

Abstract
Background
Patients of African ancestry with untreated HIV-1 infection and carrying the G1 or G2 kidney disease risk variants (Vs) at the APOL1 gene have a tenfold higher risk of developing HIV-associated nephropathy (HIVAN) compared to those with the non-risk wild type (WT) G0 variant. However, the mechanistic contribution of the APOL1 allelic state to kidney injury in HIV-1 infection remains to be elucidated.
Results
Non-risk WT APOL1 is associated with lower intracellular levels of HIV-1 in conditionally immortalized human podocytes, while the over expression of G1 or G2 risk Vs significantly increases viral accumulation. The priming of podocytes with exogenous IL-1β facilitates HIV-1 entry, via the up-regulation of DC-SIGN. The over expression of APOL1 G1 and G2 risk Vs in combination with an increase in IL-1β levels causes a greater increase in viral concentration than either condition alone. In turn, HIV-1 and exogenous IL-1β together induce a de novo secretion of endogenous IL-1β and an increase of APOL1 gene expression.
Conclusions
Our findings indicate that the presence of risk Vs of APOL1 is permissive of HIV-1 persistence in human podocytes in synergy with IL-1β, a cytokine that characterizes the inflammatory milieu of acute and chronic phases of HIV-1 infection. The elucidation of these molecular mechanisms explains, at least in part, the higher frequency of HIVAN in populations carrying the risk polymorphic genetic variant of APOL1 gene.
Keywords: APOL1, HIV-1, HIVAN, Podocyte, IL-1β, DC-SIGN
Background
HIV-1 associated nephropathy (HIVAN) is an important complication of HIV-1 infection and is characterized by collapsing focal segmental glomerulosclerosis (FSGS) and massive proteinuria [1, 2]. Different risk factors contribute to the pathogenesis of HIVAN. A longstanding high viral load is a major risk factor for the development of HIVAN [3–6]. Immune activation and chronic inflammation are additional risk factors for the onset of HIVAN together with specific genetic mutations in the host [7–10]. Indeed, a direct association between kidney risk variants (Vs) of the Apolipoprotein L1 (APOL1) gene and the development of HIVAN had been reported [11–17]. These allelic Vs have been termed G1 (Ser342Gly and Ile384Met) and G2 (del.N388/Y389), in contrast to the non-risk wild type (WT) G0 allele. APOL1 is a minor component of plasma circulating High-Density Lipoprotein (HDL) endowed with the ability to kill Trypanosoma brucei responsible for African sleeping sickness [18–22]. The emerging resistance to the non-risk WT allele by Trypanosoma brucei gambiense and rhodesiense remarkably increased the frequency of the risk Vs of APOL1 in the residents of many regions of Sub-Saharan Africa as a consequence of pathogen selection pressure [23]. Intracellular expression of APOL1 has been reported in several cell types, including podocytes, and appears to be a lipid-binding protein relevant for cellular homeostasis through endosomal trafficking regulation, for lysosomal function and autophagy ruling and for activation of innate immune response [24–28]. However, the molecular mechanisms explaining the role of APOL1 in the pathogenesis of HIVAN remain elusive.
Podocytes are epithelial cells acting in conjunction with fenestrated endothelium and glomerular basement membrane to ensure the integrity of the blood-urine barrier and glomerular filtration [29]. The challenge of studying human primary podocytes is due to their terminally differentiated phenotype. Development of the HIVAN transgenic murine model and the use of conditionally immortalized human podocytes (CIHPs) have provided in vivo and in vitro models that have greatly advanced our understanding of HIVAN physiopathology [30–32]. In particular, it has been demonstrated that viral gene products directly induce pathologic changes in the phenotype and functions of podocytes such as deregulations of several host cellular pathways that involve cell cycle, oxidative stress, and apoptosis [33–39]. Data from kidney biopsy of HIVAN patients also showed that podocytes host and accumulate HIV-1 and serve as viral reservoirs in kidney [4, 40]. Finally, in vitro experiments demonstrated that human podocytes are able to capture HIV-1 and spread the virus by trans-infecting target cells [41–43].
Interleukin-1β (IL-1β) possesses a strong pro-inflammatory effect. Its production is tightly controlled by two steps: (1) induction of pro-IL-1β gene expression and (2) caspase-1-mediated cleavage of pro-IL-1β through the activation of the inflammasome complexes [44]. Several types of viruses, including hepatitis C virus and HIV-1 have been found to induce the production of IL-1β through the induction of NLRP3 inflammasome [45, 46]. Recently, increased secretion of bioactive IL-1β was observed to be relevant for inflammatory programmed cell death (pyroptosis) of CD4pos T cells with abortive HIV-1 infection [46]. Very little is known about the possible role of IL-1β in HIVAN pathogenesis.
Herein, we demonstrate that IL-1β in human podocytes facilitates HIV-1 trafficking by greatly enhancing the uptake of the virus via the up-regulation of DC-SIGN receptor. High intracellular levels of HIV-1 in synergy with IL-1β increase the expression of APOL1 that can serve as a natural anti-viral restriction factor. Indeed, the non-risk WT APOL1 gene product inhibits HIV-1 accumulation in podocyte, while the over expression of the APOL1 G1/G2 Vs increases the amount of HIV-1 in the same cells. These findings indicate that the allelic state of APOL1 is a key in determining the equilibrium between HIV-1 degradation and accumulation in human podocytes, thus representing a critical pathogenic factor in the pathogenesis of HIVAN.
Results
APOL1 targets HIV-1 trafficking in human podocytes
It has been previously reported that human podocytes constitutively express APOL1 [26, 27]. Our experiments of confocal microscopy showed that APOL1 protein is located in proximity of the cellular membrane of CIHPs and preferentially co-localize with early endocytosed vesicles expressing Rab5 protein and not with endosomes marker EEA1 required for endosomes maturation. On the other hand, we did not observe APOL1 co-localization with either Rab7 or LAMP1 molecules which are expressed in late endosomes and lysosome compartments respectively (Fig. 1a). These findings prompted us to study a possible role of APOL1 in regulating HIV-1 entry and trafficking within human podocytes.
Fig. 1.
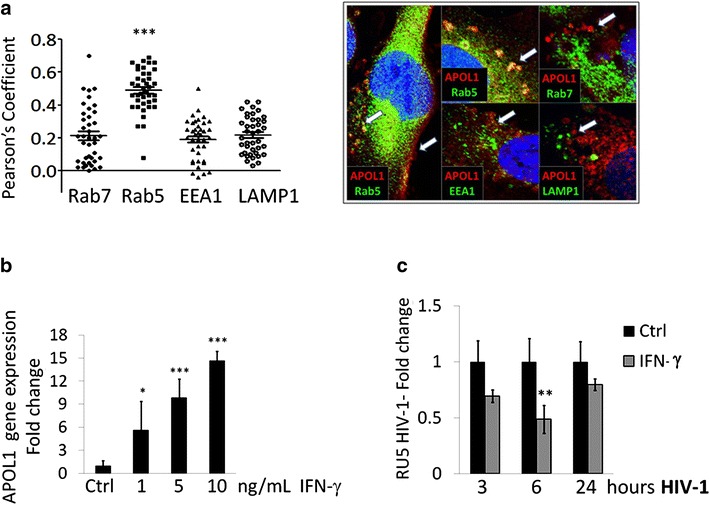
Increased expression of WT APOL1 inversely correlates to HIV-1 persistence in CIHPs. a Summary graph of statistical dot plots (left panel) showing the co-localization off WT APOL1 with Rab5, Rab7, EEA1 and LAMP1 as an average measured by Pearson’s Coefficient on data analyzed with the Olympus FLUOVIEW FV1000 confocal microscope software (N = 3). Representative fluorescent microscopic images (right panel) showing the co-localization of WT APOL1 (red) with Rab5, Rab7, EEA1 and LAMP1 (green) marked vesicles and plasma membrane in CIHPs. Nucleuses are stained with DAPI in blue. b Statistical histogram graph showing the dose-dependent response of APOL1 gene expression after IFN-γ stimulation (16 h). Results are expressed as relative fold change in IFN-γ treated CIHPs versus untreated controls (Ctrl) and normalized to GAPDH gene expression (N = 4). c Statistical histogram graph showing HIV-1 accumulation in CIHPs incubated for 16 h with IFN-γ (10 ng/mL) compared to their untreated counterparts (Ctrl). Results are expressed as relative fold change of HIV-1 DNA RU5 in IFN-γ treated cells versus Ctrl and normalized to GAPDH gene (N = 4). P values *<0.05, **<0.01; ***<0.001
Since IFN-γ is a known inducer of APOL1 expression in podocytes [27, 28], CIHPs were pre-stimulated with IFN-γ. IFN-γ increases the APOL1 gene expression in CIHPs in a concentration-dependent manner (Fig. 1b). Previously it has been observed that in vitro experiments the capture of HIV-1 in human podocytes does not generate a productive virus replication [42, 43]. Therefore, to detect HIV-1 in podocytes we measured the specific HIV-1 strong stop DNA (RU5 HIV-1). This is possible because virions harbor strong-stop DNA since the endogenous reverse transcription of HIV-1 occurs prior to infection of target cells [47].
We then determined the viral entry in the podocytes by measuring the specific HIV-1 strong stop DNA concentration. Our data then demonstrated that the amount of HIV-1 significantly decreases in CIHPs treated with IFN-γ compared to their untreated counterparts (Fig. 1c). These results suggest that the IFN-γ-mediated up-regulation of APOL1 interfere with the entry and/or early post-entry steps regulating HIV-1 trafficking in podocytes. In this regard, we previously reported that HIV-1 internalization in CIHPs is mediated by both lipids raft and DC-SIGN receptor [41, 42]. However, IFN-γ treatment did not have any effect on DC-SIGN expression in podocytes (data not shown), thus ruling out a direct contribution of this inflammatory cytokine in limiting the viral entry in CIHPs through the modulation of this lectin-type receptor.
We then analyzed whether APOL1 plays a direct role in regulating HIV-1 accumulation in human podocytes. To this end, we transiently transfected CIHPs with the non-risk WT allele of APOL1. Similar to CIHPs stimulated with IFN-γ, we found that increased expression of WT APOL1 significantly decrease the levels of HIV-1 compared with CIHPs transfected with an empty control vector (Fig. 2a). We then proceeded to assess the effect of risk G1 and G2 APOL1 allelic Vs on HIV-1 accumulation in CIHPs stably transfected with either WT or G1/G2 APOL1 Vs. In contrast with non-risk WT APOL1, the over expression of the G1 or the G2 APOL1 Vs significantly increased HIV-1 levels in CIHPs (Fig. 2b), thus demonstrating that these risk Vs of APOL1facilitate HIV-1 accumulation and persistence in human podocytes. The higher amounts of virus in CIHPs transfected with the G1 or the G2 APOL1 Vs also induced a significant higher trans-infection of co-cultured CD4pos T cells that showed a significant increase of viral replication compared to that exerted by CIHPs transfected with WT APOL1 allele (Fig. 2c).
Fig. 2.
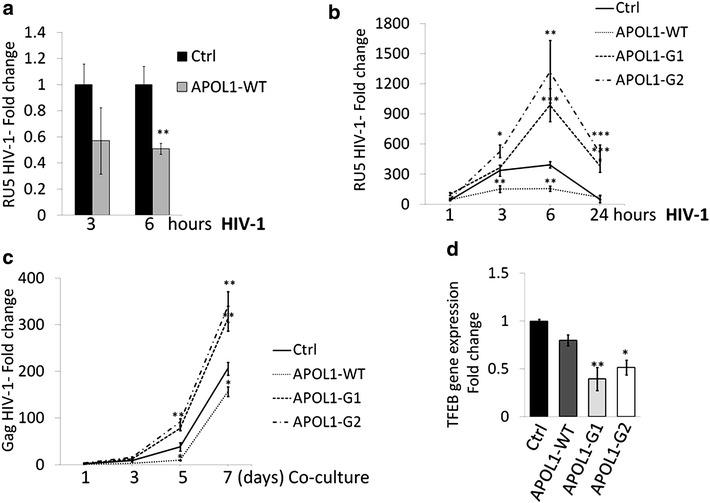
APOL1 polymorphism regulates HIV-1 persistence in CIHPs. a Statistical histogram graph showing HIV-1 accumulation in CIHPs transiently transfected with a APOL1-WT expressing vector compared to the control vector (Ctrl) following a time course incubation (3 and 6 h) with the virus. Results are expressed as relative fold change of HIV-1 DNA RU5 in APOL1-WT transfected cells compared to Ctrl and normalized to GAPDH gene (N = 4). b Time-course experiments showing the HIV-1 accumulation in CIHPs stable transfected with the control vector (Ctrl) or the specific APOL1-WT or APOL1-G1/G2 expressing vectors. Results are expressed as relative fold change of HIV-1 DNA RU5 in HIV-1 incubated CIHPs versus non HIV-1 treated cells and normalized to GAPDH gene (N = 4). c Rescue of infectious HIV-1 by CD4pos T lymphocytes co-cultured in time-course experiments (1, 3, 5 and 7 days) with HIV-1 pulsed CIHPs stable transfected with APOL1-WT or APOL1-G1 or APOL1-G2 compared with cells transfected with control vector (Ctrl). Since HIV-1 internalization in human podocytes is characterized by an abortive HIV-1 infection, cell lysates were collected and analyzed by qPCR for HIV-1 Gag gene not present in podocytes and indicative of a productive infection in CD4pos T cells. Results are expressed as fold increased of HIV-1 DNA Gag gene in PBMC co-cultured with HIV-1-pulsed CIHPs transfected with APOL1-WT or APOL1-G1 or APOL1-G2 or an empty vector (Ctrl) compared to their HIV-1 untreated counterparts and normalized to the amount of GAPDH gene (N = 4). d Statistical histogram graph showing TFEB gene expression in CIHPs transiently transfected with control vector (Ctrl) or APOL1-WT or APOL1-G1 or APOL1-G2 expressing vector. Results are expressed as fold change of transfected cells compared to Ctrl and normalized to the expression of GAPDH gene (N = 4). P values: *<0.05, **<0.01; ***<0.001
We also assessed in podocytes the transcriptional levels of TFEB, the master transcription factor that regulates lysosome biosynthesis and whose activity is regulated by APOL1 expression [28, 48]. In addition, in human macrophages APOL1-dependent expression of TFEB was associated with high HIV-1 Gag protein degradation [28, 48]. Although, we did not observe differences in TFEB gene expression in WT APOL1 transfected podocytes, over expression of G1/G2 Vs of APOL1 significantly decrease the TFEB gene expression in CIHPs compared to that of cells transfected with WT APOL1 allele (Fig. 2d). These findings suggest that the APOL1 risk Vs may promote HIV-1 accumulation in human podocytes through the disruption of endosomal/lysosomal pathway regulation.
IL-1β facilitates HIV-1 entry in human podocytes via DC-SIGN and increases viral accumulation
The establishment of inflammation together with the pathologic production of several pro-inflammatory cytokines is a hallmark of HIV-1 infection. In particular, the increased systemic levels of IL-1β have been found in the sera of HIV-1 infected patients both in acute and chronic phases of the disease [49–52]. More recently, it has also been reported that abortive infection of HIV-1 in CD4pos T cells induces an inflammatory programmed cell death (i.e. pyroptosis) caused by the release of pro-inflammatory cytokines including IL-1β [46]. Furthermore, an increased expression of inflammasome markers such as IL-1β and caspases-1 was observed in the renal cortex of HIV-transgenic mice (Tg26) [53]. This experimental evidence prompted us to analyze the direct effect of exogenous rhIL-1β on CIHPs at different time points. We found that the stimulation of CIHPs with 25 ng/mL of rhIL-1β induce a rapid and significant increase of IL-1β gene expression that reached a plateau after 6 h of incubation (Fig. 3a). This phenomenon appeared to be also dose-dependent as the maximum response was reached at the concentration of 1.0 ng/mL of rhIL-1β (Fig. 3b). Moreover, the incubation of rhIL-1β-primed CIHPs with HIV-1 also induced a significantly higher IL-1β transcription levels compare to CIHPs stimulated with rhIL-1β alone (Fig. 3c). Nevertheless, the treatment of CIHPs with HIV-1 alone did not induce any increase of the IL-1β gene expression nor any release of detectable levels of IL-1β in cell supernatant (Fig. 3d). Only the stimulation of podocytes with rhIL-1β followed by incubation with HIV-1 induced a detectable secretion of endogenous IL-1β (Fig. 3d). These data suggest that increased levels of circulating IL-1β in the glomerular milieu of HIV-1 infected patients can induce a pathologic inflammatory loop that leads to the production and secretion of endogenous IL-1β by human podocytes.
Fig. 3.
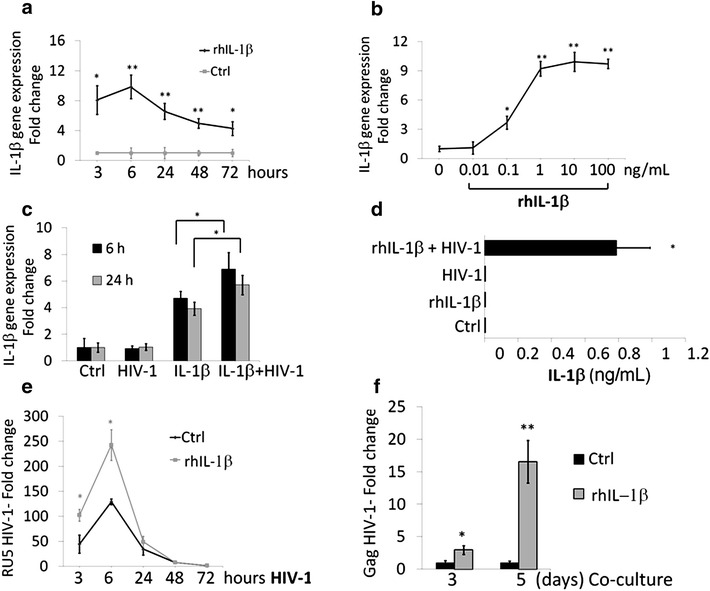
IL-1β increases HIV-1 accumulation in CIHPs. a Time and b dose response of IL-1β gene expression in unstimulated (Ctrl) and rhIL-1β treated CIHPs. Results are expressed as relative fold change in rhIL-1β treated cells versus Ctrl and normalized to GAPDH gene expression (N = 3). c Statistical histogram graph showing the IL-1β gene expression in CIHPs untreated (Ctrl), treated with rhIL-1β alone (25 ng/mL) for 16 h or incubated with HIV-1 either alone (6, 24 h) or with IL-1β (6, 24 h). Results are expressed as fold change of IL-1β gene expression in treated cells versus Ctrl and normalized to the GAPDH gene expression (N = 5). d Statistical histogram graph showing the amount of IL-1β protein secretion by CIHPs primed with rhIL-1β (25 ng/mL) and subsequently incubated with HIV-1 for 6 h compared to their counterparts incubated with rhIL-1β alone, HIV-1 alone or untreated cells (Ctrl) (N = 5). e Time course experiments showing HIV-1 accumulation in CIHPs either untreated (Ctrl) or pre-stimulated with rhIL-1β (25 ng/mL) for 16 h and followed by incubation with HIV-1 at different time points (3, 6, 24, 48, 72 h). Results are expressed as relative fold change of HIV-1 DNA RU5 in HIV-1 treated cells compared to their HIV-1 untreated counterparts for both experimental settings and normalized to GAPDH gene (N = 5). f Statistical histogram graph showing the rescue of infectious HIV-1 by CD4pos T lymphocytes co-cultured for 3 and 5 days with HIV-1-pulsed CIHPs either primed or none (Ctrl) with rhIL-1β. Since HIV-1 internalization in human podocytes is characterized by an abortive HIV-1 infection, cell lysates were collected and analyzed by qPCR for HIV-1 Gag gene not present in podocytes and indicative of a productive infection in CD4pos T cells. Results are expressed as relative fold change of DNA HIV-1 Gag copies in rhIL-1β treated compared to Ctrl and normalized to DNA copies of GAPDH gene (N = 5). P values: *<0.05, **<0.01
We then assessed the effect of rhIL-1β in viral trafficking within human podocytes by stimulating CIHPs with 25 ng/mL of rhIL-1β for 16 h followed by incubation with HIV-1. Cells were harvested at different time-points and HIV-1 strong stop DNA was measured by qPCR. The priming of CIHPs with rhIL-1β significantly increased the amount of HIV-1 compared to podocytes incubated in the absence of rhIL-1β (Fig. 3e). The higher concentration of HIV-1 in rhIL-1β-primed CIHPs also induced a significantly higher trans-infection of co-cultured CD4pos T lymphocytes that showed an increase of viral replication compared to that exerted by CIHPs not primed with rhIL-1β (Fig. 3f). Given that HIV-1 entry in CIHPs depends on DC-SIGN expression [42] we than analyzed the transcripts levels of DC-SIGN in podocytes following stimulation with rhIL-1β. The incubation with rhIL-1β induced a rapid and significant increase of DC-SIGN gene expression in CIHPs (Fig. 4a). These results indicate that the inflammatory loop triggered by rhIL-1β is permissive of HIV-1 internalization in CIHPs via the up-regulation of DC-SIGN, which represent an already reported mechanism employed by HIV-1 to enter in these cells [42].
Fig. 4.
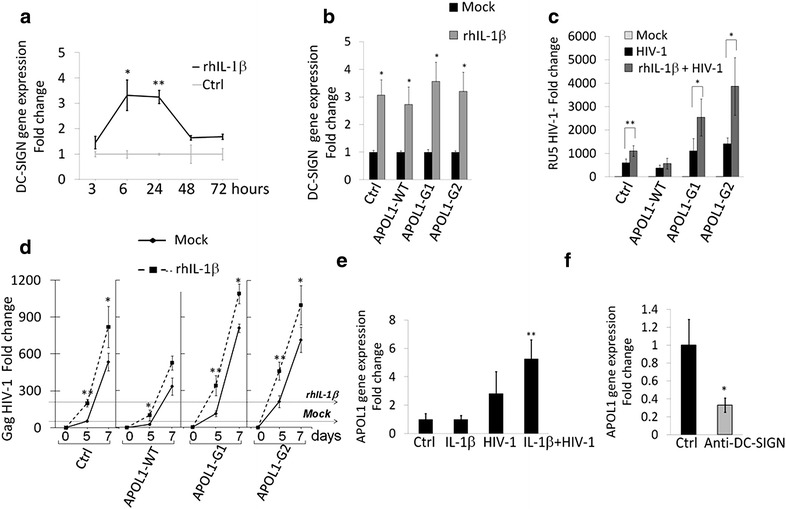
APOL1 polymorphism affects IL-1β-dependent persistence of HIV-1 in CIHPs. a Time course experiments showing DC-SIGN gene expression in CIHPs after stimulation with rhIL-1β (25 ng/mL) compared to untreated cells (Ctrl) and normalized to GAPDH gene expression (N = 3). b Statistical histogram graph showing DC-SIGN gene expression after 18 h of rhIL-1β (25 ng/mL) treatment in CIHPs stably transfected with either empty vector (Ctrl) or WT or APOL1 risk Vs. Results are expressed as relative fold change in rhIL-1β treated cells versus non treated cells (Mock) and normalized to GAPDH gene expression (N = 3). c Statistical histogram graph showing HIV-1 accumulation after rhIL-1β stimulation in CIHPs stably transfected with the control vector (Ctrl) or the specific APOL1-WT or APOL1-G1/G2 expressing vectors. Results are expressed as relative fold change of HIV-1 DNA RU5 in HIV-1 incubated CIHPs versus non HIV-1-pulsed cells (Mock) and normalized to GAPDH gene (N = 3). d Rescue of infectious HIV-1 by CD4pos T lymphocytes co-cultured in time-course experiments (0, 5 and 7 days) with HIV-1 pulsed CIHPs stable transfected with control vector (Ctrl), APOL1-WT or APOL1-G1/G2 Vs and pretreated or not (Mock) with rhIL-1β. Results are expressed as fold increased of HIV-1 DNA Gag gene in PBMC co-cultured with HIV-1 pulsed CIHPs compared to their HIV-1 untreated counterparts and normalized to the amount of GAPDH gene (N = 3). Horizontal lines represent Mock and rhIL-1β in the Ctrl sample depicting the fold change in HIV-1 replication. e APOL1 gene expression in CIHPs incubated with rhIL-1β (25 ng/mL) and subsequently treated with HIV-1 for 6 h compared to their counterparts incubated with rhIL-1β alone, HIV-1 alone or untreated (Ctrl) Results are expressed as relative fold change normalized to GAPDH gene expression (N = 5). f Statistical histogram graph showing the APOL1 gene expression in CIHPs incubated with IL-1β (25 ng/mL) and subsequently treated with HIV-1 for 6 h either in pre absence (Ctrl) or in the presence of blocking anti-DC-SIGN mAb. Results are expressed as relative fold change in treated cells normalized to GAPDH gene expression (N = 3). P values *< 0.05, **< 0.01
RhIL-1β increases expression of DC-SIGN also in CIHPs stably transfected with either WT or G1/G2 APOL1 Vs (Fig. 4b), therefore, we used this experimental model to test the direct impact of rhIL-1β priming on HIV-1 entry and trans-infection to CD4pos T lymphocytes in the context of APOL1 polymorphism. RhIL-1β priming in both G1 and G2 APOL1 risk Vs was associated with the significantly increased accumulation of HIV-1. This phenomenon was correlated with the higher trans-infection of CD4pos T lymphocytes compared to the non treated cells with rhIL-1β (Fig. 4c). Podocytes transfected with APOL1 WT and pre-treated with rhIL-1β also resulted with the significant upper trans-infection of CD4pos T lymphocytes after 5 days of co-culture (Fig. 4d). Nevertheless, over expression of APOL WT in rhIL-1β pre-stimulated cells showed lower virus concentration versus rhIL-1β-primed CIHPs transfected with empty vector. These results confirmed that only the presence of APOL1 WT, and not of APOL1 Vs, is associated with the control of HIV-1 entry and accumulation in podocytes.
In addition, we found that the incubation of CIHPs with rhIL-1β and HIV-1 together is required to raise the level of APOL1 gene expression, as neither rhIL-1β nor HIV-1 alone induced a significant increase of its transcript when compared to control experiments (Fig. 4e). To verify whether the up-regulation of APOL1 expression in rhIL-1β-primed CIHPs is dependent on HIV-1 binding to DC-SIGN receptor, cells were pre-treated with a masking anti-DC-SIGN mAb before incubation with HIV-1. We found that the blocking of DC-SIGN induced a significant decrease of APOL1 gene expression, thus reversing the up-regulation of this apolipoprotein in response to simultaneous stimulation given by rhIL-1β and HIV-1 (Fig. 4f).
Discussion
The pathogenic role of APOL1 is emerging in several kidney diseases targeting podocytes and strong association between APOL1 high-risk Vs and progressive non-diabetic kidney disease [15, 26, 54], FSGS and HIVAN [11–17] have been reported. The present study demonstrates that risk G1 and G2 variants of APOL1 enhances HIV-1 accumulation and persistence in human podocytes, and also contributes to spread infection to neighbor CD4pos T cells. This process is amplified by IL-1β, whose priming of human podocytes increases viral entry and, together with HIV-1 itself, up-regulates the APOL1 gene expression (Fig. 5).
Fig. 5.
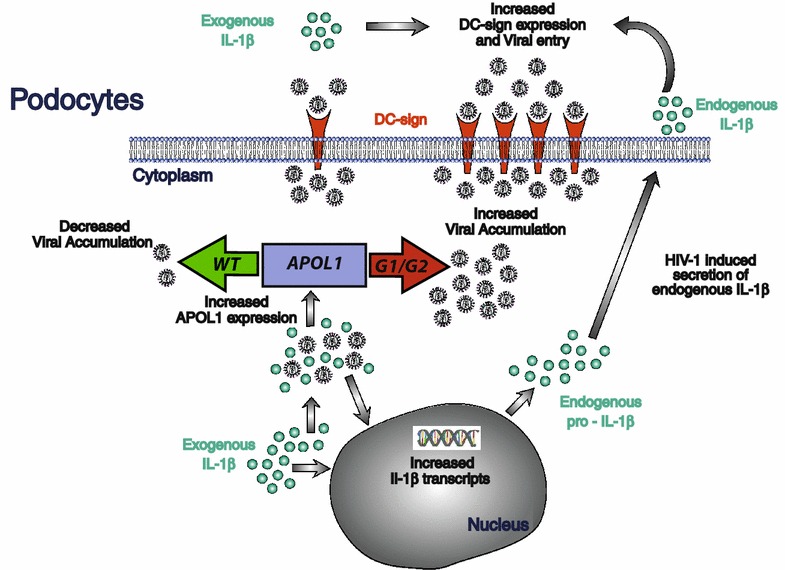
Mechanistic insights regulating HIV-1 entry and persistence in human podocytes. High levels of IL-1β both in tissues and in blood characterize acute and chronic HIV-1 infection and can prime human podocytes in the context of the inflamed microenvironment of kidney glomerula. Indeed, exogenous IL-1β enhances HIV-1 entry via the up-regulation of DC SIGN and induces the production/secretion of endogenous IL-1β. In turn, the presence of both IL-1β and HIV-1 increase the expression of APOL1 within podocytes. In this context, the non-risk WT variant of APOL1 is key to control viral accumulation within podocytes and limit HIV-1 boarding. On the other side, the alternative presence of the high-risk G1 or G2 polymorphic variants of APOL1 increase the accumulation of HIV-1 in podocytes, thus worsening the vicious pathogenic inflammatory loop in podocytes. This can explain, at least in part, the higher frequency of HIVAN in populations carrying the high-risk genetic variants of APOL1 gene
Previous in vitro studies reported that the over expression of APOL1 in podocytes induces lysosomal swelling and cell death with the G1 and G2 risk variants exerting stronger effects at a lower threshold compared to the non-risk WT APOL1 [55]. The present study first sought to examine the impact of APOL1 polymorphism in regulating HIV-1 trafficking in human podocytes. Indeed, these cells are able to capture HIV-1 that, instead of fully replicating to generate a productive infection, is rapidly degraded [42, 43]. We demonstrate here that the transfection of non-risk WT APOL1 allele in CIHPs decreases the intracellular concentration of HIV-1, thus highlighting that the WT variant of APOL1 serves as a natural anti-viral restriction factor in podocytes. Our additional experimental evidence showing that the expression of APOL1 is increased in rhIL-1β-primed podocytes following HIV-1 entry further supports this hypothesis. In this regard, it has been also shown that the anti-viral cytokine IFN-γ is able to greatly increase the expression of APOL1 [27] that, in turn, can target multiple steps of HIV-1 replication cycle in monocytes with the final result of inducing viral endocytosis and degradation within lysosome compartments [28]. Our data confirmed that also the stimulation of CIHPs with IFN-γ induce higher transcription levels of APOL1 that, as an intracellular protein, co-localize with Rab5 early endosomes. Of note, it has been already reported that Rab5 early endosomes regulate the homeostasis of endosomal pathway of podocytes [56], and that binding of HIV-1 to DC-SIGN leads to an internalization of the virus in early Rab5 positive endosomes [57]. These findings suggest that the IFN-γ-mediated up-regulation of APOL1, together with DC-SIGN-mediated uptake of the virus, likely play a major role in endocytic trafficking and degradation of HIV-1 also in human podocytes.
Another important contribution to antiviral responses of podocytes might occur as a consequence of the inflammatory processes that characterize both acute and chronic HIV-1 infection. In particular, both circulating and tissue resident immune cells (i.e. monocytes/macrophages, T CD4pos lymphocytes, gut epithelial cells, etc.) have been reported to produce high amount of IL-1β that, in turn, boost local and systemic inflammation during the course of HIV-1 disease [46, 50, 58–60]. Indeed, high levels of IL-1β have been found in the sera of HIV-1 infected patients [61–63]. Although the harmful effect of this potent cytokine in increasing viral replication and in directly damaging several immune and not immune cells both in blood and tissue have been extensively reported [46, 59, 60, 64–66], little is known about the pathogenic role of IL-1β in HIV-1 targeted podocytes. Recently, it has been shown in a murine HIVAN model (Tg26) that HIV-1 induces the activation of the inflammasome in podocytes [53]. We show here that the stimulation of podocytes with exogenous rhIL-1β induces a de novo IL-1β gene transcription. However, the secretion of bioactive endogenous IL-1β requires a second signal delivered by HIV-1 following its uptake and internalization in podocytes. In this regard, we already reported the HIV-1 entry in human podocytes is mainly mediated by DC-SIGN [42], a lectin-type surface receptor able to bind HIV-1 in a CD4-independent manner and promote trans-infection to CD4pos T cells [67]. Additionally, we also demonstrate here that in vitro stimulation of CIHPs with rhIL-1β increases the expression of DC-SIGN, thus explaining both the higher accumulation of HIV-1 in rhIL-1β-primed podocytes and the increased trans-infection of co-cultured CD4pos T cells. Hence, the priming with rhIL-1β facilitates viral entry into podocytes and triggers a harmful and vicious loop that increases viral spreading and induces a de novo secretion of endogenous IL-1β by podocytes. In this regard, it has also been reported that under inflammatory stress podocytes can serve as non-hematopoietic professional antigen-presenting cells that up-regulate the expression of MHC class-II molecules [68–70]. The elucidation of these mechanisms in the context of the inflamed glomerular milieu of HIV-1 infected patients containing high levels of IL-1β help us to better understand the pathological changes observed in kidneys that ultimately culminate in cell injuries and organ failure [36–39].
In the context of the HIV-1 driven inflammation in kidney, we demonstrate that the non-risk WT variant of APOL1 is endowed with natural antiviral activity that limits HIV-1 accumulation in podocytes. Indeed, our data showing that rhIL-1β priming in synergy with HIV-1 increases the gene expression of this apolipoprotein in podocytes via the up-regulation of DC-SIGN explain, at least in part, the low incidence of HIVAN in the general population carrying the WT APOL1. This is not the case for those populations carrying the risk G1 and G2 polymorphic variants of APOL1 that, following HIV-1 infection, experience a much higher frequency of HIVAN development [11–17]. The present study demonstrates that the over expression of these latter risk Vs of APOL1 is associated with significantly higher HIV-1 accumulation in podocytes, thus representing a relevant risk factor for the onset of HIVAN. Furthermore, the expression of APOL1 G1 and G2 risk Vs in combination with an increase in IL-1β levels causes a greater increase in viral concentration than either condition alone. Since, the risk Vs of APOL1 inhibit in podocytes the gene expression of TFEB, a master transcription factor that regulates lysosome biosynthesis [48], this strongly suggests that the increased boarding of HIV-1 in the podocytes carrying the G1 or G2 APOL1 polymorphism is due to a defective viral clearance. Furthermore, it has been previously reported that only APOL1 risk Vs could induce lysosomal leakage in podocytes effects at a lower threshold compared to the non-risk WT APOL1 [55]. The reversal from clearance to enhanced boarding of HIV-1 as a function of the non-risk WT versus risk G1 or G2 alleles state of APOL1, may explain why, of all podocytopathic kidney diseases, HIVAN shows the most striking odds ratio, reading >89 in one recent study [71].
Conclusions
The present study demonstrates that APOL1 risk polymorphic Vs have a great impact in increasing uptake, accumulation and persistence of the HIV-1 in human podocytes. The inflammatory milieu that characterizes HIV-1 infection both at systemic and tissue levels can enhance this pathologic loop via the IL-1β priming. This explains, at least in part, the higher frequency of HIVAN in populations carrying the G1 and G2 genetic variants of APOL1 gene (Fig. 5).
Methods
Cells and virus
Conditionally immortalized human podocytes (CIHPs) were developed and cultured as described in Saleem et al. [30]. CIHPs were transiently transfected with the empty vector or pcDNA3-APOL1-WT or -APOL1-G1/G2 expressing plasmid by using as a transfection reagent Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Meanwhile 48–72 h following transfection cells were used for qPCR analysis or treated with HIV-1BaL. Stable G0/G1/G2 APOL1 CIHPs were generated by retroviral infection. Briefly, the open reading frame APOL1 (G0, G1, G2) was cloned into the retroviral vector pBABE carrying resistance to puromycin. To generate retroviral particles, the viral packaging cell line HEK-GP were co-transfected with the pBABE construct of interest and the VSV gene. CIHPs were infected twice within 24 h with the viral-containing supernatant of HEK-GP cells. Selection with puromycin (1 μg/mL) was continued for a week, and comparable expression of respective sequence of the corresponding APOL1 was verified.
Human PBMCs were obtain from Buffy coats of healthy volunteers who signed consent forms in accordance with the Declaration of Helsinki and with clinical protocols approved by the Institutional Review Board of Desio Hospital, Milan, Italy. PBMCs were isolated over Ficoll Paque™ Premium density gradients (GE Healthcare Bio-Sciences AB, Little Chalfont, Buckinghamshire, UK) and cultured in RPMI 1640 medium, supplemented with 10 % FBS, 2 mM l-glutamine, 100 U/mL penicillin/streptomycin and activated with 5 μg/mL of Lectin from Phaseolus vulgaris (PHA-L) (Sigma-Aldrich, Saint Louis, MO, USA) for 3 days PHA-L and subsequently with 200 IU/mL of human recombinant IL-2 (Peprotech, Rocky Hill, NJ, USA). In co-cultivation studies activated lymphocytes were added to podocytes at a ratio of 5:1 in a RPMI complete medium supplemented with 200 U/mL of IL-2. HIV-1BAL strain was expanded in activated PBMCs collected at the peak of virus replication. HIV-1 p24 Ag concentrations in the culture supernatants were determined by ELISA (Aalto, Ratfharnham Village, Ireland) as described in [42].
Reagents
Human recombinant (h) IL-1β and interferon-γ were purchased from PeproTech. Human IL-1β secreted protein was measured in cell-free harvested supernatants by ELISA (DuoSet DY201, R&D System, Minneapolis, MN, USA). In blocking experiments CIHPs were pre-treated with rIL-1β, washed out and then incubated with control Isotype (Ctrl) or mouse anti-DC-SIGN monoclonal Ab (mAb) at the dose of 5 mg/mL (Abcam) before HIV-1 treatment.
QPCR
For cellular gene expression total RNA was extracted using RNeasy mini columns (Qiagen, Valencia, CA, USA), following manufacturer’s instructions. One μg of total RNA was used to generate cDNA templates for RT-PCR, using random primers, RNase inhibitor and High-Capacity cDNA Reverse Transcription Kit from Applied Biosystem (Foster City, CA, USA). IL-1β, TFEB, DC-SIGN, WT-1, podocin, nephrin, synaptopodin and housekeeping S18 and GAPDH genes expression were analyzed by the TaqMan® mRNA specific assays. Followed primers were used for the SYBR Green PCR Master Mix analysis (Applied Biosystem): APOL1 Fw-5′-ATCTCAGCTGAAAGCGGTGAAC-3′ and Rev-5′-TGACTTTGCCCCCTCATGTAAG-3′; 18S Fw-5′-ACTTTCGATGGTAGTCGCCGT-3′ and Rev-5′-CCTTGGATGTGGTAGCCGTTT-3′.
HIV-1 detection
Viral stock before incubation with podocytes were treated (1 h at room temperature) with 200 U/mL of RNase-free DNase (Roche, Basel, Switzerland). Before preparation of cell lysates, control and HIV-1-pulsed cells were washed three times with PBS, then treated with 0.05 % trypsin at 37 °C for 10 min to eliminate non internalized virus, and subsequently washed five times with PBS. Cell lysates were prepared as described in Malnati et al. [72] and subjected to qPCR TaqMan analysis by using HIV-1 specific primers: Gag Fw-5′-ACATCAAGCAGCCATGCAAAT-3′, Rev-5′-ATCTGGCCTGGTGCAATAGG-3′ and probe 5′-CATCAATGAGGAAGCTGCAGAATGGGATAGA-3′, LTR RU5 Fw-5′-GGCTAACTAGGGAACCCACTG-3′, Rev-5′-CTGCTAGAGATTTTCCACACTGAC-3′ and probe 5′-TGTGTGCCCGTCTGTTGTGTG-3′ [42, 73]. Amplification of GAPDH gene as a reference was used to control the amount of DNA in each sample (Applied Biosystem).
Fluorescence confocal microscopy
CIHPs were growth on the coverslip’s coated with human collagen IV then were fixed with 4 % paraformaldehyde, followed by permeabilization and blocking with 0.3 % Triton X-100 and 10 % goat serum. Immunoblotting with the followed primary antibodies were performed: Mouse monoclonal anti-APOL1, anti-DC-SIGN and rabbit polyclonal anti-podocin purchased from Abcam; Rabbit polyclonal anti-Rab5, -Rab7, -EEA, -Lamp1 purchased from Life Technolgies.
Statistical analysis
The significance of the data was assessed using Student’s t test statistical analysis. Data shown are mean ± S.D. The number of experiments is specified in the Figure legends. In the figures statistical significance (P) is indicated by asterisks (*). *P < 0.05; **P < 0.01; ***P < 0.001.
Authors’ contributions
JM and DM designed the study and wrote the manuscript. JM, FO, FP, PTXL performed experiments and analyzed data. MAS and KS provided reagents. KS and PCS analyzed data and revised the manuscript. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
This work has been supported by Italian Ministry of Health (Grant AIDS RF-ICH-2009-130413 to J.M. and RF-ICH-2009-1299677 to D.M.), by European Union (Marie Curie International Reintegration Grant 249249 to J.M.) and by intramural research and clinical funding programs of Humanitas Research Hospital assigned to D.M. This work was also supported by Grant 1R01DK098074 assigned to P.C.S. from National Institutes of Health, Bethesda, MD, USA and by the Israel Science Foundation (ISF 182/15) and the Ernest and Bonnie Beutler Fund and intramural grants of the Rambam Medical Center to K.S.
Contributor Information
Joanna Mikulak, Email: joanna.mikulak@humanitasresearch.it.
Ferdinando Oriolo, Email: Ferdinando.Oriolo@humanitasresearch.it.
Federica Portale, Email: federica.portale@gmail.com.
Paolo Tentorio, Email: Paolo.Tentorio@humanitasresearch.it.
Xiqian Lan, Email: Xlan@NSHS.edu.
Moin A. Saleem, Email: m.saleem@bristol.ac.uk
Karl Skorecki, Email: skorecki@tx.technion.ac.il.
Pravin C. Singhal, Email: PSinghal@NSHS.edu
Domenico Mavilio, Email: domenico.mavilio@unimi.it.
References
- 1.Mikulak J, Singhal PC. HIV-1 and kidney cells: better understanding of viral interaction. Nephron Exp Nephrol. 2010;115:e15–e21. doi: 10.1159/000312882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medapalli RK, He JC, Klotman PE. HIV-associated nephropathy: pathogenesis. Curr Opin Nephrol Hypertens. 2011;20:306–311. doi: 10.1097/MNH.0b013e328345359a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruggeman LA, Ross MD, Tanji N, Cara A, Dikman S, Gordon RE, Burns GC, D’Agati VD, Winston JA, Klotman ME, Klotman PE. Renal epithelium is a previously unrecognized site of HIV-1 infection. J Am Soc Nephrol. 2000;11:2079–2087. doi: 10.1681/ASN.V11112079. [DOI] [PubMed] [Google Scholar]
- 4.Tanji N, Ross MD, Tanji K, Bruggeman LA, Markowitz GS, Klotman PE, D’Agati VD. Detection and localization of HIV-1 DNA in renal tissues by in situ polymerase chain reaction. Histol Histopathol. 2006;21:393–401. doi: 10.14670/HH-21.393. [DOI] [PubMed] [Google Scholar]
- 5.Husain M, Gusella GL, Klotman ME, Gelman IH, Ross MD, Schwartz EJ, Cara A, Klotman PE. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol. 2002;13:1806–1815. doi: 10.1097/01.ASN.0000019642.55998.69. [DOI] [PubMed] [Google Scholar]
- 6.He JC, Husain M, Sunamoto M, D’Agati VD, Klotman ME, Iyengar R, Klotman PE. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1, 2 pathways. J Clin Invest. 2004;114:643–651. doi: 10.1172/JCI200421004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eustace JA, Nuermberger E, Choi M, Scheel PJ, Jr, Moore R, Briggs WA. Cohort study of the treatment of severe HIV-associated nephropathy with corticosteroids. Kidney Int. 2000;58:1253–1260. doi: 10.1046/j.1523-1755.2000.00280.x. [DOI] [PubMed] [Google Scholar]
- 8.Smith MC, Austen JL, Carey JT, Emancipator SN, Herbener T, Gripshover B, Mbanefo C, Phinney M, Rahman M, Salata RA, et al. Prednisone improves renal function and proteinuria in human immunodeficiency virus-associated nephropathy. Am J Med. 1996;101:41–48. doi: 10.1016/S0002-9343(96)00065-4. [DOI] [PubMed] [Google Scholar]
- 9.Kimmel PL, Cohen DJ, Abraham AA, Bodi I, Schwartz AM, Phillips TM. Upregulation of MHC class II, interferon-alpha and interferon-gamma receptor protein expression in HIV-associated nephropathy. Nephrol Dial Transplant. 2003;18:285–292. doi: 10.1093/ndt/18.2.285. [DOI] [PubMed] [Google Scholar]
- 10.Snyder A, Alsauskas ZC, Leventhal JS, Rosenstiel PE, Gong P, Chan JJ, Barley K, He JC, Klotman ME, Ross MJ, Klotman PE. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS. 2010;24:1107–1119. doi: 10.1097/QAD.0b013e328337b0ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papeta N, Kiryluk K, Patel A, Sterken R, Kacak N, Snyder HJ, Imus PH, Mhatre AN, Lawani AK, Julian BA, et al. APOL1 variants increase risk for FSGS and HIVAN but not IgA nephropathy. J Am Soc Nephrol. 2011;22:1991–1996. doi: 10.1681/ASN.2011040434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fine DM, Wasser WG, Estrella MM, Atta MG, Kuperman M, Shemer R, Rajasekaran A, Tzur S, Racusen LC, Skorecki K. APOL1 risk variants predict histopathology and progression to ESRD in HIV-related kidney disease. J Am Soc Nephrol. 2012;23:343–350. doi: 10.1681/ASN.2011060562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruzel-Davila E, Wasser WG, Aviram S, Skorecki K. APOL1 nephropathy: from gene to mechanisms of kidney injury. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfu391. [DOI] [PubMed] [Google Scholar]
- 16.Eggers PW, Kimmel PL. Is there an epidemic of HIV Infection in the US ESRD program? J Am Soc Nephrol. 2004;15:2477–2485. doi: 10.1097/01.ASN.0000138546.53152.A7. [DOI] [PubMed] [Google Scholar]
- 17.Lucas GM, Lau B, Atta MG, Fine DM, Keruly J, Moore RD. Chronic kidney disease incidence, and progression to end-stage renal disease, in HIV-infected individuals: a tale of two races. J Infect Dis. 2008;197:1548–1557. doi: 10.1086/587994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duchateau PN, Pullinger CR, Orellana RE, Kunitake ST, Naya-Vigne J, O’Connor PM, Malloy MJ, Kane JP. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L. J Biol Chem. 1997;272:25576–25582. doi: 10.1074/jbc.272.41.25576. [DOI] [PubMed] [Google Scholar]
- 19.Duchateau PN, Movsesyan I, Yamashita S, Sakai N, Hirano K, Schoenhaus SA, O’Connor-Kearns PM, Spencer SJ, Jaffe RB, Redberg RF, et al. Plasma apolipoprotein L concentrations correlate with plasma triglycerides and cholesterol levels in normolipidemic, hyperlipidemic, and diabetic subjects. J Lipid Res. 2000;41:1231–1236. [PubMed] [Google Scholar]
- 20.Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, Van Den Abbeele J, Pays A, Tebabi P, Van Xong H, Jacquet A, et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature. 2003;422:83–87. doi: 10.1038/nature01461. [DOI] [PubMed] [Google Scholar]
- 21.Molina-Portela MP, Samanovic M, Raper J. Distinct roles of apolipoprotein components within the trypanosome lytic factor complex revealed in a novel transgenic mouse model. J Exp Med. 2008;205:1721–1728. doi: 10.1084/jem.20071463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homble F, Vanhamme L, Tebabi P, Pays A, Poelvoorde P, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309:469–472. doi: 10.1126/science.1114566. [DOI] [PubMed] [Google Scholar]
- 23.Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, Winkler CA, Kopp J, Rotimi C, Adeyemo A, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA. 2014;111:E2130–E2139. doi: 10.1073/pnas.1400699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan G, Zhaorigetu S, Liu Z, Kaini R, Jiang Z, Hu CA. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem. 2008;283:21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma L, Shelness GS, Snipes JA, Murea M, Antinozzi PA, Cheng D, Saleem MA, Satchell SC, Banas B, Mathieson PW, et al. Localization of APOL1 protein and mRNA in the human kidney: nondiseased tissue, primary cells, and immortalized cell lines. J Am Soc Nephrol. 2014;26:339–348. doi: 10.1681/ASN.2013091017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, Friedman DJ. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2014;87:332–342. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor HE, Khatua AK, Popik W. The innate immune factor apolipoprotein L1 restricts HIV-1 infection. J Virol. 2014;88:592–603. doi: 10.1128/JVI.02828-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim YH, Goyal M, Kurnit D, Wharram B, Wiggins J, Holzman L, Kershaw D, Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001;60:957–968. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 30.Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, Xing CY, Ni L, Mathieson PW, Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 31.Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, Eckhaus M, Bryant JL, Notkins AL, Klotman PE. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci USA. 1992;89:1577–1581. doi: 10.1073/pnas.89.5.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruggeman LA, Dikman S, Meng C, Quaggin SE, Coffman TM, Klotman PE. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J Clin Invest. 1997;100:84–92. doi: 10.1172/JCI119525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barisoni L, Kriz W, Mundel P, D’Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. doi: 10.1681/ASN.V10151. [DOI] [PubMed] [Google Scholar]
- 34.Barisoni L. Podocyte biology in segmental sclerosis and progressive glomerular injury. Adv Chronic Kidney Dis. 2012;19:76–83. doi: 10.1053/j.ackd.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 35.Zhong J, Zuo Y, Ma J, Fogo AB, Jolicoeur P, Ichikawa I, Matsusaka T. Expression of HIV-1 genes in podocytes alone can lead to the full spectrum of HIV-1-associated nephropathy. Kidney Int. 2005;68:1048–1060. doi: 10.1111/j.1523-1755.2005.00497.x. [DOI] [PubMed] [Google Scholar]
- 36.Rai P, Plagov A, Lan X, Chandel N, Singh T, Lederman R, Ayasolla KR, Mathieson PW, Saleem MA, Husain M, et al. mTOR plays a critical role in p53-induced oxidative kidney cell injury in HIVAN. Am J Physiol Renal Physiol. 2013;305:F343–F354. doi: 10.1152/ajprenal.00135.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salhan D, Husain M, Subrati A, Goyal R, Singh T, Rai P, Malhotra A, Singhal PC. HIV-induced kidney cell injury: role of ROS-induced downregulated vitamin D receptor. Am J Physiol Renal Physiol. 2012;303:F503–F514. doi: 10.1152/ajprenal.00170.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shkreli M, Sarin KY, Pech MF, Papeta N, Chang W, Brockman SA, Cheung P, Lee E, Kuhnert F, Olson JL, et al. Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat Med. 2011;18:111–119. doi: 10.1038/nm.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husain M, Meggs LG, Vashistha H, Simoes S, Griffiths KO, Kumar D, Mikulak J, Mathieson PW, Saleem MA, Del Valle L, et al. Inhibition of p66ShcA longevity gene rescues podocytes from HIV-1-induced oxidative stress and apoptosis. J Biol Chem. 2009;284:16648–16658. doi: 10.1074/jbc.M109.008482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marras D, Bruggeman LA, Gao F, Tanji N, Mansukhani MM, Cara A, Ross MD, Gusella GL, Benson G, D’Agati VD, et al. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat Med. 2002;8:522–526. doi: 10.1038/nm0502-522. [DOI] [PubMed] [Google Scholar]
- 41.Mikulak J, Singhal PC. HIV-1 entry into human podocytes is mediated through lipid rafts. Kidney Int. 2010;77:72–73. doi: 10.1038/ki.2009.366. [DOI] [PubMed] [Google Scholar]
- 42.Mikulak J, Teichberg S, Arora S, Kumar D, Yadav A, Salhan D, Pullagura S, Mathieson PW, Saleem MA, Singhal PC. DC-specific ICAM-3-grabbing nonintegrin mediates internalization of HIV-1 into human podocytes. Am J Physiol Renal Physiol. 2010;299:F664–F673. doi: 10.1152/ajprenal.00629.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khatua AK, Taylor HE, Hildreth JE, Popik W. Non-productive HIV-1 infection of human glomerular and urinary podocytes. Virology. 2010;408:119–127. doi: 10.1016/j.virol.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 45.Chen I, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015;23:55–63. doi: 10.1016/j.tim.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Dornadula G, Pomerantz RJ. Endogenous reverse transcription of human immunodeficiency virus type 1 in physiological microenviroments: an important stage for viral infection of nondividing cells. J Virol. 1996;70:2809–2824. doi: 10.1128/jvi.70.5.2809-2824.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 49.Sinicco A, Biglino A, Sciandra M, Forno B, Pollono AM, Raiteri R, Gioannini P. Cytokine network and acute primary HIV-1 infection. AIDS. 1993;7:1167–1172. doi: 10.1097/00002030-199309000-00003. [DOI] [PubMed] [Google Scholar]
- 50.Sadeghi HM, Weiss L, Kazatchkine MD, Haeffner-Cavaillon N. Antiretroviral therapy suppresses the constitutive production of interleukin-1 associated with human immunodeficiency virus infection. J Infect Dis. 1995;172:547–550. doi: 10.1093/infdis/172.2.547. [DOI] [PubMed] [Google Scholar]
- 51.Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antivir Chem Chemother. 2001;12:133–150. doi: 10.1177/095632020101200301. [DOI] [PubMed] [Google Scholar]
- 52.Barqasho B, Nowak P, Tjernlund A, Kinloch S, Goh LE, Lampe F, Fisher M, Andersson J, Sonnerborg A. Group Qs: kinetics of plasma cytokines and chemokines during primary HIV-1 infection and after analytical treatment interruption. HIV Med. 2009;10:94–102. doi: 10.1111/j.1468-1293.2008.00657.x. [DOI] [PubMed] [Google Scholar]
- 53.Haque S, Lan X, Wen H, Lederman R, Chawla A, Attia M, Bongu RP, Husain M, Mikulak J, Saleem MA, Popik W, Malhotra A, Chander PN, Singhal PC. HIV promotes NLRP3 inflammasome complex activation in murine HIVAN. Am J Pathol. 2016;186(2):347–358. doi: 10.1016/j.ajpath.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol. 2011;22:2119–2128. doi: 10.1681/ASN.2011010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lan X, Jhaveri A, Cheng K, Wen H, Saleem MA, Mathieson PW, Mikulak J, Aviram S, Malhotra A, Skorecki K, Singhal PC. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol. 2014;307:F326–F336. doi: 10.1152/ajprenal.00647.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bechtel W, Helmstadter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol. 2013;24:727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joo KI, Lei Y, Lee CL, Lo J, Xie J, Hamm-Alvarez SF, Wang P. Site-specific labeling of enveloped viruses with quantum dots for single virus tracking. ACS Nano. 2008;2:1553–1562. doi: 10.1021/nn8002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Esser R, Glienke W, von Briesen H, Rubsamen-Waigmann H, Andreesen R. Differential regulation of proinflammatory and hematopoietic cytokines in human macrophages after infection with human immunodeficiency virus. Blood. 1996;88:3474–3481. [PubMed] [Google Scholar]
- 59.Maingat F, Halloran B, Acharjee S, van Marle G, Church D, Gill MJ, Uwiera RR, Cohen EA, Meddings J, Madsen K, Power C. Inflammation and epithelial cell injury in AIDS enteropathy: involvement of endoplasmic reticulum stress. FASEB J. 2011;25:2211–2220. doi: 10.1096/fj.10-175992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guha D, Klamar CR, Reinhart T, Ayyavoo V. Transcriptional regulation of CXCL5 in HIV-1-infected macrophages and its functional consequences on CNS pathology. J Interferon Cytokine Res. 2015;35:373–384. doi: 10.1089/jir.2014.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheung R, Ravyn V, Wang L, Ptasznik A, Collman RG. Signaling mechanism of HIV-1 gp120 and virion-induced IL-1beta release in primary human macrophages. J Immunol. 2008;180:6675–6684. doi: 10.4049/jimmunol.180.10.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, Wu J, Lu Y. Mechanism of HIV-1-TAT induction of interleukin-1beta from human monocytes: involvement of the phospholipase C/protein kinase C signaling cascade. J Med Virol. 2010;82:735–746. doi: 10.1002/jmv.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- 64.Poli G, Kinter AL, Fauci AS. Interleukin 1 induces expression of the human immunodeficiency virus alone and in synergy with interleukin 6 in chronically infected U1 cells: inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc Natl Acad Sci USA. 1994;91:108–112. doi: 10.1073/pnas.91.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Granowitz EV, Saget BM, Wang MZ, Dinarello CA, Skolnik PR. Interleukin 1 induces HIV-1 expression in chronically infected U1 cells: blockade by interleukin 1 receptor antagonist and tumor necrosis factor binding protein type 1. Mol Med. 1995;1:667–677. [PMC free article] [PubMed] [Google Scholar]
- 66.Kinter AL, Poli G, Fox L, Hardy E, Fauci AS. HIV replication in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. J Immunol. 1995;154:2448–2459. [PubMed] [Google Scholar]
- 67.da Silva RC, Segat L, Crovella S. Role of DC-SIGN and L-SIGN receptors in HIV-1 vertical transmission. Hum Immunol. 2011;72:305–311. doi: 10.1016/j.humimm.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goldwich A, Burkard M, Olke M, Daniel C, Amann K, Hugo C, Kurts C, Steinkasserer A, Gessner A. Podocytes are nonhematopoietic professional antigen-presenting cells. J Am Soc Nephrol. 2013;24:906–916. doi: 10.1681/ASN.2012020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baudeau C, Delarue F, He CJ, Nguyen G, Adida C, Peraldi MN, Sraer JD, Rondeau E. Induction of MHC class II molecules HLA-DR, -DP and -DQ and ICAM 1 in human podocytes by gamma-interferon. Exp Nephrol. 1994;2:306–312. [PubMed] [Google Scholar]
- 70.Coers W, Brouwer E, Vos JT, Chand A, Huitema S, Heeringa P, Kallenberg CG, Weening JJ. Podocyte expression of MHC class I and II and intercellular adhesion molecule-1 (ICAM-1) in experimental pauci-immune crescentic glomerulonephritis. Clin Exp Immunol. 1994;98:279–286. doi: 10.1111/j.1365-2249.1994.tb06138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kasembeli AN, Duarte R, Ramsay M, Mosiane P, Dickens C, Dix-Peek T, Limou S, Sezgin E, Nelson GW, Fogo AB, et al. APOL1 risk variants are strongly associated with HIV-associated nephropathy in black South Africans. J Am Soc Nephrol. 2015;26(11):2882–2890. doi: 10.1681/ASN.2014050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malnati MS, Scarlatti G, Gatto F, Salvatori F, Cassina G, Rutigliano T, Volpi R, Lusso P. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load. Nat ProtocOL. 2008;3:1240–1248. doi: 10.1038/nprot.2008.108. [DOI] [PubMed] [Google Scholar]
- 73.Saba E, Origoni M, Taccagni G, Ferrari D, Doglioni C, Nava A, Lisco A, Grivel JC, Margolis L, Poli G. Productive HIV-1 infection of human cervical tissue ex vivo is associated with the secretory phase of the menstrual cycle. Mucosal Immunol. 2013;6:1081–1090. doi: 10.1038/mi.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]