonable strategy and may have synergistic efficacy. In a melanoma model, combination blockade was related to prolonged survival, proliferation, and enhanced function of CD8+ and CD4+ cells, and increased ratio of effector T cell /Treg and MDSCs[31]. Das et al[32] also reported that combination blockade is associated with changes in plasma chemokine and cytokine compared to mono blockade. Lussier et al[33] showed that combination blockade with CTLA-4 and PD-L1 (ligand of PD-1) antibody could achieve complete control of an osteosarcoma model whereas mono blockade could not. These preclinical data suggest that combination blockade with CTLA-4 and PD-1 may have more potent efficacy, even for immune resistant cancer including PAC, than monotherapy.
Several clinical trials showed the efficacy of combination blockade therapy. A phase 3 trial of combination therapy with ipilimumab and nivolumab showed that combination therapy provided greater clinical benefits than ipilimumab monotherapy did in melanoma[34]. The same combination regimen was evaluated in a phase 1 trial for NSCLC and renal cell cancer[35,36], and phase 3 trials are currently ongoing. For other tumors including PAC, a phase 1/2 trial to evaluate the tolerability and efficacy of the same regimen tumors is currently ongoing (NCT01928394). Phase 2 trial of durvalumab (MEDI4736, anti-PD-1 IgG1 mAb) with tremelimumab (anti-CTLA-4 IgG2 mAb) for advanced PAC is also ongoing (NCT02558894).
A critical issue in this combination therapy is increased toxicity. Compared to monotherapy, combination therapy was associated with increased serious adverse events (AEs) and discontinuation of treatment due to AEs[34]. Whether this combination therapy has a good risk/benefit balance or not in PAC is unclear.
Lymphocyte-activation gene 3 (LAG-3) and T-cell immunoglobulin and mucin domain 3 (TIM3) signaling are other immune checkpoint targets. Preclinical studies showed that dual blockade of PD-1 and LAG-3[37], and PD-1 and TIM3[38] induce a synergistic effect in controlling tumor growth. These combination therapies have been evaluated in phase 1 trials such as nivolumab plus BMS-986016 (anti-LAG-3) (NCT01968109 (PAC was not included) and PDR001 (anti-PD-1) plus MBG453 (anti-TIM-3) (NCT02608268). Furthermore, recent studies implicated the B7-H3 and B7-H4 pathways in the progression of pancreatic cancer and the blockade of these signaling pathways is a novel treatment target[39]. In future, B7-H3 or B7-H4 inhibitors may become combination partners with other immune checkpoint inhibitors.
Combination therapy with direct cytotoxic T cell stimulators
The immune system is regulated both by immunosuppressive signaling and by immunoreactive signaling. Immune checkpoint signaling is an example of immunosuppressive signals. On the other hand, active costimulatory signaling activates an immune response, which is contrary to immune checkpoint signaling. This costimulatory signaling is associated with the tumor necrosis family receptors superfamily (TNFRSF). TNFRSF proteins play an important role in B and T cell development, survival, and antitumor immune response[40]. In addition, some TNFRSFs are involved in the deactivation of Tregs[41]. Therefore, TNFRSF agonists activate tumor immunity, and their combination with immune checkpoint therapy is promising.
TNFRSF include 4-1BB, OX-40, GITR, CD27, GITR, TNFRSF25, and CD40. Several antibodies that act as TNFRSF agonist have been evaluated in clinical trials, combined with immune checkpoint therapy. Urelumab (BMS-663513) and PF-05082566 are agonistic 4-1 BB-specific antibodies. In a preclinical model, a combination of a 4-1BB agonist and PD-1antagonist enhanced the antitumor effector/memory T-cell activity. This activity was observed in a poorly immunogenic melanoma model without severe toxicity[42]. Thus, the combination of uretinib with nivolumab or PF-05082566 with pembrolizumab is reasonable. These combinations have been investigated in phase 1 trials (NCT02253992 and NCT02179918).
OX-40 agonists can be good partners with both CTLA-4 antagonist and PD-L1 antagonist. The combination of OX-40 agonist with CTLA-4 antagonist enhanced therapeutic efficacy[43], whereas the combination of an OX-40 agonist with a PD-L1 antagonist, restored the functions of exhausted CD8+ T cells in preclinical models[44]. MEDI6469, an OX40specific antibody, in combination with tremelimumab or MEDI4736 (an anti- PD-L1 antibody) were investigated in a phase 1 trial (NCT02205333). This study was completed in April 2016, and the results are currently awaited.
The monoclonal CD40 agonist antibody, CP-870,893 in combination with gemcitabine was well tolerated and associated with antitumor activity in patients with PAC in a phase 1 trial[45]. The combination of tremelimumab and CP-870,893 demonstrated safety and clinical activity in patients with melanoma reported in a 2015 American Association for Cancer Research (AACR) annual meeting. As far as we know, there is no clinical trial of investigating the combination of a CD40 agonist and PD-1/PD-L1 antagonist.
Targeting TNFRSF requires close patient monitoring to avoid overstimulating the immune system. Notably, treatment with TGN1412, an antibody against the CD28 receptor, led to a cytokine storm in a phase 1 trial[46].
Combination therapy with cancer vaccines
Cancer vaccines stimulate the immune system to produce tumor-specific T and B cells[47] by increasing the presentation of TAAs to the immune system. In general, cancer vaccine therapies are well tolerated because the vaccines are very specific. The most promising vaccine is GVAX, which is a whole cell vaccine composed of two irradiated cancer cell lines and engineered to express GM-CSF. GVAX induces movement of effector T cell to the TME and PD-1/PD-L1 mediated signaling. Thus, immune checkpoint therapy and GVAX have synergistic antitumor effects. This combination was compared to single ipilimumab for advanced PAC in a randomized phase 2 trial. The combination therapy achieved CA19-9 biochemical response and prolonged the patient survival, although not significantly[48]. Multicenter phase 2 study is ongoing, in which FOLFIRINOX followed by ipilimumab with GVAX is being compared to FOLFIRINOX alone.
PD-1 blockade therapy and GVAX may be more a desirable combination than ipilimumab and GVAX, regarding efficacy and toxicity. Preclinical data showed that GVAX enhances PD-L1 expression, PD-1/PD-L1 blockade with GVAX (with cyclophosphamide) overcomes the immunosuppressive situation including Treg and CTLA4 expression on T cells[49]. In a neoadjuvant setting, the efficacy of GVAX (with cyclophosphamide) with or without nivolumab was compared in phase 1/2 trial (NCT02451982). In addition, a phase 2 trial of GVAX vaccine (with cyclophosphamide) and CRS-207 with or without nivolumab in advanced PAC is ongoing.
Moreover, triple therapy, PD-1 and CLTA-4 blockade with GVAX was better than dual blockade therapy was in a preclinical setting[50]. As far as we know, this triple therapy has not been translated to the clinical setting.
Combination therapy with radiotherapy
Radiotherapy is a good candidate combination partner for immune checkpoint therapy for two reasons. First, radiation facilitates the cross-priming of CTLs, and secondly, radiation has an abscopal effect.
Radiation enhances cross-priming of CTLs in two ways. When radiation induces tumor cell apoptosis, calreticulin is displayed on the surface of tumor cells, which acts as an “eat me” signal to DCs. This signal facilitates the cross-priming of CTLs. In addition, radiation induces the release of danger-associated molecular patterns (DAMPs) including ATP and HMGB-1. These DAMPs are endogenous immune adjuvants that stimulate DC activation, facilitating cross-priming of CTLs[51].
The abscopal effect is a phenomenon in which a primary tumor is irradiated and a response is observed at distant metastatic sites outside of the radiation field. The mechanism of this phenomenon is not understood completely, but it may be mediated by immunologic mechanisms, especially T cells[51]. The abscopal effect was reported in several types of cancer including melanoma, lymphoma, and renal cell carcinoma. In PAC, the abscopal effect was reported in a xenograft model[52].
Synergistic effects of immune checkpoint blockade and radiation were reported by Dewan et al[53]. CTLA-4 blockade acts synergistically with radiation to induce an abscopal response in preclinical models of poorly immunogenic cancers. In addition, Postow et al[54] reported a case of the abscopal effect in an advanced melanoma patient treated with ipilimumab and radiotherapy. The efficacy of radiotherapy with immune checkpoint therapy has been evaluated in melanoma, castration-resistant prostate cancer, and NSCLC. A randomized phase 3 trial compared radiation with ipilimumab to radiation alone was conducted in prostate cancer, but there was no significant difference in OS between the two arms[55]. Other trials are ongoing. Optimum radiation schedule, dose, or risk benefit balances of combination therapy remain unknown.
INFLAMMATION IN PANCREATIC CANCER
Inflammation contributes to carcinogenesis and tumor progression. It is widely known that PAC is associated with both systemic and TME inflammation. The influence of systemic inflammation has been investigated in the clinical setting. On the other hand, the importance of inflammation in the TME has been implicated mainly in preclinical models.
Multiple large studies demonstrated that elevated systemic inflammatory markers are a negative prognostic factors in various cancers. This negative impact is particularly strong in patients with PAC. We previously reported that elevated C-reactive protein and pentraxin 3 were prognostic factors in of advanced PAC[56]. In addition, CRP and hypoalbuminemia are the defining measures used by the modified Glasgow Prognostic Score (mGPS)[57]. The validation of mGPS has been examined in more than 60 studies and over 30000 patients across multiple tumor types and clinical settings[58]. Systemic inflammation is also related to malignancy-associated symptoms including cachexia, muscle loss, poor performance status, fatigue, cognitive dysfunction, and reduced quality of life[59].
On the other hand, inflammation in the TME is associated with numerous tumor-promoting effects including enhancement of proliferative signaling, resistance to apoptosis, enhancement of angiogenesis[60], and modulation of antitumor immunity to support immune evasion[61]. Inflammation in the TME consists of crosstalk between tumor, stromal, and immune cells. Crosstalk is conducted by inflammatory mediators. Thus, inflammatory mediators are considered potential treatment targets. These target include the activating oncogene (Kras), tumor suppression gene (tumor protein p53 [TP53] and mothers against decapentaplegic homolog 4 [SMAD4]), chemokines (CXCR2 ligands), cytokines (interleukin [IL]-6 and IL-1), and downstream effectors (STAT3 and nuclear factor kappa-light-chain-enhancer of activated B cells [NF-κB], Figure 1).
Figure 1.
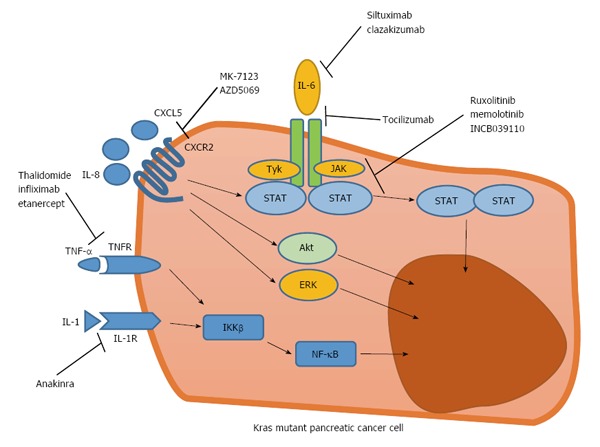
Various inflammation-associated signaling pathways activated in Kras mutant pancreatic cancer cells, as novel treatment targets. CXCL5: C-X-C motif chemokine ligand 5; ERK: Extracellular signal-regulated kinase; IL: Interleukin; JAK: Janus kinase; NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells; STAT: Signal transducer and activator of transcription; TNF: Tumor necrosis factor; TNF-R: TNF-receptor.
CXCR2 and CXCLs
Chemokines are small-molecular-weight cytokines. Chemokines and their receptors play a role as inflammatory mediators. In malignant conditions, CXCLs and CXCR2 are of particular importance. CXCR2 is a G-protein-coupled cell surface chemokine receptor commonly found on neutrophils. In normal physiology, CXCR2 signaling is important for neutrophil migration in acute inflammation or wound healing. Ligands of CXCR2 (CXCLs) are ELR motif positive chemokines. CXCLs include CXCL-1, 2, 3, 5, 6, and 8 (IL-8).
CXCR2 signal is upregulated during the primitive stage of PAC development, and this signaling maintains the feed-forward loop in PAC cells and this autocrine effect of CXCR2 signaling promotes transformation and progression of PAC[62]. In addition to inducing direct effects, CXCR2 signaling also promotes the progression of PAC indirectly to affect the TME[63].
CXCLs also play important roles. CXCL5 and CXCL8 expression in tumors are elevated[64], and overexpression of CXCL5 is associated with poor survival[65]. CXCL5 also activates several pathways including the protein kinase B (Akt), extracellular signal-regulated kinase (ErK), and STAT in human endothelial cells[65].
Moreover, Kras mutation is the most important oncogene-related factor in PAC, and Kras presents in more than 90% of PAC from the early stage[66]. Kras signaling involves various downstream effectors including Raf/Mek/Erk, PI3K/Pdk1/Akt, and the Ral guanine nucleotide exchange factor pathway[67]. Kras also plays important roles in inflammatory responses. When mutant Kras expression remains activated, the stimulation of the hedgehog signaling pathway, upregulation of inflammatory mediators IL-6, STAT3, and cyclooxygenase (COX)-2 are observed. In addition, when Kras becomes inactivated, the expression of these inflammatory mediators decreases[68]. These findings suggests that Kras mutation is related to the coordination of the inflammatory response in PAC.
Although oncogenic Kras is required for initiation, maintenance and progression in PAC, pharmacological inhibition of KRAS continues to be challenging. Recent studies have revealed the association between CXCR2 signaling, Kras mutation, and tumor progression. Activated Kras enhances CXCLs in pancreatic epithelial cells[69]. Preclinical models also showed that inhibition of CXCR2 signaling regulates Kras-induced autocrine growth of PAC and disrupts the tumor-stromal interaction, suppresses metastases and improves survival in preclinical models[62]. These findings suggest CXCR2 signaling is a surrogate target of Kras in PAC.
Interestingly, CXCR2 inhibition may enhance the efficacy of PD-1 blockade therapy. Steele et al[70] and Highfill et al[71] reported that combination therapy with anti-CXCR2 and anti-PD-1 improved survival compared to anti-PD-1 monotherapy in a preclinical model. The reason for this synergistic effect is thought to be the inhibition of CXCR2, which prevents MDSCs trafficking to the TME and increases activated CD8+ cells within the tumor.
However, no clinical trial has evaluated the efficacy of CXCR2 signaling blockade in malignant tumors. Several CXCR2 antagonists were evaluated in clinical trials for chronic obstructive pulmonary disease[72].
IL-6
IL-6 is a proinflammatory cytokine produced by various cells including macrophages, hepatocytes, and pancreatic cancer cells[73]. In PAC, IL-6 plays a multifunctional role in the development and progression of PAC by directly affecting the tumor cells as well as modulating the TME[74]. Although several molecules including mesothelin and receptor for advanced glycation end products influence IL-6 expression, the most important enhancer of IL-6 is Kras[75]. IL-6 signaling activates Janus kinase (JAK)-1 and JAK2, which leads to the phosphorylation of STAT-1 and 3, which plays an important role in tumor growth, survival, angiogenesis, and metastasis. In addition, IL-6 signaling activates the Ras-mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)-Akt signaling cascades[76], which is associated with antiapoptotic and tumorigenic functions. Furthermore, IL-6 contributes to creating a pro-tumorigenic TME by enhancing the expression of IL-10, IL-13, IL-5, IL-7, and GM-CSF[77]. In patients with PAC, high serum IL-6 was related to cachexia, progression to an advanced stage, and poor survival[78]. Thus, IL-6 signaling has emerged as a treatment target of PAC.
Siltuximab (CNTO328) is a chimeric anti-IL-6-monoclonal antibody, which binds strongly to IL-6 and neutralizes its activity and, thereby, promotes tumor cell death. The safety and efficacy of monotherapy with siltuximab were evaluated in phase 1/2 trials. Because IL-6 has a significant role in mutant Kras-driven tumorigenesis, patients with Kras-mutant tumors including PAC, NSCLC, colorectal, head, and neck cancers received siltuximab in the phase 1 expansion and phase 2 cohorts. Unfortunately, no objective response was observed in any of the 84 patients. Therefore, no further trial is planned with this drug for solid tumors[79]. However, siltuximab was approved for the treatment of human immunodeficiency virus negative and human herpes virus (HHV)-8 negative multicentric Cattleman’s disease by the United States Food and Drug Administration (FDA) in 2014[80]. In addition, siltuximab is a promising agent for the treatment of B-cell non-Hodgkin’s lymphoma and multiple myeloma.
Tocilizumab is a humanized antibody that acts as an antagonist of the IL-6 receptor while clazakizumab is a glycosylated anti-IL-6 monoclonal antibody. Both agents have shown efficacy in the treatment of rheumatoid arthritis[81]. Currently, there are no ongoing clinical trials to evaluate the efficacy of both agents in solid tumors. However, anti-IL-6 and anti-IL-6R antibodies may be useful in cancer-related cachexia. Ando et al[82] reported a case of a patient with lung cancer who had cachexia, and whose symptoms were rapidly palliated by the use of tocilizumab[82]. Clazakizumab was well tolerated in a phase 2 trial (NCT00866970), and it improved the tumor-related symptoms of patients with NSCLC[83].
In PAC, the sources of IL-6 are myeloid cells[84] and CAFs[76]. These cells are components of the TME. Recent studies revealed that a somatostatin analog SOM230 (pasireotide) inhibits protein synthesis in activated CAFs, and decreases IL-6 secretion[85]. Thus, targeting CAFs is another strategy to regulate IL-6. At the ASCO 2015 annual meeting, the results of a phase 1 trial were reported showing that the combination of SOM230 with FOLFIRI was well tolerated for gastrointestinal malignancies including PAC. In addition, combination of SOM230 LAR and gemcitabine was also well tolerated and achieved a disease control rate of 68% for patients with advanced PAC[86] .
NF-κB
NF-κB is a transcription factor that plays critical roles in inflammation, cell production and differentiation, immune responses, and cancer[87]. In cancer cells, the oncogenic role of NF-κB includes the promotion of cell proliferation, control of apoptosis, stimulation of angiogenesis, and metastasis. NF-κB transcriptional factor is constitutively activated in most patients with PAC[88]. Kras mutation in PAC induces the secretion of the cytokine IL-1α, which further leads to the ubiquitination of the TNF receptor-associated factor 6 (TRAF6) and activation of inhibitor of nuclear factor kappa-B kinase subunit-β. This further activates NF-κB to induce its target genes including the protein p62, which in turn positively regulates TRAF6 ubiquitination and promotes the constitutive NF-κB. This feed-forward loop leads to PAC development[89]. In addition, NF-κB functions as a crucial link between pancreatic inflammation and PAC.
Several previous trials examined the role of anti-TNF directed therapy in the treatment of pancreatic cancer and cachexia, and the efficacy of anti-TNF targeted therapy in PAC was controversial. Thalidomide is an immunomodulatory agent known to decrease TNF levels. A randomized and double blind phase 2 study of thalidomide demonstrated an improvement in weight and lean body mass at 8 wk compared to the placebo[90]. In contrast, a placebo-controlled randomized phase 2 study of gemcitabine and infliximab, a monoclonal antibody that blocks TNF, did not show a benefit in preserving lean body mass or survival[91]. Etanercept is a recombinant human TNF receptor that specifically binds to soluble TNF and biologically inactivates it by blocking its interaction with cell surface TNF receptors. Recently, the safety and efficacy of etanercept combined with gemcitabine were evaluated in a phase 1/2 study[92]. This combination was well-tolerated; however, etanercept did not show a significant enhancement of the activity of gemcitabine[92]. These results imply that targeting TNF alone is not sufficient for an antitumor response or reversal of cachexia.
Another proposed target is IL-1. Aanakinra is a recombinant, non-glycosylated synthetic form of the human IL-1β receptor antagonist. This agent has been approved by the FDA for the treatment of neonatal-onset multisystem inflammatory disease and rheumatoid arthritis. In a preclinical model, anakinra significantly decreased NF-κB, and its co-administration with gemcitabine reduced the tumor burden[93]. Several early clinical trials to evaluate the safety and efficacy of anakinra are currently ongoing. For PAC, treatment with anakimura plus a conventional cytotoxic regimen are under investigation (NCT02550327, NCT02021422) as well.
JAK/STAT
The JAK/STAT pathway is an emerging and promising treatment target of PAC. JAKs are a family of cytoplasmic tyrosine kinases that consist of four members, JAK1, JAK2, JAK3, and Tyk2. In addition, the STATs, which are a family of downstream transcription factors for JAKs[94], have dual roles as cytoplasmic signal transduction molecules and nuclear transcription factors. Both intrinsic and extrinsic pathways activate JAK/STAT signaling. Abnormalities of the JAK/STAT pathway contribute directly to cellular transformation, increased cell proliferation, apoptosis, and angiogenesis in cancer. In addition, STAT3 induces the expression of various cytokines, chemokines, and other mediators including IL-6 and COX-2, which are associated with cancer-promoting inflammation. Importantly, the receptors for numerous cytokines, chemokines, and mediators in turn further activate STAT3, thereby forming autocrine and paracrine feed-forward loops that result in a stable change that promotes cancer-related inflammation[95].
Among the several JAK/STAT signaling targeted drugs, the development of ruxolitinib is the most advanced. Ruxolitinib is a potent JAK1/JAK2 inhibitor and deregulator of JAK/STAT signaling. In a randomized phase 2 study, 127 patients with metastatic gemcitabine-refractory PAC were administered capecitabine plus either ruxolitinib or a placebo. In the intent-to-treat population, the addition of ruxolitinib to capecitabine did not demonstrate any significant improvement in the OS or PFS. However, in a prespecified subgroup analysis of patients whose serum CRP level was greater than that of the study population median, the OS was significantly longer in the combination arm compared to the monotherapy arm. A post hoc analysis indicated that ruxolitinib achieved a longer OS in patients with high mGPS, representing a more severe systemic inflammation. Two randomized, double-blind phase 3 trials with ruxolitinib or placebo in combination with capecitabine in patients with PAC patients who failed to respond to first-line chemotherapy (the JANUS 1 and JANUS 2 Studies) are currently ongoing. Based on the previous phase 2 trial, the selection criteria in these studies included an mGPS of 1 or 2 (NCT02119663, NCT02117479). A phase 1b study of the safety and tolerability of ruxolitinib in combination with gemcitabine with or without nab-paclitaxel in advanced solid tumors is also currently ongoing. (NCT01822756). Treatments with other novel JAK inhibitor are also being investigated.
CONCLUSION
Various research studies have revealed that immunosuppression and inflammation play critical roles in oncogenesis, development, invasion, and metastasis in PAC. Understanding the complicated crosstalk between the immune and stromal cells would certainly lead to the development of effective treatment strategies. However, single immune checkpoint therapy may not achieve the desired clinical benefits. Combination therapy with immune chokepoint blockers and other agents or anti-inflammation targeted therapy are expected to provide considerable clinical benefits to patients with PAC.
ACKNOWLEDGMENTS
We would like to express our appreciation to Ms. Rubi Mukoyama, Ms. Keiko Kondo, and Ms. Hiroko Hosoi for their valuable help in writing this paper.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: Shiro Kimbara have no conflict of interest associated with this manuscript. Shunsuke Kondo received research funding from AstraZeneca, Eli Lilly and Company, and Bayer AG.
Peer-review started: March 27, 2016
First decision: May 12, 2016
Article in press: August 1, 2016
P- Reviewer: Li SD, Srimathveeravalli G S- Editor: Ma YJ L- Editor: A E- Editor: Wang CH
References
- 1.Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46:765–781. doi: 10.1016/j.ejca.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Ribas A. Tumor immunotherapy directed at PD-1. N Engl J Med. 2012;366:2517–2519. doi: 10.1056/NEJMe1205943. [DOI] [PubMed] [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 12.Santarpia M, González-Cao M, Viteri S, Karachaliou N, Altavilla G, Rosell R. Programmed cell death protein-1/programmed cell death ligand-1 pathway inhibition and predictive biomarkers: understanding transforming growth factor-beta role. Transl Lung Cancer Res. 2015;4:728–742. doi: 10.3978/j.issn.2218-6751.2015.12.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol. 2007;601:213–223. doi: 10.1007/978-0-387-72005-0_22. [DOI] [PubMed] [Google Scholar]
- 14.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13:265–270. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28:401–409. doi: 10.1093/intimm/dxw025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 19.Mei L, Du W, Ma WW. Targeting stromal microenvironment in pancreatic ductal adenocarcinoma: controversies and promises. J Gastrointest Oncol. 2016;7:487–494. doi: 10.21037/jgo.2016.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chae YK, Pan A, Davis AA, Raparia K, Mohindra NA, Matsangou M, Giles FJ. Biomarkers for PD-1/PD-L1 Blockade Therapy in Non-Small-cell Lung Cancer: Is PD-L1 Expression a Good Marker for Patient Selection? Clin Lung Cancer. 2016 doi: 10.1016/j.cllc.2016.03.011. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 22.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laghi L, Beghelli S, Spinelli A, Bianchi P, Basso G, Di Caro G, Brecht A, Celesti G, Turri G, Bersani S, et al. Irrelevance of microsatellite instability in the epidemiology of sporadic pancreatic ductal adenocarcinoma. PLoS One. 2012;7:e46002. doi: 10.1371/journal.pone.0046002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emens LA. Chemoimmunotherapy. Cancer J. 2010;16:295–303. doi: 10.1097/PPO.0b013e3181eb5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duffy AG, Greten TF. Immunological off-target effects of standard treatments in gastrointestinal cancers. Ann Oncol. 2014;25:24–32. doi: 10.1093/annonc/mdt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jure-Kunkel M, Masters G, Girit E, Dito G, Lee F, Hunt JT, Humphrey R. Synergy between chemotherapeutic agents and CTLA-4 blockade in preclinical tumor models. Cancer Immunol Immunother. 2013;62:1533–1545. doi: 10.1007/s00262-013-1451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H, Cuillerot JM, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 28.Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH, Bagalà C, Colombi F, Cagnazzo C, Gioeni L, Wang E, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. 2014;25:1750–1755. doi: 10.1093/annonc/mdu205. [DOI] [PubMed] [Google Scholar]
- 29.Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C, Muñoz M, Quijano Y, Cubillo A, Rodriguez-Pascual J, et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer. 2013;109:926–933. doi: 10.1038/bjc.2013.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ku GY, Yuan J, Page DB, Schroeder SE, Panageas KS, Carvajal RD, Chapman PB, Schwartz GK, Allison JP, Wolchok JD. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116:1767–1775. doi: 10.1002/cncr.24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R, Dhodapkar MV, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194:950–959. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination immunotherapy with α-CTLA-4 and α-PD-L1 antibody blockade prevents immune escape and leads to complete control of metastatic osteosarcoma. J Immunother Cancer. 2015;3:21. doi: 10.1186/s40425-015-0067-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonia SJ, Gettinger SN, Chow LQM, Juergens RA, Borghaei H, Yun Shen Y, Harbison C, Chen AC, Ready N, Rizvi NA. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) and ipilimumab in first-line NSCLC: Interim phase I results. J Clin Oncol. 2014;32:5s (suppl; abstr 8023). [Google Scholar]
- 36.Hammers HJ, Plimack ER, Infante JR, Ernstoff MS, Rini BI, McDermott DF, Razak AA, Pal SK, Voss MH, Sharma P, et al. Phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC) J Clin Oncol. 2014;32:5s (suppl; abstr 4504). doi: 10.1200/JCO.2016.72.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X, Li DC, Zhu XG, Gan WJ, Li Z, Xiong F, Zhang ZX, Zhang GB, Zhang XG, Zhao H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int J Mol Med. 2013;31:283–291. doi: 10.3892/ijmm.2012.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 41.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92:475–480. doi: 10.1038/icb.2014.26. [DOI] [PubMed] [Google Scholar]
- 42.Chen S, Lee LF, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3:149–160. doi: 10.1158/2326-6066.CIR-14-0118. [DOI] [PubMed] [Google Scholar]
- 43.Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res. 2014;2:142–153. doi: 10.1158/2326-6066.CIR-13-0031-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buchan SL, Manzo T, Flutter B, Rogel A, Edwards N, Zhang L, Sivakumaran S, Ghorashian S, Carpenter B, Bennett CL, et al. OX40- and CD27-mediated costimulation synergizes with anti-PD-L1 blockade by forcing exhausted CD8+ T cells to exit quiescence. J Immunol. 2015;194:125–133. doi: 10.4049/jimmunol.1401644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–6295. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 47.Lollini PL, Cavallo F, Nanni P, Forni G. Vaccines for tumour prevention. Nat Rev Cancer. 2006;6:204–216. doi: 10.1038/nrc1815. [DOI] [PubMed] [Google Scholar]
- 48.Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA, Donehower RC, Jaffee EM, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36:382–389. doi: 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38:1–11. doi: 10.1097/CJI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591–3603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vatner RE, Cooper BT, Vanpouille-Box C, Demaria S, Formenti SC. Combinations of immunotherapy and radiation in cancer therapy. Front Oncol. 2014;4:325. doi: 10.3389/fonc.2014.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blanquicett C, Saif MW, Buchsbaum DJ, Eloubeidi M, Vickers SM, Chhieng DC, Carpenter MD, Sellers JC, Russo S, Diasio RB, et al. Antitumor efficacy of capecitabine and celecoxib in irradiated and lead-shielded, contralateral human BxPC-3 pancreatic cancer xenografts: clinical implications of abscopal effects. Clin Cancer Res. 2005;11:8773–8781. doi: 10.1158/1078-0432.CCR-05-0627. [DOI] [PubMed] [Google Scholar]
- 53.Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15:5379–5388. doi: 10.1158/1078-0432.CCR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366:925–931. doi: 10.1056/NEJMoa1112824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–712. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morizane C, Okusaka T, Morita S, Tanaka K, Ueno H, Kondo S, Ikeda M, Nakachi K, Mitsunaga S. Construction and validation of a prognostic index for patients with metastatic pancreatic adenocarcinoma. Pancreas. 2011;40:415–421. doi: 10.1097/MPA.0b013e3182021376. [DOI] [PubMed] [Google Scholar]
- 57.McMillan DC, Elahi MM, Sattar N, Angerson WJ, Johnstone J, McArdle CS. Measurement of the systemic inflammatory response predicts cancer-specific and non-cancer survival in patients with cancer. Nutr Cancer. 2001;41:64–69. doi: 10.1080/01635581.2001.9680613. [DOI] [PubMed] [Google Scholar]
- 58.McMillan DC. The systemic inflammation-based Glasgow Prognostic Score: a decade of experience in patients with cancer. Cancer Treat Rev. 2013;39:534–540. doi: 10.1016/j.ctrv.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 59.Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM, Nemunaitis JJ, Stella PJ, Pipas JM, Wainberg ZA, et al. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol. 2015;33:4039–4047. doi: 10.1200/JCO.2015.61.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 61.Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014;15:e493–e503. doi: 10.1016/S1470-2045(14)70263-3. [DOI] [PubMed] [Google Scholar]
- 62.Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK, Singh RK. CXCR2 signaling regulates KRAS(G¹²D)-induced autocrine growth of pancreatic cancer. Oncotarget. 2016;7:7280–7296. doi: 10.18632/oncotarget.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B, Tada M, Mohri D, Miyabayashi K, Asaoka Y, Maeda S, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest. 2011;121:4106–4117. doi: 10.1172/JCI42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frick VO, Rubie C, Wagner M, Graeber S, Grimm H, Kopp B, Rau BM, Schilling MK. Enhanced ENA-78 and IL-8 expression in patients with malignant pancreatic diseases. Pancreatology. 2008;8:488–497. doi: 10.1159/000151776. [DOI] [PubMed] [Google Scholar]
- 65.Li A, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, Li G, Lu X, Strieter RM, Burdick M, et al. Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am J Pathol. 2011;178:1340–1349. doi: 10.1016/j.ajpath.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuo Y, Campbell PM, Brekken RA, Sung B, Ouellette MM, Fleming JB, Aggarwal BB, Der CJ, Guha S. K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways. Mol Cancer Res. 2009;7:799–808. doi: 10.1158/1541-7786.MCR-08-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steele CW, Karim SA, Leach JD, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29:832–845. doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. 2014;6:237ra67. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER, Watz H, Lu S, Stryszak P, Rosenberg E, et al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2015;191:1001–1011. doi: 10.1164/rccm.201405-0992OC. [DOI] [PubMed] [Google Scholar]
- 73.Martignoni ME, Kunze P, Hildebrandt W, Künzli B, Berberat P, Giese T, Klöters O, Hammer J, Büchler MW, Giese NA, et al. Role of mononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. Clin Cancer Res. 2005;11:5802–5808. doi: 10.1158/1078-0432.CCR-05-0185. [DOI] [PubMed] [Google Scholar]
- 74.Roshani R, McCarthy F, Hagemann T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014;345:157–163. doi: 10.1016/j.canlet.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 75.Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, Stanger BZ, Chung I, Rhim AD, di Magliano MP. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73:6359–6374. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, Brunicardi FC, Chen C, Yao Q, Min L. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther. 2007;6:1096–1100. doi: 10.4161/cbt.6.7.4328. [DOI] [PubMed] [Google Scholar]
- 78.Holmer R, Goumas FA, Waetzig GH, Rose-John S, Kalthoff H. Interleukin-6: a villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat Dis Int. 2014;13:371–380. doi: 10.1016/s1499-3872(14)60259-9. [DOI] [PubMed] [Google Scholar]
- 79.Angevin E, Tabernero J, Elez E, Cohen SJ, Bahleda R, van Laethem JL, Ottensmeier C, Lopez-Martin JA, Clive S, Joly F, et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2014;20:2192–2204. doi: 10.1158/1078-0432.CCR-13-2200. [DOI] [PubMed] [Google Scholar]
- 80.van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fosså A, Simpson D, Capra M, Liu T, Hsieh RK, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15:966–974. doi: 10.1016/S1470-2045(14)70319-5. [DOI] [PubMed] [Google Scholar]
- 81.Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–997. doi: 10.1016/S0140-6736(08)60453-5. [DOI] [PubMed] [Google Scholar]
- 82.Ando K, Takahashi F, Motojima S, Nakashima K, Kaneko N, Hoshi K, Takahashi K. Possible role for tocilizumab, an anti-interleukin-6 receptor antibody, in treating cancer cachexia. J Clin Oncol. 2013;31:e69–e72. doi: 10.1200/JCO.2012.44.2020. [DOI] [PubMed] [Google Scholar]
- 83.Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther. 2011;11:1663–1668. doi: 10.1517/14712598.2011.627850. [DOI] [PubMed] [Google Scholar]
- 84.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, Yoshimura A, Reindl W, Sipos B, Akira S, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 85.Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F, Gayral M, Cordelier P, Delisle MB, Bousquet-Dubouch MP, et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med. 2015;7:735–753. doi: 10.15252/emmm.201404346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suleiman Y, Mahipal A, Shibata D, Siegel EM, Jump H, Fulp WJ, Springett GM, Kim R. Phase I study of combination of pasireotide LAR + gemcitabine in locally advanced or metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2015;76:481–487. doi: 10.1007/s00280-015-2814-8. [DOI] [PubMed] [Google Scholar]
- 87.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 88.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- 89.Prabhu L, Mundade R, Korc M, Loehrer PJ, Lu T. Critical role of NF-κB in pancreatic cancer. Oncotarget. 2014;5:10969–10975. doi: 10.18632/oncotarget.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gordon JN, Trebble TM, Ellis RD, Duncan HD, Johns T, Goggin PM. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. Gut. 2005;54:540–545. doi: 10.1136/gut.2004.047563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wiedenmann B, Malfertheiner P, Friess H, Ritch P, Arseneau J, Mantovani G, Caprioni F, Van Cutsem E, Richel D, DeWitte M, et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J Support Oncol. 2008;6:18–25. [PubMed] [Google Scholar]
- 92.Wu C, Fernandez SA, Criswell T, Chidiac TA, Guttridge D, Villalona-Calero M, Bekaii-Saab TS. Disrupting cytokine signaling in pancreatic cancer: a phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas. 2013;42:813–818. doi: 10.1097/MPA.0b013e318279b87f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhuang Z, Ju HQ, Aguilar M, Gocho T, Li H, Iida T, Lee H, Fan X, Zhou H, Ling J, et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-κB Activation. Clin Cancer Res. 2016;22:1432–1444. doi: 10.1158/1078-0432.CCR-14-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seidel HM, Lamb P, Rosen J. Pharmaceutical intervention in the JAK/STAT signaling pathway. Oncogene. 2000;19:2645–2656. doi: 10.1038/sj.onc.1203550. [DOI] [PubMed] [Google Scholar]
- 95.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]