

Summary
Current therapies for multiple sclerosis (MS) reduce the frequency of relapses by modulating adaptive immune responses but fail to limit the irreversible neurodegeneration driving progressive disability. Experimental autoimmune encephalomyelitis (EAE) in Biozzi ABH mice recapitulates clinical features of MS including relapsing–remitting episodes and secondary‐progressive disability. To address the contribution of recurrent inflammatory events and ageing as factors that amplify progressive neurological disease, we examined EAE in 8‐ to 12‐week‐old and 12‐month‐old ABH mice. Compared with the relapsing–remitting (RREAE) and secondary progressive (SPEAE) EAE observed in young mice, old mice developed progressive disease from onset (PEAE) associated with pronounced axonal damage and increased numbers of CD3+ T cells and microglia/macrophages, but not B cells. Whereas the clinical neurological features of PEAE and SPEAE were comparable, the pathology was distinct. SPEAE was associated with significantly reduced perivascular infiltrates and T‐cell numbers in the central nervous system (CNS) compared with PEAE and the acute phase of RREAE. In contrast to perivascular infiltrates that declined during progression from RREAE into SPEAE, the numbers of microglia clusters remained constant. Similar to what is observed during MS, the microglia clusters emerging during EAE were associated with axonal damage and oligodendrocytes expressing heat‐shock protein B5, but not lymphocytes. Taken together, our data reveal that the course of EAE is dependent on the age of the mice. Younger mice show a relapsing–remitting phase followed by progressive disease, whereas old mice immediately show progression. This indicates that recurrent episodes of inflammation in the CNS, as well as age, contribute to progressive neurological disease.
Keywords: autoimmunity, experimental autoimmune encephalomyelitis, multiple sclerosis, neuroimmunology
Abbreviations
- aEAE
acute EAE – the first neurological episode of RREAE
- CFA
complete Freund's adjuvant
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- HspB5
heat‐shock protein B5
- IBA‐1
ionized calcium‐binding adapter molecule 1
- MC
microglial cluster
- MS
multiple sclerosis
- NAGM
normal‐appearing grey matter
- NAWM
normal‐appearing white matter
- NF‐H
neurofilament heavy
- Olig2
oligodendrocyte transcription factor 2
- PEAE
progressive EAE
- PPMS
primary‐progressive MS
- PVC
perivascular cluster
- RREAE
relapsing–remitting EAE
- RRMS
relapsing–remitting MS
- SCH
spinal cord homogenate
- SPEAE
secondary‐progressive EAE
- SPMS
secondary‐progressive MS
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system (CNS) characterized by inflammation associated with reversible neurological deficits and secondary progressive neurodegeneration. That these clinical phenomena may have different underlying pathologies is supported by the findings that current therapies for MS reduce the frequency of relapses, yet fail to limit the irreversible neurodegeneration that drives progressive disability.1 Similar to other neurodegenerative disorders, the onset of progressive MS is related to age, a factor known to amplify neurodegeneration.2, 3 In younger people (<30 years) with MS, the disease typically begins with a relapsing–remitting phase. After 15–25 years following disease onset, or when disease is first manifested in people older than 40, MS is predominantly characterized by progressive neurological disability. Hence, the development of the progressive stage of MS may well be at least in part a consequence of ageing.4 Although the mechanisms that underlie age‐associated changes in the CNS are incompletely understood, they most likely involve changes in the numbers and functions of microglia, important innate immune cells in the CNS.5 With ageing, microglia change their morphology and have a diminished capacity to migrate and phagocytose debris, known to impede regeneration and repair.5 In addition, aged microglia assume a more pro‐inflammatory profile upon activation, which also augments progressive neurodegeneration.6, 7 Moreover, age also influences the reparative processes such as remyelination in the CNS,8, 9, 10 as well as the ability of the adaptive immune responses to function efficiently. For example, T cells show reduced proliferation in vitro with age, and the functions of regulatory T cells, critical for maintaining self‐tolerance, decreases at the level of activation and cytokine production.11, 12, 13
In MS, widespread microglia/macrophage activation correlates with demyelination, axonal loss/damage and oligodendrocyte damage.14 In addition, clusters of activated HLA‐DR+ microglia, also termed pre‐active lesions,15 are present in normal‐appearing white matter (NAWM) irrespective of disease duration and subtype.16 In early MS, these microglial clusters (MC) associate with axonal injury,17 whereas in established MS, they are associated with stressed oligodendrocytes but not with demyelination, leucocyte infiltration, axonal damage or blood–brain barrier disruption.16, 18 Microglia in pre‐active MS lesions exhibit an intermediate phenotype characterized by the expression of markers for both the M1 and M2 states of activation,19 indicating clear differences between their activation state compared with active lesions.20, 21 Whether the association of MC with axonal damage in early MS and with oligodendrocyte stress in established MS is a function of disease duration and/or age of the patient is unknown.
The most applicable animal model to study mechanisms contributing to disease progression in MS is experimental autoimmune encephalomyelitis (EAE). Different models of EAE have been established, but secondary progressive EAE (SPEAE) following a relapsing–remitting episode (RREAE) of approximately 3 months has only been reported for Biozzi ABH mice.22, 23 Similar to MS, the relapsing–remitting stages of EAE are strongly associated with T‐cell and B‐cell autoimmunity24 whereas the neurodegenerative component is associated with innate immunity.25
We have previously developed an EAE model in Biozzi ABH mice in which RREAE following immunization of 8‐ to 12‐week‐old mice is followed by a secondary‐progressive phase (SPEAE) 3 months later.23 To better model primary‐progressive MS, that occurs in older people, we have induced disease in old ABH mice (12 months). In contrast to young animals, old mice immediately develop progressive disease (PEAE) upon immunization. We show that the increased severity of neurological disease during PEAE in old mice is accompanied by more severe axonal damage, increased microglia activation and enhanced infiltration by CD3+ T cells, but not by an increased appearance of MC in NAWM. Similar to what has been found during MS, MC emerging during PEAE were associated with both axonal damage and oligodendrocytes expressing heat‐shock protein B5 (HspB5), but not with infiltrated lymphocytes.
In summary, we have developed a model of severe chronic EAE in old Biozzi ABH mice that is associated with widespread and exacerbated microglial activation in the CNS, and increased numbers of CD3+ T cells but not B220+ B cells. That the numbers of MC remain constant throughout the disease, in both old and younger mice and regardless of disease severity or extent of axonal damage, suggests that these clusters arise in response to factors other than axonal damage. Possible factors include oxidative stress or oligodendrocyte apoptosis, which is associated with increased expression of HspB5.
Materials and methods
Induction of EAE
Biozzi ABH (H‐2dq1) male and female mice (originally purchased from Harlan UK Ltd, Bicester, UK) were bred at Queen Mary University of London under specific pathogen‐free conditions. Their health status was uniform throughout the studies. Mice (2 weeks, 3 weeks, 8–12 weeks, 8 months and 12 months old) were immunized with an emulsion of mouse spinal cord homogenate (SCH) in complete Freund's adjuvant (CFA) as described previously.23, 26 The same batch of SCH was used for all experiments. Based on power analysis and sampling for pathology studies 10–22 mice per group were immunized for EAE. The studies were performed in three separate experiments to accommodate the different aged mice. As controls for each study a group of 8‐ to 12‐week‐old mice were immunized with SCH in CFA and an aged‐matched control group of mice were injected with CFA only. Control mice were sampled at corresponding time‐points after injection to coincide with the post‐immunization sampling days of mice with EAE.
Neurological deficits were assessed daily from day 11 onwards, and neurological signs scored on a scale from 0 to 5, whereby 0 = normal, 1 = fully flaccid tail, 2 = impaired righting reflex, 3 = hind‐limb paresis, 4 = complete hind‐limb paresis and 5 = moribund/death. EAE stages that were distinguished were acute (aEAE), first remission, relapse, second remission, post‐relapsing SPEAE23 and PEAE (see Supplementary material, Table S1).
Animal studies were performed in compliance with UK law for care of animals and approved by the Home Office under Animals (Scientific procedures) Act 1986. We adhered to the ARRIVE guidelines for animal research in EAE27 as described previously.23
Immunohistochemistry
Sections (5 μm) from paraffin‐embedded brains and spinal cords from different EAE stages (n = 8/stage; details on the clinical history of the mice used for pathology are summarized in the Supplementary material, Table S1.) or age‐matched controls were deparaffinized in xylene, rehydrated in descending grades of alcohol and washed in PBS. Endogenous peroxidase activity was blocked with 0·3% H2O2 in PBS for 30 min at room temperature. For antigen retrieval, sections were heated for 10 min in Tris/EDTA‐buffer (pH 9·0) or citrate buffer (pH 6·0), after which non‐specific binding was blocked for 1 hr using CleanVision Blocking Solution (Immunologic, Duiven, the Netherlands) for mouse antibodies, or 5% normal goat serum (Dako, Glostrup, Denmark) in PBS when using rabbit antibodies. Sections were rinsed and incubated with primary antibodies directed to IBA‐1, an ionized calcium‐binding protein 1 to detect microglia/macrophages, proteolipid protein to detect myelin integrity, the non‐phosphorylated epitope of neurofilament heavy (NF‐H) to detect neuronal damage, or Olig2 to detect oligodendrocytes (Table 1).
Table 1.
Antibodies used for immunohistochemistry
| Antigen | Species | Isotype | Dilution | Antigen retrieval | Incubation | Source |
|---|---|---|---|---|---|---|
| Primary antibodies | ||||||
| IBA‐1 | Rabbit | IgG | 1 : 10 000 | Tris/EDTA | 1 hr | Wako |
| Proteolipid protein (plpc1) | Mouse | IgG2a | 1 : 3000 | N/A | 1 hr | AbDSerotec |
| Non‐phosphorylated NF‐H (SMI‐32) | Mouse | IgG1 | 1 : 1000 | Citrate | 1 hr | BioLegend |
| HspB5 (W3/13) | Rabbit | IgG | 1 : 5000 | Citrate | O/N | Delta Crystallon BV |
| CD45R (RA3‐6B2) | Rat | IgG2a | 1 : 1500 | Citrate | 1 hr | AbDSerotec |
| CD3 (F7·2·38) | Rabbit | IgG1 | 1 : 1000 | Citrate | O/N | Dako |
| Secondary antibodies | ||||||
| Rabbit IgG‐AP | Goat | N/A | 1 : 250 | N/A | 1 hr | Southern Biotech |
| Mouse IgG2a‐HRP | Goat | N/A | 1 : 250 | N/A | 1 hr | Southern Biotech |
| Mouse IgG1‐HRP | Goat | N/A | 1 : 250 | N/A | 1 hr | Southern Biotech |
| Rabbit EnVision™ | Goat | N/A | N/A | N/A | 1 hr | Dako |
N/A, not applicable; O/N, overnight.
After washing, sections were incubated with appropriate horseradish peroxidase‐labelled secondary antibodies (Table 1). Antibody binding was visualized using 3,3′‐diaminobenzidine (Dako) and sections were rinsed in PBS. Next, the sections were treated in the microwave to remove the first antibody. Subsequently, sections were incubated with antibodies directed to IBA‐1, HspB5, CD45RA/B220 and CD3. After washing, sections were incubated with alkaline phosphatase‐labelled secondary antibodies, washed in Tris‐buffered saline and stained using Liquid Permanent Red (Dako). All antibodies were diluted in Antibody Diluent™ (Immunologic). Incubations were performed at room temperature in a humidified chamber. Sections were counterstained with haematoxylin and mounted with Aquatex (Millipore, Billerica, MA).
Quantitative analysis
Microglial clusters were identified as clusters of four or more IBA‐1+ cells in NAWM or normal‐appearing grey matter (NAGM). Perivascular clusters (PVC) were identified as clusters of four or more IBA‐1+ cells in close contact with blood vessels. The numbers of MC and PVC were assessed in each region, namely, the grey and white of the brain and spinal cord. The areas of these regions were measured using image J (National Institutes of Health, Bethesda, MD). Images were taken with a Leica DC500 (Leica Microsystems, Heidelberg, Germany) at 1·25 × magnification. The number of pixels was counted in the regions of interest and divided by the number of pixels per 1 mm2 to calculate the number of MC and PVC per 10 mm2. Sections stained for IBA‐1 were also double labelled for expression of the non‐phosphorylated epitope of NF‐H, Olig2, HspB5, CD3 and B220. The analysis of the pathology was performed by two observers blinded to the EAE stage and disease severity.
Statistical analysis
Data were analysed using graphpad prism software (GraphPad Software, San Diego, CA). Comparisons of the numbers of MC and PVC present during EAE stages were performed using Kruskal–Wallis one‐way analysis of variance. The numbers per stage and region were compared pairwise using the Mann–Whitney U‐test. Probability values (P) of 0·05 or less were considered statistically significant.
Results
Age is a critical factor that controls the disease course of EAE
To examine the impact of age on the development of EAE, Biozzi mice of different ages were immunized with SCH in CFA. In contrast to our studies showing that 8‐ to 12‐week‐old (young) mice develop RREAE followed by post‐relapsing SPEAE about 3–5 months later,23 Biozzi ABH mice aged 2 weeks did not develop EAE (Table 2; P < 0·001) and 3‐week‐old mice developed disease but with a significantly reduced incidence of disease (P < 0·01) and disease severity (P < 0·01) compared with 8‐ to 12‐week‐old mice. Of significance was the finding that 12‐month‐old mice consistently developed chronic disease, without a preceding relapsing–remitting course as observed in the 8‐ to 12‐week‐old mice (Table 2; clinical score 4·8 ± 0·2 versus 3·9 ± 0·1; P < 0·01). Although mortality was not typically observed in young mice, a markedly increased mortality rate (8/10) was found in old mice aged 12 months.
Table 2.
Age determines the clinical course of experimental autoimmune encephalomyelitis (EAE) in Biozzi ABH mice
| Age mice | No. aEAE 1 | Mortality | EAE score 2 | Day onset | No. remission | No. PEAE |
|---|---|---|---|---|---|---|
| 2 weeks | 0/11*** | 0/11 | N/A | N/A | N/A | N/A |
| 3 weeks | 3/22** | 0/16 | 0·2 ± 0·1** | 18·2 ± 2·0 | N/A | N/A |
| 8–12 weeks | 12/12 | 0/12 | 3·9 ± 0·1 | 12·5 ± 0·3 | 12/12 | 0/12 |
| 8 months | 13/13 | 1/13* | 4·1 ± 0·1 | 11·2 ± 0·5 | 10/12 | 2/12 |
| 12 months | 10/10 | 8/10*** | 4·8 ± 0·2** | 14·1 ± 0·4 | 0/8 | 8/8 |
1Biozzi mice of different ages were immunized with spinal cord homogenate in complete Freund's adjuvant and monitored until day 60 where appropriate.
2Maximum clinical score during the acute EAE.
*P < 0·05; **P < 0·01; ***P < 0·001 compared with 8‐ to 12‐week‐old mice.
Progressive EAE in old mice is associated with activated microglia and CD3+ T cells
To examine the involvement of innate or adaptive responses during the different stages of EAE, spinal cord sections from mice with aEAE and PEAE as well as age‐matched controls were stained for IBA‐1, CD3 and CD45RA/B220. As shown in Fig. 1, IBA‐1 expression in spinal cord of young control mice was significantly less prominent in both white (Fig. 1a, e, P = 0·026) and grey (Fig. 1b, e, P = 0·017) matter compared with old control mice (Fig. 1c–e). As expected, numbers of IBA‐1+ cells in mice with EAE were significantly increased in the white and grey matter compared with control mice (for all groups P < 0·005; see Supplementary material, Fig. S1). No significant difference was observed in IBA‐1 expression in the white matter during aEAE in young mice compared with PEAE in old mice (Fig. 1f,h,j), its expression was significantly increased in the grey matter during PEAE (Fig. 1g,i,j; P = 0·0001).
Figure 1.
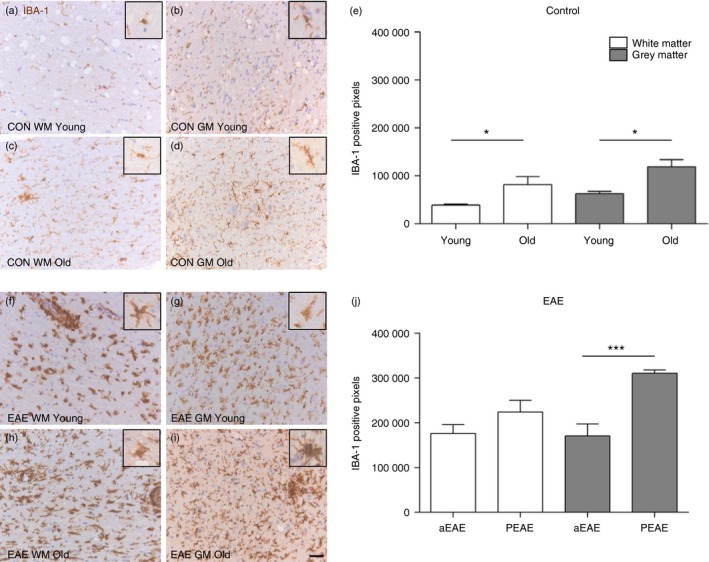
Ageing and experimental autoimmune encephalomyelopathy EAE augments IBA‐1+ microglia/macrophages in mice. Immunohistochemistry was used to detect IBA‐1+ cells in the spinal cord of young and old control mice (upper panels) and young and old mice with EAE (lower panels). Numbers of IBA‐1+ microglia/macrophages in the spinal cord are higher in old mice as compared to young mice (a–d, e). During acute EAE (aEAE) in young mice similar numbers of IBA‐1+ cells are observed in white and grey matter (f, g, j) whereas an increase is observed in the grey matter of old mice with progressive EAE (PEAE) (g, i, j). Inserts exhibit a zoomed in cell to show the morphology of a single IBA‐1+ cell. *P < 0·05; ***P < 0·001. Mean ± SEM (n = 8). Scale bar = 50 μm.
We next compared CD3 and CD45RA/B220 expression during aEAE, PEAE and SPEAE. This revealed that CD3 expression was significantly higher in white matter (Fig. 2a,b; P < 0·01) and grey matter in PEAE (P < 0·05) compared with aEAE. To address the impact of recurrent neuroinflammatory episodes, CD3 expression in the CNS during aEAE and PEAE was compared with SPEAE. This revealed that T‐cell numbers are significantly lower during SPEAE compared with aEAE (white matter P < 0·01; grey matter P < 0·01) and PEAE (white matter P < 0·01; grey matter P < 0·01).
Figure 2.
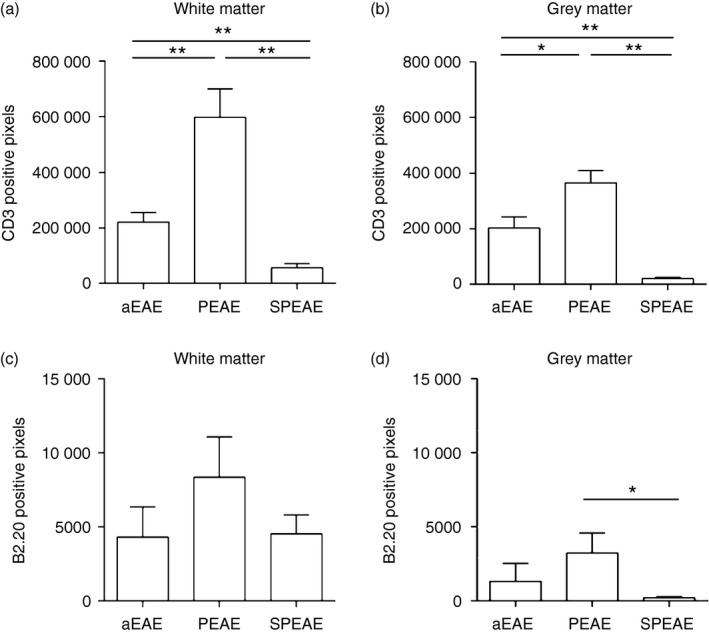
Progressive experimental autoimmune encephalomyelitis (PEAE) but not secondary‐progressive EAE (SPEAE) is associated with increased numbers of CD3+ T cells. The expression of CD3 (a, b) and B220 (c, d) was evaluated in the white and grey matter of the spinal cord of young mice with acute EAE (aEAE) and SPEAE, and in old mice with PEAE. CD3 expression was increased in the white (a) and grey (b) matter during PEAE compared with aEAE or SPEAE. B220 expression was lower than that of CD3 (note scale of x‐axis in c and d compared with a and b). B220+ cells in white matter (c) and grey matter (d) during aEAE, PEAE and SPEAE reveal a significant increase in grey matter during PEAE. *P < 0·05; **P < 0·01. Mean ± SEM (n = 6).
To examine differences between the numbers of infiltrating B cells during the different forms of EAE, the expression of the marker B220 was examined. This analysis revealed no differences in the level of B‐cell infiltration of white matter regions during aEAE, PEAE and SPEAE, but did show significantly lower numbers of CD45RA/B220+ B cells in the grey matter during SPEAE compared with PEAE (Fig. 2c; P < 0·05).
Microglial activation during PEAE is associated with exacerbated axonal damage in the white but not grey matter
Spinal cords from aEAE and PEAE were examined for co‐expression of IBA1+ microglia, and NF‐H as a marker of axonal damage. Whereas axonal damage was absent in control mice (Fig. 3a,b), such damage was clearly observed in the subpial regions of the spinal cord white matter in young mice with aEAE (Fig. 3c). In old mice with PEAE, severe axonal damage associated with numerous IBA‐1+ cells was found throughout the white matter, with only minor axonal damage in the grey matter close to the border of the white matter (Fig. 3d).
Figure 3.
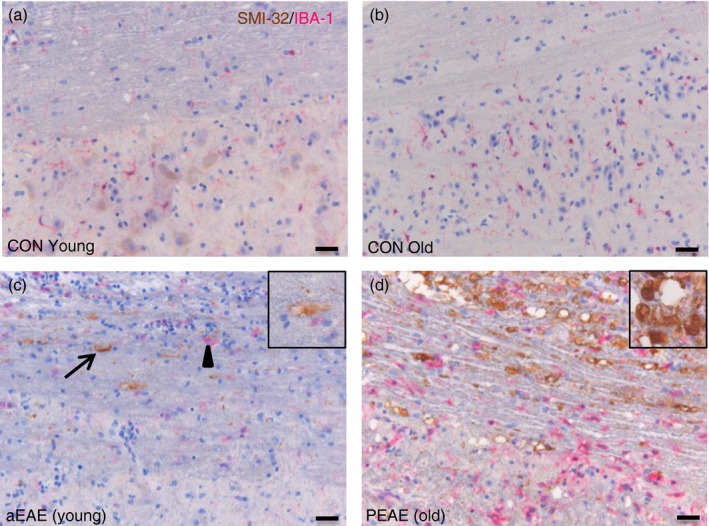
IBA‐1+ cells in progressive experimental autoimmune encephalomyelitis (PEAE) are associated with severe axonal damage. Representative images of spinal cords from young and old control (CON) mice, or mice during acute EAE (aEAE) and PEAE (n = 6 group) show IBA‐1+ cells and their association with axonal damage as determined by SMI‐32 expression. No axonal damage was observed in control mice (a, b). Axonal damage was observed in young mice during aEAE (c, arrow; insert), and was associated with IBA‐1+ cells (c, arrowhead). Old mice with PEAE (d) developed severe axonal damage (d, insert) associated with increased numbers of IBA‐1+ cells. Scale bar = 25 μm.
PVC but not MC decline with EAE progression
Given that therapies aimed at modulating adaptive immunity typically fail to limit the irreversible neurodegeneration that occurs during both progressive MS1, 3 and SPEAE,24 we examined whether the extent of lymphocyte infiltration declines with disease progression, by examining the numbers of PVC and MC in NAWM and NAGM during different forms of EAE. PVC were identified as clusters of four or more IBA‐1+ cells in close contact with blood vessels (Fig. 4a,b) whereas MC were identified as clusters of four or more IBA‐1+ cells in NAWM or NAGM and not associated with blood vessels (Fig. 4c,d). PVC and MC were assessed during aEAE, RREAE and SPEAE in 8‐ to 12‐week‐old mice and compared with PEAE in 12‐month‐old mice. Compared with aEAE and PEAE, the numbers of PVC in the CNS in SPEAE were significantly lower (Fig. 5a illustrating spinal cords, P < 0·0029; P < 0·0005; Fig. 5b illustrating brains; P < 0·0015; P < 0·0102). The difference was most prominently observed in white matter regions of the spinal cord (Fig. 5c) but was less marked in grey matter (Fig. 5d).
Figure 4.
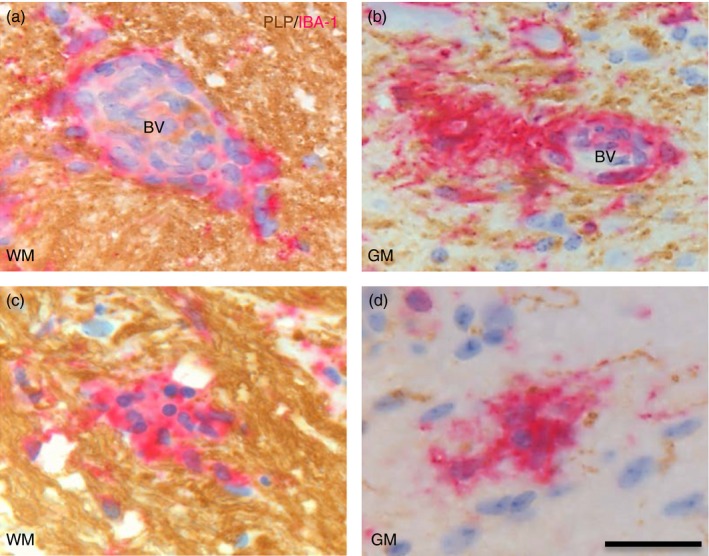
IBA‐1+ cells in perivascular clusters (PVC)and microglia clusters (MC). PVC were identified as clusters of four or more IBA‐1+ cells closely associated with blood vessels (bv) in normal appearing myelin as determined by proteolipid protein (PLP) expression (brown) in white matter (WM; a) or grey matter (GM; b). MC were identified as four or more IBA‐1+ microglia in normal‐appearing WM (c) or normal‐appearing GM (d) that were not associated with endothelial cells or erythrocytes. Scale bar = 25 μm.
Figure 5.
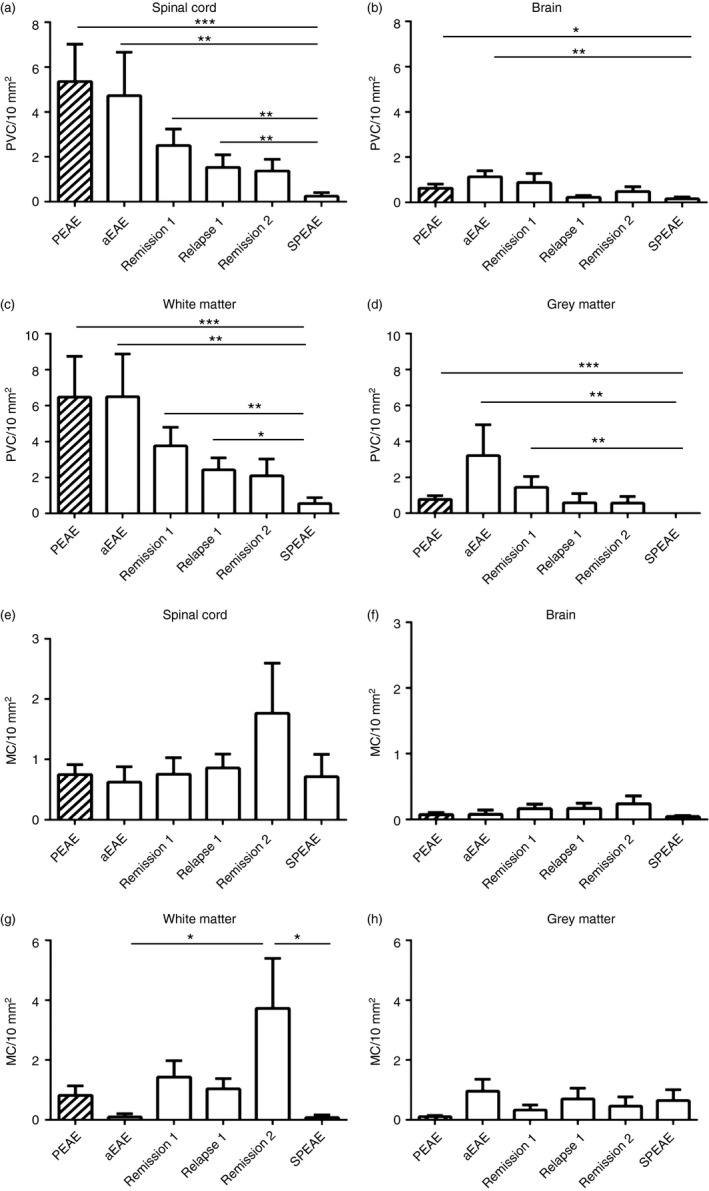
Numbers of perivascular IBA‐1+ clusters (PVC) but not of microglia clusters (MC) decline during progression of experimental autoimmune encephalomyelitis (EAE). Numbers of PVC (a–d) and MC (e–h) were quantified in the spinal cord and brain during PEAE in old mice (hatched bars) and during relapsing–remitting EAE (RREAE) and secondary‐progressive EAE (SPEAE) in young mice. Compared with RREAE (white bars) or PEAE mice the numbers of PVC in the spinal cord (a) and brain (b) were significantly reduced in SPEAE. This difference was more pronounced in spinal cord white matter (c) than grey matter (d). In contrast, numbers of MC in spinal cord (e) and brain (f), both in the white (g) and grey (h) matter, revealed higher numbers of MC during the second remission. Mean SEM. *P < 0·05; **P < 0·005; ***P < 0·001 (n = 8 per group).
In contrast to these differences in the numbers of PVC, no such differences were observed for MC (Fig. 5e,f) although more MC tended to emerge in the white matter during the second remission (Fig. 5g). MC were observed in 30 of 41 (73%) younger mice with EAE at all time‐points, and in all 12‐month‐old mice (100%), but not in control mice. Given previous data illustrating that during MS, MC are predominantly associated with axonal damage in biopsy tissues of acute MS,17 and with stressed HspB5+ oligodendrocytes in post‐mortem tissues during established MS,18 we next examined the relationship between the emergence of MC during EAE and either axonal damage or oligodendrocyte stress by staining for NF‐H and HspB5. As shown in Fig. 6(a,b), IBA‐1+ MC were observed in NAWM in the absence as well as presence of axonal damage, and they were associated with HspB5+ cells too (Fig. 6c). To confirm that the HspB5+ cells were oligodendrocytes, a double staining was performed with the oligodendrocyte marker olig2 (data not shown). In contrast, IBA‐1+ MC were not observed with the presence of CD3+ T cells or B220+ B cells (Fig. 6d–f).
Figure 6.
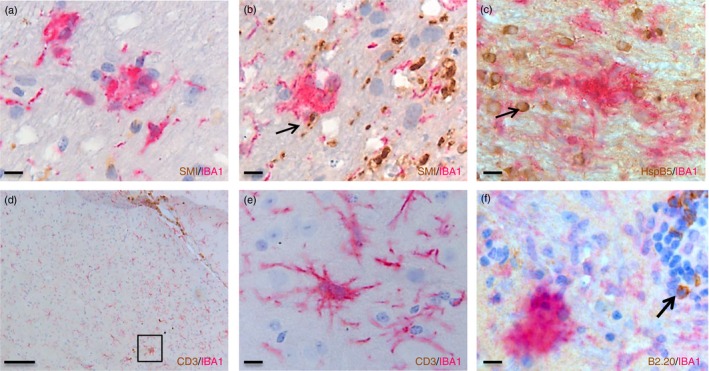
Microglia clusters during experimental autoimmune encephalomyelitis (EAE) are associated with axonal damage and heat‐shock protein B5+ (HspB5+) oligodendrocytes but not with CD3+ or B220+ lymphocytes. Clusters of IBA‐1+ cells in the absence of (a) or in close association with neurofilament heavy+ ( NF‐H+) axons (arrow in b) and HspB5+ oligodendrocytes (arrow in c) during EAE. Such clusters were not associated with CD3+ T cells (d); (e) is a zoomed‐in picture of the box in (d), or B220+ B cells (arrow in f). Scale bars a,b,c,d,f = 10 μm, e = 100 μm
Discussion
Current immune‐modulating therapies for MS reduce the frequency of clinical relapses, yet fail to impact on the irreversible neurodegeneration driving progressive forms of MS1 indicating an urgent unmet clinical need. The finding that age is an important risk factor for conversion from RRMS to SPMS and for the onset of PPMS indicates that senescence of the immune system and CNS are important contributors to disease progression.28 In support of a role for ageing in MS is the finding that paediatric MS is typically relapsing–remitting in nature and only rarely involves SPMS or PPMS.29
To study pathogenic mechanisms and to support the development of potential therapeutic strategies in MS, EAE is a widely used preclinical model, but EAE models have so far provided only limited insight into the pathophysiology of progressive forms of MS. This is in part due to the frequent focus on acute EAE in C57BL/6 mice that do not develop secondary progressive disease.30 In contrast, we have previously reported that young (8–12 weeks) Biozzi ABH mice exhibit RREAE followed by SPEAE. That this model reiterates key features of SPMS including demyelination, gliosis and neuronal and axonal loss23 indicates that this model is more suited to examine mechanisms contributing to progression of MS. To better understand the role of age and development of progressive disease we compared the EAE susceptibility of differently aged Biozzi ABH mice. In contrast to the RREAE and SPEAE that occurs in young mice, we show that juvenile mice (2 weeks old) do not develop clinical signs of EAE whereas old mice (12 months) develop monophasic neurodegenerative EAE from the onset. These different clinical forms were reflected by distinct pathology in the CNS, indicating that repeated episodes of inflammation and neurological disability observed in SPEAE can be distinguished from progressive neurological disease that developed in aged individuals.
That Biozzi mice < 2 weeks of age are fully resistant to EAE induction and 3‐week‐old mice partially resistant may in part reflect the inadequate functional development of the immune system and pathogenic T cells including their ability to produce pro‐inflammatory cytokines31 or their ability to migrate into the CNS32 at that young age. On the other hand, myelin‐reactive T‐cells from 5‐week‐old mice have been shown to be pathogenic following adoptive transfer into older mice,33 indicating that also the immaturity of the innate immune system contributes to EAE resistance in young animals.31 Compared with young (8–12 week) Biozzi mice that develop RREAE followed by SPEAE, disease induction in 12‐month‐old mice triggered marked progressive EAE from onset. During ageing, thymic involution amplifies autoreactive T cells and so the propensity to develop autoimmunity.34 Whether the progressive failure of negative selection upon ageing, facilitated by decreased expression of the autoimmune regulator or impaired generation of regulatory T cells, additionally plays a role in the exclusive development of PEAE in old mice requires further study. In addition, the age‐related decline in antigen‐presenting cell function and myeloid‐derived suppressor cells, coupled with the increased expression of MHC‐class II antigens and co‐stimulatory molecules31 may explain the resistance against EAE in young mice and the susceptibility to more severe disease in old mice.
That CD4+ cells are more resistant to apoptosis and regulatory T‐cell suppression suggests that immunosenescence in MS resembles dysfunctions associated with ageing,35 possibly explaining the higher numbers of T cells in PEAE in old mice. However, in contrast to aEAE and PEAE, perivascular infiltrates in the CNS declined with disease progression and T cells in the CNS during SPEAE were significantly reduced. Whether this is due to T‐cell exhaustion as described in EAE in SJL mice36 or increased T‐cell apoptosis in the CNS is unclear. However, SPEAE continues despite ablation of T cells indicating that disease progression is independent of peripheral T‐cell responses.24 Moreover, rather than the manifestation of adaptive immune responses, the SPEAE lesion, like those in PEAE, is dominated by activated microglia25, 26 underscoring the role of innate immunity in neurodegenerative diseases. In old mice with PEAE, this may reflect enhanced priming of microglia and an increased expression of pro‐inflammatory cytokines, as have been observed in old mice and Ercc1 mutant mice, a DNA repair‐deficient mouse model that displays features of accelerated ageing in multiple tissues including the CNS.37 Hence, atrophy of the thymus as well as alterations in peripheral immune factors are likely to play a concerted role in the now well‐recognized phenomenon of ‘inflamm‐ageing’, such as an increased propensity for tissue‐damaging inflammatory processes to develop upon ageing.11 Interestingly, in comparison to perivascular infiltrates and T‐cell numbers, the numbers of MCassociated with axonal damage and HspB5+ oligodendrocytes in NAWM and NAGM did not decline with age or during disease progression in RREAE, SPEAE and PEAE. Higher numbers of MC in the white matter were associated with remission, suggesting that such clusters, like so‐called pre‐active lesions in MS, may perform a regulatory role.38
Remyelination in the CNS is key to repair and recovery from relapses in MS and although extensive remyelination is observed in MS lesions, it is frequently insufficient to fully restore efficient conduction.39 The causes of incomplete remyelination in MS are unknown but may be a function of age40 as well as recurrent episodes of myelin damage. The resistance to EAE of young mice and increased susceptibility of old mice to PEAE may likewise also be a function of the regenerative capability of a youthful CNS. This was recently shown using lysolecithin‐induced demyelination in dual‐colour reporter mice, by revealing differences in the proliferation, recruitment and differentiation of dorsal and ventral oligodendrocyte progenitors as a factor of age.41 In line with this, heterochronic parabiosis has shown that exposure of aged mice to a youthful systemic milieu stimulates repair following toxin‐induced demyelination with lysolecithin.8 Pusic et al.42 reported this to be the effect of the production of peripheral exosomes that stimulate OPC differentiation and myelination. Whether such factors can prevent or ameliorate SPEAE and PEAE in old mice requires further investigation.
In summary this study supports the notion that ageing as well as recurrent episodes of inflammation in the CNS contribute to experimentally induced progressive neurological disease. Whereas RRMS is characterized by higher disease activity and inflammation in the CNS, pathology studies do not distinguish between PPMS and SPMS. Our observation that three different manifestations of EAE can be induced in the same mouse strain, and are dependent on the age of disease induction, underscores the notion that age and its response to insults plays a major role in the clinical manifestation of autoimmune demyelinating disease. Hence, the models described here not only provide relevant preclinical models to develop therapies for progressive forms of MS, but they also underscore the existence of different pathological features of neurodegeneration and progressive disease. Clarification of the differences between the pathological mechanisms that contribute to PEAE, SPEAE and RREAE may be useful to uncover the key factors that contribute to progressive forms of MS.
Disclosures
JMvN holds equity in Delta Crystallon BV. The other authors have no conflicts of interest to report.
Supporting information
Figure S1. Numbers of IBA‐1+ microglia/macrophages as represented by the number of positive pixels (see Materials and methods for description) in the spinal cord are higher in the white (WM) and grey (GM) matter of young mice with acute experimental autoimmune encephalomyelitis (aEAE) and progressive EAE (PEAE) in old mice compared with control age‐matched mice *** P < 0·001. Mean ± SEM (n = 8).
Table S1. Clinical history of mice with experimental autoimmune encephalomyelitis.
Acknowledgements
LAN Peferoen, M Breur, S van de Berg, R Peferoen‐Baert and G Pryce performed the experiments. LAN Peferoen, M Breur and S Amor analysed the data. S Amor JM, van Noort, D Baker, LAN Peferoen, HWGM Boddeke and P van der Valk designed the study and wrote the paper.
We gratefully acknowledge the Progressive Multiple Sclerosis Alliance for financial support.
References
- 1. Comi G. Disease‐modifying treatments for progressive multiple sclerosis. Mult Scler 2013; 19:1428–36. [DOI] [PubMed] [Google Scholar]
- 2. Duncan GW. The aging brain and neurodegenerative diseases. Clin Geriatr Med 2011; 27:629–44. [DOI] [PubMed] [Google Scholar]
- 3. Scalfari A, Neuhaus A, Daumer M, Ebers GC, Muraro PA. Age and disability accumulation in multiple sclerosis. Neurology 2011; 77:1246–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanai SA, Saini V, Benedict RH, Zivadinov R, Teter BE, Ramanathan M et al Aging and multiple sclerosis. Mult Scler 2016; 22:717–25. [DOI] [PubMed] [Google Scholar]
- 5. Schwartz M, Kipnis J, Rivest S, Prat A. How do immune cells support and shape the brain in health, disease, and aging? J Neurosci 2013; 33:17587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 2013; 39:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jurgens HA, Johnson RW. Dysregulated neuronal‐microglial cross‐talk during aging, stress and inflammation. Exp Neurol 2012; 233:40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruckh JM, Zhao J‐W, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ et al Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 2012; 10:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wong WT. Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci 2013; 7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harry GJ. Microglia during development and aging. Pharmacol Ther 2013; 139:313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E et al Inflamm‐aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000; 908:244–54. [DOI] [PubMed] [Google Scholar]
- 12. De Martinis M, Franceschi C, Monti D, Ginaldi L. Inflamm‐ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett 2005; 579:2035–9. [DOI] [PubMed] [Google Scholar]
- 13. Qi Q, Zhang DW, Weyand CM, Goronzy JJ. Mechanisms shaping the naïve T cell repertoire in the elderly – thymic involution or peripheral homeostatic proliferation? Exp Gerontol 2014; 54:71–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Popescu BFG, Pirko I, Lucchinetti CF. Pathology of multiple sclerosis: where do we stand? Continuum (Minneap Minn) 2013; 19:901–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Groot CJ, Bergers E, Kamphorst W, Ravid R, Polman CH, Barkhof F et al Post‐mortem MRI‐guided sampling of multiple sclerosis brain lesions: increased yield of active demyelinating and (p)reactive lesions. Brain 2001; 124(Pt 8):1635–45. [DOI] [PubMed] [Google Scholar]
- 16. Van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L et al Clusters of activated microglia in normal‐appearing white matter show signs of innate immune activation. J Neuroinflammation 2012; 9:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013; 125:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L et al α B‐crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol 2010; 69:694–703. [DOI] [PubMed] [Google Scholar]
- 19. Peferoen LAN, Vogel DYS, Ummenthum K, Breur M, Heijnen PDAM, Gerritsen WH et al Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J Neuropathol Exp Neurol 2015; 74:48–63. [DOI] [PubMed] [Google Scholar]
- 20. Vogel DYS, Vereyken EJF, Glim JE, Heijnen PDAM, Moeton M, van der Valk P et al Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation 2013; 10:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bsibsi M, Peferoen LAN, Holtman IR, Nacken PJ, Gerritsen WH, Witte ME et al Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN‐γ and α B‐crystallin. Acta Neuropathol 2014; 128:215–29. [DOI] [PubMed] [Google Scholar]
- 22. Van der Star BJ, Vogel DYS, Kipp M, Puentes F, Baker D, Amor S. In vitro and in vivo models of multiple sclerosis. CNS Neurol Disord Drug Targets 2012; 11:570–88. [DOI] [PubMed] [Google Scholar]
- 23. Al‐Izki S, Pryce G, O'Neill JK, Butter C, Giovannoni G, Amor S et al Practical guide to the induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH mouse. Mult Scler Relat Disord 2012; 1:29–38. [DOI] [PubMed] [Google Scholar]
- 24. Pryce G, O'Neill JK, Croxford JL, Amor S, Hankey DJ, East E et al Autoimmune tolerance eliminates relapses but fails to halt progression in a model of multiple sclerosis. J Neuroimmunol 2005; 165:41–52. [DOI] [PubMed] [Google Scholar]
- 25. Hampton DW, Anderson J, Pryce G, Irvine K‐A, Giovannoni G, Fawcett JW et al An experimental model of secondary progressive multiple sclerosis that shows regional variation in gliosis, remyelination, axonal and neuronal loss. J Neuroimmunol 2008; 15:200–11. [DOI] [PubMed] [Google Scholar]
- 26. Baker D, O'Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL. Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol 1990; 28:261–70. [DOI] [PubMed] [Google Scholar]
- 27. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group . Animal research: reporting in vivo experiments: the ARRIVE guidelines. J Gene Med 2010; 12:561–3. [DOI] [PubMed] [Google Scholar]
- 28. Tutuncu M, Tang J, Zeid NA, Kale N, Crusan DJ, Atkinson EJ et al Onset of progressive phase is an age‐dependent clinical milestone in multiple sclerosis. Mult Scler 2013; 19:188–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bar‐Or A, Antel JP. Central nervous system inflammation across the age span. Curr Opin Neurol 2016; 29:381–7. [DOI] [PubMed] [Google Scholar]
- 30. Delarasse C, Smith P, Baker D, Amor S. Novel pathogenic epitopes of myelin oligodendrocyte glycoprotein induce experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunology 2013; 140:456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hertzenberg D, Lehmann‐Horn K, Kinzel S, Husterer V, Cravens PD, Kieseier BC et al Developmental maturation of innate immune cell function correlates with susceptibility to central nervous system autoimmunity. Eur J Immunol 2013; 43:2078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Djikić J, Nacka‐Aleksić M, Pilipović I, Kosec D, Arsenović‐Ranin N, Stojić‐Vukanić Z et al Age‐related changes in spleen of Dark Agouti rats immunized for experimental autoimmune encephalomyelitis. J Neuroimmunol 2015; 15:123–35. [DOI] [PubMed] [Google Scholar]
- 33. Smith ME, Eller NL, McFarland HF, Racke MK, Raine CS. Age dependence of clinical and pathological manifestations of autoimmune demyelination. Implications for multiple sclerosis. Am J Pathol 1999; 155:1147–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coder BD, Wang H, Ruan L, Su D‐M. Thymic involution perturbs negative selection leading to autoreactive T cells that induce chronic inflammation. J Immunol 2015; 194:5825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thewissen M, Linsen L, Somers V, Geusens P, Raus J, Stinissen P. Premature immunosenescence in rheumatoid arthritis and multiple sclerosis patients. Ann N Y Acad Sci 2005; 1051:255–62. [DOI] [PubMed] [Google Scholar]
- 36. Targoni OS, Baus J, Hofstetter HH, Hesse MD, Karulin AY, Boehm BO et al Frequencies of neuroantigen‐specific T cells in the central nervous system versus the immune periphery during the course of experimental allergic encephalomyelitis. J Immunol 2001; 166:4757–64. [DOI] [PubMed] [Google Scholar]
- 37. Raj DDA, Jaarsma D, Holtman IR, Olah M, Ferreira FM, Schaafsma W et al Priming of microglia in a DNA‐repair deficient model of accelerated aging. Neurobiol Aging 2014; 35:2147–60. [DOI] [PubMed] [Google Scholar]
- 38. Bsibsi M, Holtman IR, Gerritsen WH, Eggen BJL, Boddeke E, van der Valk P et al α‐B‐crystallin induces an immune‐regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol 2013; 72:970–9. [DOI] [PubMed] [Google Scholar]
- 39. Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H et al Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006; 129(Pt 12):3165–72. [DOI] [PubMed] [Google Scholar]
- 40. Goldschmidt T, Antel J, König FB, Brück W, Kuhlmann T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 2009; 72:1914–21. [DOI] [PubMed] [Google Scholar]
- 41. Crawford AH, Tripathi RB, Richardson WD, Franklin RJM. Developmental origin of oligodendrocyte lineage cells determines response to demyelination and susceptibility to age‐associated functional decline. Cell Rep 2016; 15:761–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pusic AD, Kraig RP. Youth and environmental enrichment generate serum exosomes containing miR‐219 that promote CNS myelination. Glia 2014; 62:284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Numbers of IBA‐1+ microglia/macrophages as represented by the number of positive pixels (see Materials and methods for description) in the spinal cord are higher in the white (WM) and grey (GM) matter of young mice with acute experimental autoimmune encephalomyelitis (aEAE) and progressive EAE (PEAE) in old mice compared with control age‐matched mice *** P < 0·001. Mean ± SEM (n = 8).
Table S1. Clinical history of mice with experimental autoimmune encephalomyelitis.