

Summary
Japanese encephalitis virus (JEV) is a re‐emerging zoonotic flavivirus that poses an increasing threat to global health and welfare due to rapid changes in climate and demography. Although the CCR2–CCL2 axis plays an important role in trafficking CD11b+ Ly‐6Chi monocytes to regulate immunopathological diseases, little is known about their role in monocyte trafficking during viral encephalitis caused by JEV infection. Here, we explored the role of CCR2 and its ligand CCL2 in JE caused by JEV infection using CCR2‐ and CCL2‐ablated murine models. Somewhat surprisingly, the ablation of CCR2 and CCL2 resulted in starkly contrasting susceptibility to JE. CCR2 ablation induced enhanced resistance to JE, whereas CCL2 ablation highly increased susceptibility to JE. This contrasting regulation of JE progression by CCR2 and CCL2 was coupled to central nervous system (CNS) infiltration of Ly‐6Chi monocytes and Ly‐6Ghi granulocytes. There was also enhanced expression of CC and CXC chemokines in the CNS of CCL2‐ablated mice, which appeared to induce CNS infiltration of these cell populations. However, our data revealed that contrasting regulation of JE in CCR2‐ and CCL2‐ablated mice was unlikely to be mediated by innate natural killer and adaptive T‐cell responses. Furthermore, CCL2 produced by haematopoietic stem cell‐derived leucocytes played a dominant role in CNS accumulation of Ly‐6Chi monocytes in infected bone marrow chimeric models, thereby exacerbating JE progression. Collectively, our data indicate that CCL2 plays an essential role in conferring protection against JE caused by JEV infection. In addition, blockage of CCR2, but not CCL2, will aid in the development of strategies for prophylactics and therapeutics of JE.
Keywords: CCL2, CCR2, Japanese encephalitis, Ly‐6Chi monocytes, neuroinflammation
Abbreviations
- CNS
central nervous system
- dpi
days post‐infection
- HIV
human immunodeficiency virus
- HSC
hematopoietic stem cell
- HSV
herpes simplex virus
- JEV
Japanese encephalitis virus
- KO
knock‐out
- WNV
West Nile virus
Introduction
Japanese encephalitis virus (JEV) is a re‐emerging zoonotic flavivirus that is transmitted by mosquitos from birds and/or swine to humans. It contains a single‐stranded, positive‐sense RNA (~11 kb, monopartite, linear) genome in a capsid. JEV poses an increasing threat to global health and welfare, with approximately 67, 900 cases reported annually.1, 2, 3 JEV is now spreading to previously unaffected regions, such as Indonesia, Pakistan and northern Australia, due to rapid changes in climate and demography.1, 2 Considerable progress in uncovering the pathogenesis of JE, acute neuroinflammation caused by JEV, has been made in infected patients and murine models.4, 5, 6 The incubation period of JE ranges from 5 to 15 days and the disease is fatal in 25 to 30% of cases, mostly in infants. A high proportion of surviving patients have serious neurological and psychiatric sequelae.3 Hence, JE may have higher mortality than the acute neuroinflammation caused by infection with West Nile virus (WNV), which results in 3–5% mortality (1, 100 deaths/29, 000 symptomatic infections).7 Although JEV is thought to infect and kill neurons directly in the central nervous system (CNS), CNS invasion by JEV induces activation of microglia/glia and infiltration of peripheral leucocytes. This leads to indirect neuronal killing via uncontrolled secretion of pro‐inflammatory cytokines [such as interleukin‐6 (IL‐6) and tumour necrosis factor‐α (TNF‐α)] and soluble mediators that can induce neuronal death.8, 9 Therefore, adequate CNS infiltration and activation of peripheral immune cells play an important role in protecting hosts from JE progression without tissue injury.
During the progression of viral encephalitis, CNS trafficking of peripheral immune cells is mediated through a multistep process governed by chemokines and chemokine receptors.10 Indeed, viral encephalitis caused by infection by flaviviruses including JEV and WNV is characterized by infiltration of peripheral leucocytes, such as monocytes and T cells, in the perivascular space and parenchyma. This can result in beneficial or detrimental outcomes, depending on the encephalitis context.11, 12, 13 For example, infection of CCR5‐deficient mice was uniformly fatal with drastically decreased infiltration of T cells and monocytes in the CNS following WNV infection.14 A similar increase in susceptibility was observed in a homozygous individual with a complete loss‐of‐function mutation, CCR5Δ32.15 Individual inhibition of CXCR3, CCR2 and CCR5 with corresponding antagonists also produced no changes in viral titre and survival following infection with virulent Semliki Forest virus, whereas simultaneous blockage of CXCR3 and CCR2 resulted in significantly reduced mortality in response to viral encephalitis caused by Semliki Forest virus, rather than an increase in mortality.16
CCR2 is an extensively studied chemokine receptor that regulates the progression of viral encephalitis. CCR2 is considered critical to CD11b+ Ly‐6Chi monocyte trafficking and is thought to be expressed on subsets of activated T cells, dendritic cells and natural killer (NK) cells.17 The primary and specific ligand for CCR2 is CCL2, but this receptor can also bind to CCL7 and CCL12/CCL13.10, 17 Trafficking of CD11b+ Ly‐6Chi monocytes governed by CCR2 appears to have a critical role during viral encephalitis such as WNV infection,18, 19 although whether these cells function to protect or promote pathogenesis is unclear. Indeed, the CCR2–CCL2 axis can provide beneficial or detrimental results, depending on the context of immunopathological diseases caused by sterile and non‐sterile stimuli.10, 17 These various results mainly depend on the trafficking of CD11b+ Ly‐6Chi monocytes, which affects progression of neuroinflammation. Therefore, CCR2‐deficient mice showed highly increased susceptibility to neuroinflammation caused by WNV infection with early decreased CNS infiltration of CD11b+ Ly‐6Chi monocytes,18 whereas the recruitment of Ly‐6Chi monocytes exacerbates intracerebral haemorrhage, depending on CCR2.20 CCR2, but not its ligand CCL2, also exhibited an important role in regulating the progression of encephalitis caused by infection with coronavirus via an intracerebral route.21, 22 Collectively, results indicate that the role of the CCR2–CCL2 axis in progression of viral encephalitis is complicated, depending on the context of the viral encephalitis.
CCL2 has also been detected in the plasma and cerebrospinal fluid of patients infected with JEV.23, 24, 25 However, little is known regarding the role of the CCR2–CCL2 axis and monocyte trafficking during viral encephalitis caused by JEV infection in murine models. Uncovering the role of CCR2/CCL2 in JE progression may be a prerequisite for developing prophylactic and therapeutic strategies because CCR2/CCL2 blockers remain a possibility in developing therapeutic strategies for immunopathological diseases. In this study, we explored the role of CCR2 and its ligand CCL2 in JE progression following JEV infection. Somewhat surprisingly, CCR2 and its cognate ligand CCL2 differentially regulated JE progression. CCR2‐deficient mice showed enhanced resistance to JE, whereas CCL2 ablation provided highly increased susceptibility to JE. Subsequently, we investigated pathological features as well as innate NK cell and T‐cell adaptive immune responses in CCR2‐ and CCL2‐ablated mice. Starkly contrasting regulation of JE severity by CCR2 and CCL2 was closely associated with CNS infiltration by peripheral leucocytes, including CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Chi granulocytes. However, innate NK cell and T‐cell immune responses appeared to provide no contribution to JE progression. Our results also revealed that CCL2 produced by leucocytes derived from haematopoietic stem cells (HSCs) played a dominant role in the CNS accumulation of Ly‐6Chi monocytes, thereby exacerbating JE progression. These results suggest that blockage of CCR2, but not CCL2, will allow for the development of strategies for prophylactic and therapeutic treatment of JE.
Materials and methods
Ethics statement
All animal experiments described in the present study were conducted at Chonbuk National University according to guidelines set by the Institutional Animal Care and Use Committees of Chonbuk National University. All experiments were pre‐approved by the Ethical Committee for Animal Experiments of Chonbuk National University (permission code 2013‐0028), which was fully accredited by the Korean Association for Laboratory Animal Sciences, adopted by the Council of the Korean Government for Animal Care. All experimental protocols requiring biosafety were approved by Institutional Biosafety Committees of Chonbuk National University.
Animals, cells and viruses
C57BL/6 (H‐2b) mice (4–6 weeks old) were purchased from Samtako (O‐San, Korea). CCR2 and CCL2 knockout (KO) mice (H‐2b) were obtained from Jackson Laboratories (Bar Harbor, ME). A JEV Beijing‐1 strain was obtained from Green Cross Research Institute (Suwon, Korea) and propagated in the mosquito cell line (C6/36) using Dulbecco's modified Eagle's medium supplemented with 2% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 U/ml).26 The C6/36 cultures were infected with JEV Beijing‐1 at a multiplicity of infection of 0·1, and were incubated in a humidified CO2 incubator for 1 hr at 28°C. After absorption, the inoculum was removed, and 7 ml of a maintenance medium containing 2% fetal bovine serum was added. Approximately 6–7 days post‐infection (dpi), cultures of the host cells showing 80–90% cytopathic effects were harvested. The virus stocks were titrated by conventional plaque assay or focus‐forming assay and were stored in aliquots at −80° until use. Mice were infected intradermally with JEV [3·0 × 107 or 5·0 × 107 plaque‐forming units(pfu)/mouse] via the footpad route.
Antibodies and reagents
The monoclonal antibodies used for flow cytometric analysis and other experiments were obtained from eBioscience (San Diego, CA) or BD Biosciences (San Diego, CA). These include fluorescein isothiocynate (FITC) ‐conjugated anti‐CD3ε (154‐2C11), CD4 (RM4‐5), CD8 (53–67), phycoerythrin (PE)–conjugated anti‐mouse CD11b (M1/70), interferon‐γ (IFN‐γ; XMG1.2), granzyme B (NGZB), peridinin chorophyll protein complex (PerCP) –conjugated anti‐mouse IFN‐γ (XMG1.2), Ly‐6C (HK 1.4), PE‐Cyanine dye (Cy7)‐conjugated anti‐mouse NK1.1 (PL136), allophycocyanin (APC)–conjugated anti‐mouse Ly‐6G (1A8), TNF‐α (MP6‐XT22), and biotin‐conjugated anti‐mouse CD49b (DX5). The peptides of defined I‐Ab‐restricted epitopes JEV NS1132–145 (TFVVDGPETKECPD) and NS3563–574 (WCFDGPRTNAIL), and H‐2Db‐restricted epitope JEV NS4B215–223 (SAVWNSTTA) were chemically synthesized at Peptron Inc. (Daejeon, Korea). The JEV‐specific primers for detecting viral RNA (JEV10 564–10 583 forward, 5′‐CCC TCA GAA CCG TCT CGG AA‐3′ and JEV10, 862–10, 886 reversqe, 5′‐CTA TTC CCA GGT GTC AAT ATG CTG T‐3′) and primers specific for the chemokine ligand and receptor (Table 1) were synthesized at Bioneer Corp. (Daejeon, Korea) and used for PCR amplification of target genes.
Table 1.
Specific primers for the expression of chemokines and their receptors used in real‐time quantitative RT‐PCR
| Gene name | Primer sequence (5′–3′)a | Position cDNA | GenBank ID |
|---|---|---|---|
| CCL3 | FP: CCA AGT CTT CTC AGC GCC AT | 158–177 | NM_011337.2 |
| RP: GAA TCT TCC GGC TGT AGG AGA AG | 206–228 | ||
| CCL4 | FP: TTC TGT GCT CCA GGG TTC TC | 128–147 | NM_013652.2 |
| RP: GAG GAG GCC TCT CCT GAA GT | 388–407 | ||
| CCL5 | FP: CCC TCA CCA TCA TCC TCA CT | 77–96 | NM_013653.3 |
| RP: CTT CTT CTC TGG GTT GGC AC | 275–294 | ||
| CCL7 | FP: CCA GCA AGC AGC TCA ACA TT | 978–998 | NM_013654 |
| RP: GCC GAT GAA GGC ATA CAA GA | 1049–1069 | ||
| CCL12 | FP: GGG AAG CTG TGA TCT TCA GG | 235–255 | NM_011331.2 |
| RP: GGG AAC TTC AGG GGG AAA TA | 293–313 | ||
| CCL17 | FP: CAG GGA TGC CAT CGT GTT TC | 223–243 | NM_011332 |
| RP: CAC CAA TCT GAT GGC CTT CTT | 292–313 | ||
| CXCL2 | FP: ATC CAG AGC TTG AGT GTG ACG C | 194–215 | NM_009140.2 |
| RP: AAG GCA AAC TTT TTG ACC GC | 264–283 | ||
| CXCL9 | FP: TGC ACG ATG CTC CTG CA | 137–154 | NM_019494.1 |
| RP: AGG TCT TTG AGG GAT TTG TAG TGG | 175–199 | ||
| CXCL11 | FP: AAG CTC GCC TCA TAA TGC AG | 323–343 | NM_019494.1 |
| RP: CAC AGT CAG ACG TTC CCA | 389–407 | ||
| CCR1 | FP: ACC TTC GGC AGC TGT TTC A | 1397–1416 | NM_009912 |
| RP: TCC ACA GAG AGG AAG GGC AG | 1446–1466 | ||
| CCR2 | FP: TGT TAC CTC AGT TCA TCC ACG G | 150–171 | NM_009915.2| |
| RP: CAG AAT GGT AAT GTG AGC AG | 446–465 | ||
| CCR4 | FP: TCC TGA CGG ACG TGT ACC T | 325–344 | NM_009916 |
| RP: CAG ACC TAG TCC AAA AAC CCA C | 412–434 | ||
| CCR5 | FP: TGG ATT TTC AAG GGT CAG TTC C | 106–128 | NM_009917 |
| RP: GAG CCG CAA TTT GTT TCA CA | 181–201 | ||
| CXCR2 | FP: CAT CTT ATA CAA CCG GAG CGC C | 358–380 | NM_009909.3| |
| RP: TAG TAA GGA GAT GGC TAT GCA CAC | 635–659 | ||
| CXCR3 | FP: TGA GAC AAC TGA GGC CTC CTA | 1160–1181 | NM_009910 |
| RP: TCT TGC TCC CCA GTT GAT G | 1254–1274 | ||
| β–actin | FP: TGG AAT CCT GTG GCA TCC ATG AAA C | 885–909 | NM_007393.3 |
| RP: TAA AAC GCA GCT CAG TAA CAG TCC G | 1209–1233 |
FP, forward primer; RP, reverse primer.
Analysis of leucocytes in spleen, blood and brain
Spleen, blood and brain tissues were collected from C57BL/6, CCL2 KO and CCR2 KO mice infected with JEV (5·0 × 107 pfu/mouse) 2, 3, 5 and 7 dpi. Splenocytes and blood cells were used for leucocyte analysis after lysing red blood cells with hypotonic solution (NH4Cl3). To obtain leucocytes from the brain of JEV‐infected mice, mice were perfused with 30 ml of HBSS on 2, 3, 5 and 7 dpi via cardiac puncture of the left ventricle. Brains were then harvested and homogenized by gently pressing them through a 100‐mesh tissue sieve. They were then digested with 25 μg/ml of collagenase type IV (Worthington Biochem, Freehold, NJ), 0·1 μg/ml trypsin inhibitor Na‐p‐tosyl‐l‐lysine chloromethyl ketone, 10 μg/ml DNase I (Amresco, Solon, OH), and 10 mm HEPES in Hanks’ balanced salt solution for 1 hr at 37° with shaking. Cells were separated by Optiprep density gradient (18/10/5%) centrifugation at 800 × g for 30 min (Axis‐Shield, Oslo, Norway), after which cells were collected from the 18% to 10% interface and washed twice with PBS. Prepared cells were counted and stained for CD11b, Ly‐6G, Ly‐6C, CD3, CD4 and CD8 with directly conjugated antibodies (eBioscience) for 30 min at 4°. Finally, cells were fixed with 10% formaldehyde. Data collection and analysis were performed with a FACS Calibur flow cytometer (Becton Dickson Medical Systems, Sharon, MA) and the flowjo (Tree Star, San Carlos, CA) software.
Quantitative real‐time RT‐PCR for viral burden and chemokines/receptors
Viral burden, chemokine ligand (CCL3, CCL4, CCL5, CCL7, CCL12, CCL17, CXCL2, CXCL9 and CXCL11) and chemokine receptor (CCR1, CCR2, CCR4, CCR5, CXCR2 and CXCR3) expression in inflammatory and lymphoid tissues were determined by conducting quantitative SYBR Green‐based real‐time RT‐PCR (real‐time qRT‐PCR). Mice were infected with JEV (5·0 × 107 pfu/mouse), and tissues including brain, spinal cord and spleen were harvested at 2, 3, 4 and 5 dpi. Total RNAs extracted from tissues using easyBLUE (iNtRON, Inc., Daejeon, Korea) were employed for real‐time qRT‐PCR using a CFX96 Real‐Time PCR Detection system (Bio‐Rad Laboratories, Hercules, CA). Following reverse transcription of total RNAs with High‐Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster, CA), the reaction mixture contained 2 μl of template cDNA, 10 μl of 2 × SYBR Primix Ex Taq, and 200 nM primers, with a final volume of 20 μl. The reactions were denatured at 95°C for 30 seconds and then subjected to 45 cycles of 95°C for 5 seconds and 60°C for 20 seconds. After the reaction cycle was completed, the temperature was increased from 65°C to 95°C at a rate of 0·2°C/15 seconds, and the fluorescence was measured every 5 seconds to construct a melting curve. A control sample containing no template DNA was run with each assay, and all determinations were performed at least in duplicate to ensure reproducibility. The authenticity of amplified product was determined by melting curve analysis. The relative ratio of viral RNA in infected samples to uninfected samples was determined. All data were analysed using the Bio‐Rad CFX manager version 2·1 analysis software (Bio‐Rad Laboratories).
Analysis and activation of NK cells
The activity of NK cells was assessed by their capacity to produce IFN‐γ and granzyme B following brief stimulation with PMA and ionomycin (Sigma Aldrich, St. Louis, MO). Splenocytes were prepared from C57BL/6, CCR2 KO and CCL2 KO mice 2 dpi and stimulated with PMA (50 ng/ml) plus ionomycin (750 ng/ml) to induce expression of IFN‐γ and granzyme B in the presence of monensin (2 μM) for 1 and 2 hr, respectively. After stimulation, cells were surface‐stained by FITC anti‐mouse‐CD3ε, PE‐Cy7 anti‐mouse NK1.1, biotin‐conjugated anti‐mouse pan‐NK cell (CD49b) (DX5) antibodies, and streptavidin‐APC for 30 min at 4°. Cells were then washed twice with FACs buffer containing monensin. After fixation, the cells were permeabilized with 1 × permeabilization buffer (eBioscience) and intracellularly stained with PE anti‐mouse IFN‐γ (XMF1.2) and granzyme B (16G5) antibodies in permeailization buffer for 30 min at room temperature. Finally, cells were washed twice with PBS and analysis was performed with a FACS Calibur flow cytometer (Becton Dickson Medical Systems, Sharon, MA).
JEV‐specific CD4+ and CD8+ T‐cell responses
To monitor CD4+ and CD8+ T‐cell responses specific for JEV, the surviving mice were killed at day 7 pi and splenocytes were prepared. Erythrocytes were depleted by treating the single‐cell suspensions with NH4Cl–Tris buffer for 5 min at 37°C. Splenocytes were cultured in a 96‐well culture plate (5 × 105 cells/well) with synthetic peptide epitopes (NS1132–145, NS3563–575 and NS4B215–225) for 12 hr in the presence of PE anti‐mouse CD154 and 6 hr to analyse CD4+ and CD8+ T‐cell responses, respectively.27, 28 Monensin at a concentration of 2 μM was added to the antigen‐stimulated cells 6 hr before harvest. Cells were washed twice with PBS and surface‐stained for FITC‐anti‐CD4 or anti‐CD8 antibodies for 30 min at 4°C. They were then washed twice with PBS containing monensin. After fixation, the cells were washed twice with permeabilization buffer (eBioscience) and then stained with PE‐ or PerCP Cy5.5‐anti‐mouse IFN‐γ and APC‐anti‐mouse TNF‐α in permeabilization buffer for 30 min at room temperature. Finally, cells were washed twice with PBS and fixed using a fixation buffer. Sample analysis was performed with a FACSCalibur flow cytometer.
Generation of CCL2 KO bone marrow chimeric mice and viral infection
C57BL/6 mice (7–8 weeks old) and CCL2 KO mice (7–8 weeks old) were γ‐irradiated with one dose of 950 rads. Within 12 hr, mice were reconstituted with 107 donor bone marrow (BM) cells derived from CCL2 KO or C57BL/6 mice. The recipient mice were given sulfamethoxazole and trimethoprim in drinking water for 10 days after irradiation. Mice were infected with JEV (5·0 × 107 pfu/mouse) 4–6 weeks after irradiation.
Statistical analysis
All data were expressed as the mean ± standard deviation, and statistically significant differences between groups were analysed by unpaired two‐tailed Student's t‐test for leucocyte population analysis and in vitro experiments or analysis of variance and post‐hoc testing for multiple comparisons of the mean. The statistical significance of viral burden and in vivo cytokine gene expression were evaluated by the Mann–Whitney U test or unpaired two‐tailed Student's t‐test. Kaplan–Meier survival curves were analysed by the log rank test. P‐values ≤ 0·05 were considered significant. All data were analysed using the prism software (GraphPadPrism4, San Diego, CA).
Results
CCL2, but not its receptor, is essential to protect hosts from JE
The inflammatory role of CCR2/CCL2 has been complicated by various results in different immunopathological contexts.10, 17 Because CCR2/CCL2 blockers remain interesting for developing therapeutic strategies for immunopathological diseases, uncovering the role of CCR2/CCL2 in these diseases may be a prerequisite for developing prophylactic and therapeutic strategies. However, the role of CCR2/CCL2 signals in viral encephalitis caused by neurotrophic flaviviruses such as JEV and WNV is not fully elucidated. To investigate the role of CCR2 and its ligand, CCL2, in flavivirus‐induced encephalitis, the impact of CCR2 and CCL2 molecules in JE progression were assessed by evaluating the susceptibility of CCR2 KO and CCL2 KO mice to JEV infection. Somewhat surprisingly, CCR2 KO and CCL2 KO mice were observed to induce completely contrasting regulation of JE. While CCR2 KO mice showed increased resistance to JE compared with wild‐type C57BL/6 mice (P = 0·1170 for 3·0 × 107 pfu; P = 0·0526 for 5·0 × 107 pfu), CCL2 KO mice were highly susceptible to JE (Fig. 1a). Notably, CCL2 ablation resulted in marked increases in mortalities, with 80% and 90% for CCL2 KO versus 40% and 55% for C57BL/6 mice following JEV infection (3·0 × 107 and 5·0 × 107 pfu, respectively). Likewise, CCR2 KO mice showed delayed signs of neurological disorder with a lower frequency of occurrence, while CCL2 KO mice displayed more rapid and frequent signs of neurological disorder compared with C57BL/6 mice (Fig. 1b). CCL2 KO mice, but not CCR2 KO mice, also showed more apparent loss of body weight depending on infection date (Fig. 1c). Collectively, these results clearly indicate that ablation of CCR2 and its cognate ligand CCL2 differentially affects the outcome of neuroinflammatory disease caused by JEV.
Figure 1.
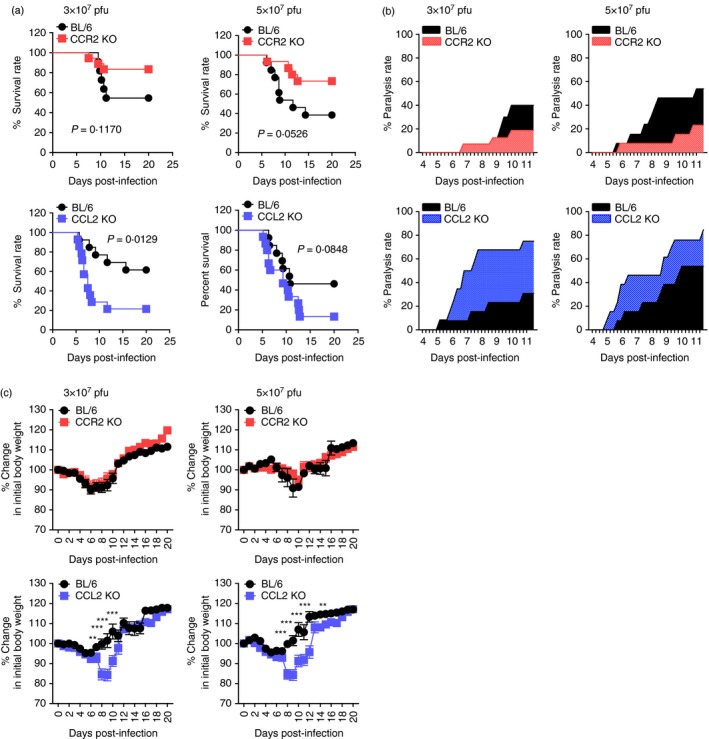
Contrasting susceptibility of CCR2‐ and CCL2‐deficient mice to Japanese encephalitis (JE). C57BL/6, CCR2 knockout (KO), and CCL2 KO mice (4‐ to 5‐week‐old, n = 13–20) were intradermally infected with Japanese encaphalitis virus (JEV) (3·0 or 5·0 × 107 pfu) via the footpad route. (a) Survival rate. Surviving mice were recorded daily up to 20 days post‐infection (dpi), and the proportion of surviving inoculated mice was presented. (b) Ratio of mice showing neurological disorders during JE progression. Mice infected with JEV were examined every 6 hr from 4 to 11 dpi, and the ratio of inoculated mice showing neurological disorders was recorded. (c) Change in body weight. Data are expressed as the mean percentage ± SD of body weight relative to time of challenge. *P < 0·05; **P < 0·01; ***P < 0·001 compared with levels of the indicated group.
CCL2 ablation enhances viral burden in neural tissues
CCR2–CCL2 signalling appears to be critical for recruiting immune cells to mediate virus clearance.10 Therefore, determination of viral burden in lymphoid and CNS tissues is important to further understand the neuroinflammation caused by JEV. Both CCR2 KO and CCL2 KO mice contained comparable levels of viral burden in the primary lymphoid tissue, the spleen (Fig. 2). However, CCL2 KO mice exhibited 103‐fold to 104‐fold elevated viral burden in the brain and spinal cord compared with C57BL/6 mice. CCR2 KO mice showed no significantly altered levels of viral burden in the CNS. These results indicate that viral burden in the CNS during JE progression is affected by CCL2 ablation, rather than ablation of the cognate receptor CCR2.
Figure 2.
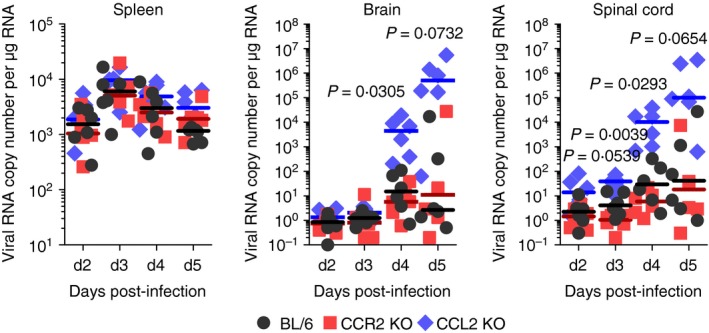
Enhanced viral burden in inflamed tissues of CCL2 knockout (KO) mice. C57BL/6, CCR2 KO, and CCL2 KO mice were intradermally infected with Japanese encephalitis virus (JEV) via the footpad route. Viral burden in spleen, brain, and spinal cord was determined by real‐time quantitative RT‐PCR using extracted total RNA at the indicated days post‐infection (dpi). Viral RNA load was expressed by viral RNA copy number per microgram of total RNA. Each symbol represents the level of an individual mouse; horizontal line indicates median of each group (n = 5–6). *, P < 0·05; **, P < 0·01; ***, P < 0·001 compared between BL/6 and CCL2 KO mice.
Local chemokine expression is associated with accumulation of leucocytes in the CNS
CCR2/CCL2‐mediated recruitment of leucocytes affects tissue pathology in neuroinflammatory conditions such as ischaemic brain injury, traumatic brain injury and viral encephalitis.10, 17 CCR2/CCL2 signalling is believed to involve CCR2+ Ly‐6Chi monocytes that play an important role in CNS inflammation as precursors of macrophages and microglia.19, 29, 30 The debatable role of CD11b+ Ly‐6Chi monocytes has been raised regarding their differential contribution to immunopathology depending on the neuroinflammation context.18, 22, 31, 32, 33, 34 To further characterize JE in CCR2‐ and CCL2‐ablated environments, the infiltration of myeloid‐derived immune cells was assessed, including Ly‐6Chi monocytes, Ly‐6Ghi granulocytes, and lymphoid T‐cell subsets. The results revealed that nearly identical percentages of CD11b+ Ly‐6Chi and CD11b+ Ly‐6Cint monocytes were retained in the spleen of C57BL/6, CCR2 KO and CCL2 KO mice. Only splenic CD11b+ Ly‐6Ghi granulocytes were observed in CCR2 KO and CCL2 KO mice with moderately higher frequency compared with C57BL/6 mice (Fig. 3a). Unlike results derived from the spleen, both CCR2 KO and CCL2 KO mice showed apparently decreased frequency of CD11b+ Ly‐6Chi monocytes in the blood compared with C57BL/6 mice, whereas CD11b+ Ly‐6Ghi granulocytes were detected only in the blood of CCL2 KO mice with highly increased frequency following JEV infection. The absolute number of CD11b+ Ly‐6Chi monocytes, CD11b+ Ly‐6Cint monocytes and CD11b+ Ly‐6Ghi granulocytes followed the frequency trends of those myeloid‐derived subpopulations in the spleen and blood (Fig. 3b,c). Notably, CCL2 KO displayed markedly increased accumulation of CD11b+ Ly‐6Ghi granulocytes in the blood compared with other C57BL/6 and CCR2 KO mice (Fig. 3c). Regarding lymphoid T‐cell subsets, CCL2 KO mice showed prolonged presence and a higher number of CD4+ and CD8+ T cells in the spleen (Fig. 3d).
Figure 3.
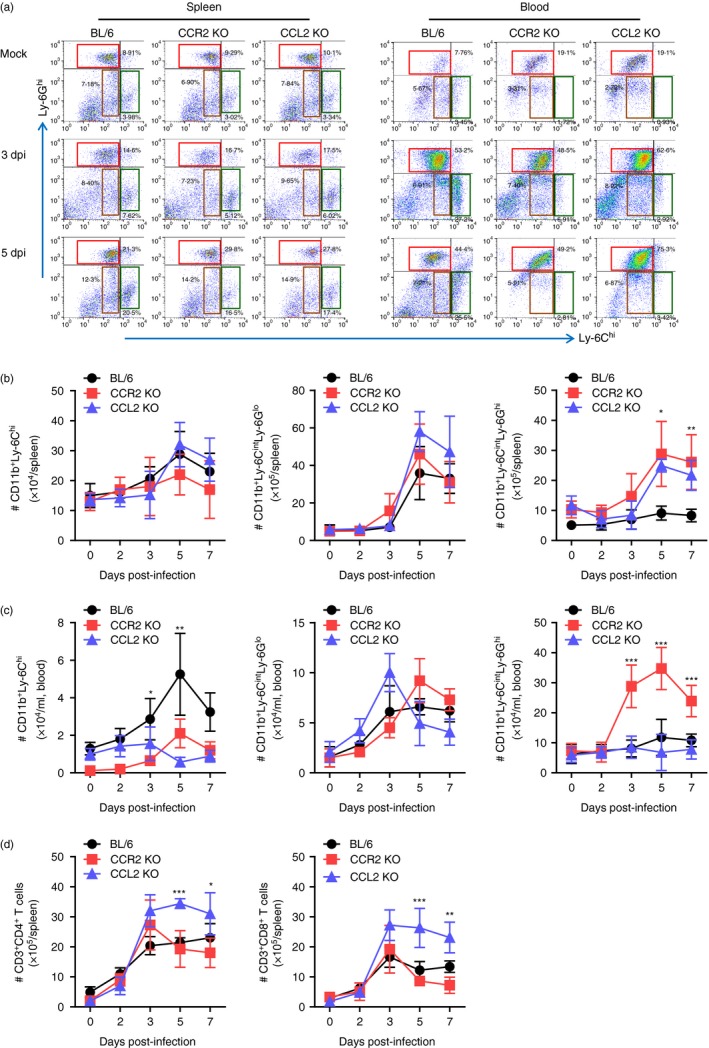
Reduced accumulation of CD11b+ Ly‐6Chi monocytes in spleen and blood of CCR2 knockout (KO) and CCL2 KO mice. (a) Frequency of Ly‐6Chi monocytes and Ly‐6Ghi granulocytes in spleen and blood. Frequency of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in spleen and blood were determined by flow cytometric analysis at 3 and 5 days post‐infection (dpi). (b,c) Accumulated number of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in spleen and blood. Accumulated number of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in spleen (b) and blood (c) was kinetically determined by flow cytometric analysis from 2 to 7 dpi. Left graph, Ly‐6Chi monocytes; Middle graph, Ly‐6Cint monocytes; Right graph, Ly‐6Ghi granulocytes. (d) Accumulated number of CD3+ CD4+ and CD3+ CD8+ T cells in spleen. Values in representative dot‐plots denote average percentage of indicated cell population after gating on CD11b+ cells. Data in graph represent the mean ± SD of values derived from at least four mice per group. *P < 0·05; **P < 0·01; ***P < 0·001 compared between C57BL/6 and CCR2 KO or CCL2 KO mice.
Meanwhile, CCL2 KO mice contained CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes in the CNS with highly increased frequency following JEV infection, whereas CCR2 KO mice showed significantly decreased frequency of CD11b+ Ly‐6Chi monocytes compared with C57BL/6 mice (Fig. 4a). Consistent with this, CCL2 KO mice showed a prolonged course and higher number of CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes in the brain, whereas CCR2 KO mice contained a significantly decreased number of CD11b+ Ly‐6Chi monocytes in the brain compared with C57BL/6 mice (Fig. 4b). Also, CCL2 KO mice had highly increased accumulation of CD4+ and CD8+ T cells in the CNS until 7 dpi compared with C57BL/6 and CCR2 KO mice (Fig. 4c). In summary, highly increased CNS infiltration of CD11b+ Ly‐6Chi monocytes, CD11b+ Ly‐6Ghi granulocytes, and CD4+/CD8+ T cells in CCL2 KO mice was closely associated with increased susceptibility to JE. In contrast, CCR2 KO mice showed less CNS accumulation of CD11b+ Ly‐6Chi monocytes, resulting in increased resistance to JE.
Figure 4.
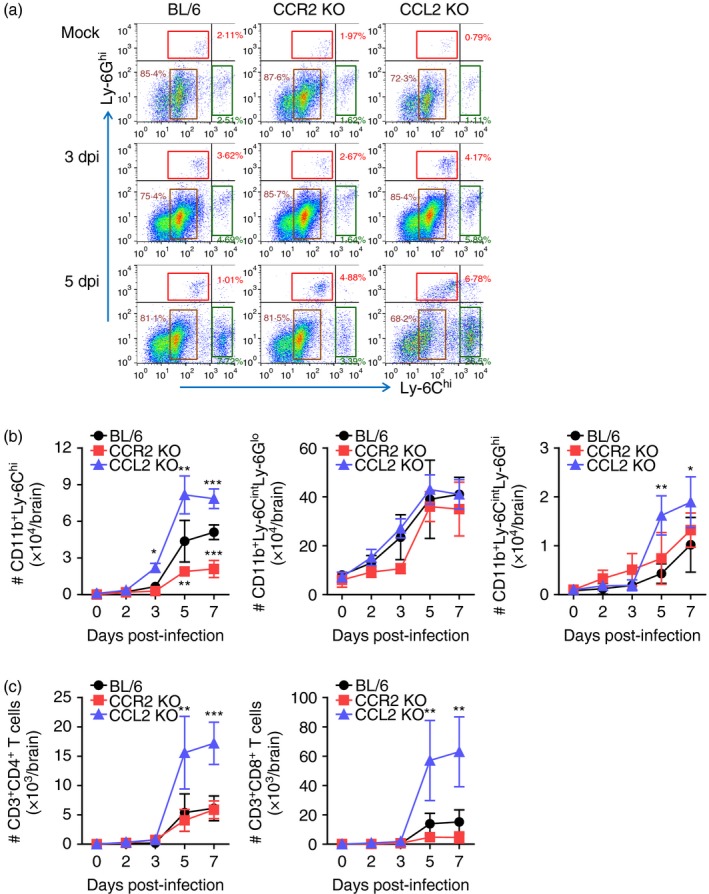
Enhanced accumulation of Ly‐6Chi monocytes in central nervous system (CNS) of CCL2 knockout (KO) mice. (a) Frequency of Ly‐6Chi monocytes and Ly‐6Ghi granulocytes in the brain. Frequency of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in the brain were determined by flow cytometric analysis at 3 and 5 days post‐infection (dpi). (b) Accumulated number of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in the brain. Accumulated number of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in the brain was kinetically determined by flow cytometric analysis from 2 to 7 dpi. Left graph, Ly‐6Chi monocytes; Middle graph, Ly‐6Cint monocytes; Right graph, Ly‐6Ghi granulocytes. (c) Accumulated number of CD3+ CD4+ and CD3+ CD8+ T cells in the brain. Values in representative dot‐plots denote average percentage of indicated cell population after gating on CD11b+ cells. Data in graph represent the mean ± SD of values derived from at least four mice per group. *P < 0·05; **P < 0·01; ***P < 0·001 compared between C57BL/6 and CCR2 KO or CCL2 KO mice.
In terms of severe neuroinflammation manifested by highly increased CNS infiltration of leucocytes in CCL2 KO mice, the expression of chemokines and their receptors within the CNS is required to further explain the severity of encephalitis. Therefore, expression of chemokines and their receptors in the brains of CCR2 KO and CCL2 KO mice following JEV infection was investigated. As expected, CCL2 KO mice showed highly increased expression levels of all tested CC and CXC chemokines except CCL17, whereas CCR2 KO mice showed lower expression levels of chemokines including CCL3, CCL4, CCL5, CCL7 and CXCL2 following JEV infection (Fig. 5a). Similarly, CCL2 KO mice showed highly increased CNS expression levels of CC and CXC chemokine receptors including CCR1, CCR2, CCR4, CCR5 and CXCR2 (Fig. 5b). But, the CXCR3 receptor was expressed at lower levels in CCL2 KO mice compared with C57BL/6 mice. Also, CCR2 KO mice displayed significantly decreased expression levels of CC chemokine receptors including CCR1 and CCR2. These results suggest that highly increased CNS accumulation of Ly‐6Chi monocytes, Ly‐6Ghi granulocytes, and T cells in CCL2 KO mice was coupled to the increased expression of CC/CXC chemokines and their receptors. In contrast, CCR2 KO mice showed decreased expression of CC chemokines and their receptors in the CNS, resulting in less CNS accumulation of leucocytes.
Figure 5.
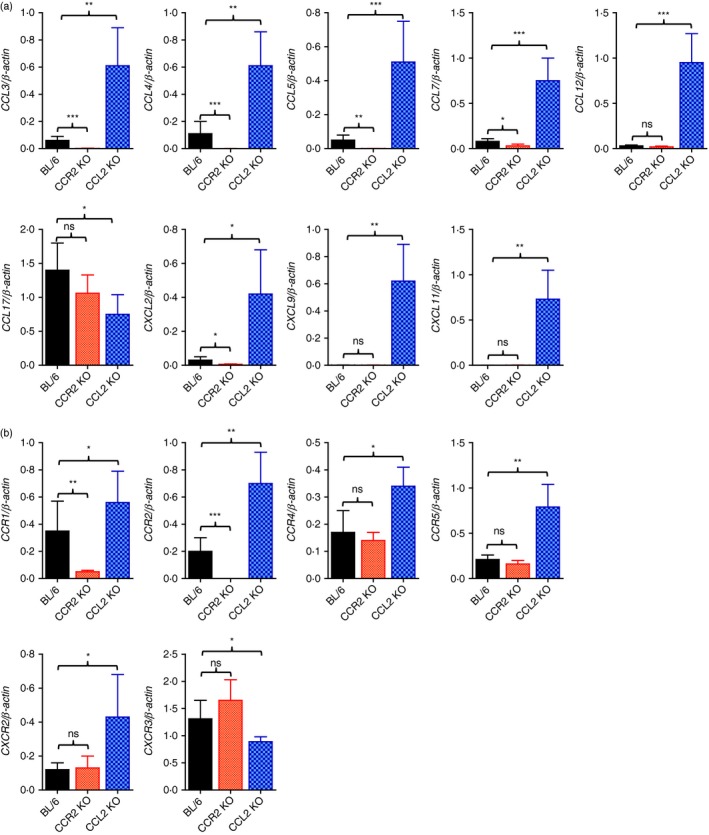
CCL2 ablation provides highly enhanced expression of chemokines and their receptors in inflamed tissues. Expression of chemokines and their receptors in the central nervous system (CNS) was determined by real‐time quantitative RT‐PCR using total RNA extracted from the CNS at 5 days post‐infection (dpi). (a) Chemokine expression in the CNS. (b) Expression of chemokine receptors in the CNS. Data represent the mean ± SD of values derived from at least four mice per group. *P < 0·05; **P < 0·01; ***P < 0·001 compared with the levels of the indicated group.
Dispensability of NK cell innate immune response in JE regulation of CCR2‐ and CCL2‐ablated mice
Antiviral innate NK cell activation and adaptive antigen‐specific T‐cell responses are believed to play an important role in regulating JE progression through the control and clearance of JEV in extraneural and neural tissues.35 To characterize the immunological parameters associated with impaired control of JEV replication in CCL2 KO mice, we examined and compared NK and adaptive T‐cell responses in CCR2 KO, CCL2 KO and wild‐type C57BL/6 mice. Analysis of splenic NK cells revealed that CCL2 KO mice showed basally and moderately higher frequencies and numbers of CD3− NK1.1+ DX5+ NK cells compared with C57BL/6 mice, whereas CCR2 KO mice contained slightly lower frequencies and numbers of CD3− NK1.1+ DX5+ NK cells (Fig. 6a,b). Following JEV infection, the frequencies of CD3− NK1.1+ DX5+ NK cells were decreased in C57BL/6, CCR2 KO and CCL2 KO mice, as demonstrated in previous studies.5, 13 CCL2 KO mice showed an apparently decreased number of NK cells, resulting in a comparable total number of NK cells with C57BL/6 mice following JEV infection (Fig. 6b). CCL2 KO mice also showed enhanced activity of NK cells upon JEV infection when evaluated by enumeration of NK cells producing IFN‐γ and granzyme B in response to stimulation of PMA plus ionomycin. CCL2 KO mice thereby contained an enhanced total number of NK cells producing IFN‐γ and granzyme B (Fig. 6c,d). Ultimately, these results indicate that the promoted responses of NK cells in CCL2‐ablated mice failed to control JEV replication, which suggests that NK cells provide no effective contribution to the regulation of JE progression.
Figure 6.
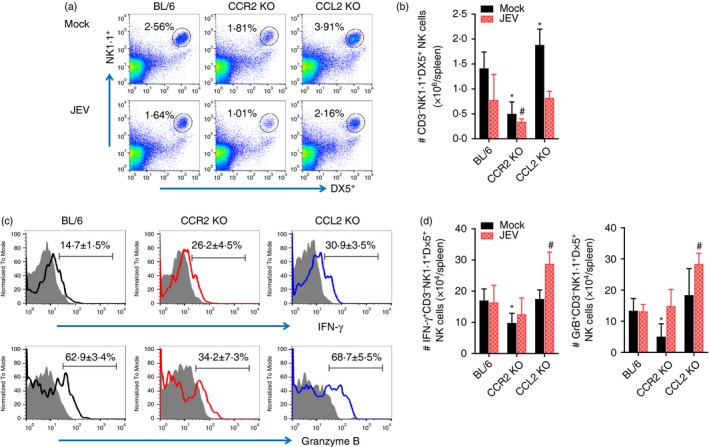
Enhanced activation of innate natural killer (NK) cells in CCL2 knockout (KO) mice. (a,b) Frequency and absolute number of NK cells. Frequency and number of CD3− NK1·1+ DX5+ NK cells were determined by flow cytometric analysis using collagenase‐treated spleen at 2 days post‐infection (dpi). Values in representative dot‐plots denote average percentage of NK cells after gating on CD3‐negative cells. (c) NK cell activation. Activation of NK cells was evaluated by enumerating NK cells producing interferon‐γ (IFN‐γ) and granzyme B (GrB) using intracellular cytokine staining at 2 dpi. Values in representative histograms denote average percentage of cells producing IFN‐γ or GrB in CD3− NK1.1+ DX5+ NK cells. Bar graph shows the mean ± SD of values derived from at least four mice per group. *P < 0·05; **P < 0·01; ***P < 0·001 compared with levels of the mock‐infected BL/6 group. #P<0.05 compared with levels of JEV‐infected BL/6 group.
JEV‐specific T‐cell responses in CCL2 and CCR2‐ablated mice
In addition to NK cells, adaptive immune responses specific for JEV antigen are required for the regulation of JE progression through peripheral control of JEV replication.36, 37 CCL2 not only has chemotactic properties, but is also expressed at T helper type 1 (Th1) or Th2 polarized inflammatory sites to facilitate the polarization of both types of inflammation.38, 39 CCL2 appeared to have immunosuppressive activity in certain settings, resulting in reduced expression of IL‐12 and TNF‐α and greater expression of IL‐10.40 Therefore, an understanding of JEV‐specific T‐cell responses may be required to further explain the contrasting results of CCR2 KO and CCL2 KO mice during JE progression. To examine JEV‐specific‐CD4+ and CD8+ T‐cell responses, splenocytes prepared from surviving C57BL/6, CCR2 KO and CCL2 KO mice 7 dpi were stimulated with corresponding JEV epitope peptides specific for CD4+ and CD8+ T cells, respectively. The results revealed that CCL2 ablation induced comparable CD4+ T‐cell responses with wild‐type C57BL/6 mice when CD4+ T‐cell responses were evaluated by intracellular CD154 and IFN‐γ staining in response to stimulation with two epitope peptides (NS1132–145 and NS3563–574) derived from JEV (Fig. 7a–d). CCR2 KO mice showed significantly decreased CD4+ T‐cell responses, rather than increased JEV‐specific CD4+ T‐cell responses. Similarly, JEV‐specific CD8+ T‐cell responses displayed comparable responses in wild‐type C57BL/6 and CCL2 KO mice, whereas CCR2 KO mice showed decreased JEV‐specific CD8+ T‐cell responses compared with wild‐type C57BL/6 mice (Fig. 7e,f). Because CCR2 KO mice showed enhanced resistance to JE and adequate JEV‐specific T‐cell responses require time to develop, JEV‐specific CD4+ and CD8+ T‐cell responses may not be key players to control JE progression in CCR2 and CCL2‐ablated mice.
Figure 7.
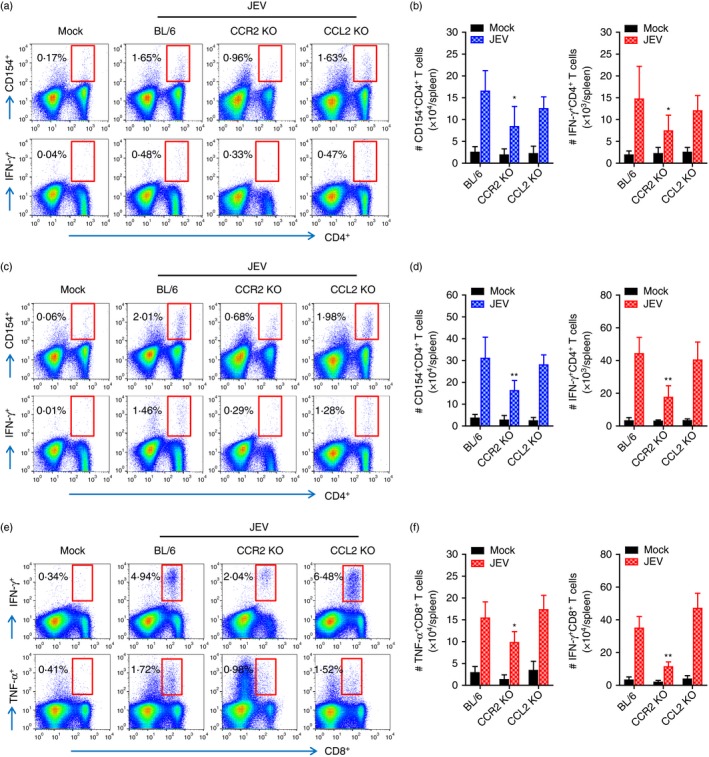
Japanese encephalitis virus (JEV) ‐specific T‐cell immunity in C57BL/6, CCR2 knockout (KO), and CCL2 KO mice. Splenocytes prepared from surviving mice at 7 days post‐infection (dpi) were stimulated with JEV epitope peptide of CD4+ T cells or CD8+ T cells for 12 or 6 hr, respectively. Frequency and absolute number of JEV‐specific CD4+ and CD8+ T cells were determined by intracellular CD154 and cytokine [interferon‐γ (IFN‐γ) and tumour necrosis factor‐α (TNF‐α)] staining combined with surface CD4 and CD8 staining. (a–d) JEV‐specific CD4+ T‐cell response. Values in representative dot‐plots denote average percentage of CD154+ or IFN‐γ + cells in CD4+ T cells derived from at least four mice. (a, b) NS1132–145; (c, d) NS3563–574. (e, f) CD8+ T‐cell response specific for JEV NS4B215–223 epitope. Values in representative dot‐plots denote the mean percentage of IFN‐γ + or TNF‐α + cells in CD8+ T cells derived from at least four mice. Bar graph show the mean ± SD of values derived from at least four mice per group. *P < 0·05; **P < 0·01; ***P < 0·001 compared with levels of the indicated group.
Dominant role of CCL2 produced from resident cells to alleviate JE
Our results support that CCL2 ablation exacerbates JE progression by providing highly enhanced CNS infiltration of myeloid‐derived leucocytes, including CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes, as well as enhancing viral burden, rather than impairing innate NK and adaptive T‐cell responses. CCL2 is believed to be involved in recruiting various cell types, including monocytes, T cells and other cells that express CCR2.41 CCL2 can be produced by myeloid‐derived leucocytes and resident epithelial and stromal cells through various stimuli in immunopathological diseases such as neuroinflammation and cancer.41 Therefore, we were interested in testing which cell types are dominant in regulating JE progression and CNS infiltration of Ly‐6Chi monocytes and Ly‐6Ghi granulocytes, focusing on resident cells and myeloid cells derived from HSCs. To this end, BM chimeric models of wild‐type C57BL/6 and CCL2 KO mice were used. CCL2 produced by myeloid cells derived from HSCs played a dominant role in exacerbating JE progression because CCL2 KO recipients of wild‐type C57BL/6 BM donor cells (WT‐KO) showed comparable susceptibility to JE with CCL2 KO recipients of CCL2 KO BM donor cells (KO‐KO), whereas wild‐type C57BL/6 recipients of CCL2 KO BM donor cells (KO‐WT) exhibited almost identical resistance to JE as those of the WT‐WT BM chimeric model (Fig. 8a). Both WT‐WT and KO‐WT BM chimeric models showed delayed and lower frequency of neurological disorders, and also experienced less change in body weight after JEV infection compared with WT‐KO and KO‐KO chimeric models (Fig. 8b,c). These results indicate that CCL2 produced from γ‐irradiation‐resistant resident epithelial and stromal cells plays a dominant role in ameliorating JE progression.
Figure 8.
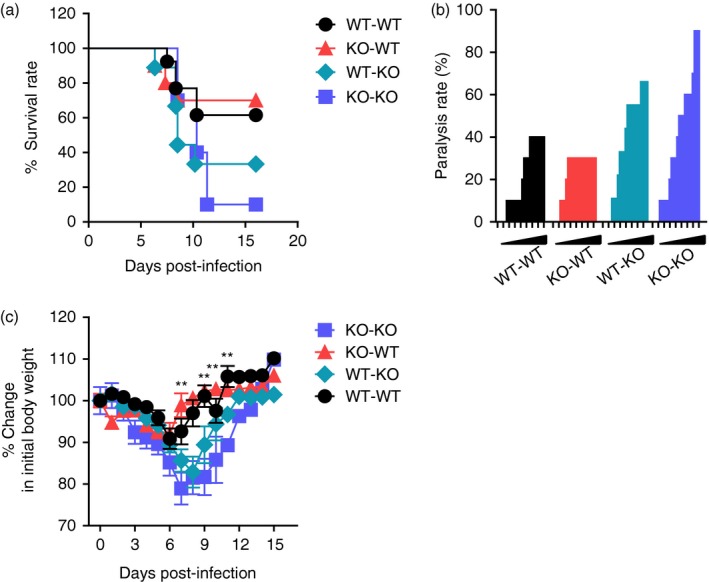
CCL2 produced from resident central nervous system (CNS) cells alleviates Japanese encephalitis (JE) progression. Bone marrow (BM) cells from wild‐type (WT) or CCL2 knockout (KO) mice were grafted to lethally irradiated WT or CCL2 KO recipient mice, which were intradermally infected with Japanese encephalitis virus (JEV; 5·0 × 107 pfu) via footpad route. (a) Susceptibility of CCL2 KO BM chimeric models to JE. Infected recipient mice (n = 10–11) were examined daily up to 16 days post‐infection (dpi) to record survival rate. (b) Ratio of mice showing neurological disorder during JE progression. CCL2 KO BM chimeric models infected with JEV were examined every 6 hr from 4 to 11 dpi, and ratio of inoculated mice showing neurological disorder was recorded. (c) Change in body weight. Data are expressed as the mean percentage ± SD of body weight relative to time of challenge. *P < 0·05; **P < 0·01; ***P < 0·001 compared between KO‐WT and WT‐KO BM chimeric model.
CCL2 produced from HSC‐derived leucocytes plays a dominant role in CNS accumulation of leucocytes
To better understand the dominant role of CCL2 produced by myeloid cells derived from HSCs in exacerbating JE, CNS infiltration of myeloid‐derived leucocytes was assessed in CCL2 KO BM chimeric models during JE progression. The results revealed no significant change in the frequency of splenic CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes between four CCL2 KO BM chimeric models (Fig. 9a). Following JEV infection, the frequency of CD11b+ Ly‐6Chi monocytes in the blood of wild‐type C57BL/6 recipients of CCL2 KO BM donor cells (KO‐WT) showed no significant difference from those of the WT‐WT BM chimeric model with higher levels. The WT‐KO BM chimeric model showed the lowest frequency of CD11b+ Ly‐6Chi monocytes in blood compared with the levels found in the KO‐KO BM chimeric model. Instead, the KO‐KO BM chimeric model showed higher frequency of CD11b+ Ly‐6Ghi granulocytes in blood than other BM chimeric models. With regard to CNS‐infiltrated leucocytes, CCL2 KO recipients of wild‐type C57BL/6 BM donor cells (WT‐KO) showed a markedly higher frequency of CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes in the brain compared with WT‐WT and KO‐WT BM chimeric models. The KO‐KO BM chimeric model also showed the highest frequency of CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes in the CNS. The accumulated number of splenic CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes was not changed between CCL2 KO BM chimeric models, whereas WT‐KO and KO‐KO BM chimeric models contained fewer CD11b+ Ly‐6Chi monocytes in blood but not fewer CD11b+ Ly‐6Ghi granulocytes (Fig. 9b,c). The WT‐KO and KO‐KO BM chimeric models showed a high number of CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes in the CNS compared with WT‐WT and KO‐WT BM chimeric models (Fig. 9d). Ultimately, these results support that CNS infiltration of CD11b+ Ly‐6Chi monocytes and CD11b+ Ly‐6Ghi granulocytes was preferentially mediated by CCL2 produced from leucocytes derived from HSCs, resulting in close association with JE exacerbation.
Figure 9.
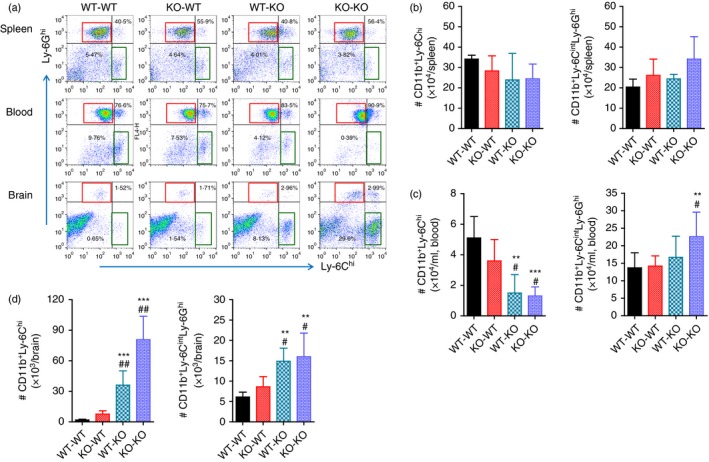
CCL2 produced from haematopoietic stem cells (HSCs) plays a dominant role in central nervous system (CNS) accumulation of leucocytes. Leucocytes from the spleen, blood and brain of CCL2 knockout (KO) bone marrow (BM) chimeric models infected with Japanese encephalitis virus (JEV; 5·0 × 107 pfu) were prepared at 5 days post‐infection (dpi). (a) Frequency of Ly‐6Chi monocytes and Ly‐6Ghi granulocytes in the spleen, blood and brain. Frequency of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes was determined by flow cytometric analysis. (b–d) Accumulated number of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes in the spleen, blood and brain. Accumulated number of Ly‐6Chi/Ly‐6Cint monocytes and Ly‐6Ghi granulocytes was enumerated by flow cytometric analysis at 5 dpi. Left graph, Ly‐6Chi monocytes; Right graph, Ly‐6Ghi granulocytes. Values in representative dot‐plots denote average percentage of indicated cell population after gating on CD11b+ cells. Data in graph represent the mean ± SD of values derived from at least four mice per group. **P < 0·01; ***P < 0·001 compared with levels of wild‐type (WT) ‐WT BM chimeric model. # P < 0·05; ## P < 0·01 compared with levels of KO‐WT BM chimeric model.
Discussion
Our data clearly indicate that the starkly contrasting results of JE severity in CCR2‐ and CCL2‐ablated mice were closely associated with CNS infiltration of peripheral leucocytes, including Ly‐6Chi monocytes and Ly‐6Ghi granulocytes. Infiltration of the CNS by Ly‐6Chi monocytes and Ly‐6Ghi granulocytes in CCR2‐ and CCL2‐ablated mice appeared to be coupled to trends of CC and CXC chemokines and chemokine receptor expression. Our results also revealed that CCL2 produced from HSC‐derived leucocytes played a dominant role in CNS accumulation of Ly‐6Chi monocytes, thereby exacerbating JE progression. These results ultimately suggest that CCL2 is essential to provide protection against JE progression, and blockage of CCR2 by an antagonist provides an advantage in developing effective prophylactic and therapeutic strategies for JE.
Central nervous system infiltration of CD11b+ Ly‐6Chi monocytes is a typical feature of CNS inflammation caused by sterile and non‐sterile insults.29, 42 These cells migrate into the injured brain, where they are believed to differentiate into dendritic cells, macrophages and microglia.19, 29, 30 However, the role of Ly‐6Chi monocytes in immunopathology of the CNS is debatable, depending on the encephalitis context. CNS infiltration of CD11b+ Ly‐6Chi monocytes provides significant damage and destruction to the CNS in several models of CNS diseases,31, 32 whereas CNS infiltration of CD11b+ Ly‐6Chi monocytes plays an important role in conferring protection against neuroinflammation, such as WNV‐induced encephalitis.18, 22, 33, 34 Our results clearly favour the former notion because CCR2‐ablated mice showed enhanced resistance to JE along with decreased CNS infiltration of Ly‐6Chi monocytes, rather than high susceptibility to JE. In contrast, CCL2‐ablated mice contained highly increased CNS infiltration by Ly‐6Chi monocytes, resulting in markedly increased mortality following JEV infection. The role of CCR2 in regulating Ly‐6Chi monocyte egression from BM has been critically demonstrated under homeostatic and inflammatory conditions.43, 44 Genetic ablation of CCR2 results in severe monocytopenia that provides partial or entire loss of these cells in inflamed tissues, as per the results demonstrated in this study. CCR2‐mediated recruitment of Ly‐6Chi monocytes was reported to play a critical role in conferring protection from viral encephalitis caused by WNV,18 which is closely related to members of the Flaviviridae family along with JEV. Also, higher mortality and increased pathogen burden were seen in several pathogen infection models, such as Toxoplasma,45 herpes simplex virus type 246 and coronavirus,21, 22 when Ly‐6Chi monocytes were depleted. These facts are in contrast with our data showing that CCR2‐ablated mice contained no changes in the viral burden of extraneural and neural tissues. However, our results provide a similar conclusion that CCR2‐mediated recruitment of Ly‐6Chi monocytes is detrimental to the progression of diseases, such as lung injury caused by influenza virus infection47 and herpes simplex virus encephalitis.48 On the other hand, Ly‐6Chi monocytes are direct targets for pathogens such as HIV,49 Listeria 50 and Toxoplasma,45 which suggests that infected monocytes are directly responsible for dissemination of infection in a ‘Trojan horse’ fashion into the CNS, thereby facilitating disease progression. Similarly, CD11b+ Ly‐6Chi monocytes were observed to facilitate JEV replication.26 Therefore, highly increased levels of viral burden in the CNS of CCL2‐ablated mice may be strengthened by elevated CNS infiltration of Ly‐6Chi monocytes.
Our results also indicate that recruitment of Ly‐6Chi monocytes into inflamed sites does not completely depend on the CCR2–CCL2 axis. Indeed, other chemokine receptors and chemokines are probably responsible for recruiting Ly‐6Chi monocytes in CCL2‐ablated mice because chemokine responses can be redundant. CCR2 can bind to CCL7 and CCL12, as well as other chemokine receptors, such as CCR5 and CXCR3, which may participate in the infiltration of Ly‐6Chi monocytes into the CNS.10 In support of this, CCL7 and CCL12 expression was increased in the CNS of CCL2‐ablated mice following JEV infection. Hence, these chemokines may be in part responsible for the enhanced CNS infiltration of Ly‐6Chi monocytes in CCL2‐ablated mice. Paradoxically, transgenic expression of CCL2 in the CNS is detrimental to CNS diseases caused by intracranial infection with coronavirus, rather than showing a beneficial role in progressing viral encephalitis.51 Even though viral encephalitis is induced in CCL2 transgenic mice through a different virus strain and inoculation route, transgenic expression of CCL2 in the CNS provides ineffective clearance of the virus and a highly increased mortality, along with increased CNS infiltration of leucocytes. This fact may indicate that CNS infiltration of peripheral leucocytes is coupled to viral burden, no matter how the viral encephalitis is induced. Considering that CCL2 function appears redundant and can be replaced by the activity of other chemokines,10, 17 it was interesting that lacking a single chemokine CCL2 drove the dramatic phenotype of progressing viral encephalitis caused by JEV. The complicated network of chemokine responses in progressing JE remains to be defined at the molecular level to uncover which steps play a critical role in regulating JE.
One interesting finding of this study was that ablation of CCR2 or CCL2 induced accumulation of CD11b+ Ly‐6Ghi granulocytes in extraneural and neural tissues. Notably, CCL2 ablation appeared to provide more apparent accumulation of Ly‐6Ghi granulocytes in the blood and brain. In line with this finding, deficiencies of CCL2 and CCR2 were observed to induce infiltration of CD11b+ Ly‐6Ghi granulocytes into inflamed tissues in a few disease models, such as herpetic stromal keratitis46 and arthritis caused by Chikungunya virus.52 Although Ly‐6Ghi granulocytes are a type of innate immune cell involved in immunopathology, their role in viral diseases such as viral encephalitis remains obscure. Because CD11b+ Ly‐6Ghi granulocytes contain myeloid‐derived suppressor cells that were recently described as a regulatory cell population in various inflammatory reactions,53 it is possible that gradual increases in Ly‐6Ghi granulocytes are involved in the resolution of disease progression. In contrast, dominant infiltration of Ly‐6Ghi granulocytes could exacerbate diseases by preventing replacement of inflammatory monocytes with M2 macrophages that have high efferocytosis activity, as efferocytosis is a key driver of inflammation resolution.54 Our results revealed that CCL2 ablation induces over‐expression of CXCL2 (formerly called MIP‐2), which is a strong chemoattractant for Ly‐6Ghi granulocytes. Conceivably, this over‐expression of CXCL2 in the CNS of CCL2‐ablated mice is secondary to CNS entry of the infectious virus from the periphery through Ly‐6Chi monocytes. Therefore, CNS accumulation of CD11b+ Ly‐6Ghi granulocytes in CCL2‐ablated mice is probably a secondary phenomenon caused by over‐expression of CXCL2 and depending on viral entry during JE progression. Ultimately, this dysregulated CNS accumulation of peripheral innate immune cells may exacerbate JE progression.
Another intriguing result in this study was that CNS infiltration of Ly‐6Chi monocytes from peripheral sites is dominantly governed by CCL2 produced from HSC‐derived leucocytes, exacerbating JE progression. A similar study demonstrated the dominant role of CCL2 produced by HSC‐derived leucocytes in recruiting Ly‐6Chi monocytes into inflamed sites in mucosal infection with herpes simplex virus type 1.55 CCL2 is secreted by a variety of cell types, including endothelial, fibroblast, epithelial, smooth muscle, mesangial, astrocytic, monocytic, and microglial cells, either constitutively or after induction by oxidative stress, cytokines, or growth factors.41 CCL2 production can also be induced by stimulation with type IFNs (IFN‐I) alone through an IFN‐responsive element in the CCL2 upstream promoter.56 Hence, pro‐inflammatory cytokines and IFN‐I produced by a facilitated inflammatory reaction in the CNS of CCL2‐ablated recipients could affect peripheral leucocytes derived from HSCs. This promotes CCL2 production by HSC‐derived leucocytes and subsequently facilitates recruitment of Ly‐6Chi monocytes to the CNS. This speculation is based on results that decipher the detailed pathway to establish orchestrated mobilization of Ly‐6Chi monocytes through CCL2 produced from HSC‐derived leucocytes.55 However, it was interesting that CCL2 produced from tissue‐resident cells in the CNS restrict the progression of JE because WT recipients of CCL2 KO BM donor cells (KO‐WT) exhibited comparable resistance with the WT‐WT BM chimeric model. Considering that CCL2 produced by resident microglia and glia cells causes the recruitment of TNF‐ and inducible nitric oxide synthase‐expressing macrophages and myeloid dendritic cells,57 it is likely that CCL2 produced by resident cells may contribute to control viral dissemination in the CNS. Collectively, although the detailed role of CCL2 produced from HSC‐derived leucocytes in CNS recruitment of Ly‐6Chi monocytes remains to be defined, our results provide insights into the role of CCL2 produced from HSC‐derived leucocytes in exacerbating JE progression.
Enhanced production of IFN‐γ by NK cells and antigen‐specific Th1 CD4+ T cells is believed to play a role in controlling viral replication in extraneural tissues and the CNS during JE.35, 36 In addition, the cytolytic function of infected target cells by antigen‐specific CD8+ T cells is thought to play a crucial role in recovery from JE.37 Our data are inconsistent with these notions because CCR2‐ablated mice that showed enhanced resistance to JE displayed reduced responses of NK and antigen‐specific CD4+ CD8+ T cells. Although we did not define why CCR2‐ablated mice exhibited reduced T‐cell responses, such reduced adaptive T‐cell responses may be derived from decreased antigen burden in CCR2‐ablated mice. Because NK‐cell‐depleted mice show changes in viral burden or survival,58 our data support that NK cell responses do not significantly contribute to host survival. This notion is also consistent with the absence of a protective role of NK cells and immune escape from NK cell attack by up‐regulation of MHC I in flavivirus infection models.59, 60 Therefore, it is unlikely that IFN‐γ production and cytolytic function by NK cells is dominant in regulating JE progression. Adaptive CD4+ and CD8+ T‐cell responses, which require time to develop, are also unlikely to play a role in regulating early viral replication in extraneural and neural tissues. In general, infected CCR2 KO and CCL2 KO mice began showing neurological signs, including paresis and rigidity, from 4 dpi before protective adaptive immune responses are developed. Hence, mice surviving at 7 dpi were used to determine adaptive CD4+ and CD8+ T‐cell responses specific for JEV antigen. In conclusion, our data demonstrate that the contrasting susceptibility of CCR2‐ and CCL2‐ablated mice to JE progression probably depends on CNS infiltration of Ly‐6Chi monocytes. However, our data did not provide defined results indicating how the ablation of a single chemokine CCL2 induced highly increased expression of CXC and CC chemokines in the CNS, resulting in CNS infiltration of Ly‐6Chi monocytes and enhanced susceptibility to JE. Although our data showed that CCL2 produced from HSC‐derived leucocytes plays an important role in CNS infiltration of Ly‐6Chi monocytes, the detailed cascade pathway to induce CNS infiltration of Ly‐6Chi monocytes in CCL2‐ablated mice remains to be investigated at the molecular and cellular levels.
Authors' contributions
JHK and AMP designed and performed the experiments, analysed the results, drafted the figures and manuscript. JYC, SBK, EU and FMAH participated in experiments, analysed the results and drafted the figures. SYP, JHL and KK contributed to reagent/materials/analysis tools and provided critical conceptual guidance. SKE designed the study, interpreted the data and edited the manuscript.
Disclosures
There are no financial conflicts of interest to declare.
Acknowledgements
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MISP) (2013R1A4A1069486). The funder had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript. We thank Dr. Yoon‐Young Choi, Center for University Research Facility (CURF) at Chonbuk National University, for sorting and analysing cells by FACS Aria.
References
- 1. Le Flohic G, Porphyre V, Barbazan P, Gonzalez JP. Review of climate, landscape, and viral genetics as drivers of the Japanese encephalitis virus ecology. PLoS Negl Trop Dis 2013; 7:e2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daep CA, Munoz‐Jordan JL, Eugenin EA. Flaviviruses, an expanding threat in public health: focus on dengue, West Nile, and Japanese encephalitis virus. J Neurovirol 2014; 20:539–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Centers for Disease Control and Prevention . Japanese encephalitis surveillance and immunization – Asia and the Western Pacific, 2012. MMWR Morb Mortal Wkly Rep 2013; 62:658–62. [PMC free article] [PubMed] [Google Scholar]
- 4. Ghosh D, Basu A. Japanese encephalitis – a pathological and clinical perspective. PLoS Negl Trop Dis 2009; 3:e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim SB, Choi JY, Kim JH, Uyangaa E, Patil AM, Park SY et al Amelioration of Japanese encephalitis by blockage of 4‐1BB signaling is coupled to divergent enhancement of type I/II IFN responses and Ly‐6C(hi) monocyte differentiation. J Neuroinflammation 2015b; 12:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dubot‐Peres A, Sengvilaipaseuth O, Chanthongthip A, Newton PN, de Lamballerie X. How many patients with anti‐JEV IgM in cerebrospinal fluid really have Japanese encephalitis? Lancet Infect Dis 2015; 15:1376–7. [DOI] [PubMed] [Google Scholar]
- 7. Center for Disease Control . West Nile activity – human disease cases reported. 2005–2009 (2009) Available at: http://www.cdc.gov/ncidod/dvbid/westnile/sur&control.htm. (Accessed: 4th September 2014).
- 8. Chen CJ, Ou YC, Lin SY, Raung SL, Liao SL, Lai CY et al Glial activation involvement in neuronal death by Japanese encephalitis virus infection. J Gen Virol 2010; 91:1028–37. [DOI] [PubMed] [Google Scholar]
- 9. Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P et al Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 2007; 55:483–96. [DOI] [PubMed] [Google Scholar]
- 10. Michlmayr D, Lim JK. Chemokine receptors as important regulators of pathogenesis during arboviral encephalitis. Front Cell Neurosci 2014; 8:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hosking MP, Lane TE. The role of chemokines during viral infection of the CNS. PLoS Pathog 2010; 6:e1000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 2012; 12:623–35. [DOI] [PubMed] [Google Scholar]
- 13. Han YW, Choi JY, Uyangaa E, Kim SB, Kim JH, Kim SB et al Distinct dictation of Japanese encephalitis virus‐induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PLoS Pathog 2014; 10:e1004319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med 2005; 202:1087–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA et al CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 2006; 203:35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Michlmayr D, McKimmie CS, Pingen M, Haxton B, Mansfield K, Johnson N et al Defining the chemokine basis for leukocyte recruitment during viral encephalitis. J Virol 2014; 88:9553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bose S, Cho J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch Pharm Res 2013; 36:1039–50. [DOI] [PubMed] [Google Scholar]
- 18. Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol 2011; 186:471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B et al Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med 2008; 205:2319–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hammond MD, Taylor RA, Mullen MT, Ai Y, Aguila HL, Mack M et al CCR2+ Ly6Chi inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J Neurosci 2014; 34:3901–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Held KS, Chen BP, Kuziel WA, Rollins BJ, Lane TE. Differential roles of CCL2 and CCR2 in host defense to coronavirus infection. Virology 2004; 329:251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benjamin PC, William AK, Thomas EL. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol 2001; 167:4585–92. [DOI] [PubMed] [Google Scholar]
- 23. Winter PM, Dung NM, Loan HT, Kneen R, Wills B, le Thu T et al Proinflammatory cytokines and chemokines in humans with Japanese encephalitis. J Infect Dis 2004; 190:1618–26. [DOI] [PubMed] [Google Scholar]
- 24. Singh A, Kulshreshtha R, Mathur A. Secretion of the chemokine interleukin‐8 during Japanese encephalitis virus infection. J Med Microbiol 2000; 49:607–12. [DOI] [PubMed] [Google Scholar]
- 25. Gupta N, Lomash V, Rao PV. Expression profile of Japanese encephalitis virus induced neuroinflammation and its implication in disease severity. J Clin Virol 2010; 49:4–10. [DOI] [PubMed] [Google Scholar]
- 26. Kim JH, Choi JY, Kim SB, Uyangaa E, Patil AM, Han YW et al CD11chi dendritic cells regulate Ly‐6Chi monocyte differentiation to preserve immune‐privileged CNS in lethal neuroinflammation. Sci Rep 2015a; 5:17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M et al Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat Med 2005; 11:1118–24. [DOI] [PubMed] [Google Scholar]
- 28. Chattopadhyay PK, Yu J, Roederer M. Live‐cell assay to detect antigen‐specific CD4+ T‐cell responses by CD154 expression. Nat Protoc 2006; 1:1–6. [DOI] [PubMed] [Google Scholar]
- 29. Terry RL, Getts DR, Deffrasnes C, van Vreden C, Campbell IL, King NJ. Inflammatory monocytes and the pathogenesis of viral encephalitis. J Neuroinflammation 2012; 9:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330:841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med 2000; 192:1075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 2000; 192:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ben‐Nathan D, Huitinga I, Lustig S, Van Rooijen Kobiler D. West Nile virus neuroinvasion and encephalitis induced by macrophage depletion in mice. Arch Virol 1996; 141:459–69. [DOI] [PubMed] [Google Scholar]
- 34. Iijima N, Mattei LM, Iwasaki A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc Natl Acad Sci U S A 2011; 108:284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Larena M, Regner M, Lobigs M. Cytolytic effector pathways and IFN‐γ help protect against Japanese encephalitis. Eur J Immunol 2013; 43:1789–98. [DOI] [PubMed] [Google Scholar]
- 36. Kumar P, Sulochana P, Nirmala G, Chandrashekar R, Haridattatreya M, Satchidanandam V. Impaired T helper 1 function of nonstructural protein 3‐specific T cells in Japanese patients with encephalitis with neurological sequelae. J Infect Dis 2004; 189:880–91. [DOI] [PubMed] [Google Scholar]
- 37. Larena M, Regner M, Lee E, Lobigs M. Pivotal role of antibody and subsidiary contribution of CD8+ T cells to recovery from infection in a murine model of Japanese encephalitis. J Virol 2011; 85:5446–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chensue SW, Warmington KS, Ruth JH, Sanghi PS, Lincoln P, Kunkel SL. Role of monocyte chemoattractant protein‐1 (MCP‐1) in Th1 (mycobacterial) and Th2 (schistosomal) antigen‐induced granuloma formation: relationship to local inflammation, Th cell expression, and IL‐12 production. J Immunol 1996; 157:4602–8. [PubMed] [Google Scholar]
- 39. Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nat Immunol 2001; 2:102–7. [DOI] [PubMed] [Google Scholar]
- 40. Zisman DA, Kunkel SL, Strieter RM, Tsai WC, Bucknell K, Wilkowski J et al MCP‐1 protects mice in lethal endotoxemia. J Clin Invest 1997; 99:2832–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein‐1 (MCP‐1): an overview. J Interferon Cytokine Res 2009; 29:313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rustenhoven J, Scotter EL, Jansson D, Kho DT, Oldfield RL, Bergin PS et al An anti‐inflammatory role for C/EBPδ in human brain pericytes. Sci Rep 2015; 5:12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 2006; 7:311–7. [DOI] [PubMed] [Google Scholar]
- 44. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP et al Critical roles for CCR2 and MCP‐3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 2007; 117:902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neal LM, Knoll LJ. Toxoplasma gondii profilin promotes recruitment of Ly6Chi CCR2+ inflammatory monocytes that can confer resistance to bacterial infection. PLoS Pathog 2014; 10:e1004203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim B, Sarangi PP, Lee Y, Deshpande Kaistha S, Lee S, Rouse BT. Depletion of MCP‐1 increases development of herpetic stromal keratitis by innate immune modulation. J Leukoc Biol 2006; 80:1405–15. [DOI] [PubMed] [Google Scholar]
- 47. Dessing MC, van der Sluijs KF, Florquin S, van der Poll T. Monocyte chemoattractant protein 1 contributes to an adequate immune response in influenza pneumonia. Clin Immunol 2007; 125:328–36. [DOI] [PubMed] [Google Scholar]
- 48. Boivin N, Menasria R, Gosselin D, Rivest S, Boivin G. Impact of deficiency in CCR2 and CX3CR1 receptors on monocytes trafficking in herpes simplex virus encephalitis. J Gen Virol 2012; 93:1294–304. [DOI] [PubMed] [Google Scholar]
- 49. Cassol E, Alfano M, Biswas P, Poli G. Monocyte‐derived macrophages and myeloid cell lines as targets of HIV‐1 replication and persistence. J Leukoc Biol 2006; 80:1018–30. [DOI] [PubMed] [Google Scholar]
- 50. Drevets DA, Dillon MJ, Schawang JS, Van Rooijen N, Ehrchen J, Sunderkotter C et al The Ly‐6Chigh monocyte subpopulation transports Listeria monocytogenes into the brain during systemic infection of mice. J Immunol 2004; 172:4418–24. [DOI] [PubMed] [Google Scholar]
- 51. Trujillo JA, Fleming EL, Perlman S. Transgenic CCL2 expression in the central nervous system results in a dysregulated immune response and enhanced lethality after coronavirus infection. J Virol 2013; 87:2376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poo YS, Nakaya H, Gardner J, Larcher T, Schroder WA, Le TT et al CCR2 deficiency promotes exacerbated chronic erosive neutrophil‐dominated chikungunya virus arthritis. J Virol 2014; 88:6862–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schouppe E, Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA. Modulation of CD8+ T‐cell activation events by monocytic and granulocytic myeloid‐derived suppressor cells. Immunobiology 2013; 218:1385–91. [DOI] [PubMed] [Google Scholar]
- 54. Ariel A, Serhan CN. New lives given by cell death: macrophage differentiation following their encounter with apoptotic leukocytes during the resolution of inflammation. Front Immunol 2012; 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Uyangaa E, Kim JH, Patil AM, Choi JY, Kim SB, Eo SK. Distinct upstream role of type I IFN signaling in hematopoietic stem cell‐derived and epithelial resident cells for concerted recruitment of Ly‐6chi monocytes and NK cells via CCL2‐CCL3 cascade. PLoS Pathog 2015; 11:e1005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Conrady CD, Zheng M, Mandal NA, van Rooijen N, Carr DJ. IFN‐α‐driven CCL2 production recruits inflammatory monocytes to infection site in mice. Mucosal Immunol 2013; 6:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dogan RNE, Elhofy A, Karpus WJ. Production of CCL2 by central nervous system cells regulates development of murine experimental autoimmune encephalomyelitis through the recruitment of TNF‐ and iNOS‐expressing macrophages and myeloid dendritic cells. J Immunol 2008; 180:7376–84. [DOI] [PubMed] [Google Scholar]
- 58. Lobigs M, Mullbacher A, Wang Y, Pavy M, Lee E. Role of type I and type II interferon responses in recovery from infection with an encephalitic flavivirus. J Gen Virol 2003; 84:567–72. [DOI] [PubMed] [Google Scholar]
- 59. Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol 2006; 80:119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lobigs M, Müllbacher A, Regner M. MHC class I up‐regulation by flaviviruses: immune interaction with unknown advantage to host or pathogen. Immunol Cell Biol 2003; 81:217–23. [DOI] [PubMed] [Google Scholar]