

Summary
Defects in rapid clearance of apoptotic cells lead to an accumulation of dead cells (late apoptotic or secondary necrotic cells), which results in an aberrant immune response. However, little is known about whether and how macrophages (Mφs) cooperate with dendritic cells (DCs) in the presentation of dead‐cell‐associated antigens in this process. By transferring high numbers of dead cells to mimic a failure of apoptotic cell clearance in vivo, we found that Mφs and neutrophils were the predominant phagocytes in the uptake of dead cells in the spleen. Moreover, both Mφs and DCs were required for an optimal CD4+ T‐cell response triggered by dead‐cell‐associated antigens. Importantly, although Mφs alone had a poor capacity for antigen presentation, they could transfer phagocytosed antigens to DCs for potent antigen presentation to enhance T‐cell responses. Finally, we found that exosomes released from Mφs acted as a transmitter to convey antigens to DCs partially in a ceramide‐dependent manner, since treatment with the neutral sphingomyelinase inhibitor GW4869 and spiroepoxide resulted in a significant reduction of T‐cell proliferation in vitro and in vivo. These findings point to a novel pathway of cross‐talk between Mφs and DCs, which will be helpful to explain possible mechanisms for autoimmune diseases characterized by increased rates of apoptosis.
Keywords: antigen transfer, dead‐cell‐associated antigen, dendritic cells, exosomes, macrophages
Abbreviations
- BSA
bovine serum albumin
- CLL
clodronate liposome
- DCs
dendritic cells
- ESCRT
endosomal sorting complex required for transport
- FBS
fetal bovine serum
- Mφs
macrophages
- nSMase 2
sphingomyelinase 2
- OVA
ovalbumin
- SLE
systemic lupus erythematosus
Introduction
The mutual interplay of immune cells has been suggested to play a critical role in optimal innate and adaptive immunity.1, 2, 3, 4, 5 In recent years, much attention has been paid to the role of cross‐talk between macrophages (Mφs) and dendritic cells (DCs) in maintaining homeostasis or inducing a protective immune response under physiological or pathological conditions, respectively.6, 7, 8 During the development of multicellular organisms, a controlled and programmed form of cell death is indispensable. Therefore, phagocytes must constantly remove apoptotic cells for normal homeostasis and tissue turnover, as the conversion of excessive apoptotic cells to secondary necrotic dead cells would induce inflammation or autoimmunity.9, 10
Both DCs and Mφs contribute to the uptake of apoptotic cells and dead cells.11 Interestingly, communication between these two cell types has been suggested to be important in responding to early apoptotic cells to generate a suppressive microenvironment.12, 13 However, failure to immediately clear apoptotic cells leads to an accumulation of dead cells, which can be presented by DCs to autoreactive lymphocytes to induce autoimmune diseases such as systemic lupus erythematosus.14 Hence, DCs are suggested to be critically involved in the pathogenesis of autoimmunity by presenting dead‐cell‐associated antigens. Like DCs, Mφs also take up dead cells but even more efficiently15; however, few studies have investigated the role of dying‐cell‐derived antigen presentation on Mφs, especially presentation by MHC II molecules, although Mφs have been shown to dominate anti‐tumour immunity by cross‐presenting dead‐cell‐associated antigens to initiate a CD8+ T‐cell immune response.16 Hence, investigating whether dead cells activate Mφs to trigger a CD4+ T‐cell immune response would be of great interest, and it also raises questions of whether and how Mφs process dead‐cell‐associated antigens and subsequently present them to CD4+ T cells. In fact, the complementing specialties of Mφs (antigen uptake) and DCs (antigen presentation) prompt an interesting possibility that these two cell types may be collaborating in the induction of aberrant T‐cell responses to dead‐cell‐associated antigens, which might be responsible for development of some autoimmune diseases. However, to date, whether and how these two cell types cross‐talk to prime the CD4+ T‐cell response has not been definitively elucidated in dead‐cell‐induced immune responses.
Extracellular vesicles have been demonstrated to be involved in numerous physiological processes and act as mediators of cellular cross‐talk in autoimmunity.17 These vesicles, which vary from 30 to 100 nm in size, are known as exosomes.18 However, whether exosomes participate in cross‐talk between DCs and Mφs in some autoimmune diseases induced by excessive apoptotic cells remains unclear. In this study, we injected mice intravenously (i.v.) with late apoptotic cells to mimic the failure of immediate clearance of apoptotic cells, leading to an accumulation of dead cells, and observed whether and how Mφs and DCs cooperate with each other in the CD4+ T‐cell response to dead‐cell‐associated antigens. Our results showed that splenic Mφs and neutrophils were predominant phagocytes in the uptake of dead cells, and both Mφs and DCs were required for augmentation of the CD4+ T‐cell response in this process. Importantly, after phagocytosis of antigens, exosomes containing antigens from Mφs were secreted partially in a ceramide‐dependent manner and acted as transmitters to convey antigens to DCs, leading to an enhanced T‐cell response. Here, we provide evidence that the exosomes released by Mφs taking up dead‐cell‐associated antigens acted as a vehicle between DCs and Mφs, which may be responsible for the disordered immune response resulting in the induction of autoimmunity.
Materials and methods
Mice
C57BL/6 (CD45.2) mice, C57BL/6‐Tg (Itgax‐cre,‐EGFP) mice, ovalbumin (OVA) ‐specific MHC II‐restricted T‐cell receptor‐transgenic mice (OT‐II mice), as well as transcription factor Batf3‐deficient (Batf3 −/−) mice, were purchased from the Jackson Laboratory (Bar Harbor, ME). OT‐II × CD45.1 F1 mice were prepared by crossing OT‐II mice with B6.SJL‐Ptprc a Pep3 b/BoyJ mice (congenic CD45.1 mice on a B6 background). These mice were bred and housed in a specific pathogen‐free barrier unit. All experiments using mice were in accordance with requirements and regulations of the Institutional Animal Care and Use Committee.
Cell preparations
Splenic single‐cell suspensions were prepared by digestion for 30 min at 37° with 1 mg/ml collagenase type IV (Gibco, Grand Island, USA). Splenocytes were resuspended in PBS (SH30256.01B; Thermo Fisher Scientific, South Logan, Utah) for subsequent experiments. To prepare dead‐cell‐associated antigens, thymocytes from CD45.2 or CD45.1 mice were resuspended in RPMI‐1640 medium (SH30809.01B; Thermo Fisher Scientific) containing 1% BSA (Sigma‐Aldrich, St Louis, MO) and 20 mg/ml OVA (Sigma‐Aldrich) protein and then irradiated with 40 Gy X‐ray, followed by 24 hr of culture at 37° to induce late apoptotic cells (OVA/cells).16 Dead cells were washed extensively before injection to remove the soluble free OVA.
Tracking of circulating dead cells
Thymocytes from CD45.1 mice were labelled with 2·5 μm CFSE (Cell Tracker Green; Molecular Probes, Eugene, OR) or 10 μm eFluor‐450 (Cell Tracker; Molecular Probes) for 15 min at room temperature, and then late apoptosis was induced as above. Dead cells (4 × 107) in 200 μl PBS were injected i.v. into CD45.2 mice. Spleens were resected 1 hr after injection, and cryosections were prepared for staining with anti‐CD11c clone N418 (45‐0114‐82; eBioscience Inc., San Diego, CA), anti‐F4/80 clone BM8 (17‐4801‐82; eBioscience), anti‐CD169 clone MOMA‐1 (MCA947F; AbD Sectero and eBioscience), anti‐MARCO clone ED31 (MCA1849FT; AbD Sectero and eBioscience, inc, San Diego, CA) and anti‐B220 clone RA3‐6B2 (45‐0452‐82; eBioscience) for confocal microscopic observation. Splenic cells were also stained with anti‐CD45.1 clone A20 (47‐0453‐82; eBioscience), anti‐CD45.2 clone 104 (45‐0454‐82; eBioscience), anti‐CD11b clone M1/70 (12‐0112‐82; eBioscience), anti‐F4/80, anti‐Gr‐1 clone RB6‐8C5 (45‐5931‐80; eBioscience), and anti‐Ly6C clone HK1.4 (48‐5932‐82; eBioscience) for assessment by flow cytometry.
Clodronate liposome preparation and injection
Clodronate liposomes (CLLs) were prepared as described elsewhere.19 Briefly, 1% of the dichloromethylene‐bisphosphonate (Cl2MBP) could be encapsulated in the liposomes, and the final Cl2MBP–liposome suspension (4 ml) contained approximately 20 mg of Cl2MBP. In our study, to deplete splenic DCs and/or Mφs, C57BL/6 mice were i.v. injected with 200 μl of CLL per mouse (i.e. 50 mg/kg) or an equivalent amount of PBS‐loaded control liposomes. The depletion efficiency and the rate of turnover were tested by FACS at the indicated time‐points.
Immunohistochemistry
Spleens were obtained from mice administered with CLL or not, followed by injection with or without CFSE‐labelled dead cells. Cryosections (6‐μm thickness) of the spleen were fixed in cold acetone and blocked with 1% anti‐FcγRII/III clone 93 (14‐0161‐86; eBioscience). Sections were stained with the corresponding monoclonal antibodies, mounted with FluorSave (Calbiochem, Beyotime, Shanghai, China) and observed by fluorescence microscopy (Leica SD AF).
Antigen transfer assay in vitro
C57BL/6 mice were injected i.v. with 4 × 107 eFluor450‐labelled (Ag‐Mφ) or unlabelled (no‐Ag‐Mφ) dead cells. One hour later, 3 × 105 splenic F4/80hi CD11bint Mφs were sorted and incubated with 3 × 105 DCs purified from naive C57BL/6‐Tg (Itgax‐cre, ‐EGFP) mice (naive DC) for 1, 3 or 5 hr. Live CD11c‐GFP+ DCs positive for eFluor‐450 were considered to have internalized dead‐cell‐associated antigens from Mφs and were analysed by FACS at various time‐points.
Antigen‐specific CD4+ T‐cell proliferation assay in vivo and in vitro
For the in vivo assay, eFluor‐450 labelled CD4+ Vβ5+ Vα2+ T cells from OT‐II × CD45.1 F1 mice were adoptively transferred i.v. into CD45.2 mice followed by injection with 4 × 107 OVA/cells in 200 μl of PBS after 24 hr. Three days later, the splenic cells were stained with anti‐CD45.1, anti‐CD4 clone GK1.5 (47‐0041‐82; eBioscience), anti‐Vβ5+ clone MR9‐4 (139506; Biolegend, San Diego, CA) and anti‐Vα2+ clone B20.1 (47‐5812‐80; eBioscience) antibodies to examine antigen‐specific CD4+ T‐cell proliferation by FACS. For the in vitro assay, F4/80hi CD11bint Mφs or CD11chi DCs purified from C57BL/6 mice injected with OVA/cells or not were co‐cultured for 3 days with eFluor‐450‐labelled CD4+ Vβ5+ Vα2+ OT‐II T cells in 96‐well round‐bottomed plates. Proliferation was assessed by calculation of the dilution of eFluor‐450 or ki67 clone So1A15 (25‐5698‐82; eBioscience) staining by FACS.
Transwell assay
Approximately 1 × 105 naive DCs from naive mice and 3 × 105 F4/80hi CD11bint Mφs from mice injected with OVA/cells or not in 100 μl culture medium were seeded in the lower or upper chamber of 96‐well 0·4‐μm Transwell plates, and 2 × 105/100 μl OT‐II T cells were added to the lower chamber. Three days later, CD4+ T cells were harvested, and the proliferation of OT‐II T cells was analysed by FACS.
Exosome preparation
The mouse Mφ cell line RAW264.7 was maintained at 37° in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM)/Low Glucose (SH30021.01B; Thermo Fisher Scientific) supplemented with 10% exosome‐free fetal bovine serum (FBS), 25 mm HEPES, 100 units/ml penicillin and 100 μg/ml streptomycin. The FBS used in the cell culture media for exosome isolation was ultracentrifuged at 100 000 g for 16 hr to remove any contaminating exosomes. RAW264.7 cells (15 × 106) were treated with GW4869 or DMSO in a 175‐cm2 flask for 48 hr. The collected supernatants were differentially centrifuged at 300 g for 10 min, 1200g for 20 min and 10 000 g for 30 min to remove whole cells and cell debris. The final supernatant was then ultracentrifuged at 100 000 g for 60 min.20 The resulting pellets were treated with 0·08% pronase or not (Merck‐Calbiochem) in PBS for 20 min, ultracentrifuged, treated the same way again, and 10 ml exosome‐free FBS was added to quench the pronase activity, then washed with 10 ml ice‐cold PBS and centrifuged at 100 000 g for 60 min twice.21 The resulting exosome pellets were resuspended in DMEM containing exosome‐free 10% FBS for functional assays, or the exosome fractions were resuspended in 100 μl PBS and measured for its protein content using the Micro BCA protein assay kit (Shanghai BoCai, Shanghai, China). Exosomes from RAW264.7 cells treated or not with OVA‐FITC were termed EXO or EXN, respectively.
Western blot analysis
Exosomes (40 μg) were resuspended in radioimmunoprecipitation assay buffer with protease inhibitors. The suspension was mixed with Laemmli buffer, heated at 95° for 5 min. Gels for SDS–PAGE were calibrated with Precision Plus protein standards. Anti‐CD9 clone EPR2949 (1 : 500, Abcam, Cambridge, UK), anti‐CD63 clone MX‐49.129.5 (1 : 200, Abcam), anti‐CD81 clone EPR4244 (1 : 500, Abcam), anti‐OVA clone 6C8 (1 : 500, Abcam), and anti‐β‐actin clone AC‐15 (1 : 2000, Sigma‐Aldrich) were used as primary antibodies. Secondary antibodies (horseradish peroxidase‐labelled anti‐mouse or anti‐rabbit) were used at a dilution of 1 : 2000. Bound antibodies were visualized by chemiluminescence using the ECL Plus Western Blotting detection system, and luminescent images were analysed on a LuminoImager.
Inhibition of exosome secretion by GW4869 and spiroepoxide
The inhibitor GW4869 (Sigma‐Aldrich) and spiroepoxide (Santa Cruz Biotechnology, Inc., Dallas, TX) were used to block the secretion of exosomes.22, 23, 24 For in vitro experiments, 10 μm GW4869 or 5 μm spiroepoxide was added into the culture system individually or together. In some experiments, DCs were stained with anti‐CD80 clone 16‐10‐A1 (12‐0801‐82; eBioscience), anti‐CD86 clone GL1 (12‐0862‐83; eBioscience) and anti‐MHC II clone M5/114.15.2 (12‐5321‐83; BioLegend). T cells were stained with anti‐CD25 clone PC61 (102038; BioLegend), anti‐CD44 clone IM7 (45‐0441‐82; eBioscience), anti‐CD62L clone MEL‐14 (12‐0621‐83; eBioscience) and anti‐CD69 clone H1.2F3 (11‐0691‐82; eBioscience). For in vivo experiments, 1·25 mg/kg GW4869 or spiroepoxide was intraperitoneally injected into C57BL/6 mice individually or together once a day for 6 days.25
Statistical analysis
Data are presented as the mean ± SD. A two‐tailed Student's t‐test was used for data analysis. The null hypothesis was rejected, and differences were assessed to be significant at a P value of < 0·05.
Results
Mφs and neutrophils are predominant phagocytes for uptake of dead cells in the spleen
Dead cells were prepared, and detection by FACS showed that > 70% of cells were AnnexinV+ 7‐AAD+ (Fig. 1a), which indicated that they were late‐stage apoptotic cells or secondary necrotic cells, as described previously.26 To observe the distribution of dead cells in vivo, we injected i.v. CFSE‐labelled dead cells into C57BL/6 mice. By immunohistofluorescence analysis, we found that CFSE+ cells were extensively located in the red pulp and white pulp, as indicated by CD11c, F4/80 and B220 staining, as well as in the marginal zone (MZ) as shown by CD169, MARCO and B220 staining (Fig. 1b). This distribution pattern of dead cells in the spleen was similar to that of early apoptotic cells, which were shown to localize to the MZ and red pulp primarily upon entering the spleen from circulation in previous studies.27, 28 To examine which cell type could take up dead cells in the spleen, we first defined distinct splenic cell populations that might carry out phagocytosis, by using a strategy described previously.29, 30 As shown in the left panel of Fig. 1(c), at least five populations in the spleen were identified by using antibodies against CD11c and CD11b, which were further confirmed by the use of other antibodies against F4/80, MHC II as shown in the right panel of Fig. 1(c). Here, CD11chi cells were subdivided into CD11chi CD11blo myeloid DCs (gate: R1) and CD11chi CD11bneg to low lymphoid DCs (gate: R2); meanwhile, CD11cint CD11b− F4/80lo cells were considered plasmacytoid DCs (gated as R3). The fourth population was CD11bint F4/80hi MHCIIint red pulp Mφs (gate: R4), and Ly6C+ Gr‐1+ cells were neutrophils and monocytes (gate: R5). Furthermore, these two cell types in Gate R5 were differentiated by antibodies against CD11b, Ly6C and Gr‐1 into Gr‐1hi CD11bhi neutrophils and Ly6Chi CD11bint monocytes (Fig. 1d). Hence, we found that CFSE+ cells were preferentially phagocytosed by F4/80hi CD11bint Mφs and CD11bhi Gr‐1hi neutrophils at the ratios of 33 ± 6·5% and 26·2 ± 5·2%, respectively, whereas < 5% of CFSE+ dead cells were taken up by lymphoid DCs (Fig. 1e). To clarify whether CFSE+ cells were internalized into the cytosol of phagocytes and did not remain on the surface of phagocytes, we sorted CD45.2+ CFSE+ cells and observed them under a confocal microscope. As shown in Fig. 1(f), CFSE+ fragments were indeed localized in the cytosol of sorted cells. Hence, the selectivity of phagocytosis suggested that Mφs and neutrophils were the relative predominant phagocytes engulfing dead cells in the spleen, although multiple cell types were involved in this process.
Figure 1.

Circulating dead cells were dominantly taken up by macrophages (Mφs) and neutrophils in the spleen. (a) Thymocytes were irradiated with 40 Gy X‐ray followed by culture at 37° for 24 hr. Treated (right panel) and untreated (left panel) cells were stained with Annexin V and 7‐AAD to detect late apoptotic cells. (b) CFSE‐labelled late apoptotic cells were injected intravenously into C57BL/6 mice. One hour later, cryosections of spleen were stained with anti‐CD11c (red), anti‐F4/80 (red), anti‐CD169 (red) or anti‐MARCO (red) antibody, and anti‐B220 (blue) staining was used as the background. Co‐localization of splenic Mφs or dendritic cells (DCs) with dead cells was observed under a microscope. Scale bars, 50 μm. A higher magnification on a portion of images for the co‐localization was shown (200 ×). (c) Splenic cells were stained with anti‐CD11c, anti‐CD11b, anti‐MHC II, anti‐F4/80, anti‐Ly6C and anti‐Gr‐1. Phenotypic definition of splenic cell populations depended on the expression of these surface markers. Histograms represent the expression profile for a given marker on cells that belong to the indicated gate. Fluorescence minus one (FMO) control is shown by the dotted histograms. (d) Gate R5 was further analysed by the expression of Ly6C, Gr‐1 and CD11b. (e) CD45.2+ mice were injected intravenously with CFSE‐labelled late apoptotic cells from CD45.1+ mice, whereas control mice received live cells or PBS alone. Splenic cells were prepared 1 hr after injection, stained with the indicated antibodies and analysed by flow cytometry. CD45.2+ CFSE + cells were identified as cells that have taken up dead cells, and their distribution among different immune phagocytic cells was further analysed by CD11b versus CD11c staining. Overlays showed CFSE + cells (blue) and total live CD45.2+ cells (yellow) as indicated. (f) CD45.2+ CFSE + cells were sorted on a FACSAria II instrument and observed under a confocal microscope. Data shown are representative of at least three to five mice in each group corresponding to three independent experiments.
Lack of Mφs affects uptake of dead‐cell‐associated antigens and antigen‐specific CD4+ T‐cell responses
To evaluate whether Mφs were involved in the immune response triggered by dead cell materials, we sought to deplete Mφs by systemic CLL treatment, an approach that has been used to efficiently deplete phagocytes.31, 32 In our study, to prepare an Mφ‐specific depletion window, C57BL/6 mice were treated with CLL or PBS‐loaded control liposomes. Splenic cells were analysed by FACS or by confocal microscopy to dynamically evaluate the deletion rate and turnover time‐point of phagocyte populations. As shown in Fig. 2(a), we found that approximately 50% of myeloid DCs, 90% of lymphoid DCs and 99% of CD11bint F4/80hi Mφs were depleted 1 day after CLL injection. On day 5 after injection of CLL, all DC subsets have repopulated completely, whereas most of the CD11bint F4/80hi Mφs and two MZ‐related Mφ subsets (MZ Mφ and marginal metallophilic Mφ) were still absent (Fig. 2a and b). Here, to better separate DC subsets and macrophages, we analysed them according to the strategy33 (see Supplementary material, Fig S1): CD11chi cells were used as conventional DCs, which were all highly expressed MHC II, then CD11bint F4/80hi cells from the remaining CD11cneg to low cells were used as Mφs. The data suggested that a specific window of Mφ depletion was indeed created. This technique was applied for the following experiments. First, we further confirmed the contribution of Mφs in trapping of dead cells when Mφs were completely depleted on day 5 after administration with CLL. As expected, the ratio of CFSE+ cells in the spleen declined to 0·071% in the Mφ‐depleted spleen compared with control (Fig. 2c), which indicated impaired trapping of antigens in the spleen after selective depletion of Mφs. Given the relative predominant phagocytosis of dead cells by Mφs, it became imperative to examine the function of this population in the immune response induced by dead‐cell‐associated antigens. The data showed that antigen‐specific CD4+ T‐cell proliferation was significantly impaired on day 5 and day 12 after the selective depletion of splenic Mφs by CLL treatment (Fig. 2d and e). Hence, the results demonstrated that impaired antigens trapping in the Mφ‐depleted spleen resulted in a decreased CD4+ T‐cell response, suggesting that splenic Mφs were involved in an optimal CD4+ T‐cell response to dead‐cell‐associated antigens.
Figure 2.

CD4+ T‐cell proliferation induced by dead‐cell‐associated antigens was impaired by macrophage (Mφ) depletion. (a) C57BL/6 mice received clodronate liposome (CLL) or PBS‐loaded control liposomes (PBSL) intravenously (i.v.) in a 200‐μl final volume. Splenic cells were prepared on days 1, 5 and 12 after CLL injection. The disappearance of splenic phagocyte populations and the kinetics of their repopulation were analysed by flow cytometry. (b) The spleen cryosections were stained with anti‐CD169 and anti‐MARCO antibodies specific for marginal metallophilic Mφ (MMM) and marginal zone Mφ (MZM), respectively, at each time‐point as described in (a). Scale bars, 50 μm. (c) CD45.2 mice received CLL or PBSL i.v. in a 200‐μl final volume followed by injection of CFSE‐labelled dead cells from CD45.1 mice after 5 days. One hour later, splenic cells were prepared and stained with the indicated antibodies. The amount of engulfed dead cells was assessed by FACS. CD45.2+ CFSE + cells were identified as cells that have taken up dead cells, and their distribution among F4/80hi macrophages was further analysed by CD11b, CD11c and F4/80 staining. Overlays show F4/80hi CFSE + cells (blue) from CD45.2+ CD11cneg to low CFSE + cells and live CD11cneg to low CD45.2+ cells (yellow) as indicated. (d) CD45.2 mice were injected i.v. with CLL or PBSL in a 200‐μl final volume. eFluor‐450 dye‐labelled CD62L− Vβ5+ CD4+ CD45.1+ OT‐II T cells (1 × 106) were transferred i.v. into CLL‐ or PBSL‐treated mice at different time‐points followed by 4 × 107 ovalbumin (OVA)/cells transferred i.v. after 1 day. Three days later, the proliferation of adoptively transferred CD4+ T cells was detected by flow cytometry. (e) Relative proliferation ratio was further analysed using a column graph. Data are shown as the mean ± SD of three to five mice in each group and are representative of three independent experiments. Two‐tailed Student's t‐test was used for data analysis. *P < 0·05, ***P < 0·0001.
Mφs participate in CD4+ T‐cell response by transferring dead‐cell‐associated antigens to DCs
Dendritic cells are well‐known as the most potent professional antigen‐presenting cells and play a central role in modulating innate and adaptive immunity.34 However, as shown in Fig. 2(d, e), when Mφs were absent from the spleen, the CD4+ T‐cell response to dead‐cell‐associated antigens only reached approximately 50% of the normal level, although DCs had already completely recovered at that time‐point (CLL 5 day). These data indicated that DCs alone were insufficient to induce a fully activated T‐cell expansion reaction to the infusion of dead‐cell‐associated antigens, hinting that splenic Mφs were also involved in optimizing the response. This finding prompted us to further monitor the contribution of DCs in the immune response to dead‐cell‐associated antigens by using transcription factor Batf3 −/− mice, in which splenic CD8α + DCs are absent but the development and function of other DC subsets and helper T cells are quite normal.35 Not surprisingly, we found that the expansion of CD4+ OT‐II T cells was significantly decreased by almost 70% in Batf3 −/− mice compared with that of the control (Fig. 3a). These data demonstrated that DCs were essential for the induction of the CD4+ T‐cell response to dead‐cell‐associated antigens.
Figure 3.
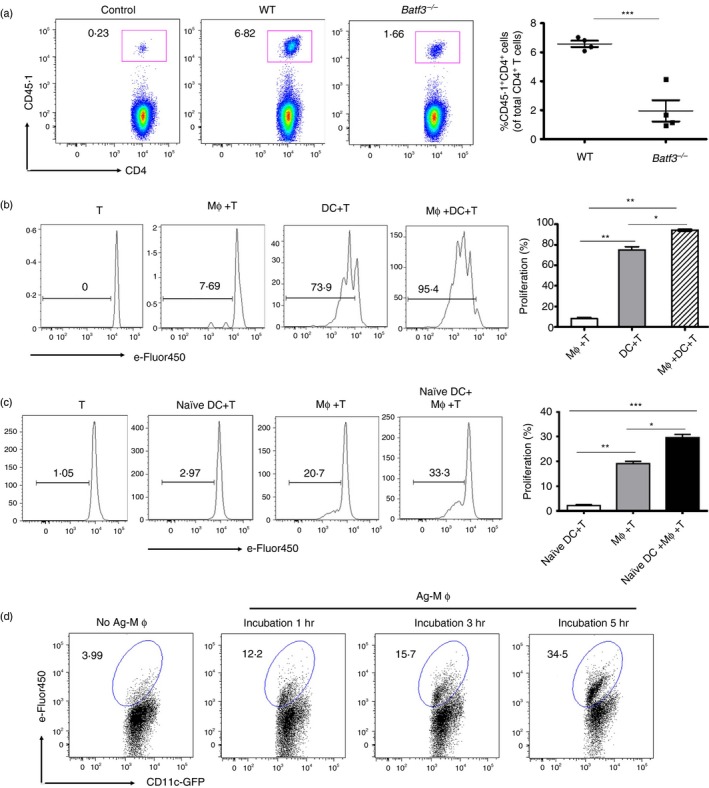
Splenic dendritic cells (DCs) acquired dead‐cell‐associated antigens from macrophages (Mφs) to amplify antigen‐specific CD4+ T‐cell proliferation. (a) Batf3 −/− or C57BL/6 mice were administered intravenously (i.v.) with 2 × 106 CD62L+ Vβ5+ CD4+ CD45.1+ OT‐II T cells, followed by 4 × 107 ovalbumin (OVA)/cells after 1 day. Three days later, the expansion of CD45.1+ CD4+ T cells was analysed by FACS. (b) C57BL/6 mice were i.v. injected with 2 × 107 OVA/cells. One hour later, splenic Mφs and DCs were sorted and co‐cultured with 5 × 105 eFluor‐450‐labelled CD4+ Vβ5+ Vα2+ OT‐II T cells individually or together. Three days later, the proliferation of CD45.1+ CD4+ OT‐II T cells was analysed with diluted eFluor‐450 dye. (c) Proliferation of co‐cultures of 1 × 106 eFluor‐450‐labelled CD45.1+ OT‐II T cells with 2·5 × 105 DCs from naive mice (naive DCs), 2·5 × 105 Mφs from OVA/cells injected mice (Ag‐Mφs) or a combination of naive DCs and Ag‐Mφs. (d) Ag‐Mφs or no‐Ag‐Mφs were incubated with naive DCs. The kinetics of GFP + eFlour‐450+ DCs were determined by FACS. Data are shown as the mean ± SD of three to five mice in each group and are representative of three independent experiments. Two‐tailed Student's t‐test was used for data analysis. *P < 0·05, **P < 0·01, ***P < 0·001.
Based on the contribution of splenic DCs and Mφs in the immune response to dead‐cell‐associated antigens, the question was raised of how Mφs participated in this process, directly presented dead‐cell‐associated antigens to T cells or acted as an accessory tool to support presentation by DCs. To test the first possibility, splenic CD11bint F4/80hi Mφs and CD11chi DCs based on the CD11c, CD11b and F4/80 staining were purified (according to the strategy, see Supplementary material, Fig S1) from mice injected i.v. with dead‐cell‐associated antigens, and co‐cultured with OT‐II T cells separately or together for 72 hr. The results showed that Mφs alone weakly induced antigen‐specific CD4+ T‐cell proliferation compared with DCs (Fig. 3b), which was consistent with the conventional view that Mφs had a poor antigen‐presenting capacity due to their lower expression of MHC II and co‐stimulatory molecules.6 However, when OT‐II T cells were co‐cultured with Mφs and DCs together, the proliferation of T cells increased significantly compared with that with Mφs or DCs separately. Hence, we investigated the second possibility mentioned above, that splenic Mφs might be involved in the augmentation of the T‐cell response to dead‐cell‐associated antigens through the help of DCs. Our results showed that naive DCs from mice without the OVA/cell injection could not stimulate T‐cell proliferation, whereas naive DCs cultured together with Mφs from mice injected with OVA/cells induced stronger proliferation of CD4+ T cells (Fig. 3c). The data further suggested that a cooperation between Mφs and DCs occurred to facilitate a stronger CD4+ T‐cell response to dead cell materials. Previous studies showed that Mφs could transfer antigens to DCs to generate effector T cells in different microenvironments.6, 8 Therefore, we speculated that splenic Mφs might transfer dead‐cell‐associated antigens to DCs to enhance the CD4+ T‐cell response in this process. To test this hypothesis, splenic Mφs from mice injected with eFluor‐450‐labelled dead cells were incubated with naive DCs from naive CD11c‐cre GFP mice for various time intervals. The results showed that the ratio of eFluor‐450+ DCs that contained cell‐associated antigens gradually increased over time (Fig. 3d), which indicated that DCs could acquire antigens from Mφs. In summary, these results demonstrated that Mφs could transfer dead‐cell‐associated antigens to DCs for an enhanced CD4+ T‐cell response, even though Mφs themselves possessed a poor antigen‐presenting ability.
Exosomes containing antigens shed by Mφs act as transmitters between Mφs and DCs
To investigate how splenic Mφs from mice injected with OVA/cells conveyed antigens to splenic DCs from naive mice in vitro, we cultured them with naive OT‐II T cells together or separately. As shown in Fig. 4(a), the combination of naive DCs and Mφs from mice injected with OVA/cells could stimulate the proliferation of OT‐II T cells, which was consistent with results shown in Fig. 3(c). However, we also found that the increased proliferation was significantly reduced when fixed Mφs were used in the co‐culture system, suggesting that Mφs transferred antigens to DCs in a soluble manner. To further confirm this finding, we performed a Transwell assay, and found that antigen–Mφs from mice exposed to dead‐cell‐associated antigens could stimulate OT‐II T‐cell proliferation, which demonstrated that the transfer of antigens by Mφs to DCs was closely related to the ‘secretion’ of soluble factors that contain antigens (Fig. 4b).
Figure 4.

Macrophages (Mφs) transferred antigens to dendritic cells (DCs) through secretion of exosomes. Ovalbumin (OVA)/cells (4 × 107) were intravenously (i.v.) injected into CD45.2+ mice. One hour later, Ag‐Mφs were purified by FACS (a,b,c). Splenic DCs were sorted from naive mice (a,b,c and e). CD62L+ Vβ5+ CD4+ T cells from OT‐II mice were labelled with eFluor‐450 dye (a,b,c and e). (a) Mφs from OVA/cell‐injected mice were treated with (Mφ‐fixed) or without (Mφ) paraformaldehyde and co‐cultured with OT‐II T cells and DCs for 3 days. The proliferation of T cells was analysed by flow cytometry. (b) Ag‐Mφs or no‐Ag‐Mφs were loaded in the upper chamber of a Transwell apparatus, and CD4+ T/DCs were added in the lower chamber. After 3 days of culture, the proliferation of CD4+ T cells was analysed by flow cytometry. (c) RAW264.7 cells were incubated with OVA‐FITC (1·25 mg/ml) at 37° for 40 min, and the uptake of ovalbumin (OVA) ‐FITC by RAW264.7 cells was visualized by confocal microscopy. (d) Exosomal proteins secreted from 15 × 106 RAW264.7 cells incubated with OVA (EXO) or without OVA (EXN) were analysed by immunoblotting for the presence of OVA and CD9. (e) Naive CD45.1+ OT‐II cells were cultured with exosomes or a combination of DCs with exosomes for 3 days, and the proliferation of T cells was evaluated using Ki67 staining. Two‐tailed Student's t‐test was used for data analysis. **P < 0·01, ***P < 0·001.
Recent accumulating evidence suggests that exosomes can function as intercellular transmitters to convey their contents, including proteins, microRNAs or antigens, thereby drawing attention as a cell–cell communication tool.36, 37 Here, we speculated that antigens might have been encapsulated into exosomes, which were subsequently released into the extracellular space and captured by DCs. To address this possibility, we first incubated RAW264.7 mouse Mφ cells with OVA‐FITC and observed that these cells could internalize the fluorescence‐labelled protein (Fig. 4c). Exosomal proteins secreted from RAW264.7 cells incubated with or without OVA were then analysed by immunoblotting. The results demonstrated that the exosome fractions could be verified by the exosome‐specific surface marker CD9. Moreover, OVA also could be detected in these exosomes derived from cell culture supernatants treated with OVA‐FITC (Fig. 4d), suggesting that exosomes released from Mφs included the antigens that were phagocytosed. To further examine whether exosomes containing OVA (EXO) from Mφs could be captured by DCs to induce T‐cell proliferation, we cultured OT‐II T cells with exosomes and DCs, separately or together. As expected, EXO plus DCs induced significant proliferation of OT‐II T cells (Fig. 4e). We also found that EXO alone induced a weak CD4+ T‐cell proliferation, which has been explained by the fact that exosomes could carry MHC II and co‐stimulatory molecules to directly stimulate the T‐cell response.38 In addition, purified exosomes were treated by 0·08% pronase, we excluded the effect of OVA sticking to the outside of exosomes on OVA packages of exosomes (see SUPplementary material, Fig S2a) and T‐cell proliferation in the presence of DCs (see Supplementary material, Fig S2b). Together, these results indicated that the exosomes containing antigens released by Mφs could be captured by DCs, leading to antigen presentation to T cells.
Mφs release exosomes partially through ceramide‐dependent pathway
The ceramide‐dependent secretory pathway has been suggested to be used by certain cell types to transfer tumour‐suppressive microRNA‐containing exosomes to attenuate tumour cell proliferation22 or to transfer exosomes containing antiviral molecules to mediate interferon‐α‐induced antiviral activity.24 Hence, we considered that the ceramide‐dependent pathway might be also involved in the exosome‐mediated antigen transfer machinery in our system. To address this hypothesis, we cultured the RAW264.7 cell line in medium containing a series of concentrations of the neutral sphingomyelinase 2 (nSMase 2) inhibitor, GW4869 and spiroepoxide, which is known to inhibit ceramide biosynthesis. As a result of this treatment, the amount of exosomal protein purified from the supernatant of RAW264.7 cell cultures (each containing the same number of cells) was inhibited in a dose‐dependent manner (Fig. 5a). Furthermore, reductions of exosome marker CD9 and OVA were observed by immunoblotting of exosomes purified from the supernatant of the same number of inhibitor‐treated RAW264.7 cells when compared with DMSO‐treated cells (Fig. 5b). However, levels of OVA and exosomal surface markers such as CD9, were unchanged in the exosomes from inhibitor‐treated cells, as shown by Western blotting when the same amounts of exosomal proteins were loaded (Fig. 5c). These data showed that the treatment of GW4869 or spiroepoxide reduced the amount of exosomes released into the conditioned medium but did not alter the exosomal protein composition, indicating partial involvement of a ceramide‐dependent secretory pathway in the exosome release from antigen‐exposed Mφs.
Figure 5.
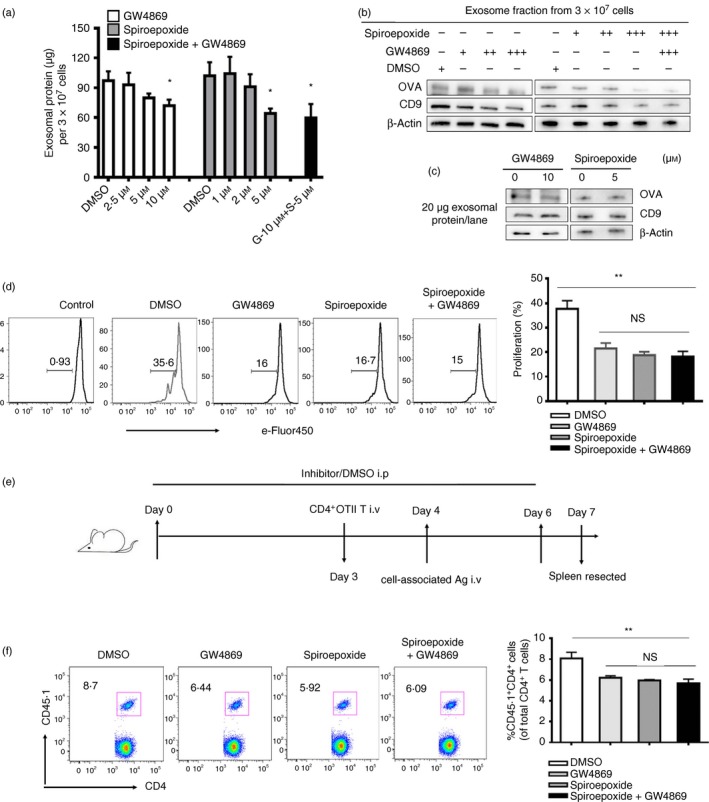
Inhibition of exosome secretion decreased the proliferation of antigen‐specific CD4+ T cells both in vitro and in vivo. (a) RAW264.7 cells were incubated with control (DMSO) or with 2·5 μm (+), 5 μm (++), 10 μm (+++) GW4869 or/and 1 μm (+), 2 μm (++), 5 μm (+++) spiroepoxide, and purified exosomes from supernatants of equal cell numbers of control‐ or inhibitor‐treated macrophages (Mφs) were quantified by a BCA assay. (b) Purified exosomes secreted by equal number of DMSO‐treated or inhibitor‐treated RAW264.7 cells were analysed by immunoblotting for the presence of exosomal marker CD9 and ovalbumin (OVA). (c) Equal amounts of exosomal proteins (quantified by BCA assay) secreted by either control‐ or inhibitor‐treated RAW264.7 cells cultured with OVA were analysed by immunoblotting for the presence of CD9 and OVA protein. (d) eFluor‐450 dye‐labelled naive CD4+ OT‐II T cells, naive dendritic cells (DCs) from naive mice and Mφs from OVA/cell‐injected mice were co‐cultured in the presence of DMSO (solvent), or GW4869 (10 μm) or/and spiroepoxide (5 μm) for 3 days. The proliferation of OT‐II T cells was analysed by flow cytometry. (e) CD45.2 mice were intraperitoneally injected with GW4869 or/and spiroepoxide (1·25 mg/kg) or DMSO once a day for 6 days, and the mice were intravenously administered 2 × 106 CD45.1+ CD4+ Vβ5+ Vα2+ T cells on the fourth day followed by injection of OVA/cells after 1 day. Three days after the OVA/cells injection, proliferation of CD45.1+ CD4+ T cells was analysed by FACS (f). Data are shown as the mean ± SD of three to five mice per group and representative of three independent experiments. Two‐tailed Student's t‐test was used for data analysis. *P < 0·05, **P < 0·01.
Based on the secretory mechanism of exosomes from Mφs, we further examined whether the process of releasing antigen‐carrying exosomes by Mφs was indeed involved in the CD4+ T‐cell response induced by dead‐cell‐associated antigens by using inhibitory functional assays in vitro and in vivo. First, we found that the proliferation of OT‐II T cells decreased significantly if GW4869 or/and spiroepoxide were added to the co‐culture system in vitro (Fig. 5d). Subsequently, we performed the in vivo experiment as schematically depicted in Fig. 5(e) and found that the proliferation of antigen‐specific CD4+ T cells was decreased in mice treated with GW4869 and/or spiroepoxide (Fig. 5f). However, the decrease of T‐cell proliferation was not enlarged with the increase of the dose of inhibitor in vitro (see Supplementary material, Fig S3a) and in vivo (see Supplementary material, Fig S3b), even when the two inhibitors were administered together (Fig. 5). In addition, we also excluded the effect of inhibitor on antigen presentation and T‐cell activation (see Supplementary material, Fig S4). Taken together, these results suggested that Mφs transferred antigens to DCs mediated by antigen‐carrying exosomes at least partially via a ceramide‐dependent pathway.
Discussion
Dendritic cells and Mφs are well‐established as major components of the phagocyte system and play critical roles in the clearance of apoptotic cells to induce immune tolerance and maintain homeostasis.39, 40 However, whether the two cell types also act as primary scavengers for dead cells (late apoptotic or necrotic cells) has been unclear. In our study, we found that dead cells were predominantly taken up by Mφs and neutrophils in vivo, confirming the Mφ‐mediated phagocytosis observed in vitro by a previous study.41 However, the finding was different in that neutrophils were held off to engulf apoptotic cells,14 which may be related to different characteristics of secretory signals and recognition molecules between early apoptotic and late apoptotic cells. Moreover, in addition to phagocytosis of dead cells, the function of neutrophils should be further studied. Here, we pondered the role that splenic Mφs would play if a massive number of dead cells needed to be phagocytosed. To explore this issue, we created a Mφ‐specific depletion window with CLL and found impaired antigen‐trapping in the spleen after selective depletion of Mφs. Amazingly, the CD4+ T‐cell response to dead‐cell‐associated antigens was significantly impaired in Mφ‐depleted mice, which seemed contrary to conventional views that Mφs have poor antigen presentation ability.6 These findings prompted us to explore further how Mφs participate in the dead‐cell‐associated antigen‐induced immune response.
Dendritic cells are known as the most potent professional antigen‐presenting cells and play a central role in modulating innate and adaptive immunity.34 In our study, we found that DCs were also required for the CD4+ T‐cell response to dead cell infusion by CLL treatment and the Batf3 −/− mouse model. In addition, DCs alone still display a strong antigen presentation although T‐cell proliferation was inferior to that for co‐cultures of DCs and Mφs together (Fig. 3b). These data indicated that DCs were the major presenter and essential for the induction of CD4+ T‐cell response to dead‐cell‐associated antigens. However, our data also showed that Mφs were the relative predominant phagocytes engulfing dead cells in the spleen compared with other immune cell types (including DCs) in Fig 1. Moreover, the issue was confirmed as shown in Fig. 2(c) that the ratio of CFSE+ cells in the spleen declined by about 50% in the Mφ‐depleted spleen compared with PBS‐loaded control liposomes. Comprehensive analysis of these data give us a suggestion that DCs have stronger ability to present antigens, although they have inferior ability to uptake antigens compared with Mφs, and Mφs have stronger ability to uptake antigens, although they were regarded as poor mediators of T‐cell activation compared with DCs. However, our data also showed that Mφs played a partial role in the CD4+ T‐cell response to dead‐cell‐associated antigens (Figs 2d, e and 3b), it raised a question of how Mφs were involved in this process. Based on the contribution of DCs and Mφs in antigen presentation and antigen uptake, we speculated that there was cooperation between the two cell populations. We first excluded the possibility of directly presenting antigens by Mφs in Fig. 3(b) and previous studies.6 Then we performed the following experiment (Fig. 3c and d) to test that Mφs might act as an accessory tool to support antigen presentation by DCs. These results implied that DCs could acquire dead‐cell antigens from Mφs and present them to induce T‐cell proliferation. Next, to study what factors were involved in this process, we performed the following experiment.
Cellular interaction is mediated by direct cell contact or soluble factors. By using fixed Mφs and the Transwell assay, we found that Mφs could release certain soluble factors containing antigens, which were captured by DCs to induce a T‐cell response. However, the question of what factors specifically were involved in this process needed to be answered. Exosomes, formed by the fusion of multivesicular bodies with the plasma membrane followed by budding,18 have been demonstrated to act as a mediator of cellular cross‐talk in autoimmunity,42 as well as to function as cell–cell communication cargo to regulate immune responses.17 Until now, whether exosomes can act as mediators between Mφs and DCs in the immune response to dead cell materials has been unknown. In our study, by addition of OVA into a culture of the mouse Mφ cell line RAW264.7, we first found that Mφ‐derived exosomes contained the uptaken exogenous antigen OVA (Fig. 4e). This finding was similar to that of a previous study, in which 29 mycobacterial proteins could be identified in exosomes released from Mφs incubated with Mycobacterium tuberculosis culture filtrate protein.43 Subsequently, we found that exosomes could induce significant proliferation of OT‐II T cells in the presence of DCs, although exosomes alone could also weakly stimulate the CD4+ T‐cell response. This observation might reflect the direct stimulation of T cells by EXO considering its ability to carry surface MHC II molecules.38 The results clearly demonstrated that the Mφs released exosomes containing antigens that could be captured by DCs, leading to antigen presentation to augment the T‐cell response. However, how DCs acquired the antigen‐containing exosomes remained unclear.
The biogenesis of exosomes appears to be associated with the endosomal sorting complex required for transport (ESCRT) machinery44; however, it can still be generated in the absence of key subunits of ESCRT.22, 45 Moreover, exosome secretion may be triggered by sphingolipid ceramide, which has a role in the budding of intralumenal vesicles.46 Some cell types, such as tumour cells and human Mφs, have been shown to release exosomes and exosomal contents by the ceramide‐dependent pathway in vivo and in vitro.22, 23, 24 However, whether Mφs transferred uptaken antigens to DCs through the ceramide‐dependent exosome‐releasing pathway was unclear. By treatment with an inhibitor of nSMase 2, GW4869, we found that the release of exosomal proteins from the murine Mφ cell line was decreased in a dose‐dependent manner. To further verify the function of exosomes as transmitters in the immune response to dead‐cell‐associated antigens, we tested the effect of GW4869 in our system. We found that the OT‐II T‐cell response to dead‐cell‐associated antigens was decreased both in vitro and in vivo after treatment with GW4869, suggesting that dead‐cell‐associated antigens could be incorporated into exosomes and released partially at least via a ceramide‐dependent pathway. Although another structurally unrelated inhibitor of nSMase, spiroepoxide, was used to inhibit exosome release, we found the effect of spiroepoxide on exosome release and their function in vitro and in vivo were similar to that of GW4869, even when they were combined. It is considered that exosome secretion via a ceramide‐dependent pathway might arrive at saturation, suggesting that some other pathway, such as the ESCRT pathway, might be involved in this process, or interaction between DCs and Mφs was also mediated by other factors. In summary, exosomes released by Mφs via a ceramide‐dependent pathway played a partial role in this experiment.
In addition, little is known about how dead‐cell‐associated antigens are sorted into exosomes for their secretion, and the precise mechanism involved in the actual cellular transfer requires further investigation. Although many studies have reported that exosomes participate in mediating autoimmune diseases,17, 40 the detailed mechanism remains to be explored. Hence, our study provides a novel insight into the potential function of secretory exosomes containing late apoptotic cell‐associated antigens as a transmitter between Mφs and DCs. Such exosome‐mediated cross‐talk may play an important role in the process of autoimmune diseases such as systemic lupus erythematosus, which is caused by an aberrant T‐cell immune response to excessive apoptotic cells proceeding to secondary necrosis. However, the issue needs further investigation in future work using an appropriate disease model.
In summary, we demonstrated splenic Mφs and neutrophils as the predominant phagocytes engulfing dead cells. Moreover, both Mφs and DCs were required for a robust CD4+ T‐cell response to dead‐cell‐associated antigens. Interestingly, Mφs could transfer antigens to DCs, leading to an enhanced T‐cell response to dead‐cell‐associated antigens. Finally, we identified that exosomes containing antigens were released from Mφs partially in a ceramide‐dependent manner, and served as transmitters to convey antigens to DCs. Therefore, our findings have provided insight into the novel pathways of cellular cross‐talk between Mφs and DCs, which will be helpful in illuminating the mechanisms of the immune response in autoimmune diseases characterized by accumulation of dead cells.
Author contribution
Y.X. and H.T. designed the experiments; Y.X. performed the experiments; W.S. assisted with the experiments and revised the manuscript. Y.L., C.Y., L.K., M.W., J.H. and H.H. performed and analysed the animal experiments. Y.X. and H.T. wrote the manuscript.
Disclosure
There are no conflicts of interest.
Supporting information
Figure S1. The strategy of analysing and purifying splenic dendritic cells and macrophages. Splenic single‐cell suspension was prepared and analysed by FACS. First, CD11chi dendritic cells were separated based on the expression of CD11c, then isolated CD11bint F4/80hi macrophages, from the CD11cneg to low cells, based on the expression of CD11b and F4/80. Overlay (the upper right panel) showed CD11chi cells (blue) and live cells (red) stained by CD11c and MHC II. Overlay (the bottom right panel) showed F4/80hi cells (blue) from CD11cneg to low cells and live cells (red) stained by CD11c and CD11b as indicated.
{kind=link}
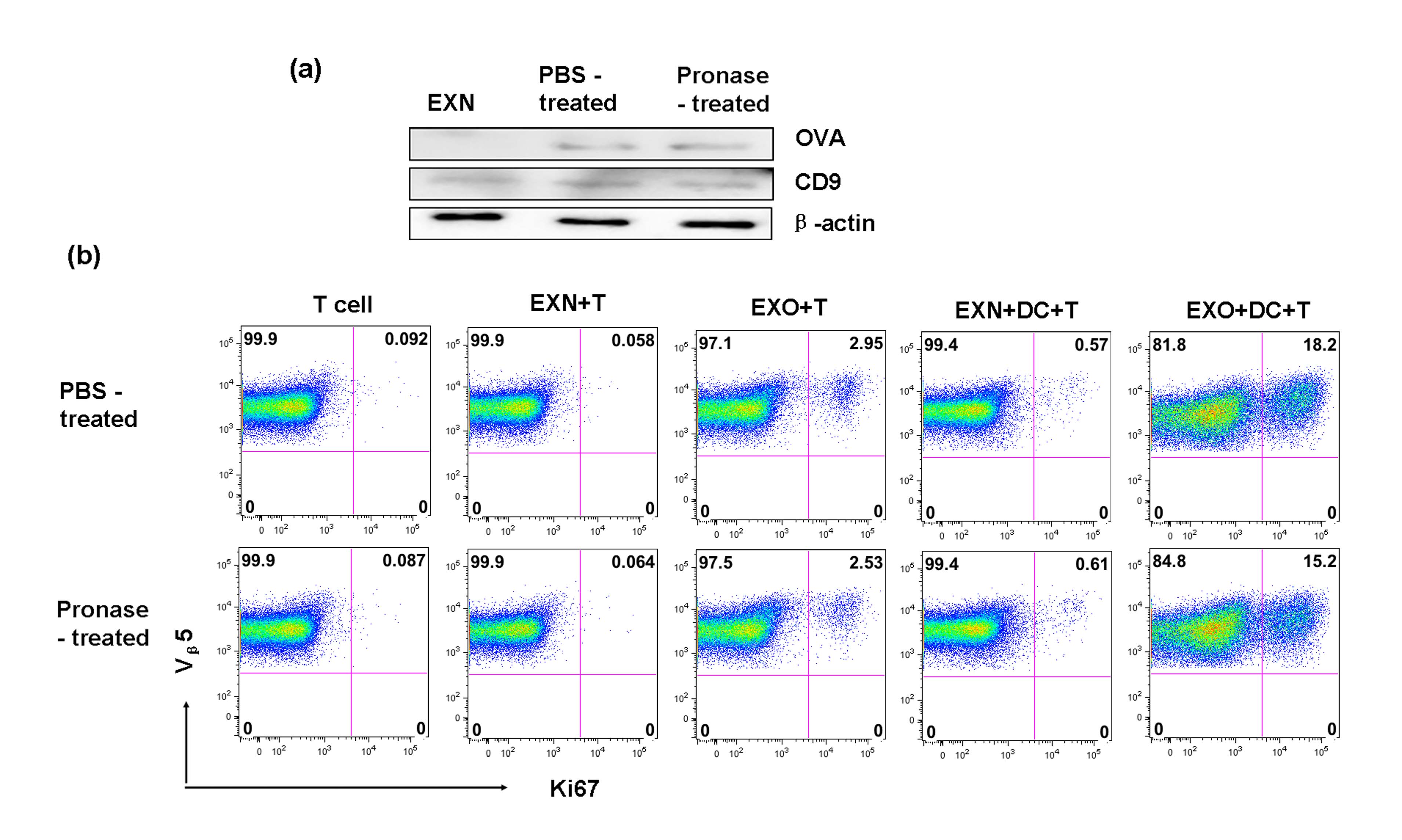
Figure S2. Pronase treatment had little effect on ovalbumin (OVA) contents from exosome and T‐cell proliferation in the presence of dendritic cells. Exosomal proteins secreted from 15 × 106 RAW264.7 cells incubated with OVA (EXO) or not (EXN), were purified according to the method as described in the Material and methods. Then purifed exosomes were treated with 0.08% pronase (Merck‐Calbiochem) in PBS or PBS alone for 20 min, ultracentrifuged, treated the same way again, and an equal volume of exosome‐free fetal bovine serum was added to quench the pronase activity. The purified exosomes were then washed with PBS, and separated into two parts: one part were analysed by immunoblotting for the presence of OVA and CD9 (a), the other were filtered with 0.22‐μm mesh for functional analysis (b). (b) Naive CD45.1+ OT‐II T cells were cultured with exosomes (pronase treated or not) or a combination of dendritic cells with exosomes for 3 days, and the proliferation of T cells was evaluated using Ki67 staining. Experiments were repeated at least three times with similar results.
{kind=link}
Figure S3. T‐cell proliferation was decreased after treatment with GW4869, but the decrease was not dependent on the dose of GW4869 eFluor‐450 dye‐labelled naive CD4+ OT‐II T cells, naive DCs from naive mice and macrophages (Mφs) from ovalbumin (OVA)/cell‐injected mice were co‐cultured in the presence of GW4869 (10 μm, 20 μm) or DMSO (solvent) for 3 days. The proliferation of OT‐II T cells was analysed by flow cytometry. (b) CD45.2 mice were intraperitoneally injected with GW4869 (1.25 mg/kg, 2.5 mg/kg, 5 mg/kg ) or DMSO once a day for 6 days, and the mice were intravenously injected with 1 × 106 CD45.1+ CD4+ Vβ5+ Vα2+ T cells on the fourth day followed by injection of OVA/cells after 1 day. Three days after the OVA/cells injection, the proliferation of CD45.1+ CD4+ T cells was analysed on FACS. Data are shown as the mean ± SD of three to five mice per group and representative of three independent experiments. *P < 0.05, NS, not significant.
{kind=link}
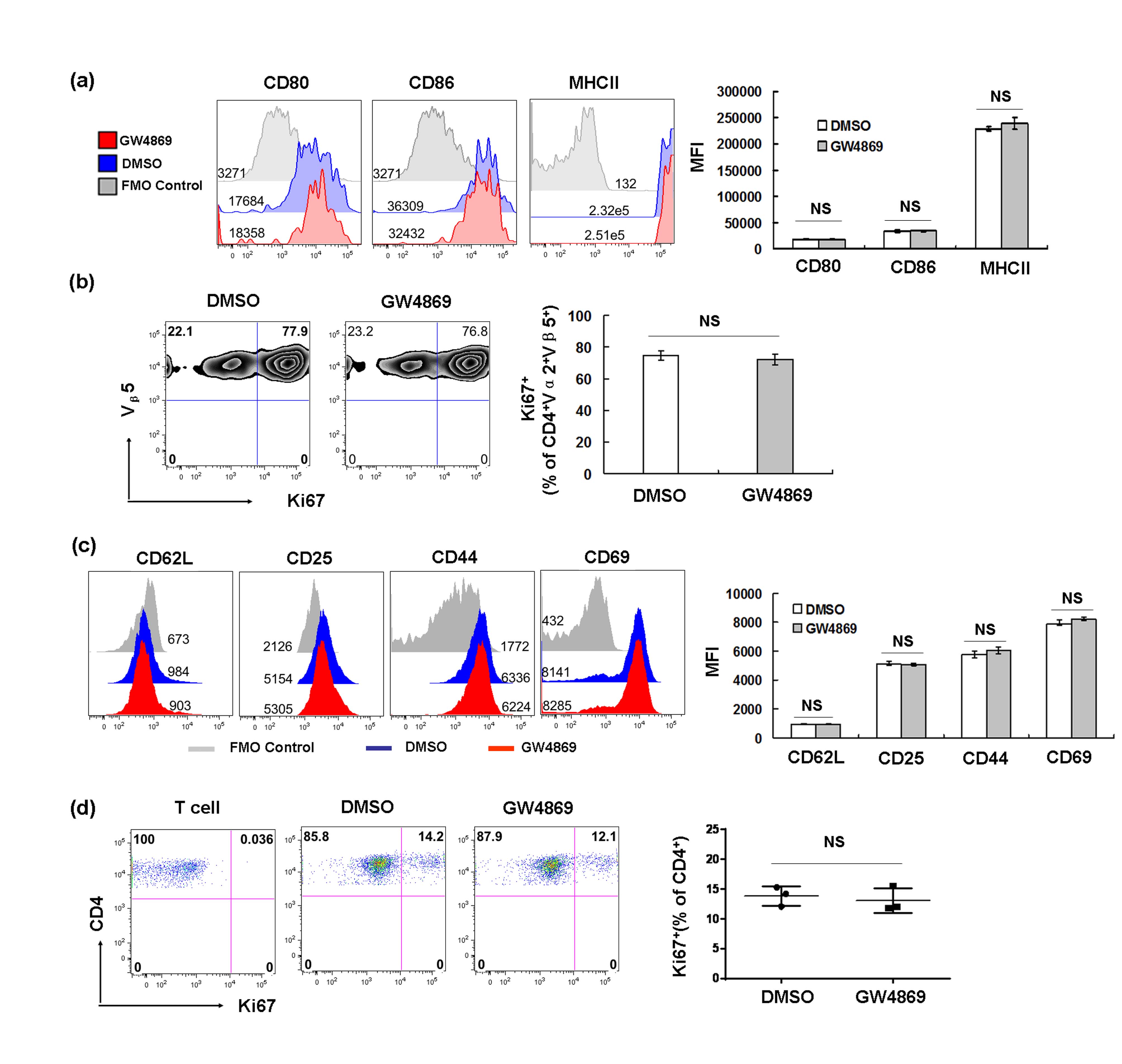
Figure S4. The addition of GW4869 to the T‐cell proliferation had no effects on antigen presentation/T‐cell activation (a) GW4869 (10 μm) was added into a co‐culture system of purified OT‐II T cells (1 × 105/well) and dendritic cells (2.5 × 104/well) in the presence of 10 μg ovalbumin (OVA) protein. After 3 days, dendritic cell maturation based on CD80, CD86 and MHCII expression was analysed by FACS, and (b) the proliferation of T cells was analysed by Ki67 staining. (c) FACS‐sorted naive CD4+ T cells (1 × 105/well) were stimulated by anti‐CD3 (2 μg/ml) and anti‐CD28 (10 μg/ml) in the presence of DMSO or GW4869 (10 μm). One day after stimulation, T‐cell activation based on the expressions of CD62L, CD25, CD44 and CD69 were analysed by FACS. (d) Three days after stimulation, the proliferation of T cells was analysed by Ki67 staining. Experiments were repeated at least three times with similar results. NS, no significant.
{kind=link}
Acknowledgments
We thank Dr Qing‐Sheng Mi (Henry Ford Immunology Program, Henry Ford Hospital) for his help in preparing the manuscript. We thank Professor Qiuling Zhang (Taishan Medical University) for her help in the maintenance of mice. This work was supported by grants from the National Natural Science Foundation of China (Nos. 81302603, 81471553, 81172882, 81272315, 81172791, 81202308 and 81273212), the National Basic Research Programme of China (2015CB943203), the National Science and Technology Major Projects (2014ZX09508003), and the National Science Foundation of Shandong Provice, China (ZR2011CM037, J13LK01).
References
- 1. Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce TH2 and tolerogenic responses. Nat Immunol 2010; 11:647–55. [DOI] [PubMed] [Google Scholar]
- 2. Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y et al Microbiota‐dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014; 343:1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Vries VC, Pino‐Lagos K, Nowak EC, Bennett KA, Oliva C, Noelle RJ. Mast cells condition dendritic cells to mediate allograft tolerance. Immunity 2011; 35:550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stefanovic‐Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL et al Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity‐associated increases in CD11c+ cells in adipose tissue and liver. Diabetes 2012; 61:2330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang H, Cao W, Kasturi SP, Ravindran R, Nakaya HI, Kundu K et al The T helper type 2 response to cysteine proteases requires dendritic cell‐basophil cooperation via ROS‐mediated signaling. Nat Immunol 2010; 11:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Backer R, Schwandt T, Greuter M, Oosting M, Jungerkes F, Tuting T et al Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc Natl Acad Sci U S A 2009; 107:216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Y, Zhang R, Zhang H, Liu J, Yang Z, Xu P et al Microparticles released by Listeria monocytogenes‐infected macrophages are required for dendritic cell‐elicited protective immunity. Cell Mol Immunol 2012; 9:489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 2014; 40:248–61. [DOI] [PubMed] [Google Scholar]
- 9. Szondy Z, Garabuczi E, Joos G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol 2014; 5:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol 2010; 6:280–9. [DOI] [PubMed] [Google Scholar]
- 11. Xu W, Roos A, Daha MR, van Kooten C. Dendritic cell and macrophage subsets in the handling of dying cells. Immunobiology 2006; 211:567–75. [DOI] [PubMed] [Google Scholar]
- 12. McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 2011; 117:5403–12. [DOI] [PubMed] [Google Scholar]
- 13. Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M et al Tolerance to apoptotic cells is regulated by indoleamine 2,3‐dioxygenase. Proc Natl Acad Sci U S A 2012; 109:3909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Biermann MH, Veissi S, Maueroder C, Chaurio R, Berens C, Herrmann M et al The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: dendritic cells as potential targets. Expert Rev Clin Immunol 2014; 10:1151–64. [DOI] [PubMed] [Google Scholar]
- 15. Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN et al Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell 2004; 15:1089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M et al CD169‐positive macrophages dominate antitumor immunity by crosspresenting dead cell‐associated antigens. Immunity 2011; 34:85–95. [DOI] [PubMed] [Google Scholar]
- 17. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 2014; 14:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I et al Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles 2013; 2:20677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods 1994; 174:83–93. [DOI] [PubMed] [Google Scholar]
- 20. Cai Z, Yang F, Yu L, Yu Z, Jiang L, Wang Q et al Activated T cell exosomes promote tumor invasion via Fas signaling pathway. J Immunol 2012; 188:5954–61. [DOI] [PubMed] [Google Scholar]
- 21. Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen‐bearing dendritic cells. Science 2006; 312:1672–6. [DOI] [PubMed] [Google Scholar]
- 22. Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 2010; 285:17442–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R et al MicroRNAs bind to Toll‐like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012; 109:E2110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H et al Exosomes mediate the cell‐to‐cell transmission of IFN‐α‐induced antiviral activity. Nat Immunol 2013; 14:793–803. [DOI] [PubMed] [Google Scholar]
- 25. Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T et al MicroRNA‐containing T‐regulatory‐cell‐derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014; 41:89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Munoz LE, Maueroder C, Chaurio R, Berens C, Herrmann M, Janko C. Colourful death: six‐parameter classification of cell death by flow cytometry – dead cells tell tales. Autoimmunity 2012; 46:336–41. [DOI] [PubMed] [Google Scholar]
- 27. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell‐associated antigens. J Clin Invest 2007; 117:2268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y et al The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo . J Exp Med 2002; 195:1289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E et al Distinct dendritic cell subsets differentially regulate the class of immune response in vivo . Proc Natl Acad Sci U S A 1999; 96:1036–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duriancik DM, Hoag KA. The identification and enumeration of dendritic cell populations from individual mouse spleen and Peyer's patches using flow cytometric analysis. Cytometry A 2009; 75:951–9. [DOI] [PubMed] [Google Scholar]
- 31. van Rooijen N, Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol 2010; 605:189–203. [DOI] [PubMed] [Google Scholar]
- 32. van Rooijen N, Kors N, Kraal G. Macrophage subset repopulation in the spleen: differential kinetics after liposome‐mediated elimination. J Leukoc Biol 1989; 45:97–104. [DOI] [PubMed] [Google Scholar]
- 33. Steinman RM, Idoyaga J. Features of the dendritic cell lineage. Immunol Rev 2010; 234:5–17. [DOI] [PubMed] [Google Scholar]
- 34. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2011; 30:1–22. [DOI] [PubMed] [Google Scholar]
- 35. Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U et al The transcription factor BATF controls the global regulators of class‐switch recombination in both B cells and T cells. Nat Immunol 2011; 12:536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo . Blood 2007; 110:3234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giri PK, Kruh NA, Dobos KM, Schorey JS. Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis‐infected and culture filtrate protein‐treated macrophages. Proteomics 2010; 10:3190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ et al B lymphocytes secrete antigen‐presenting vesicles. J Exp Med 1996; 183:1161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramirez‐Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ et al The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol 2013; 14:917–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell 2010; 140:619–30. [DOI] [PubMed] [Google Scholar]
- 41. Hirt UA, Leist M. Rapid, noninflammatory and PS‐dependent phagocytic clearance of necrotic cells. Cell Death Differ 2003; 10:1156–64. [DOI] [PubMed] [Google Scholar]
- 42. Beyer C, Pisetsky DS. The role of microparticles in the pathogenesis of rheumatic diseases. Nat Rev Rheumatol 2009; 6:21–9. [DOI] [PubMed] [Google Scholar]
- 43. Cheng Y, Schorey JS. Exosomes carrying mycobacterial antigens can protect mice against Mycobacterium tuberculosis infection. Eur J Immunol 2013; 43:3279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat Rev Mol Cell Biol 2014; 11:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stuffers S, Sem Wegner C, Stenmark H, Brech A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009; 10:925–37. [DOI] [PubMed] [Google Scholar]
- 46. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F et al Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008; 319:1244–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The strategy of analysing and purifying splenic dendritic cells and macrophages. Splenic single‐cell suspension was prepared and analysed by FACS. First, CD11chi dendritic cells were separated based on the expression of CD11c, then isolated CD11bint F4/80hi macrophages, from the CD11cneg to low cells, based on the expression of CD11b and F4/80. Overlay (the upper right panel) showed CD11chi cells (blue) and live cells (red) stained by CD11c and MHC II. Overlay (the bottom right panel) showed F4/80hi cells (blue) from CD11cneg to low cells and live cells (red) stained by CD11c and CD11b as indicated.
Figure S2. Pronase treatment had little effect on ovalbumin (OVA) contents from exosome and T‐cell proliferation in the presence of dendritic cells. Exosomal proteins secreted from 15 × 106 RAW264.7 cells incubated with OVA (EXO) or not (EXN), were purified according to the method as described in the Material and methods. Then purifed exosomes were treated with 0.08% pronase (Merck‐Calbiochem) in PBS or PBS alone for 20 min, ultracentrifuged, treated the same way again, and an equal volume of exosome‐free fetal bovine serum was added to quench the pronase activity. The purified exosomes were then washed with PBS, and separated into two parts: one part were analysed by immunoblotting for the presence of OVA and CD9 (a), the other were filtered with 0.22‐μm mesh for functional analysis (b). (b) Naive CD45.1+ OT‐II T cells were cultured with exosomes (pronase treated or not) or a combination of dendritic cells with exosomes for 3 days, and the proliferation of T cells was evaluated using Ki67 staining. Experiments were repeated at least three times with similar results.
Figure S3. T‐cell proliferation was decreased after treatment with GW4869, but the decrease was not dependent on the dose of GW4869 eFluor‐450 dye‐labelled naive CD4+ OT‐II T cells, naive DCs from naive mice and macrophages (Mφs) from ovalbumin (OVA)/cell‐injected mice were co‐cultured in the presence of GW4869 (10 μm, 20 μm) or DMSO (solvent) for 3 days. The proliferation of OT‐II T cells was analysed by flow cytometry. (b) CD45.2 mice were intraperitoneally injected with GW4869 (1.25 mg/kg, 2.5 mg/kg, 5 mg/kg ) or DMSO once a day for 6 days, and the mice were intravenously injected with 1 × 106 CD45.1+ CD4+ Vβ5+ Vα2+ T cells on the fourth day followed by injection of OVA/cells after 1 day. Three days after the OVA/cells injection, the proliferation of CD45.1+ CD4+ T cells was analysed on FACS. Data are shown as the mean ± SD of three to five mice per group and representative of three independent experiments. *P < 0.05, NS, not significant.
Figure S4. The addition of GW4869 to the T‐cell proliferation had no effects on antigen presentation/T‐cell activation (a) GW4869 (10 μm) was added into a co‐culture system of purified OT‐II T cells (1 × 105/well) and dendritic cells (2.5 × 104/well) in the presence of 10 μg ovalbumin (OVA) protein. After 3 days, dendritic cell maturation based on CD80, CD86 and MHCII expression was analysed by FACS, and (b) the proliferation of T cells was analysed by Ki67 staining. (c) FACS‐sorted naive CD4+ T cells (1 × 105/well) were stimulated by anti‐CD3 (2 μg/ml) and anti‐CD28 (10 μg/ml) in the presence of DMSO or GW4869 (10 μm). One day after stimulation, T‐cell activation based on the expressions of CD62L, CD25, CD44 and CD69 were analysed by FACS. (d) Three days after stimulation, the proliferation of T cells was analysed by Ki67 staining. Experiments were repeated at least three times with similar results. NS, no significant.