

Abstract
Inference of the biological roles of lncRNAs in breast cancer development remains a challenge. Here, we analyzed RNA-seq data in tumor and normal breast tissue samples from 18 breast cancer patients and 18 healthy controls and constructed a functional lncRNA-mRNA co-expression network. We revealed two distinctive co-expression patterns associated with breast cancer, reflecting different underlying regulatory mechanisms: (1) 516 pairs of lncRNA-mRNAs have differential co-expression pattern, in which the correlation between lncRNA and mRNA expression differs in tumor and normal breast tissue; (2) 291 pairs have dose-response co-expression pattern, in which the correlation is similar, but the expression level of lncRNA or mRNA differs in the two tissue types. We further validated our findings in TCGA dataset and annotated lncRNAs using TANRIC. One novel lncRNA, AC145110.1 on 8p12, was found differentially co-expressed with 127 mRNAs (including TOX4 and MAEL) in tumor and normal breast tissue and also highly correlated with breast cancer clinical outcomes. Functional enrichment and pathway analyses identified distinct biological functions for different patterns of co-expression regulations. Our data suggested that lncRNAs might be involved in breast tumorigenesis through the modulation of gene expression in multiple pathologic pathways.
Breast cancer is the most common invasive cancer and the leading cause of cancer death in women. It is a genetic disease of aberrant gene expression –the result of dysregulation of gene networks that maintain normal cellular functions and identity. In humans, only ~1.2% of the genome is protein-coding, and substantial fractions of the genome (~80%) can be transcribed into noncoding RNAs (ncRNAs) with no protein-coding capacity1. Recent research shows that ncRNAs, rather than transcriptional noise as previously believed, are capable of transacting a wide repertoire of regulatory functions2, suggesting the potential role of ncRNAs in shaping the genetic susceptibility of disease3.
Long noncoding RNAs (lncRNAs) are a class of ncRNAs with transcript size >200 nucleotides. They structurally resemble mRNAs but display a more tissue-specific expression pattern4. It is reported that lncRNAs might play widespread roles in gene regulation and other cellular processes, including acting as host genes for miRNAs, preventing miRNA, mRNA and proteins from binding with their intended targets, acting as molecular scaffolds, and serving as guides to direct proteins to their chromosomal targets5. Increasing evidence indicates that lncRNAs play an important role in cancer development and progression6. For example, the expression of HOTAIR is associated with metastasis and poor prognosis of breast cancer7. Despite the growing body of knowledge of lncRNAs, it remains a challenge to identify cancer-related lncRNAs on a genomic scale and further characterize their potential biological function in breast cancer development.
Co-expression analyses of protein-coding RNAs and lncRNAs have been reported to study the potential function of lncRNAs in biological processes and cancers8,9, including breast cancer10,11. However, these reports provide limited understanding of lncRNAs for the following reasons. First, most studies investigated co-expression of lncRNAs and mRNAs across a variety of tumor subtypes or states, but much less commonly between tumor and normal breast tissue. Second, in few studies, pathologically normal tissue adjacent to tumor, which was used as a baseline control, represented a suboptimal control as its expression profile has been shown to be altered in response to the adjacent tumor and was, to some extent, similar to that of tumor tissue12,13. Consequently, important breast cancer-associated lncRNAs may not be identifiable when using adjacent normal tissue as a baseline for comparison. Finally, most reports analyzed the co-expression of differentially expressed lncRNAs and mRNAs only, thus may fail to detect significant differences of the lncRNA-mRNA relationship between tumor and normal tissue but without dramatic changes of lncRNA or mRNA expression levels.
In this study, using RNA-sequencing data and normal breast tissue from healthy women as a desirable baseline control, we investigated the expression of lncRNAs in 18 breast cancer tumors and 18 normal tissue controls. Clinical information of the 18 breast cancer patients and the 18 healthy controls was summarized in Supplementary Table S1. We identified novel breast cancer-associated lncRNAs that were differentially expressed in breast tumor and normal tissue. Employing a genome-wide analytic approach, we further investigated co-expression of lncRNAs and mRNAs in breast tumor and normal tissue, and revealed two distinct co-expression patterns associated with breast cancer. Pathway analyses suggested different biological functions in tumorigenesis for each co-expression pattern. Overall, our study highlights the importance of lncRNAs in the carcinogenesis of breast cancer, and provides a valuable resource for lncRNA studies in cancer.
Results
Overview of the analysis pipeline
The analysis pipeline is outlined in Fig. 1. We first performed differential expression (DE) analysis by comparing lncRNA or mRNA expression in tumor and normal breast tissue; we then analyzed co-expression of lncRNAs and mRNAs in tumor and normal breast tissue and revealed two distinct co-expression patterns associated with breast cancer; finally, we validated our findings using an external database, The Cancer Genome Atlas (TCGA) dataset, and further annotated lncRNAs using various bioinformatics resources and inferred their functional enrichment based on Gene Ontology (GO)/KEGG terms. Co-expression of lncRNAs and mRNAs in tumor and normal breast tissue was analyzed considering two scenarios: (1) differential co-expression in which the correlation between lncRNA and mRNA expression differs in tumor and normal breast tissue; and (2) dose-response co-expression in which the correlation is similar in tumor and normal breast tissue, but the expression level of lncRNA or mRNA differs in two tissue types.
Figure 1. Flowchart of the analysis pipeline.

We performed alignment and quantification on each RNA-seq sample, and then performed differential expression (DE) analysis to identify breast cancer-associated lncRNAs and mRNAs, as well as co-expression analysis between lncRNAs and mRNAs to infer potential function of lncRNAs considering two possible underlying mechanisms. Finally, our findings were validated using external TCGA dataset and other available bioinformatics resources including TANRIC functional annotation and pathway analysis.
Differential expression of lncRNAs and mRNAs in tumor and normal breast tissue
We analyzed genome-wide expression of 7,450 lncRNA and 22,362 mRNA transcripts in tumor and normal breast tissue. We first investigated whether these transcripts could distinguish tumor from normal tissue. Unsupervised Principal Components Analysis (PCA) of lncRNAs demonstrated a clear separation of tumor from normal breast tissue (Supplementary Fig. S1a), similar to that of mRNAs (Supplementary Fig. S1b), illustrating the vast differences in their transcriptomic profiles. We also noted that lncRNAs distinguish tumor from normal tissue with three times fewer transcripts than mRNAs. This finding is in line with previous reports showing that lncRNAs display higher expression variation than mRNAs14.
We further performed a differential expression analysis to examine the differences in lncRNA and mRNA expression profiles between tumor and normal breast tissue. By the criteria of FDR adjusted P value < 0.01 and a two-fold change, we identified a total of 598 lncRNAs differentially expressed between tumor and normal breast tissue (Supplementary Fig. S2a; Supplementary Table S2). We found 348 lncRNAs (58.2%) down-regulated in tumor tissue (Fig. 2a). Similar reduction of expression levels have been reported for microRNAs in human cancer15, substantiating a common pattern of dysregulation of non-coding RNAs in carcinogenesis3. By the same criteria 2,980 mRNAs were identified to be differentially expressed between tumor and normal breast tissue (Supplementary Fig. S2b; Supplementary Table S3), consisting of 1,609 up-regulated (54.0%) and 1,371 down-regulated mRNAs (Fig. 2b).
Figure 2. Expression differences of lncRNAs and mRNAs in breast tumor and normal breast tissue.

Hierarchical clustering analysis of (a) differentially expressed lncRNAs; and (b) differentially expressed coding mRNAs between 18 breast tumor and 18 normal breast tissue samples (|fold change| ≥2 and FDR-adjusted P < 0.01). In the heatmap, columns represent samples, and rows represent each gene. Colors ranged from green (low expression) to red (high expression), represent the relative expression levels of lncRNAs and mRNAs.
The top ranked lncRNA in differential expression analysis, RP11-118E18.2, shows higher expression in breast tumor compared to normal tissue (FC = 16.7, FDR P = 5.49 × 10−20). Little is known regarding its biological function, but evidence from TANRIC16 shows differential expression of RP11-118E18.2 between carriers and non-carriers of mutations in clinically actionable genes including TP53, NAV3, MUC5B and MAP1A. Additionally, RP11-118E18.2 shows differential expression associated with ER status (P = 0.00026) and PR status (P = 0.001), PAM50 (P = 0.00046) and breast cancer therapy based on molecular signatures (P = 0.01) (Supplementary Fig. S3). However, the function remains largely unknown for most of the differentially expressed lncRNAs identified in our study, which is consistent with a previous report by Reiche et al.10. We also observed consistent direction of differential expression in tumor and normal tissue for majority of known cancer-related lncRNAs in two studies, though our study in general had larger fold changes than the study by Reiche et al.10 (Supplementary Table S4).
Enrichment analysis of differentially expressed mRNAs demonstrated that these genes are involved in cancer-related pathways, such as cell cycle, PPAR signaling pathway, apoptosis, and transcriptional dysregulation (Supplementary Fig. S4; Supplementary Table S5). We further employed IPA software to predict upstream transcriptional regulators that are either “inhibited” or “activated” based on the entire set of the 2,980 mRNAs that were differentially expressed in tumor and normal breast tissue17. The top list of regulator genes are enriched for cancer-related genes including TGFB1 (P = 9.20 × 10−29)18, TNF (P = 1.43 × × 10−21)19, TP53 (P = 1.83 × 10−21)20 and ER (P = 1.15 × 10−18)21, supporting the biological functions of the differentially expressed mRNAs relevant to breast cancer.
Co-expression of lncRNAs and mRNAs in tumor and normal breast tissue
Differential co-expression analysis
We identified 516 lncRNA-mRNA pairs that were significantly and differentially co-expressed between tumor and normal breast tissue (Supplementary Table S6), of which 26 pairs (5.0%) were located on the same chromosome (cis-acting) and the remaining 490 pairs (95.0%) on different chromosomes (trans-acting), suggesting that most of the lncRNAs regulate mRNA expression in trans. These findings are in agreement with a recent study by Guttman et al., which reported that 92% of the lncRNAs were trans-acting in their study22. Details of regulation patterns for each chromosome are displayed in Supplementary Table S7.
Among those 516 lncRNA-mRNA pairs, we only detected 131 unique lncRNAs and 294 unique mRNAs. Seventy-five lncRNAs (57.3%) showed differential co-expression with just a single mRNA, while 56 (42.7%) showed differential co-expression with at least two mRNAs. On the other hand, 212 mRNAs (72.1%) showed differential co-expression with just a single lncRNA, while 82 (27.9%) showed differential co-expression with at least two lncRNAs. The top lncRNAs and mRNAs with the largest numbers of associations in differential co-expression are listed in Table 1.
Table 1. Top lncRNAs and mRNAs with the largest numbers of associations in differential and dose-response co-expression analysis.
| lncRNA | lncRNA Chromosome | #mRNAs | mRNA | mRNA Chromosome | # lncRNAs | |
|---|---|---|---|---|---|---|
| Differential co-expression | AC145110.1 | 8 | 127 | TOX4 | 14 | 25 |
| RP11-136K7.2 | 5 | 36 | CETN1 | X | 19 | |
| TINCR | 19 | 26 | KIN | 10 | 15 | |
| RP11-680F20.12 | 11 | 18 | CETN2 | X | 11 | |
| CTB-131K11.1 | 17 | 17 | TMEM41A | 3 | 9 | |
| Dose-response co-expression | RP11-161M6.2 | 16 | 17 | SLC19A3 | 2 | 4 |
| ADIPOQ-AS1 | 3 | 11 | ACSM5 | 16 | 2 | |
| CTD-2363C16.1 | 5 | 11 | AIFM2 | 10 | 2 | |
| CTD-2541J13.2 | 18 | 11 | AK021888 | 5 | 2 | |
| LINC00341 | 14 | 10 | ANKRD20A2 | 2 | 2 |
Of interest, AC145110.1 was found differentially co-expressed with 127 mRNAs in tumor and normal breast tissue, acting as a master regulator. Pathway analyses suggested these mRNAs are enriched in biological functions related to “cellular growth and proliferation”, “cell–to–cell signaling and interaction”, and “Hematological System development and function” (Supplementary Fig. S5). TANRIC analysis shows the lncRNA AC145110.1 is correlated with disease stage of breast cancer (P = 0.0029), ER status (P = 0.0159), and HER2 status (P = 0.019) in the TCGA samples (Supplementary Fig. S6). Additionally, the TCGA samples show differential expression of AC145110.1 between carriers and non-carriers of mutations in 19 clinically actionable genes. Taken together, these results suggest that AC145110.1 may play an important role in cancer development through long-range regulation of expression for multiple cancer-related genes.
Dose-response co-expression analysis
Using the criteria described in the methods, we identified a total of 291 lncRNA-mRNA pairs that were dose-response co-expressed between tumor and normal breast tissue (Supplementary Table S8), of which 115 pairs (39.5%) located on the same chromosome and 176 pairs (60.5%) on different chromosomes, representing cis- and trans- acting regulation, respectively. Similar to the differential co-expression results, these findings are consistent with the recent findings that lncRNAs mostly regulate mRNA expression in trans22. Details of regulation patterns for each chromosome are displayed in Supplementary Table S9.
Those 291 lncRNA-mRNA pairs are represented by 149 unique lncRNAs and 262 unique mRNAs. Of which, 111 lncRNAs (74.5%) showed dose-response co-expression with a single mRNA, while the remaining 38 (25.5%) with at least two mRNAs. On the other hand, 235 mRNAs (89.7%) showed dose-response co-expression with just a single lncRNA, while 27 mRNAs (10.3%) with at least two lncRNAs. The top lncRNAs and mRNAs with the largest numbers of associations in dose-response co-expression are listed in Table 1.
Validation in TCGA dataset
Differentially expressed mRNAs
We validated the differentially expressed mRNAs identified in our study using TCGA dataset of 848 breast tissue samples, consisting of 744 breast tumors and 104 adjacent normal breast tissue samples from women of European ancestry. Out of 14,371 mRNAs we analyzed in TCGA dataset, we identified 263 mRNAs differentially expressed between TCGA tumor and adjacent normal breast tissue with FDR adjusted P values < 0.01 and a two-fold change. Of these, 207 were also differentially expressed in our data. We found 99.5% of these mRNAs showed consistent direction of differential expression across two datasets (Fig. 3a). Of note, the fold change for a specific mRNA was generally larger in our dataset than in TCGA.
Figure 3. Validation in TCGA.

(a) Validation of mRNA expression differences in TCGA. X-axis represents Log2FC between 18 breast tumor and 18 normal tissue samples from our dataset; Y-axis represents Log2FC between 744 breast tumors and 104 adjacent normal tissue samples from TCGA dataset; 207 differentially expressed mRNAs in both datasets are shown. (b) and (c) Validation of lncRNA-mRNA co-expression in breast tumors in TCGA. Our dataset consists of 18 breast tumor samples; and TCGA dataset consists of 692 breast tumors. (b) Directions of the lncRNA-mRNA associations in two datasets were compared in 183 lncRNA-mRNA pairs identified in differential co-expression analysis; (c) Directions of the lncRNA-mRNA associations were compared in 139 lncRNA-mRNA pairs identified in dose-response co-expression analysis. Red arrows indicate positive correlations, and Green arrows indicate negative correlation between lncRNAs and mRNAs.
Co-expression of lncRNAs and mRNAs in breast tumors
We further validated the co-expression of lncRNAs and mRNAs in 692 breast tumor samples from the TCGA dataset. After quality control, only 6,556 lncRNAs and 15,074 mRNAs were retained for co-expression analysis in TCGA tumor tissue samples. Of the 516 differentially co-expressed lncRNA-mRNA pairs identified in our data, only 183 pairs were available to analyze in TCGA data, and 56.8% of these overlapping pairs had consistent direction of co-expression correlations in TCGA and our datasets (Fig. 3b). Of the 291 lncRNA-mRNA pairs we identified in our dose-response co-expression analysis, we were only able to analyze 139 pairs in TCGA data, and 84.2% of these overlapping pairs had consistent direction of co-expression correlations in TCGA and our datasets (Fig. 3c). These results may reflect larger variability in differential co-expression, when compared to dose-response co-expression between tumor and normal breast tissue. While high concordance rate for dose-response co-expression attested the validity of our data, the lower concordance rate for differential co-expression might be due to the different distribution of tumor characteristics (e.g. subtypes) in our dataset and TCGA dataset, as it is conceivable that differential co-expression is likely more sensitive to changes in tumor characteristics.
Functional characterization of the identified lncRNAs
To better understand the function of the lncRNAs identified in our dataset, we employed an analytic pipeline to assess two aspects of the identified lncRNAs: (1) annotation of their co-expressed mRNAs; (2) their co-localization with known breast cancer risk loci.
To infer biological function of lncRNAs, their co-expressed mRNAs were subjected to Gene Ontology (GO), KEGG, and BioCarta annotations. The 294 mRNAs showing differential co-expression with lncRNAs were enriched for functions including “Glycosaminoglycan biosynthesis”, “Glycosphingolipid biosynthesis”, and “One carbon pool by folate” (Fig. 4a). It is intriguing to note the third function is methylation-related, suggesting that epigenetic mechanism might play an important role in lncRNA-mRNA differential co-expression regulation between tumor and normal breast tissue. Interrogation of 262 mRNAs that were co-expressed with lncRNAs in dose-response fashion reveals cancer-related pathways including “PPAR signaling pathway”, “AMPK signaling pathway”, “VEGF hypoxia and angiogenesis”, and “adipocytokine signaling pathway” (Fig. 4b). GO analysis showed similar results for functional enrichment (Supplementary Table S10).
Figure 4. Functional enrichment for the mRNAs identified from lncRNA-mRNA co-expression analysis in our data.
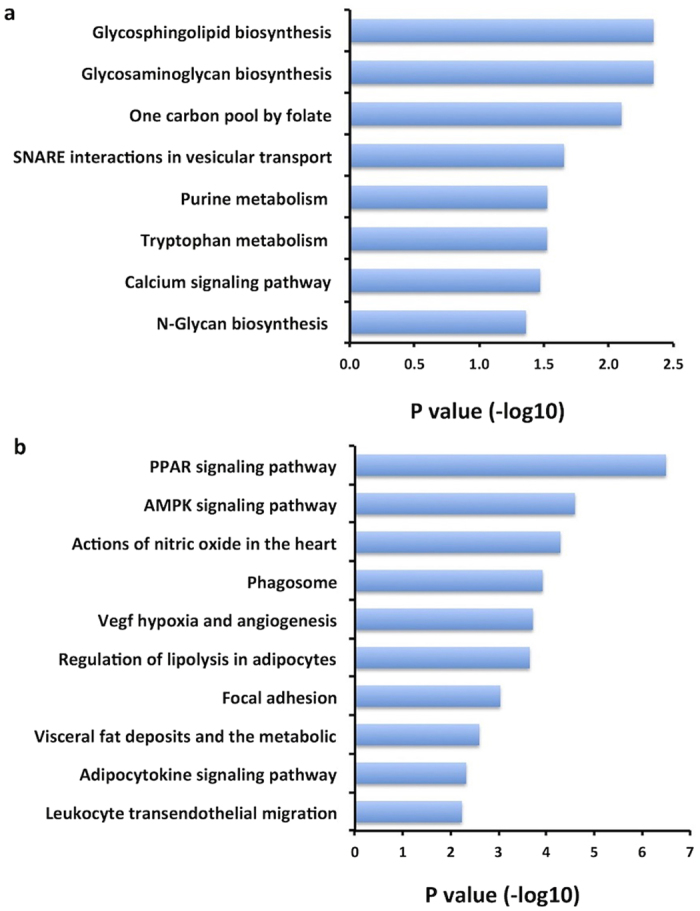
(a) mRNAs co-expressed with lncRNAs in differential co-expression network; (b) mRNAs co-expressed with lncRNAs in dose-response co-expression network.
Secondly, we investigated co-localization of lncRNAs with known breast cancer risk loci. We identified 368 SNPs from the NHGRI-EBI Catalog of published GWAS of breast cancer23 (Nov, 2015 accessed). The majority of these genetic loci fall into non-coding regions24. Mapping these loci to the genomic regions, we found 44 loci located within 43 lncRNA regions (Supplementary Table S11). Of those, three risk loci (rs9832625, rs11836164 and rs2823779) are located in lncRNAs that were identified in either differential expression or co-expression analysis in our study (Table 2). Of note, lncRNA LINC00478 harbors SNP rs2823779, a risk locus associated with toxicity after radiotherapy in breast cancer patients25. This lncRNA was found differentially co-expressed with four mRNAs in our study, including TOX4 and CETN1.
Table 2. Significant differential expression and co-expression results for lncRNAs co-localized with known breast cancer risk SNPs in our data.
| SNP | Position | Near Gene(s) | LncRNA | lncRNA-DE analysis |
mRNA | Dose-response Co-expression analysis |
Differential Co-expression analysis |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Log2 Fold Change | FDR P-value | βa | P value b | β_ Tumorc | β_ Normalc | Interaction P valued | |||||
| rs9832625 | 3p24.1 | RBMS3 | ENSG00000235904.1|RBMS3-AS3 | −2.66 | 2.61E-04 | — | — | — | — | — | — |
| rs11836164 | 12p12.1 | SSPN - ITPR2 | ENSG00000255750.1|RP11-283G6.5 | −1.2 | 1.53E-03 | — | — | — | — | — | — |
| rs2823779 | 21q21.1 | LINC00478, MIR99AHG | ENSG00000215386.6|LINC00478 | −1.69 | 2.88E-04 | ||||||
| TOX4 | — | — | 1.48 | 4.26 | 2.73E-11 | ||||||
| CETN1 | — | — | −0.08 | −1.82 | 8.67E-11 | ||||||
| KIAA1253 | — | — | 5.16 | 3.02 | 1.25E-10 | ||||||
| CETN2 | — | — | 0.98 | 3.19 | 2.80E-10 | ||||||
aβ refers to the change of mRNA expression level corresponding to each unit increase of lncRNA expression level in tumor and normal breast tissue, which was estimated from generalized linear model 3.
bP value was estimated from generalized linear model 3.
cβ refers to the change of mRNA expression level corresponding to each unit increase of lncRNA expression level in tumor and normal breast tissue, respectively, which was estimated from generalized linear model 2.
dP value for interaction term (lncRNA· tissue type) was estimated from generalized linear model 2.
Cross Validation
Overlap between the identified lncRNAs
We compared the lncRNAs identified in differential expression, differential and dose-response co-expression, and assessed the overlap specific to each comparison (Supplementary Fig. S7). We observed 119 lncRNAs overlapping in dose-response co-expression and differential expression, which was as expected due to our significance criteria used for dose-response co-expression. Interestingly, few lncRNAs overlap between differential and dose-response co-expression, indicating that most lncRNAs might exclusively involve in distinct co-expression regulations with different underlying mechanisms. We also observed the small overlap of lncRNAs between differential expression and differential co-expression. Intriguingly, a significant proportion of lncRNAs identified in differential co-expression were not differentially expressed between tumor and normal breast tissue. This may be due to the very stringent P value threshold we used in differential co-expression. It is also possible that lncRNAs regulate mRNA expression differently in tumor and normal tissue but without significant changes of their own expression levels.
Overlap with conventional co-expression analysis
Previous studies commonly used a conventional approach that only analyzed the co-expression of lncRNAs and mRNAs that are both differentially expressed between tumor and normal tissue26,27. We employed a genome-wide approach to analyze co-expression of lncRNAs and mRNAs in tumor and normal breast tissue and applied a stringent significance threshold to guard against false positives. Out of the 516 lncRNA-mRNA pairs identified in our differential co-expression, only 8 pairs had both differentially expressed lncRNAs and mRNAs (Table 3). The overlap was more pronounced in dose-response co-expression analysis. Out of the 291 pairs identified in our dose-response co-expression, 162 pairs had both differentially expressed lncRNAs and mRNAs (Supplementary Table S12). These results suggested that our approach might be more robust to identify co-expression, especially differential co-expression pattern, between lncRNAs and mRNAs that were associated with breast cancer.
Table 3. Differential co-expression of lncRNAs and mRNAs by conventional approach.
| lncRNA ID | lncRNA Name | lncRNA Chromosome | mRNA ID | mRNA Chromosome | β_ Tumora | β_ Normala | log2FC (lncRNA) b | log2FC (mRNA) b |
|---|---|---|---|---|---|---|---|---|
| ENSG00000263069.1 | CTD-2047H16.4 | 17 | CETN2 | X | 9.60 | 5.06 | 1.01 | 1.11 |
| ENSG00000215386.6 | LINC00478 | 21 | CETN2 | X | 0.98 | 3.19 | −1.69 | 1.11 |
| ENSG00000267078.1 | RP11-666A8.9 | 17 | EPPK1 | 8 | −0.63 | 2.99 | 1.37 | 2.09 |
| ENSG00000253187.2 | HOXA-AS4 | 7 | FAM89A | 1 | −4.36 | −2.48 | −2.32 | −2.89 |
| ENSG00000229970.2 | AC007128.1 | 7 | KIAA1724 | 2 | −3.47 | −5.69 | 3.09 | 1.07 |
| ENSG00000261716.1 | RP11-196G18.22 | 1 | RP11-426E5.2 | 10 | −0.06 | −7.32 | 1.22 | −4.90 |
| ENSG00000261716.1 | RP11-196G18.22 | 1 | ST6GALNAC3 | 1 | 0.43 | −4.40 | 1.22 | −1.91 |
| ENSG00000253187.2 | HOXA-AS4 | 7 | ST6GALNAC6 | 9 | −0.38 | 1.84 | −2.32 | −1.04 |
aβ refers to the change of mRNA expression level corresponding to each unit increase of lncRNA expression level in tumor and normal breast tissue, respectively, which was estimated from generalized linear model 2.
bFC refers to the fold change of expression level in breast tumor versus normal tissue for mRNAs and lncRNAs, respectively.
Co-expression patterns of known breast cancer-related lncRNAs
We further investigated known breast cancer-related lncRNAs in our study. We found HOTAIR and HOTAIRM1 were differently expressed between tumor and normal breast tissue based on our criteria (Supplementary Table S13). This finding is consistent with previous research28. For co-expression analysis, only two of these lncRNAs, MALAT1 and XIST, were found differentially co-expressed with three mRNAs (TOX4, ALG14, and C12orf32) in our data (Table 4). No known breast cancer-related lncRNAs were found significant in our dose-response co-expression analysis.
Table 4. Significant results for known breast cancer-related lncRNAs in our data.
| lncRNA ID | lncRNA/Reference | Position | lncRNA-DE analysis |
mRNA-DE Analysis |
Dose-response Co-expression Analysis |
Differential Co-expression Analysis |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log2FC a | FDR adjusted P value | mRNA | position | Log2FCa | FDR adjusted P value | βb,c | P valueb,d | β in Tumorb,e | β in Normalb,e | P value Interactionb,f | |||
| ENSG00000251562.3 | MALAT129 | 11 | 0.70 | 3.80E-02 | ALG14 | 1p21.3 | −0.12 | 8.18E-01 | — | — | 1.24 | −0.65 | 2.86E-10 |
| TOX4 | 14q11.2 | 0.39 | 4.60E-01 | — | — | −4.05 | −7.27 | 1.46E-10 | |||||
| ENSG00000229807.5 | XIST60 | Xq13.2 | −0.04 | 8.90E-01 | C12orf32 | 12p13.33 | 1.5 | 1.88E-05 | — | — | 2.30 | −0.16 | 1.29E-10 |
| ENSG00000228630.1 | HOTAIR29 | 12q13.13 | 3.64 | 4.02E-08 | — | — | — | — | — | — | — | — | — |
| ENSG00000233429.5 | HOTAIRM160 | 7p15.2 | −1.19 | 5.64E-06 | — | — | — | — | — | — | — | — | — |
aFC refers to the fold change of expression level in breast tumor versus normal tissue for mRNAs and lncRNAs, respectively.
b“-” refers to non-significant in our study.
cβ refers to the change of mRNA expression level corresponding to each unit increase of lncRNA expression level in tumor and normal breast tissue, which was estimated from generalized linear model 3.
dP value was estimated from generalized linear model 3.
eβ refers to the change of mRNA expression level corresponding to each unit increase of lncRNA expression level in tumor and normal breast tissue, respectively, which was estimated from generalized linear model 2.
fP value for interaction term (lncRNA· tissue type) was estimated from generalized linear model 2.
Discussion
In past decades, transcriptomic studies have focused on the analysis of protein-coding transcripts to characterize their patterns and potential functional roles. The recent development of next-generation sequencing technology has greatly accelerated the discovery and characterization of a new class of non-coding RNA transcripts, lncRNAs. Increasing evidence suggests that lncRNAs have key regulatory functions in chromatin remodeling and gene expression in the progress of disease, including cancer3. Indeed, a few dysregulated lncRNAs, such as HOTAIR and DSCAM-AS1, have been linked to breast cancer29. However, the role of lncRNAs in breast cancer development remains largely unknown. Using high-throughput RNA sequencing technology and a computational approach, we systematically evaluated genome-wide expression of lncRNAs and co-expression between lncRNAs and mRNAs in tumor and normal breast tissue. Our study identified novel breast cancer-associated lncRNAs and inferred their potential biological and pathological roles in gene regulation and cancer development.
Although emerging evidence indicates that lncRNAs play a key role in many biological processes such as cell differentiation, immune response, and tumorigenesis, the functions of lncRNAs are not well understood30. In order to functionally characterize lncRNAs, a “guilt by association” strategy is commonly used to construct a co-expression network of lncRNAs and mRNAs31. In the conventional approach, differential expression analysis is first performed in tumor and normal tissue and only the identified differentially expressed lncRNAs and mRNAs are analyzed in the following co-expression network11,26. While this approach significantly reduces the number of tests, it might fail to detect some important, cancer-related co-expression relationship between lncRNAs and mRNAs that shows no significant change in expression level of either RNA. In this study, we employed a genome-wide approach and systematically investigated lncRNA-mRNA co-expression in a serial of statistical models using stringent significance threshold. We considered two possible co-expression patterns related to breast cancer, that is, differential and dose-response co-expression networks. Indeed, compared to the conventional approach, our genome-wide approach identified significantly more lncRNA-mRNA co-expression relationship associated with breast cancer. Of particular interest, the conventional approach only identified less than 2% of differential co-expression pairs in our genome-wide approach, while this proportion is 55% for dose-response co-expression pairs. These results suggested our genome-wide approach is more robust to identify breast cancer-associated lncRNA-mRNA co-expression, especially for differential co-expression network.
Most previous studies investigated transcriptome profiling in tumor and adjacent normal tissue from cancer patients, in which histologically normal tissue adjacent to the tumor was commonly used as a baseline control because of its ready availability. However, adjacent normal tissue represents a suboptimal control, because its molecular profile has been shown to be altered in response to the adjacent tumor and is, to some extent, similar to that of tumor tissue13. Consequently, lncRNA expression or lncRNA-mRNA co-expression pattern is more similar or having smaller difference in tumor and adjacent normal tissue when compared to that in tumor and normal tissue from healthy controls. As a result, important breast cancer-associated lncRNAs or lncRNA-mRNA co-expression network may not be identifiable when using adjacent normal tissue as a baseline for comparison. Furthermore, comparing tumor tissue from cancer patients to normal breast tissue from healthy women will allow the identification of novel breast cancer- associated lncRNAs and lncRNA-mRNA co-expression that are influenced by individuals’ different genetic background and environmental risk factors, reflecting individuals’ susceptibility to the disease that is also biologically important to breast cancer development. This subset of molecular changes is unidentifiable when comparing tumor to adjacent non-tumorous or contralateral normal breast tissue from the same patients, because the germline genetic variation and environmental exposures are identical in those matched types of tissue. Indeed, we not only identified more significant findings but also observed the fold change for a specific transcript was generally larger between tumor and normal tissue in our dataset than that between tumor and adjacent normal tissue in TCGA. To our knowledge, our study is the first one that used normal breast tissue from healthy women as a more desirable baseline to identify breast cancer-related lncRNAs and lncRNA-mRNA co-expression network on a genome-wide scale. Our findings were not only consistent with previous studies on several known breast cancer-related lncRNAs and mRNAs, such as HOTAIR7, BRCA232, MMP9 and MMP1133, but more importantly, our study was able to identify novel breast cancer-related lncRNAs and lncRNA-mRNA co-expression networks. For examples, we observed a novel lncRNA, RP11-118E18.2, was significantly over-expressed in breast tumors (FC = 17, FDR P = 5.49 × 10−20). Although its biological mechanism has not been elucidated, validation by TANRIC confirmed its associations with multiple breast cancer clinical outcomes. We also found that two lncRNAs, CAHM and KCNQ1DN, are down-regulated in breast tumors compared to healthy normal controls in our study. These two lncRNAs has been found down-regulated in colorectal cancer34 and glioblastoma tumors35 but no previous studies linked them to breast cancer. For breast cancer-associated lncRNA-mRNA co-expression pattern, we found lncRNA AC145110.1 was differentially co-expressed with multiple mRNAs in tumor and normal breast tissue, including CETN1 and MAEL. CETN1 is a cancer testis antigen that is highly expressed in prostate and pancreatic cancer36. Knockdown of CETN1 inhibits breast cancer cell proliferation37. MAEL is also a cancer testis gene regulated by DNA methylation. It interacts with stress gene in cancer cells and promotes hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition38. This implies lncRNA AC145110.1 is likely to be a hub of the lncRNA-mRNA regulatory network and may thus be involved in the important process of breast cancer development.
Our study revealed two lncRNA-mRNA co-expression patterns associated with breast cancer, suggesting distinct underlying regulation mechanisms. It is noteworthy that there is little overlap of lncRNAs involved in differential and dose-response co-expression networks, indicating most lncRNAs were exclusively involved in one of the two co-expression regulations. GO and pathway analyses of genes involved in differential lncRNA-mRNA co-expression identified biological functions enriched in metabolic processes including folate metabolism; while genes involved in dose-response co-expression were enriched in signal transduction pathways. Although both co-expression regulation patterns show functional enrichments and pathways implicated in breast tumorigenesis, it is conceivable that differential co-expression might be more functionally “disruptive” compared to dose-response co-expression as the former leads to different lncRNA-mRNA correlations in breast tumor and normal tissue. The lncRNA-mRNA correlation is similar in tumor and normal breast tissue in dose-response co-expression, suggesting similar regulatory mechanism in two tissue types. As mRNA expression changes in response to lncRNA expression level in a dose-response fashion, we could speculate that the continuing changes in lncRNA or mRNA expression level would gradually introduce changes in molecular phenotypes and eventually lead to breast cancer, resulting in the observed differences in lncRNA or mRNA expression in tumor and normal breast tissue. Moreover, in both differential and dose-response co-expression analyses, we noticed the existence of the phenomenon that one lncRNA was co-expressed with multiple mRNAs as well as multiple lncRNAs were co-expressed with one mRNA (Table 1). This phenomenon appears more pronounced in differential co-expression than in dose-response co-expression, indicating a more complex regulatory relationship between lncRNAs and mRNAs in differential co-expression network.
Regulatory mechanisms underlying the co-expression of lncRNAs and mRNAs may involve competing endogenous RNA (ceRNA), transcription factors, DNA methylation, and copy number variation39. In our study, we observed the enrichment of DNA methylation-related pathways in differential co-expression, suggesting that epigenetics might be an important regulatory mechanism underlying the lncRNA-mRNA relationship for breast carcinogenesis. Recent studies showed that the majority of lncRNAs in human are produced from divergently transcribed protein-coding genes and that the divergent lncRNA/mRNA pairs exhibit coordinated changes in transcription40,41, representing cis-acting co-expression of lncRNAs and neighboring mRNAs. LncRNAs may also show a functional role in gene expression by targeting distant (trans-acting) coding genes42,43. While both cis- and trans-acting co-expression of lncRNAs and mRNAs were identified in association with breast cancer in our study, the proportion of trans-acting co-expression was significantly higher in differential co-expression than in dose-response co-expression network (95% vs. 60%), suggesting more complex, long-range mechanisms involved in differential co-expression regulation.
Given the importance of lncRNAs in biology and disease, there is great interest in defining functions of lncRNAs previously discovered. Our study provided a functional characterization of known breast cancer-related lncRNAs through lncRNA-mRNA co-expression. We found two breast cancer-related lncRNAs, MALAT1 and XIST, were significantly and differentially co-expressed with mRNAs (ALG14, TOX4, and C12orf32) in tumor and normal breast tissue via trans-acting mechanism. MALAT1 has been shown to be differentially expressed in multiple tumors, including breast cancer, prostate cancer, and lung cancer44. It was differentially co-expressed with TOX4 and ALG4 in our study. TOX4 plays an important role in DNA damage response and cell cycle45, functions implicated in tumorigenesis. It is epigenetically regulated in breast cancer46. Of note, TOX4 is the top mRNA with the largest number of associations in differential co-expression analysis, suggesting it is regulated by multiple lncRNAs. The other mRNA, ALG14, may involve in glycosylation and lipid metabolism47, but its role in carcinogenesis remains unknown. Xist has been identified as an important mediator of X inactivation48. It was differentially co-expressed with C12orf32 (also known as RHNO1) in our study. Consistent with a previous report49, C12orf32 found overexpressed in tumors in our data. This gene may also involve in the DNA-damage response50 and has been suggested as a novel anticancer molecular drug target (e.g. siRNA drugs)49. Our study provides a new source of functional annotating and prioritizing lncRNAs with potentially functional importance for downstream experimental validation.
The majority of breast cancer genetic susceptibility loci identified from genome-wide association studies (GWAS) fall into non-coding regions24. One post-GWAS challenge is to functionally characterize these genetic loci in the development of breast cancer. It is possible that genetic variants mapping to lncRNAs could play an important role in regulating gene expression levels via lncRNA-mRNA co-expression network. Our study found three breast cancer risk loci (rs9832625, rs11836164 and rs2823779) mapped to lncRNAs that were either differentially expressed or co-expressed with mRNAs in tumor and normal breast tissue. Of note, rs2823779 mapped to lncRNA LINC00478. The latter was differentially co-expressed with four mRNAs in the lncRNA-mRNA regulatory network, including cancer-related genes CETN136,37 and TOX446. In addition, intron of LINC00478 encodes a miRNA cluster comprising let-7c, miR-99a, and miR-125b that might regulate HER2 signaling in breast cancer progression51. Our study attested the important roles of those mRNAs and lncRNAs in breast tumorigenesis.
We acknowledge a number of limitations in our study. First, the study power was limited due to the moderate sample size, especially for lncRNA-mRNA co-expression analyses. However, we took multiple testing into consideration and applied a stringent significance threshold to guard against false positive results. Furthermore, we validated our findings using external resources, including TCGA dataset and other public bioinformatics resources. Future studies with larger sample sizes are needed to validate the reported findings. Second, we employed a computational and bioinformatics approach to infer lncRNA functions. We integrated expression profiles of mRNAs and lncRNAs into co-expression models to study lncRNA characteristics in tumor and normal breast tissue. Although we identified a set of mRNAs that were co-expressed with lncRNAs, the detailed mechanism of gene regulation remains unknown. Experimental validation of lncRNA roles in “wet” lab is warranted. Third, we considered all breast cancer cases as one group in our study due to limited sample size. However, it is possible that breast cancer-associated lncRNAs and lncRNA-mRNA co-expression regulation are specific to histological and molecular subtypes. Subgroup analyses by breast cancer subtypes are warranted in future studies. Finally, we only considered one possible mechanism for the function of lncRNAs in gene regulation, that is, through correlations of gene expression levels between lncRNAs and mRNAs. LncRNAs can work through multiple other mechanisms such as chromatin remodeling, promoter demethylation, microRNA silencing, and acting as molecular scaffolds5,42,43. Thus future research is needed to take into account these possible mechanisms for a more complete understanding of lncRNA functions.
In conclusion, using normal breast tissue as a desirable baseline control and a genome-wide analytic approach, we identified a number of tissue-specific lncRNAs associated with breast cancer and further inferred their biological functional through lncRNA-mRNA co-expression analyses. Our findings suggest a complex and extensive role of lncRNAs in breast cancer development through regulating gene expression. Further work is needed to validate our findings and to understand the detailed molecular mechanism of specific lncRNAs implicated in breast cancer.
Methods
Study subjects and breast tissue samples
The study includes 18 breast cancer cases and 18 healthy controls of women of European Ancestry between ages 32 to 80 (Supplementary Table 1). Cases are patients with pathologically confirmed primary breast cancer diagnosed at one of three hospitals in Indianapolis, Indiana, between 1998 and 2009: Indiana University (IU) Hospital, Eskenazi Hospital (previously known as Wishard Hospital), and IU Simon Cancer Center (IUSCC). Controls are randomly selected from a pool of healthy women who donated both blood and normal breast tissue samples to the Susan G. Komen Tissue Bank at IUSCC between 2005 and 2009, and were free of breast cancer up to the time of donation. The participants completed a questionnaire on medical histories and health-related exposures at the time of donation. Controls are matched to cases based on ancestry and age (within 2 years). Breast tissue (untreated tumor or normal) biospecimens were collected from each case and control under standard operating procedures at IUSCC. All breast tissue samples were snap-frozen immediately after removal and stored in liquid nitrogen until processed, and were determined to be of high quality through histological and molecular quality control tests. Tumor samples were pathologically verified for high tumor content. Information concerning demographics, clinical data, and personal risk factors, including age, race, reproductive history, family history of breast cancer, are either extracted from medical records (for cases) or obtained through the questionnaires (for controls). Signed informed consent was obtained from each case or control prior to tissue samples collection. The study was approved by Indiana University institutional review board. The study was carried out in accordance with the approved guidelines.
RNA-sequencing data
Total RNA was extracted from freshly frozen breast tissue (tumor or normal) samples using the Qiagen miRNeasy Mini Kit. Construction of cDNA libraries and subsequent RNA sequencing of paired-end reads (2 × 50 nt reads) were performed according to the standard Illumina protocol using the HiSeq2000 sequencing systems. The raw sequencing output was 25–35 million reads per sample. Quality control (QC) filtering was first performed on raw RNA-seq data to remove adapter sequences and poor quality bases using the FastqMCF clipping algorithm52. RNA-seq reads were then mapped by Bowtie v1.0.053 to GENCODE lncRNA reference (release 17) and UCSC GRCh37/hg19 knownGene reference, respectively, for lncRNA and mRNA annotations. Transcript abundances were quantified using NGSUtils54. Samples were further filtered based on percentage of genes detected (less than 50%) and percentage of reads mapped to the reference (less than 25%). Extreme outliers were further identified and filtered from the dataset using principal component analysis (PCA). Low expression lncRNAs were filtered from the dataset based on counts per million (CPM) threshold of 1. After these steps, a total of 7,450 lncRNAs and 22,362 mRNAs were retained and used in further analyses.
Statistical Analysis
All data analyses were performed using R and Bioconductor, unless otherwise noted. Details of analysis methods are described as follows.
Differential Expression
Differential expression (DE) analyses were performed using edgeR v2.6.12 implemented in the Bioconductor package55 to identify differentially expressed mRNAs or lncRNAs between tumor and normal breast tissue. Biological coefficients of variation between the samples were estimated using an empirical Bayes approach under the assumption that the data follows a negative binomial distribution. Differential expression between tumor and normal breast tissue was analyzed using a generalized linear model to regress RNA (lncRNA or mRNA) expression on tissue type, adjusting for age and sequencing batch. We referred to it as Model 1: YRNA = β0 + β1XTissue Type + β2XBatch + β3XAge. The false discovery rate (FDR) by Benjamini and Hochberg (BH) procedure56 was applied to correct for multiple testing. Statistical significance was defined as FDR P value <0.01 and a two-fold change (FC) of expression level between comparison of tumor and normal breast tissue. The heat map and locus-by-locus volcano plot were performed using R package.
Co-expression Analysis
Co-expression of lncRNAs and mRNAs in tumor and normal breast tissue was analyzed using a generalized linear model to regress mRNA expression on lncRNA expression, adjusting for age and sequencing batch. In our study, we were specifically interested in breast cancer-associated co-expression patterns that differ in tumor and normal breast tissue. We considered two scenarios: (1) differential co-expression in which the correlation between lncRNA and mRNA expression differs in tumor and normal breast tissue; and (2) dose-response co-expression in which the correlation is similar in tumor and normal breast tissue, but the expression level of lncRNA or mRNA differs in two tissue types. Accordingly, we constructed two generalized linear models to analyze the data:
Model 2: YmRNA = β0 + β1XlncRNA + β2XTissue Type + β3XlncRNA·XTissue Type + β4XBatch + β5XAge;
Model 3: YmRNA = β0 + β1XlncRNA + β2XTissue Type + β3XBatch + β4XAge.
A lncRNA is considered to be differentially co-expressed with a mRNA in tumor and normal breast tissue if its correlation differs in the two tissue types. In order to reduce false positives, Bonferroni correction was applied to control for multiple testing and a stringent P value threshold (P < 3 × 10−10 for the interaction term in Model 2) was applied to declare statistical significance. Statistically significant differential co-expression between a lncRNA and a mRNA was defined as P value for interaction term β3 < 3 × 10−10 from model 2. A lncRNA is considered to be dose-response co-expressed with a mRNA if its correlation is similar in tumor and normal breast tissue but the expression level of either the lncRNA or the mRNA differs in the two tissue types. Statistically significant dose-response co-expression was defined when all the following criteria are met: |β3| < 0.01 from model 2, P value for β1 < 3 × 10−10 from model 3, and either lncRNA or mRNA is significantly and differentially expressed in model 1.
Validation in TCGA Dataset
(1) Differentially expressed mRNAsRNA-seq data from 848 individuals was downloaded from TCGA, including 744 breast tumors and 104 non-tumorous adjacent-normal breast tissue samples. All samples were collected from individuals who self-reported as women of European ancestry. This dataset consisted of called gene counts for 20,531 mRNAs. We filtered out low expression transcripts based on percentage of samples (less than 50%) and CPM cutoff of 1. A total of 14,371 mRNAs were remained after filtering and used in the differential expression analysis by edgeR. The false discovery rate (FDR) method by the Benjamini and Hochberg (BH) procedure56 was applied to correct for multiple testing. Statistical significance was defined as FDR P value <0.01 and a two-fold change of expression level between comparison of tumor and adjacent normal breast tissue. (2) Co-expression patternsBeing approved, raw RNA-seq data and the corresponding clinical data (Biotab format) for 692 breast invasive carcinoma (BRCA) patients was acquired from TCGA. We chose patient barcodes as unique identifiers to build the connection of transcriptome data with clinical information. Sequencing alignment, expression qualification, and QC filtering were performed as previously described for our data. Finally, a total of 6,556 lncRNAs and 15,074 mRNAs were retained for co-expression analysis in tumor tissue from TCGA. We used a generalized linear model to regress mRNA expression on lncRNA expression, adjusting for age and sequencing batch. Bonferroni correction was used to control for multiple testing.
Functional Validation using Bioinformatics Resources
We used an open-access resource, TANRIC16, to interactively explore biological and clinical function of the lncRNAs in breast cancer based on TCGA dataset of 837 breast tumor samples and 105 adjacent normal samples. Analysis of variance (ANOVA) was used to examine if a lncRNA was differentially expressed across tumor subtypes or tumor stages. Student t-test was used to assess statistical significance of the difference in lncRNA expression between mutated and wild-type of a particular gene. To identify biological pathways that are significantly enriched among the differentially expressed mRNAs and the mRNAs that are co-expressed with lncRNA in tumor and normal breast tissue, we performed a hypergeometric test using consensusPathDB57 to calculate the enrichment significance based on annotation files from GO58, KEGG59, and BioCarta (www.biocarta.com). Functional enrichment analysis was also performed using Ingenuity Pathway Analysis (IPA) software (www.ingenuity.com). We also identified the top list of transcriptional regulators that explain the observed differential gene expression using Upstream Regulator Analytic tool17 implemented in IPA software. We further investigated whether the identified lncRNAs from our data contained any known breast cancer risk loci identified from previous genome-wide association studies (GWAS). GWAS Catalog23 was used to retrieve breast cancer-associated single nucleotide polymorphisms (SNPs).
Additional Information
How to cite this article: Wu, W. et al. Tissue-specific Co-expression of Long Non-coding and Coding RNAs Associated with Breast Cancer. Sci. Rep. 6, 32731; doi: 10.1038/srep32731 (2016).
Supplementary Material
Acknowledgments
This research was supported by NIH R01 grant R01CA194030 from the National Cancer Institute and Career Catalyst Research grant CCR15333233 from Susan G. Komen®. CH is supported by Indiana University Simon Cancer Center Breast Cancer Program and Cancer Prevention and Control Program. We thank the technical staff at Q Squared Solutions Expression Analysis LLC for their collaboration and scientific support in performing the RNA-sequencing for the study. We acknowledge Indiana CTSI Specimen Storage Facility (SSF), which is supported by NCRR Clinical and Translational Sciences Award (U54-RR025761) and NCRR construction award (C06-RR020128-01). We also thank the Indiana University Simon Cancer Center at Indiana University School of Medicine for the use of the Tissue Procurement & Distribution Core, which provided breast tumor specimens. Samples from the Susan G. Komen Tissue Bank at the Indiana University Simon Cancer Center were used in this study. We thank contributors, including Indiana University who collected samples used in this study, as well as donors and their families, whose help and participation made this work possible.
Footnotes
The authors declare no competing financial interests.
Author Contributions C.H. directed the study. E.K.W., Y.L. and C.H. designed the experiments. A.M.V.S. contributed to sample collection. W.W. and E.K.W. performed the RNA-seq data processing, the analysis pipeline, and prepared figures and tables. Y.H., X.R. and H.D. contributed to additional data processing. J.H., J.C. and Y.L. provided valuable insight, interpretations and advice. W.W., E.K.W. and C.H. wrote the manuscript with inputs from the other authors. All authors read and approved the final manuscript.
References
- Carninci P. et al. The transcriptional landscape of the mammalian genome. Science. 309, 1559–1563, 10.1126/science.1112014 (2005). [DOI] [PubMed] [Google Scholar]
- Mercer T. R., Dinger M. E. & Mattick J. S. Long non-coding RNAs: insights into functions. Nature reviews. Genetics. 10, 155–159, 10.1038/nrg2521 (2009). [DOI] [PubMed] [Google Scholar]
- Wapinski O. & Chang H. Y. Long noncoding RNAs and human disease. Trends in cell biology. 21, 354–361, 10.1016/j.tcb.2011.04.001 (2011). [DOI] [PubMed] [Google Scholar]
- Brunner A. L. et al. Transcriptional profiling of long non-coding RNAs and novel transcribed regions across a diverse panel of archived human cancers. Genome biology. 13, R75, 10.1186/gb-2012-13-8-r75 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu A., Singhal U. & Chinnaiyan A. M. Long noncoding RNAs in cancer: from function to translation. Trends in cancer. 1, 93–109, 10.1016/j.trecan.2015.08.010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb E. A., Brown C. J. & Lam W. L. The functional role of long non-coding RNA in human carcinomas. Molecular cancer. 10, 38, 10.1186/1476-4598-10-38 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X. et al. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 10.1038/onc.2015.340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X. et al. Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic acids research. 41, e35, 10.1093/nar/gks967 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes & development. 25, 1915–1927, 10.1101/gad.17446611 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiche K. et al. Long non-coding RNAs differentially expressed between normal versus primary breast tumor tissues disclose converse changes to breast cancer-related protein-coding genes. PloS one. 9, e106076, 10.1371/journal.pone.0106076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paci P., Colombo T. & Farina L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC systems biology. 8, 83, 10.1186/1752-0509-8-83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A. et al. Gene expression abnormalities in histologically normal breast epithelium of breast cancer patients. International journal of cancer. Journal international du cancer. 122, 1557–1566, 10.1002/ijc.23267 (2008). [DOI] [PubMed] [Google Scholar]
- Graham K., Ge X., de Las Morenas A., Tripathi A. & Rosenberg C. L. Gene expression profiles of estrogen receptor-positive and estrogen receptor-negative breast cancers are detectable in histologically normal breast epithelium. Clinical cancer research: an official journal of the American Association for Cancer Research. 17, 236–246, 10.1158/1078-0432.CCR-10-1369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornienko A. E. et al. Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome biology. 17, 14, 10.1186/s13059-016-0873-8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J. et al. MicroRNA expression profiles classify human cancers. Nature. 435, 834–838, 10.1038/nature03702 (2005). [DOI] [PubMed] [Google Scholar]
- Li J. et al. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer research. 75, 3728–3737, 10.1158/0008-5472.CAN-15-0273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A., Green J., Pollard J. Jr. & Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 30, 523–530, 10.1093/bioinformatics/btt703 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R., Akhurst R. J. & Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nature genetics. 29, 117–129, 10.1038/ng1001-117 (2001). [DOI] [PubMed] [Google Scholar]
- Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 9, 361–371, 10.1038/nrc2628 (2009). [DOI] [PubMed] [Google Scholar]
- Bieging K. T., Mello S. S. & Attardi L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 14, 359–370, 10.1038/nrc3711 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. & Gustafsson J. A. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 11, 597–608, 10.1038/nrc3093 (2011). [DOI] [PubMed] [Google Scholar]
- Guttman M. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 477, 295–300, 10.1038/nature10398 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 42, D1001–1006, 10.1093/nar/gkt1229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K. et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature genetics. 45, 353–361, 361e351–352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett G. C. et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother Oncol. 111, 178–185, 10.1016/j.radonc.2014.02.012 (2014). [DOI] [PubMed] [Google Scholar]
- Tsoi L. C. et al. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome biology. 16, 24, 10.1186/s13059-014-0570-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F. et al. Microarray Profiling and Co-Expression Network Analysis of LncRNAs and mRNAs in Neonatal Rats Following Hypoxic-ischemic Brain Damage. Scientific reports. 5, 13850, 10.1038/srep13850 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima R., Akhade V. S., Pal D. & Rao S. M. Long noncoding RNAs in development and cancer: potential biomarkers and therapeutic targets. Mol Cell Ther. 3, 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschner T. & Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA biology. 9, 703–719, 10.4161/rna.20481 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T. R., Dinger M. E. & Mattick J. S. Long non-coding RNAs: insights into functions. Nature reviews Genetics. 10, 155–159 (2009). [DOI] [PubMed] [Google Scholar]
- Liao Q. et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic acids research. 39, 3864–3878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongor L. et al. A genome-wide approach to link genotype to clinical outcome by utilizing next generation sequencing and gene chip data of 6,697 breast cancer patients. Genome medicine. 7, 104, 10.1186/s13073-015-0228-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q. W. et al. Expression and clinical significance of matrix metalloproteinase-9 in lymphatic invasiveness and metastasis of breast cancer. PloS one. 9, e97804, 10.1371/journal.pone.0097804 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S. K. et al. CAHM, a long non-coding RNA gene hypermethylated in colorectal neoplasia. Epigenetics : official journal of the DNA Methylation Society. 9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Z. et al. A novel imprinted gene, KCNQ1DN, within the WT2 critical region of human chromosome 11p15.5 and its reduced expression in Wilms' tumors. Journal of biochemistry. 128, 847–853 (2000). [DOI] [PubMed] [Google Scholar]
- Kim J. J. et al. CETN1 is a cancer testis antigen with expression in prostate and pancreatic cancers. Biomark Res. 1, 22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuangta X. et al. Knockdown of CETN1 inhibits breast cancer cells proliferation. J Buon. 19, 656–661 (2014). [PubMed] [Google Scholar]
- Yuan L. et al. Proteomic analysis reveals that MAEL, a component of nuage, interacts with stress granule proteins in cancer cells. Oncol Rep. 31, 342–350 (2014). [DOI] [PubMed] [Google Scholar]
- Ponting C. P., Oliver P. L. & Reik W. Evolution and functions of long noncoding RNAs. Cell. 136, 629–641 (2009). [DOI] [PubMed] [Google Scholar]
- Sigova A. A. et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci USA 110, 2876–2881, 10.1073/pnas.1221904110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili M. N. et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome biology. 16, 20, 10.1186/s13059-015-0586-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana M. et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 147, 358–369 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C., Qu K., Zhong F. L., Artandi S. E. & Chang H. Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Molecular cell. 44, 667–678, 10.1016/j.molcel.2011.08.027 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto R., Mayeda A., Yoshida M. & Nakagawa S. MALAT1 long non-coding RNA in cancer. Biochimica et biophysica acta. 10.1016/j.bbagrm.2015.09.012 (2015). [DOI] [PubMed] [Google Scholar]
- Bounaix Morand du Puch C. et al. TOX4 and its binding partners recognize DNA adducts generated by platinum anticancer drugs. Arch Biochem Biophys. 507, 296–303 (2011). [DOI] [PubMed] [Google Scholar]
- Chung W. et al. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PloS one. 3, e2079, 10.1371/journal.pone.0002079 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirkan A. et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. Plos genetics. 8, e1002490 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh C. A. et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 521, 232–236, 10.1038/nature14443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-W. et al. Involvement of C12orf32 overexpression in breast carcinogenesis. Int J Oncol. 37, 861–867 (2010). [DOI] [PubMed] [Google Scholar]
- Cotta-Ramusino C. et al. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science (New York, N Y). 332, 1313–1317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey S. T., Westerling T. & Brown M. Loss of estrogen-regulated microRNA expression increases HER2 signaling and is prognostic of poor outcome in luminal breast cancer. Cancer research. 75, 436–445, 10.1158/0008-5472.CAN-14-1041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E. ea-utils: "Command-line tools for processing biological sequencing data", http://code.google.com/p/ea-utils (2011).
- Langmead B., Trapnell C., Pop M. & Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, R25, 10.1186/gb-2009-10-3-r25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese M. R. & Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics. 29, 494–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26, 139–140, 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. & Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological). 57, 289–300, 10.2307/2346101 (1995). [DOI] [Google Scholar]
- Kamburov A., Wierling C., Lehrach H. & Herwig R. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic acids research. 37, D623–628, 10.1093/nar/gkn698 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 25, 25–29, 10.1038/75556 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Sato Y., Furumichi M. & Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research. 40, D109–114, 10.1093/nar/gkr988 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie L. et al. Long non-coding RNAs: versatile master regulators of gene expression and crucial players in cancer. American journal of translational research. 4, 127–150 (2012). [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.