

Summary
Macrophage colony‐stimulating factor 1 (CSF‐1) plays a critical role in the differentiation of mononuclear phagocytes from bone marrow precursors, and maturing monocytes and macrophages exhibit increased expression of the CSF‐1 receptor, CSF1R. The expression of CSF1R is tightly regulated by transcription factors and epigenetic mechanisms. We previously showed that prostaglandin E2 and subsequent activation of protein kinase A (PKA) inhibited CSF1R expression and macrophage maturation. Here, we examine the DNA methylation changes that occur at the Csf1r locus during macrophage maturation in the presence or absence of activated PKA. Murine bone marrow cells were matured to macrophages by incubating cells with CSF‐1‐containing conditioned medium for up to 6 days in the presence or absence of the PKA agonist 6‐bnz‐cAMP. DNA methylation of Csf1r promoter and enhancer regions was assayed by bisulphite pyrosequencing. DNA methylation of Csf1r decreased during normal macrophage maturation in concert with an increase in Csf1r mRNA expression. Treatment with the PKA agonist inhibited Csf1r mRNA and protein expression, and increased DNA methylation at the Csf1r promoter. This was associated with decreased binding of the transcription factor PU.1 to the Csf1r promoter. Treatment with the PKA agonist inhibited the responsiveness of macrophages to CSF‐1. Levels of endogenous PKA activity decreased during normal macrophage maturation, suggesting that attenuation of this signalling pathway contributes to the increase in CSF1R expression during macrophage maturation. Together, these results demonstrate that macrophage maturation is accompanied by Csf1r hypomethylation, and illustrates for the first time the ability of PKA to increase Csf1r DNA methylation.
Keywords: colony‐stimulating factor‐1 receptor, DNA methyltransferase, fms, macrophage colony‐stimulating factor, protein kinase A
Abbreviation
- ChIP
chromatin immunoprecipitation
- CREB
cAMP response element binding
- CSF1R
CSF‐1 receptor
- CSF
colony‐stimulating factor
- DNMT
DNA methyltransferase
- Epac
exchange protein activated by cAMP
- FIRE
fms intronic regulatory element
- IL
interleukin
- LPS
lipopolysaccharide
- PGE2
prostanglandin E2
- PKA
protein kinase A
- TET
ten–eleven translocation
- TNF
tumour necrosis factor
- VASP
vasodilator‐stimulated phosphoprotein
Introduction
Circulating monocytes and tissue‐recruited macrophages are key defenders of host defence1 that differentiate from myeloid precursors in a highly regulated process.2 Macrophage colony‐stimulating factor 1 (CSF‐1) is an essential factor for monocyte/macrophage maturation.3 Mice with a natural mutation of Csf1 or with knockout of the Csf1 gene exhibit substantial reductions in tissue‐derived mononuclear phagocytes.4 CSF‐1 has furthermore been shown to increase macrophage proliferation, differentiation and activation (reviewed in refs 5, 6, 7). Impairment in CSF‐1 expression is associated with a variety of diseases including osteoporosis,8 atherosclerosis9 and Alzheimer's disease,10 demonstrating the critical importance of mature monocytes and macrophages in tissue homeostasis and disease prevention.
The actions of CSF‐1 are mediated by its receptor, CSF1R (also known as CSF‐1R, M‐CSF‐R, fms, CD115), whose expression is essential for mononuclear cell development and maturation.11 CSF1R is expressed at low levels in haematopoietic stem cells12, 13 and is selectively up‐regulated during monocyte differentiation.14 Increased expression and activation of CSF1R not only drives the differentiation of myeloid progenitors into mature mononuclear phagocytes, but can even reprogramme granulocytes and pre‐B and mature B lymphocytes into expressing markers of monocyte/macrophage lineage.15, 16, 17 As a reflection of the critical importance of CSF1R, expression of CSF1R is regulated at both the transcriptional and post‐transcriptional levels.18, 19 Studies have shown that the transcription factor PU.1 occupies the Csf1r promoter even when Csf1r mRNA is expressed at low levels in haematopoietic stem cells.14, 20 Committed monocytes and macrophages exhibit enhanced expression of Csf1r mRNA due to transcription factor binding of the fms intronic regulatory element (FIRE), and remodelling of chromatin including histone acetylation and methylation.20 In addition, other epigenetic changes including DNA methylation are also observed, especially in non‐myelopoietic cells such as fibroblasts and T cells, which are hypermethylated at the Csf1r locus.16 Transcription of Csf1r mRNA is therefore a consequence of a highly coordinated interplay of transcription factors and epigenetic modifications focused at the Csf1r promoter and enhancer regions.
Prostaglandin E2 (PGE2) is a pleiotropic lipid mediator that inhibits many macrophage functions including phagocytosis and bacterial killing.21 Studies decades ago showed that PGE2,22, 23 and other cAMP‐elevating agents,24 also inhibit CSF‐1‐induced macrophage differentiation, proliferation and expression of urokinase plasminogen activator, but the operative mechanism for this was unknown. We recently showed that PGE2, via signalling through the E prostanoid 2 receptor with subsequent increase in cAMP and protein kinase A (PKA) activity, inhibited monocyte/macrophage maturation, which was associated with a decrease in CSF1R expression.25 How CSF1R expression is inhibited by increased PKA activity is unknown. As DNA methylation remains one of the most fundamental epigenetic mechanisms and has been shown to vary significantly within the Csf1r locus between different cell types,16 we investigated how DNA methylation of Csf1r changes during normal macrophage maturation, and how DNA methylation is affected by PKA signalling. We found that DNA methylation of Csf1r decreased during macrophage maturation, and that exogenous activation of PKA resulted in an increase in the DNA methylation at the Csf1r promoter, which was associated with a decrease in PU.1 transcription factor binding and Csf1r mRNA expression. Our studies therefore identify an ability for PKA, one of the most fundamental canonical signalling molecules, to affect the DNA methylation of a key gene involved in monocyte/macrophage maturation.
Materials and methods
Cell culture and maturation of bone‐marrow‐derived monocytes/macrophages
Murine macrophages were matured from bone marrow precursors in vitro with the addition of recombinant CSF‐1, or conditioned medium from CSF‐1‐secreting L929 cells, for up to 6 days.26, 27 This well‐established approach results in relatively homogeneous primary macrophages that are not conditioned by a specific tissue microenvironment. Bone marrow cells were isolated under aseptic conditions by flushing the marrow cavities of femurs and tibiae of C57BL/6 mice purchased from Jackson Laboratory (Bar Harbor, ME). All animal studies were approved by the University of Michigan Committee on the Use and Care of Animals. Bone marrow cells were cultured for up to 6 days in RPMI‐1640 (Thermo Fisher Scientific, Waltham, MA) containing 30% conditioned medium from L929 cells as previously described.26, 27 L929, a murine fibrosarcoma cell line that synthesizes CSF‐1, was cultured to confluence in Dulbecco's modified Eagle medium (Thermo Fisher Scientific) with 10% fetal bovine serum (Hyclone, Logan, UT) supplemented with 100 U/ml of penicillin/streptomycin. To examine the effects of PKA activation, bone marrow cells were treated with or without the selective PKA agonist 6‐bnz‐cAMP (500 μm; Axxora, Farmingdale, NY) at day 0 and again at day 3. In some experiments, cells were treated at day 0 with 8‐pCPT‐2′‐O‐Me‐cAMP (500 μm; Axxora), a selective agonist for the alternative cAMP effector, exchange protein activated by cAMP (Epac), or with IBMX (250 μm; BIOMOL, Farmingdale, NY), an inhibitor of the cAMP‐degradative enzyme phosphodiesterase.
For small interfering RNA (siRNA) experiments, cells were treated with 50 nm of control siRNA or combinations of either DNA methyltransferase (DNMT)‐3L siRNA alone, DNMT3L siRNA with DNMT3a siRNA (at 50 nm each), or DNMT3L siRNA with DNMT3b siRNA (at 50 nm each) (all from Qiagen, Valencia, CA) in OptiMEM (Thermo Fisher Scientific) at day 0 and again at day 1. In other experiments, cells were treated at day 0 and again at day 1 with 50 nm of control siRNA or siRNA against the ten–eleven translocation (TET) ‘demethylases’ TET2 or TET3 (all from Dharmacon, Lafayette, CO). Because cells were treated in OptiMEM, 20 ng/ml of CSF‐1 (PeproTech, Rocky Hill, NJ) was added to the medium to induce macrophage differentiation.
Flow cytometry
Cells were suspended in PBS with 2 mm EDTA and 0·5% fetal calf serum. Fc receptor‐mediated and non‐specific antibody binding were blocked by the addition of excess CD16/CD32 (BD Biosciences, San Jose, CA). Staining was performed at 4°C in the dark for 15 min. The following monoclonal antibodies were used at appropriate dilutions for staining: CD11b, (BD Biosciences Pharmingen), CSF1R (BioLegend, San Diego, CA), and F4/80 (eBioscience, San Diego, CA). A FACSCalibur flow cytometer (BD Biosciences) was used for flow cytometric characterization of cell populations, and data were analysed using flowjo software (TreeStar, Ashland, OR).
RT‐PCR
RNA was isolated from cells using Trizol (Thermo Fisher Scientific), made into cDNA, and amplified by quantitative real‐time PCR on the StepOnePlus Real‐Time PCR System (Applied Biosystems, Foster City, CA) using the SYBR‐green Master Mix (Applied Biosystems) and primers against murine Csf1r and β‐actin. The primers were as follows: for Csf1r: CGAGGGAGACTCCAGCTACA (forward) and AGAAGTCGAGACAGGCCTCA (reverse); for β‐actin: CTGCCTGACGGCCAAGTC (forward) and CAAGAAGGAAGGCTGGAAAAGAG (reverse). Primers and probes for DNMT1 (Mm01151063_m1), DNMT3a (Mm00432881_m1), DNMT3b (Mm01240113_m1), DNMT3L (Mm00457635_m1), TET1 (Mm01169087_m1), TET2 (Mm00524395_m1) and TET3 (Mm00805756_m1) were obtained from Applied Biosystems. Relative gene expression was determined by the comparative CT method (ΔΔCT) with β‐actin used as a reference gene.
Bisulphite sequencing
DNA was isolated from cells using the Dneasy kit (Qiagen), and bisulphite converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA) as per the manufacturers’ instructions. After bisulphite conversion, regions of the murine Csf1r promoter, FIRE, and 1 kb pair upstream of FIRE (FIRE –1 kb) were amplified with the following primers: for Csf1r promoter: AAGGGGAAGAGGAGTTAGTG (forward) and CCCCTTTTCTCCCCTTACCAT (reverse); for FIRE –1 kb: GATGAAGAATGGTTAGGATTAGGGTATTA (forward) and CCCTAAAAATCACAAAACACATCTTTAAAT (reverse); for FIRE: GATAATGGTTAGGAGGTTAGGGAAGTA (forward) and AACTCAAACCCCCTATCAAATC (reverse). All reverse primers were biotin‐labelled – which allowed the amplified products to be isolated with sepharose beads, denatured, and annealed to sequencing primers for pyrosequencing on the Pyromark Q24 (Qiagen). The sequencing primers for Csf1r promoter were: AGAGGAGTTAGTGTAATAGATA, for FIRE –1 kb: AAGGGAAGGTAGTGA, and for FIRE: GGGAAGTAGAAGTGAGA.
Chromatin immunoprecipitation
Bone marrow cells (1 × 107) were treated with or without the PKA agonist 6‐bnz‐cAMP for 3 days in L929 conditioned medium. To perform chromatin immunoprecipitation (ChIP), cells were incubated for 5 min in 1% formaldehyde to crosslink DNA with protein. Nuclear material was isolated using non‐ionic detergent buffers, and chromatin was sheared to 200‐ to 500‐bp fragments by 20 min of sonication using a Covaris sonicator (Covaris, Inc, Woburn, MA). Chromatin was incubated overnight with antibody to PU.1 (1 : 25, #2266 Cell Signaling, Danvers, MA) or isotype control. Chromatin was immunoprecipitated using the ActiveMotif ChIP‐IT Express Kit (ActiveMotif, Carlsbad, CA) according to the manufacturer's protocol and reverse cross‐linked. DNA was cleaned using the DNA cleanup kit (Qiagen), and isolated DNA was amplified by real‐time PCR using primers targeting a CpG dense and a non‐CpG dense region of the Csf1r promoter. The primer sequences for the non‐CpG dense region (primer set 1) were CAGGAACAGACTTGAAGCGT (forward) and CCGCTGAACCAGGTCTTCTTA (reverse) and for the CpG dense region (primer set 2) were ATCCCCTGGAGGCTATGGAG (forward) and TCTTTGCAACACTCCCCCAG (reverse).
Immunoblotting
Cell lysates were collected in lysis buffer (PBS containing 1% Nonidet P‐40, 0·5% sodium deoxycholate, 0·1% SDS) supplemented with protease inhibitor (Roche, Basel, Switzerland) and phosphatase inhibitor (EMD Chemicals, Gibbstown, NJ) cocktails. Proteins were resolved by SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with antibodies against the PKA‐dependent phosphoproteins phosphorylated vasodilator stimulated phosphoprotein (VASP, 1 : 1000, Cell Signaling) or phosphorylated cAMP response element binding (CREB, 1 : 1000 Cell Signaling), and the loading control α‐tubulin (1 : 1000, Sigma‐Aldrich, St Louis, MO). Membranes were incubated with the appropriate horseradish peroxidase‐conjugated secondary antibodies and visualized using enhanced chemiluminescence reagent (GE Healthcare, Chalfont St Giles, UK). Densitometry was performed on visualized bands using image J from the National Institutes of Health (Bethesda, MD).
cAMP assay
Bone marrow cells were cultured in L929 supernatant for 6 days; cells were lysed in 0·1 m HCl and cAMP levels were assayed using a colorimetric kit from Enzo (Farmingdale, NY).
Interleukin‐6, interleukin‐10 and tumour necrosis factor‐α ELISA
Bone marrow cells were cultured in L929 supernatant for 6 days in the presence or absence of 500 μm PKA agonist (6‐bnz‐cAMP). Non‐adherent cells were washed, and adherent cells were re‐plated at equal number for 1 hr stimulation with 20 ng/ml of recombinant CSF‐1 (Peprotech, Rocky Hill, NJ) followed by 24 hr of stimulation with 100 ng/ml of lipopolysaccharide (LPS) (Sigma). Cell supernatants were cleared of cells by centrifugation, and supernatant interleukin‐6 (IL‐6), IL‐10 and tumour necrosis factor‐α (TNF‐α) levels were assayed by ELISA (R&D Systems, Minneapolis, MN).
Statistical analysis
Data were analysed on graphpad prism 6·0 (GraphPad Prism Software, San Diego, CA) using analyses of variance or Student's t‐test, as appropriate, with P < 0·05 defined as statistically significant. Data are expressed as mean ± SE measurement.
Results
Maturation of macrophages was associated with a decrease in DNA methylation at the Csf1r promoter and FIRE
Transcription of Csf1r has been previously shown to occur at low levels in haematopoietic stem cells14 and to be selectively up‐regulated in mature macrophages, with its up‐regulation mediated by transcription factor assembly at the promoter and FIRE enhancer region.20 Alterations in histone modifications have been shown to contribute to the increased expression of CSF1R during macrophage maturation.20 There are six CpG loci closely clustered within a 100‐bp region in the promoter/exon 2 region of Csf1r, and a relatively high density of CpG loci clustered within 200‐bp regions located in the FIRE enhancer as well as 1 kb upstream of FIRE (FIRE –1 kb) (Fig. 1a). Although methylation‐sensitive restriction enzymes were used to show on average that DNA methylation was low in the promoter and FIRE regions of both myeloid progenitors and mature macrophages,16 we used bisulphite sequencing to more sensitively quantify the DNA methylation levels at each of the CpG loci within the promoter, FIRE and FIRE –1 kb during normal macrophage maturation. DNA methylation levels decreased at each of the CpG sites within the promoter (Fig. 1b) and FIRE –1 kb (Fig. 1c) regions as macrophages matured over time. Certain CpG sites in the FIRE enhancer region also exhibited decreased methylation during macrophage maturation, although baseline levels of methylation were lower in the FIRE versus FIRE –1 kb and promoter regions (Fig. 1d).
Figure 1.

The DNA methylation of Csf1r is decreased during macrophage maturation. Cells from murine bone marrow were matured in L929 conditioned medium and DNA was collected from cells at days 0, 3 and 5. Bisulphite sequencing, as described in the Materials and methods, was performed to assess the DNA methylation of CpG sites within the Csf1r gene, as illustrated (a). DNA methylation of CpG sites were analysed within the gene promoter (b), 1 kb upstream of fms intronic regulatory element (FIRE –1 kb) (c), and FIRE (d) regions (n = 3). *P < 0·05.
Activation of PKA inhibited Csf1r mRNA expression and increased DNA methylation at the Csf1r promoter
We previously demonstrated that PGE2 signalled through PKA to inhibit macrophage maturation and that treatment of bone marrow cells at day 0 with a PKA agonist, 6‐bnz‐cAMP, resulted in decreased numbers of CSF1R‐expressing F4/80+ CD11b+ mature macrophages.25 To determine whether PKA operated at the transcriptional or post‐transcriptional level to inhibit expression of CSF1R, bone marrow cells were treated at day 0 with the PKA agonist 6‐bnz‐cAMP, and CSF1R protein and mRNA levels were assayed at days 3 and 6 during macrophage maturation. As expected, cell surface expression of CSF1R increased between days 3 and 6 during macrophage maturation, and expression was inhibited in the presence of a PKA agonist (Fig. 2a). Examination of mRNA levels indicated that the increase in CSF1R protein expression during maturation and its inhibition with a PKA agonist were accompanied by corresponding changes in Csf1r mRNA (Fig. 2b).
Figure 2.

Expression of Csf1r mRNA was decreased in bone marrow cells treated with a protein kinase A (PKA) agonist. Cells from murine bone marrow were matured in L929 conditioned medium for 3 or 6 days in the presence or absence of the PKA agonist 6‐bnz‐cAMP (500 μm). (a) Macrophage colony stimulating factor 1 receptor (CSF1R) cell surface protein expression was assayed by flow cytometry; the mean fluorescence intensity from a representative experiment of three independent experiments is shown. (b) Csf1r mRNA levels were assayed by RT‐PCR (n = 3). *P < 0·05.
Treatment with the PKA agonist was associated with DNA hypermethylation of Csf1r promoter and decreased PU.1 binding
We next examined the effect of activating PKA on the DNA methylation of the Csf1r promoter, FIRE –1 kb and FIRE regions. Treatment with the PKA agonist resulted in increased Csf1r DNA methylation, particularly within the promoter (Fig. 3a). A mild increase in DNA methylation was also noted in the FIRE –1 kb region (Fig. 3b), and no change in DNA methylation was observed at the FIRE enhancer region (Fig. 3c). To determine whether the increase in DNA methylation was specific to the activation of PKA, bone marrow cells were treated with 8‐pCPT‐2’‐O‐Me‐cAMP, a cAMP analogue that specifically activates the alternative cAMP effector, the cAMP‐activated guanine nucleotide exchange protein Epac, but not PKA. Just as we had previously shown that inhibition of macrophage maturation by cAMP was a specific consequence of the activation of PKA and not Epac,25 treatment with the Epac agonist did not increase the DNA methylation of Csf1r promoter as was observed with the PKA agonist (Fig. 3d).
Figure 3.
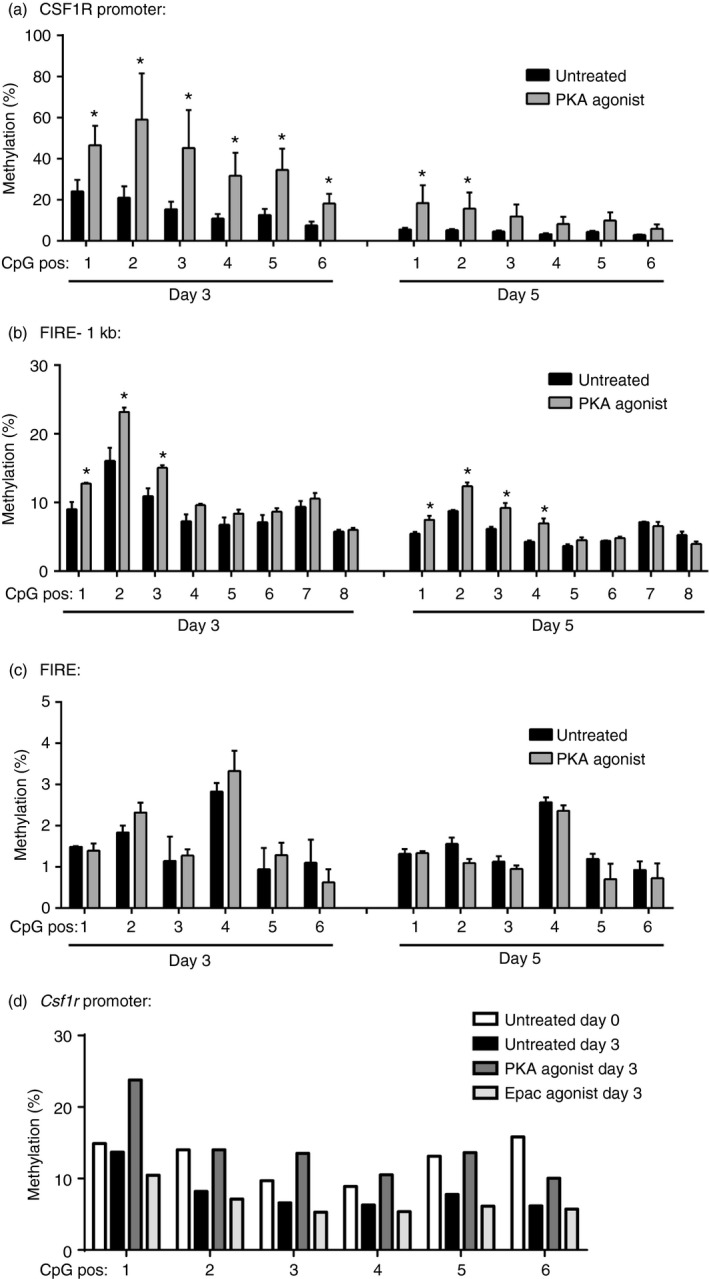
Treatment of bone marrow cells with a protein kinase A (PKA) agonist was associated with an increase in Csf1r DNA methylation. (a–c) Cells from murine bone marrow were matured in L929 conditioned medium for 3 or 5 days in the presence or absence of the PKA agonist 6‐bnz‐cAMP (500 μm). The DNA methylation of CpG sites within the Csf1r promoter (a), FIRE –1 kb (b), and FIRE (c) were assayed by pyrosequencing (n = 3). *P < 0·05. (d) Bone marrow cells were treated with either the PKA agonist 6‐bnz‐cAMP (500 μm), the exchange protein activated by cAMP (Epac) agonist 8‐pCPT‐2′‐O‐Me‐cAMP (500 μm), or vehicle control and DNA methylation within the Csf1r promoter was assayed. Shown is a representative result from two independent experiments.
PU.1 is a critical transcription factor involved in monocyte/macrophage differentiation28, 29 and it up‐regulates the expression of CSF1R.30, 31 We next sought to determine whether binding of PU.1 to the Csf1r promoter was associated with the changes in DNA methylation observed after treatment with a PKA agonist. Treatment of bone marrow cells with the PKA agonist did not significantly affect expression of PU.1 (data not shown). We next performed ChIP using an antibody specific to PU.1 and observed that there was decreased PU.1 binding to the Csf1r promoter in cells treated with the PKA agonist (Fig. 4). The decrease in PU.1 binding was specific to the region where DNA hypermethylation was observed; by comparison, an upstream region without DNA hypermethylation showed no difference in PU.1 binding between cells treated with or without PKA. These results demonstrate that activation of PKA leads to both DNA hypermethylation and decreased PU.1 binding at specific regions within the Csf1r promoter.
Figure 4.

Treatment of cells with the protein kinase A (PKA) agonist resulted in decreased PU.1 binding at the Csf1r promoter. Cells from murine bone marrow were matured in L929 conditioned medium for 3 days in the presence or absence of the PKA agonist 6‐bnz‐cAMP (500 μm). Chromatin was isolated and immunoprecipitated using an antibody to PU.1 or isotype control. Real‐time PCR was performed using primers illustrated in (a), and the relative enrichment from cells treated with or without the PKA agonist is shown for primer set 1 (b) and primer set 2 (c) (n = 3). *P < 0·05.
Macrophage maturation was associated with a decrease in endogenous PKA activity
Protein kinase A can be activated by a myriad of signals and, given the profound ability of an exogenous PKA agonist to inhibit macrophage maturation, we sought to determine whether normal macrophage maturation was associated with a decrease in endogenous levels of PKA activity. Phosphorylation of the PKA substrates VASP and CREB are commonly used readouts of intracellular PKA activity, and levels of phosphorylated VASP decreased over time during normal macrophage maturation (Fig. 5a). A similar decrease in phosphorylated CREB was also observed (Fig. 5b) suggesting that diminished activity of PKA accompanies normal macrophage maturation. PKA is activated by cAMP, and intracellular levels of cAMP were likewise lower at days 3 and 5 during macrophage maturation compared with day 0 (Fig. 5c). Treatment of bone marrow cells with the phosphodiesterase inhibitor IBMX, which inhibits cAMP degradation, resulted in both higher levels of cAMP and decreased expression of CSF1R (Fig. 5d,e). This finding inversely links cAMP levels to expression of CSF1R, indicating that the increased expression of CSF1R during macrophage maturation is attributable to the decrease in endogenous levels of cAMP and PKA activity.
Figure 5.

Macrophage maturation was associated with a decrease in endogenous cAMP levels and protein kinase A (PKA) activity. Cells from murine bone marrow were matured to macrophages in L929 conditioned medium over 5–6 days. Levels of phosphorylated vasodilator‐stimulated phosphoprotein (VASP) (a) and phosphorylated cAMP responsive element binding (CREB) protein (b) were assayed by immunoblot. Representative blot (n = 3) is shown. (c) Levels of cAMP were assayed from the lysates of maturing cells at days 0, 3 and 5. Results are normalized to control levels (n = 3). Bone marrow cells were given IBMX (250 μm), an inhibitor of the cAMP‐degradative enzyme phosphodiesterase, for 6 days and levels of cAMP (d) and CSF1R mRNA (e) were assayed (n = 3). *P < 0·05.
Treatment of bone marrow cells with a PKA agonist inhibited macrophage maturation and cytokine generation
To determine the functional consequence of inhibiting macrophage maturation on generation of immunoregulatory cytokines – a property of mature macrophages – adherent bone marrow cells treated with or without 6‐bnz‐cAMP (500 μm) for 6 days were re‐plated, washed and treated for 1 hr with CSF‐1 (20 ng/ml) followed by 24 hr of LPS (100 ng/ml), and IL‐6, IL‐10 and TNF‐α levels were assayed. Cells treated with the PKA agonist for 6 days demonstrated markedly reduced ability to synthesize IL‐6, IL‐10 and TNF‐α in response to CSF‐1 in the absence or presence of LPS (Fig. 6). As previously reported, treatment with the PKA agonist did not affect cell viability,25 suggesting that the impaired responsiveness to CSF‐1 and LPS is a consequence of lower CSF1R expression and generally impaired bone marrow macrophage maturation, and not due to decreased cell numbers or apoptosis.
Figure 6.

Inhibition of macrophage maturation with a protein kinase A (PKA) agonist attenuated the generation of immunoregulatory cytokines in response to macrophage colony stimulating factor 1 (CSF‐1) and lipopolysaccharide (LPS). Bone‐marrow‐derived monocytes/macrophages (BMDM) were matured in L929 conditioned medium for 5 days in the presence or absence of the PKA agonist 6‐bnz‐cAMP (500 μm). Cells were then re‐plated and treated for 1 hr ± MCSF (20 ng/ml) and then subsequently for 24 hr ± LPS (100 ng/ml) and supernatants were assayed for levels of interleukin‐6 (IL‐6) (a), IL‐10 (b), and tumour necrosis factor‐α (TNF‐α) (c) (n = 3 for each experiment).
Expression of TET and DNMT proteins during macrophage maturation
The DNMTs are responsible for the addition of methyl groups to DNA and consist of three catalytically active isoforms – DNMT1, DNMT3a and DNMT3b – and DNMT3L, which is catalytically inactive, but facilitates the actions of DNMT3a and DNMT3b. TET enzymes were recently discovered to add hydroxyl groups to methylated cytosine,32, 33 resulting in base‐excision repair and active demethylation. Given that macrophage maturation is associated with a decrease in Csf1r methylation, and that methylation is increased when PKA is activated, we examined the expression of various TET and DNMT isoforms during macrophage maturation in the presence or absence of a PKA agonist. Concurrent with a decrease in Csf1r methylation, TET2 and TET3 expression increased during macrophage maturation, which was inhibited in the presence of a PKA agonist (Fig. 7a–c). In parallel, expression of DNMT1, DNMT3b and DNMT3L decreased during macrophage maturation, but only DNMT3L expression increased in the presence of the PKA agonist (Fig. 7d–g).
Figure 7.

Protein kinase A (PKA) altered expression of DNA methyltransferases (DNMTs) and Ten–Eleven Translocation (TET) proteins during macrophage maturation. Bone marrow cells were treated with or without the PKA agonist 6‐bnz‐cAMP (500 μm) during macrophage maturation and levels of (a) TET1, (b) TET2, (c) TET3, (d) DNMT1, (e) DNMT3a, (f) DNMT3b and (g) DNMT3L mRNA were assayed by RT‐PCR; n = 6, *P < 0.05 relative to Day 0; #P < 0.05 between treatments with and without the PKA agonist.
Silencing TET and DNMT expression did not significantly alter Csf1r expression during macrophage maturation or during treatment with a PKA agonist
As TET2 and TET3 expression increased during macrophage maturation and their actions are associated with demethylation, we sought to determine whether silencing TET2 or TET3 would result in persistent hypermethylation of Csf1r and decreased Csf1r mRNA levels during maturation. Bone marrow cells were treated with control, TET2 or TET3 siRNA at both day 0 and day 1, and levels of Csf1r mRNA were determined at day 3. Neither silencing of TET2 nor TET3 inhibited the increase of Csf1r expression at day 3 of maturation (Fig. 8a,b).
Figure 8.

Silencing of the Ten–Eleven Translocation proteins TET2, TET3, or DNA methyltransferase 3L (DNMT3L) did not affect expression of Csf1r in the presence or absence of protein kinase A (PKA). Bone marrow cells were treated with control small interfering RNA (siRNA) or siRNA targeted against TET2 or TET3, as described in Materials and methods, and levels of TET2 and TET3 mRNA (a) were assayed by RT‐PCR (n = 3). *P < 0·05 relative to control siRNA. **P < 0·01 relative to control siRNA. (b) Levels of Csf1r mRNA were assayed by RT‐PCR in cells treated with TET2 or TET3 siRNA (n = 3). *P < 0·05 relative to control siRNA. NS, non‐significant. (c–d) Bone marrow cells were treated with control siRNA or siRNA targeted against DNMT3L in the presence or absence of the PKA agonist 6‐bnz‐cAMP (500 μm), as described in the Materials and methods. Levels of DNMT3L mRNA (c, n = 3, *P < 0·05 relative to control siRNA) and Csf1r (d, n = 4, *P < 0·05 relative to day 0) were assayed by RT‐PCR.
DNMT3L was the only DNMT isoform whose expression decreased with time as early as 3 days and also increased in the presence of a PKA agonist, making it the best candidate DNMT whose dynamic alterations in expression might explain both the hypomethylation of Csf1r observed during macrophage maturation and the hypermethylation of Csf1r seen during treatment with a PKA agonist. We therefore used siRNA to silence expression of DNMT3L to determine if this is sufficient to abolish the actions of the PKA agonist. Although we were able to achieve a robust knockdown of DNMT3L expression with siRNA (Fig. 8c), the PKA agonist was still able to decrease expression of CSF1R, suggesting that the increase in DNMT3L by PKA is not responsible for Csf1r hypermethylation (Fig. 8d). As DNMT3L itself is enzymatically inactive and binds to DNMT3a and DNMT3b to stimulate their activity,34 we also examined levels of Csf1r mRNA when DNMT3a and DNMT3L, and when DNMT3b and DNMT3L were silenced together. Neither the combined silencing of DNMT3a and DNMT3L nor the combined silencing of DNMT3b and DNMT3L was sufficient to inhibit the ability of the PKA agonist to suppress Csf1r expression (data not shown).
Discussion
CSF1R is the major receptor for CSF‐1, and up‐regulation of CSF1R is essential for monocyte/macrophage development and differentiation.11 Prostaglandin E2 and other cAMP‐elevating agents were described many years ago to inhibit CSF‐1‐mediated macrophage responses,22, 23, 24 and we recently showed that through PKA signalling, these mediators inhibit CSF1R expression and macrophage maturation.25 Here, we show that activation of PKA led to an increase in the DNA methylation of the Csf1r gene at both the promoter and FIRE –1 kb regions. This was associated with a decrease in PU.1 binding to the gene promoter and a decrease in Csf1r mRNA levels. Macrophage maturation was accompanied by both a decrease in basal levels of Csf1r DNA methylation and a decrease in intrinsic PKA activity, suggesting that endogenous PKA activity in bone marrow progenitors play a role in suppressing the expression of Csf1r. These results demonstrate how PKA signalling may influence the dynamic changes in DNA methylation of key genes crucial to monocyte/macrophage differentiation.
Protein kinase A was one of the first‐described canonical signalling kinases, and has since been shown to modulate a range of diverse cellular functions including proliferation, differentiation and survival,35, 36 often through the activation of other downstream mediators and transcription factors such as CREB. Our results demonstrated that PKA is also capable of inducing DNA methylation, in this case, at the Csf1r gene locus. A previous study noted that PKA activation increased DNA methylation and histone methylation in embryonic stem cells.37 Ours is the first to show that PKA activation affected DNA methylation in bone marrow cells.
The regulation of CSF1R expression is complex, and levels of Csf1r mRNA levels are dependent on the dynamic interaction of transcription factors, chromatin remodelling, and DNA methylation changes at the Csf1r promoter and enhancer regions.13, 18 Transcription factors such as PU.1 have been identified to sit poised on the Csf1r promoter even in myeloid progenitors, which express low levels of Csf1r mRNA,14 and through chromatin remodelling and transcription factor binding at the FIRE enhancer region, transcription of Csf1r is up‐regulated in mature macrophages.20 These studies have suggested that DNA methylation plays a minor role in regulating Csf1r expression in haematopoietic stem cells as methylation levels at the Csf1r promoter and FIRE enhancer regions were low in early myeloid progenitors.16 Our study, however, suggests that the DNA methylation levels of Csf1r in bone marrow cells are readily measurable and that DNA methylation levels in fact decrease over time with macrophage maturation. Heterogeneity in bone marrow cells and our use of a more sensitive, quantitative assay for DNA methylation at specific CpG loci may explain differences between our data and other studies.
Our results showed that decreased DNA methylation levels are associated with increased Csf1r mRNA expression during maturation and that hypermethylation in the presence of increased PKA activity is associated with its decreased expression. We recognize that we cannot ascertain whether DNA methylation changes are responsible for Csf1r expression, or whether DNA methylation changes are merely a consequence of changes in transcription factors and chromatin modifications that regulate Csf1r expression. Treatment of cells with the PKA agonist inhibited PU.1 binding to the Csf1r promoter, and examination of other possible chromatin modifications induced by the PKA agonist was beyond the scope of our study. Expression of PU.1 was not significantly affected by treatment with the PKA agonist, although it has been reported that increased cAMP levels affect PU.1 transcription38 and decrease PU.1‐DNA binding in other experimental contexts.39, 40, 41 Precedent for the ability of DNA methylation to regulate Csf1r expression, however, was best exemplified in fibroblasts, T cells and leukaemic cells that exhibited high levels of Csf1r methylation, which correlated strongly with inhibition of Csf1r expression.16, 42, 43
We were unable to determine the relative importance of increased Csf1r methylation versus inhibited demethylation in response to activation of PKA, as expression of various DNMT and TET enzymes all changed with time and with PKA treatment. TET2 and TET3, which participate in DNA hydroxymethylation and lead to demethylation,32, 33 were observed to increase with maturation, and decrease by the PKA agonist. Silencing of TET2 and TET3 during normal macrophage maturation, however, still resulted in an increase in Csf1r expression, suggesting that progressive hypomethylation of Csf1r cannot be explained purely by the increase in TET2 or TET3 during normal maturation. Notably, we were only able to effectively silence TET2 and TET3 by ~50%, and complete knockdown of these genes may be required to conclusively rule out a role for these enzymes in mediating Csf1r hypomethylation. In addition, TET2 and TET3 expression was not observed to increase until day 5, indicating that these enzymes may still have a role in later stages of macrophage maturation. Among the DNMT isoforms, only DNMT3L showed both a progressive loss of expression during maturation and an increase in expression with addition of the PKA agonist, suggesting its potential involvement in Csf1r demethylation during maturation and hypermethylation in the presence of active PKA. Targeted silencing of DNMT3L, however, did not affect Csf1r mRNA levels. Furthermore, combined silencing of both DNMT3L and its interacting partners DNMT3a/DNMT3b did not affect the ability of the PKA agonist to inhibit Csf1r expression. These data initially suggest that these enzymes are not required for the hypermethylation of Csf1r observed after the addition of a PKA agonist. However, an alternative hypothesis is that hypomethylation of Csf1r during normal maturation and hypermethylation in the presence of a PKA agonist may be the consequence of a more complex interplay between transcription factors and histone‐modifying enzymes that recruit DNMTs and TETs. PKA may interfere with PU.1 binding, which may indirectly result in Csf1r hypermethylation. Furthermore, the simultaneous silencing of DNMT and over‐expression of TET isoforms may be what is necessary to inhibit the hypermethylation of Csf1r by the PKA agonist. Further studies would also be needed to determine whether PKA induces global hypermethylation or hypomethylation of genes across the genome in macrophages and myeloid progenitors. Of note, we previously reported that PGE2 increased expression of DNMT3a in fibroblasts, resulting in global changes to DNA methylation in these cells.44 Changes in DNMT and TET isoform expression in myeloid progenitors may therefore affect the DNA methylation of other genes that may be important to macrophage maturation.
We treated cells with a PKA agonist to examine the effects of PKA activity on Csf1r methylation, but we also observed that macrophage maturation is itself associated with a decrease in endogenous PKA activity. PKA is traditionally activated by cAMP, which can be increased by various mediators including PGE2, which has been shown to inhibit a variety of macrophage functions including phagocytosis, bacterial killing and cytokine generation.21, 45 PGE2 also inhibits macrophage differentiation25 and haematopoiesis in general.46, 47 Our results demonstrating that PKA activation alters the DNA methylation profile of a key gene in macrophage maturation may partly explain how PGE2 inhibits haematopoiesis. However, we also previously showed that PGE2 levels do not decline during macrophage maturation,25 so the decline in endogenous cAMP levels and PKA activity during normal maturation is likely to reflect changes in some cAMP‐coupled receptor ligand other than PGE2. Epac is another intracellular cAMP effector molecule, and a selective Epac agonist failed to alter Csf1r DNA methylation levels. It is interesting to note that Epac expression has been reported to increase during monocyte/macrophage differentiation, and that Epac becomes functional in mature macrophages.48 Taken together with our contrasting results showing decreased endogenous PKA activity during macrophage differentiation, these findings suggest that there may be a coordinated switch in the effector molecules engaged by cAMP – from PKA to Epac – during macrophage differentiation. Finally, it should be mentioned that we did not examine whether PGE2 or PKA post‐transcriptionally regulates CSF1R expression, and a recent study suggests that PGE2 may activate CSF1R through phosphorylation.49
Mononuclear cell maturation in vitro was accomplished herein through a well‐established method described in the literature.26, 27 Freshly obtained bone marrow cells were not pre‐sorted in our studies and one might speculate that the effects of PKA activation on Csf1r methylation may be due to the expansion of haematopoietic stem cells or other non‐myeloid cell types rather than direct actions on mononuclear phagocytes. However, as adherence is characteristic of macrophages, wash steps and adherence purification during maturation ensured that macrophages were the predominant cell type in our studies. Although our studies were performed in vitro, we have previously shown that the ability of PKA to inhibit macrophage development is applicable in vivo in thioglycollate‐induced peritonitis, 25 with important consequences as demonstrated in mouse models of infection and asthma.50 Furthermore, the decrease in endogenous PKA activity observed during normal maturation indicates that mediators that regulate this signalling pathway are relevant for physiological homeostasis.
CSF‐1 activates many different macrophage functions, including phagocytosis, production of reactive oxygen species, chemotaxis and microbial killing,51 and understanding regulation of CSF1R and its promoter DNA methylation has functional implications beyond differentiation. We observed that inhibition of CSF1R expression by PKA was associated with diminished cytokine expression in the presence of LPS. Other studies have furthermore demonstrated that loss of CSF1R expression may contribute to oncogenic transformation in leukaemia.42, 52, 53 In conclusion, we show that the progressive increase in CSF1R expression during monocyte/macrophage maturation is accompanied by progressive demethylation at the Csf1r gene and that PKA inhibition of Csf1r mRNA expression correlated with DNA hypermethylation. PGE2, which activates PKA, can inhibit the maturation of macrophages; we now show for the first time that this intracellular signalling pathway is accompanied by modulation of DNA methylation and DNA methylation machinery. These studies demonstrate how epigenetic levels change during haematopoiesis and their regulation by PKA, and in a broader context, provide an illustration of how intracellular signalling pathways are capable of regulating DNA methylation patterns during differentiation and development.
Author contributions
ZZ and SKH designed the study, ZZ, AMS, and SKH performed the experiments, and ZZ, MPG, and SKH wrote the paper.
Disclosures
None.
Acknowledgements
This study was supported by research funding from the American Lung Association to Zbigniew Zaslona (senior postdoctoral fellowship), and the National Heart, Lung, and Blood Institute to Marc Peters‐Golden (HL058897) and Steven K. Huang (HL127203).
References
- 1. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol 2005; 5:953–64. [DOI] [PubMed] [Google Scholar]
- 2. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity 2014; 41:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Becker S, Warren MK, Haskill S. Colony‐stimulating factor‐induced monocyte survival and differentiation into macrophages in serum‐free cultures. J Immunol 1987; 139:3703–9. [PubMed] [Google Scholar]
- 4. Dai XM, Zong XH, Sylvestre V, Stanley ER. Incomplete restoration of colony‐stimulating factor 1 (CSF‐1) function in CSF‐1‐deficient Csf1op/Csf1op mice by transgenic expression of cell surface CSF‐1. Blood 2004; 103:1114–23. [DOI] [PubMed] [Google Scholar]
- 5. Chitu V, Stanley ER. Colony‐stimulating factor‐1 in immunity and inflammation. Curr Opin Immunol 2006; 18:39–48. [DOI] [PubMed] [Google Scholar]
- 6. Hamilton JA. Colony‐stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008; 8:533–44. [DOI] [PubMed] [Google Scholar]
- 7. Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y et al Biology and action of colony‐stimulating factor‐1. Mol Reprod Dev 1997; 46:4–10. [DOI] [PubMed] [Google Scholar]
- 8. Wei S, Dai XM, Stanley ER. Transgenic expression of CSF‐1 in CSF‐1 receptor‐expressing cells leads to macrophage activation, osteoporosis, and early death. J Leukoc Biol 2006; 80:1445–53. [DOI] [PubMed] [Google Scholar]
- 9. Di Gregoli K, Johnson JL. Role of colony‐stimulating factors in atherosclerosis. Curr Opin Lipidol 2012; 23:412–21. [DOI] [PubMed] [Google Scholar]
- 10. Flanagan AM, Lader CS. Update on the biologic effects of macrophage colony‐stimulating factor. Curr Opin Hematol 1998; 5:181–5. [DOI] [PubMed] [Google Scholar]
- 11. Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S et al Targeted disruption of the mouse colony‐stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002; 99:111–20. [DOI] [PubMed] [Google Scholar]
- 12. Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C et al Multilineage gene expression precedes commitment in the hemopoietic system. Genes Dev 1997; 11:774–85. [DOI] [PubMed] [Google Scholar]
- 13. Tagoh H, Melnik S, Lefevre P, Chong S, Riggs AD, Bonifer C. Dynamic reorganization of chromatin structure and selective DNA demethylation prior to stable enhancer complex formation during differentiation of primary hematopoietic cells in vitro . Blood 2004; 103:2950–5. [DOI] [PubMed] [Google Scholar]
- 14. Tagoh H, Himes R, Clarke D, Leenen PJ, Riggs AD, Hume D et al Transcription factor complex formation and chromatin fine structure alterations at the murine c‐fms (CSF‐1 receptor) locus during maturation of myeloid precursor cells. Genes Dev 2002; 16:1721–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borzillo GV, Ashmun RA, Sherr CJ. Macrophage lineage switching of murine early pre‐B lymphoid cells expressing transduced fms genes. Mol Cell Biol 1990; 10:2703–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tagoh H, Schebesta A, Lefevre P, Wilson N, Hume D, Busslinger M et al Epigenetic silencing of the c‐fms locus during B‐lymphopoiesis occurs in discrete steps and is reversible. EMBO J 2004; 23:4275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sasmono RT, Ehrnsperger A, Cronau SL, Ravasi T, Kandane R, Hickey MJ et al Mouse neutrophilic granulocytes express mRNA encoding the macrophage colony‐stimulating factor receptor (CSF‐1R) as well as many other macrophage‐specific transcripts and can transdifferentiate into macrophages in vitro in response to CSF‐1. J Leukoc Biol 2007; 82:111–23. [DOI] [PubMed] [Google Scholar]
- 18. Bonifer C, Hume DA. The transcriptional regulation of the colony‐stimulating factor 1 receptor (csf1r) gene during hematopoiesis. Front Biosci 2008; 13:549–60. [DOI] [PubMed] [Google Scholar]
- 19. Chambers SK, Gilmore‐Hebert M, Wang Y, Rodov S, Benz EJ Jr, Kacinski BM. Posttranscriptional regulation of colony‐stimulating factor‐1 (CSF‐1) and CSF‐1 receptor gene expression during inhibition of phorbol‐ester‐induced monocytic differentiation by dexamethasone and cyclosporin A: potential involvement of a destabilizing protein. Exp Hematol 1993; 21:1328–34. [PubMed] [Google Scholar]
- 20. Krysinska H, Hoogenkamp M, Ingram R, Wilson N, Tagoh H, Laslo P et al A two‐step, PU.1‐dependent mechanism for developmentally regulated chromatin remodeling and transcription of the c‐fms gene. Mol Cell Biol 2007; 27:878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aronoff DM, Canetti C, Serezani CH, Luo M, Peters‐Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP‐1. J Immunol 2005; 174:595–9. [DOI] [PubMed] [Google Scholar]
- 22. Hume DA, Gordon S. The correlation between plasminogen activator activity and thymidine incorporation in mouse bone marrow‐derived macrophages. Opposing actions of colony‐stimulating factor, phorbol myristate acetate, dexamethasone and prostaglandin E. Exp Cell Res 1984; 150:347–55. [DOI] [PubMed] [Google Scholar]
- 23. Hamilton JA, Vairo G, Knight KR, Cocks BG. Activation and proliferation signals in murine macrophages. Biochemical signals controlling the regulation of macrophage urokinase‐type plasminogen activator activity by colony‐stimulating factors and other agents. Blood 1991; 77:616–27. [PubMed] [Google Scholar]
- 24. Vairo G, Argyriou S, Bordun AM, Whitty G, Hamilton JA. Inhibition of the signaling pathways for macrophage proliferation by cyclic AMP. Lack of effect on early responses to colony stimulating factor‐1. J Biol Chem 1990; 265:2692–701. [PubMed] [Google Scholar]
- 25. Zaslona Z, Serezani CH, Okunishi K, Aronoff DM, Peters‐Golden M. Prostaglandin E2 restrains macrophage maturation via E prostanoid receptor 2/protein kinase A signaling. Blood 2012; 119:2358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boltz‐Nitulescu G, Wiltschke C, Holzinger C, Fellinger A, Scheiner O, Gessl A et al Differentiation of rat bone marrow cells into macrophages under the influence of mouse L929 cell supernatant. J Leukoc Biol 1987; 41:83–91. [DOI] [PubMed] [Google Scholar]
- 27. Stanley ER, Heard PM. Factors regulating macrophage production and growth. Purification and some properties of the colony stimulating factor from medium conditioned by mouse L cells. J Biol Chem 1977; 252:4305–12. [PubMed] [Google Scholar]
- 28. DeKoter RP, Walsh JC, Singh H. PU.1 regulates both cytokine‐dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J 1998; 17:4456–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 1994; 265:1573–7. [DOI] [PubMed] [Google Scholar]
- 30. Ross IL, Yue X, Ostrowski MC, Hume DA. Interaction between PU.1 and another Ets family transcription factor promotes macrophage‐specific Basal transcription initiation. J Biol Chem 1998; 273:6662–9. [DOI] [PubMed] [Google Scholar]
- 31. Yue X, Favot P, Dunn TL, Cassady AI, Hume DA. Expression of mRNA encoding the macrophage colony‐stimulating factor receptor (c‐fms) is controlled by a constitutive promoter and tissue‐specific transcription elongation. Mol Cell Biol 1993; 13:3191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES‐cell self‐renewal and inner cell mass specification. Nature 2010; 466:1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liao HF, Tai KY, Chen WS, Cheng LC, Ho HN, Lin SP. Functions of DNA methyltransferase 3‐like in germ cells and beyond. Biol Cell 2012; 104:571–87. [DOI] [PubMed] [Google Scholar]
- 35. Huang SK, Wettlaufer SH, Chung J, Peters‐Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac‐1. Am J Respir Cell Mol Biol 2008; 39:482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmitt JM, Stork PJ. PKA phosphorylation of Src mediates cAMP's inhibition of cell growth via Rap1. Mol Cell 2002; 9:85–94. [DOI] [PubMed] [Google Scholar]
- 37. Yamamizu K, Fujihara M, Tachibana M, Katayama S, Takahashi A, Hara E et al Protein kinase A determines timing of early differentiation through epigenetic regulation with G9a. Cell Stem Cell 2012; 10:759–70. [DOI] [PubMed] [Google Scholar]
- 38. Pongubala JM, Atchison ML. Activating transcription factor 1 and cyclic AMP response element modulator can modulate the activity of the immunoglobulin κ 3′ enhancer. J Biol Chem 1995; 270:10304–13. [DOI] [PubMed] [Google Scholar]
- 39. Shackelford R, Adams DO, Johnson SP. IFN‐γ and lipopolysaccharide induce DNA binding of transcription factor PU.1 in murine tissue macrophages. J Immunol 1995; 154:1374–82. [PubMed] [Google Scholar]
- 40. Park SY, Lee SW, Baek SH, Lee CW, Lee WS, Rhim BY et al Suppression of PU.1‐linked TLR4 expression by cilostazol with decrease of cytokine production in macrophages from patients with rheumatoid arthritis. Br J Pharmacol 2013; 168:1401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saeki K, Saeki K, Yuo A. Distinct involvement of cAMP‐response element‐dependent transcriptions in functional and morphological maturation during retinoid‐mediated human myeloid differentiation. J Leukoc Biol 2003; 73:673–81. [DOI] [PubMed] [Google Scholar]
- 42. Felgner J, Kreipe H, Heidorn K, Jaquet K, Zschunke F, Radzun HJ et al Increased methylation of the c‐fms protooncogene in acute myelomonocytic leukemias. Pathobiology 1991; 59:293–8. [DOI] [PubMed] [Google Scholar]
- 43. Follows GA, Tagoh H, Richards SJ, Melnik S, Dickinson H, de Wynter E et al c‐FMS chromatin structure and expression in normal and leukaemic myelopoiesis. Oncogene 2005; 24:3643–51. [DOI] [PubMed] [Google Scholar]
- 44. Huang SK, Scruggs AM, Donaghy J, McEachin RC, Fisher AS, Richardson BC et al Prostaglandin E2 increases fibroblast gene‐specific and global DNA methylation via increased DNA methyltransferase expression. FASEB J 2012; 26:3703–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim SH, Serezani CH, Okunishi K, Zaslona Z, Aronoff DM, Peters‐Golden M. Distinct protein kinase A anchoring proteins direct prostaglandin E2 modulation of Toll‐like receptor signaling in alveolar macrophages. J Biol Chem 2011; 286:8875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gentile P, Byer D, Pelus LM. In vivo modulation of murine myelopoiesis following intravenous administration of prostaglandin E2. Blood 1983; 62:1100–7. [PubMed] [Google Scholar]
- 47. Hoggatt J, Mohammad KS, Singh P, Hoggatt AF, Chitteti BR, Speth JM et al Differential stem‐ and progenitor‐cell trafficking by prostaglandin E2. Nature 2013; 495:365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bryn T, Mahic M, Enserink JM, Schwede F, Aandahl EM, Tasken K. The cyclic AMP‐Epac1‐Rap1 pathway is dissociated from regulation of effector functions in monocytes but acquires immunoregulatory function in mature macrophages. J Immunol 2006; 176:7361–70. [DOI] [PubMed] [Google Scholar]
- 49. Digiacomo G, Ziche M, Dello Sbarba P, Donnini S, Rovida E. Prostaglandin E2 transactivates the colony‐stimulating factor‐1 receptor and synergizes with colony‐stimulating factor‐1 in the induction of macrophage migration via the mitogen‐activated protein kinase ERK1/2. FASEB J 2015; 29:2545–54. [DOI] [PubMed] [Google Scholar]
- 50. Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz‐Tennenbaum S, Osterholzer JJ et al Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol 2014; 193:4245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev Comp Immunol 2004; 28:509–54. [DOI] [PubMed] [Google Scholar]
- 52. Boultwood J, Rack K, Kelly S, Madden J, Sakaguchi AY, Wang LM et al Loss of both CSF1R (FMS) alleles in patients with myelodysplasia and a chromosome 5 deletion. Proc Natl Acad Sci U S A 1991; 88:6176–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rosenbauer F, Wagner K, Kutok JL, Iwasaki H, Le Beau MM, Okuno Y et al Acute myeloid leukemia induced by graded reduction of a lineage‐specific transcription factor, PU.1. Nat Genet 2004; 36:624–30. [DOI] [PubMed] [Google Scholar]