

Abstract
Background
Visceral obesity and metabolic syndrome are commonly associated with non-alcoholic fatty liver disease (NAFLD). The progression of steatosis to NASH depends on a number of metabolic and patient-related factors. The mechanisms of genetic predisposition towards the development of NASH and related fibrosis remain unclear. In this study, our aim was to utilize mitotyping and identify mitochondrial haplotypes that may be associated with NAFLD.
Methods
We examined mitochondrial haplotypes along with patatin-like phospholipase domain containing 3 (PNPLA3) rs738409 genotype to determine their association with NAFLD phenotypes. Whole blood samples were obtained from 341 patients (BMI > 35) undergoing weight reduction surgery after written consent. Liver biopsies were centrally reviewed by a single pathologist based on predetermined pathologic protocol (41.9 % Non-NASH NAFLD, 30.4 % NASH, 27.5 % controls). A 1,122 bp of the mitochondrial control loop was sequenced for each sample and classified into haplogroups.
Results
The presence of haplogroup L exhibits protection against the development of NASH and pericellular fibrosis. The alleles of PNPLA3 locus showed differential distribution in cohorts with NAFLD, NASH and pericellular fibrosis. Heterozygosity at this locus is independently associated with higher risk of having NASH and pericellular fibrosis.
Conclusion
Mitochondrial genetics play an important role in NASH probably by modulation of oxidative stress and the efficiency of oxidative phosphorylation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-016-0324-0) contains supplementary material, which is available to authorized users.
Keywords: PNPLA3 rs738409, Hepatic fibrosis, Obesity, Ethnicity
Background
Non-alcoholic fatty liver disease (NAFLD) is a leading cause of chronic liver disease in the United States and worldwide [1, 2]. The prevalence rate of NAFLD is reported between 10 and 35 % in the general population with the prevalence rates being substantially higher in the obese and diabetic individuals [3].
NAFLD is characterized by accumulation of fat, mainly triglycerides, in the absence of excessive alcohol consumption, viral infection, hereditary disorders, or use of steatogenic medications [4, 5]. In its progressive form, NAFLD manifests as non-alcoholic steatohepatitis (NASH) defined as steatosis with ballooning degeneration and/or presence of Mallory-Denk bodies. NASH can ultimately lead to fibrosis followed by cirrhosis and hepatocellular carcinoma (HCC) [6–8].
Both visceral obesity and metabolic syndrome are commonly associated with NASH [9]. However, not all patients with NASH are obese [10]. The progression of steatosis to NASH has been associated with a number of other metabolic factors, including the quality of nutrition and patient’s lifestyle [11] as well as genetic predisposition [12]. The mechanisms of genetic predisposition to the development of NASH remain unclear, but certainly involve the variation in individual susceptibility to NAFLD [12]. A number of nuclear loci have already been identified as potential contributors to development of NASH [13].
In 2008, a genome-wide association study in a population comprising Hispanic, African American and European American subjects showed that a common genetic variant in the patatin-like phospholipase domain containing 3 protein (PNPLA3) gene is associated with liver fat accumulation [14]. The isoleucine to methionine substitution at position 148 in the patatin-like phospholipase domain containing 3 protein (PNPLA3; I148M variant, rs738409) has since then been shown to be associated with liver fat accumulation and severity of NAFLD [15–18]. The interaction between PNPLA3 and BMI has been supported by several population based studies [19–21]. The studies have shown that body mass index interacts with PNPLA3 and increases susceptibility to NAFLD. However, PNPLA3 function and the effect of the I148M substitution are not yet completely understood.
PNPLA3 protein plays anabolic and catabolic role in lipid metabolism. Purified PNPLA3 protein has been shown to have both hydrolase and lysophosphatidic acid transacetylase activities, with the hydrolase activity predominating [22–24]. However, its in-vivo function and physiological relevance remain controversial. In-vitro studies have shown the 148 M mutation to be a loss-of-function mutation abolishing the hydrolase activity [22, 24] while 148 M mutation is a gain of function mutation for lysophosphatidic acid transacetylase (LPAAT) activity [23]. Consistent with a hydrolase activity hypothesis, a relative reduction in hepatic very low-density lipoprotein secretion with the 148 M mutation has been shown in vitro and in humans [25]. Contrary to the hydrolase activity, wild-type PNPLA3 was also found to have lysophosphatidic acid transacetylase activity and the 148 M mutation was found to be a gain of function of the lipogenic activity [26]. In agreement, rats on a high-fat diet treated with pnpla3 antisense oligonucleotides showed decreased fatty acid esterification into hepatic triglycerides and reduced insulin resistance [27]. In a recent study of obese adolescents with hepatic steatosis, the I148M variant has been shown to modulate the association between oxidized metabolites derived from linoleic acid and cytokeratin 18 fragment, a robust biomarker of liver injury [28]. Recently, a cell specific PNPLA3 function linking PNPLA3 to liver fibrosis has been shown [29, 30]. The authors show retinyl-palmitate lipase activity of PNPLA3 in human hepatic stellate cells with I148M being a loss of function mutation. Thus, the genotypic effect of PNPLA3 protein is complex.
In this study, our central hypothesis is that mitochondrial haplotypes along with a susceptible nuclear genetic background may be involved in predisposing to NAFLD and NASH. With this in mind, we examined mitochondrial haplotypes along with patatin-like phospholipase domain containing 3 (PNPLA3) rs738409 genotype and determined their association with NAFLD and NASH.
Methods
Patient cohort
After informed written consent, blood samples were obtained from 341 patients (BMI > 35) undergoing weight reduction surgery and immediately flash frozen in liquid nitrogen followed by storage in −80 °C. All patients had routine liver biopsies taken which were read by a central pathologist. Clinical data from the time of enrollment were collected and de-identified in compliance with HIPAA regulations. This study has been approved by Internal Review Board of Inova Fairfax Hospital (Federal Assurance FWA00000573).
For the purpose of this study, metabolic syndrome was defined by the presence of three or more of the five following features: total cholesterol ≥200 mg/dl of blood, LDL ≥130 mg/dl, triglyceride ≥150 mg/dl, HDL < 50 mg/dl in women or less than 40 mg/dl in men, and BMI >30. None of the included subjects reported to have excessively consumed alcohol (>10 g/day in women and >20 g/day in men) in the past 5 years. Other chronic liver diseases were excluded by negative serology for hepatitis B and C, no history of toxic exposure and no other cause of chronic liver disease. Ethnicity was recorded as self-reported.
Liver biopsy
Liver biopsies were centrally reviewed by a single pathologist based on predetermined grading system [31]. Histological features such as portal inflammation, lymphoplasmacytic lobular inflammation, polymorphonuclear lobular inflammation, Kupffer cell hypertrophy, apoptotic bodies, focal parenchymal necrosis, glycogen nuclei, hepatocellular ballooning, and Mallory-Denk bodies were evaluated in the H & E sections. Steatosis (Non-NASH NAFLD) was defined as fat with/without lobular inflammation and/or portal inflammation. The severity of steatosis was evaluated according to the percentage of liver parenchyma occupied by fat: 0 = none, 1 ≤ 5 %, 2 = 6–33 %, 3 = 34–66 %, and 4 > 66 %. Non-alcoholic steatohepatitis (NASH) was defined as steatosis, lobular inflammation, and ballooning degeneration with or without Mallory Denk bodies, and with or without fibrosis. Severity of pericellular and portal fibrosis was determined by Masson trichome staining of the biopsy. The scoring was as follows: 0 = no fibrosis, 1 = mild fibrosis, 2 = moderate fibrosis, 3 = marked fibrosis. A score of ≥ 3 was considered as advanced hepatic fibrosis and a score of <3 as mild/no hepatic fibrosis. Patients with hepatic steatosis or NASH were considered to have NAFLD. Patients with normal liver histology were considered as controls.
Isolation of total DNA
Total DNA was extracted from whole blood cells using QIAamp® kits in accordance with manufacturer’s instructions (Qiagen, USA). DNA was then quantified and quality assessed by spectrophotometer (GeneQuant 1300, General Electric). Additionally, to assess the integrity of extracted DNA, agarose gel electrophoresis was carried out. The gel was inspected for evidence of poor DNA quality visible as degradation/smearing. Each DNA sample was diluted 1:10 prior to its use as PCR template.
Primer design and PCR
Amplification of the 1,122 base pair region of the mitochondrial control loop was achieved by using 4 pairs of primers (Additional file 1: Table S1) with 100 basepair overlap of resultant amplicons.
The PCR reaction was set up for 25 μl, containing 12.5 μl HotStar Taq Master Mix Kit (Qiagen®), 2.5 μl (250 nM) of each primer, 1 μl of 1:10 diluted DNA, and 6.5 μl molecular grade water. The thermal cycling was as follows: initial denaturation at 95 °C for 15 min; 30 cycles of 95 °C for 1 min; annealing for 1 min; 72 °C for 1 min. For sequencing, 20 μl of each PCR product was sent for Sanger sequencing to GenScript (Piscataway, NJ).
Sequence assembly and mitochondrial haplotypes identification
Chromatograms of forward and reverse fragments for each primer pair were examined using Chromas Lite® (Technelysium, Brisbane, Australia). The assembly and editing of the fragments was carried out using Sequencher® (version 5.2.4), a consensus sequence of 1,122 bp of the mitochondrial control loop was generated for each DNA sample. All DNA sequences were uploaded into MitoTool software [32] and classified into 15 haplogroups. The success rate of sequencing and subsequent alignment was 96 %.
PNPLA3 SNP rs738409 genotyping
Genotyping for the PNPLA3 SNP rs738409 was completed using predesigned TaqMan probe (Applied Biosystems, CA). Genotyping of rs738409 (C_7241_10) was carried out on CFX96 PCR instrument (Biorad, USA) according to the manufacturer’s protocol (50 °C for 2 min, 95 °C for 10 min, and then 40 cycles of 95 °C for 15 s and 60 °C for 1.5 min). After PCR amplification, a post-PCR plate reads were carried out to generate allelic discrimination plot. The allele assignments were independently verified by RFLP analysis [33].
Statistical analysis
To assess the association of SNP rs738409 genotype and mitochondrial haplotypes, a number of statistical procedures were employed. The pairwise comparison cohorts consisted of subjects with normal liver histology (controls), Non-NASH NAFLD, NASH and NAFLD with pericellular fibrosis. For continuous variables, p values were determined by t-test and for categorical variables, p values were determined by chi-square test. Linear regression models were tested to assess the association between genotype and continuous phenotypes, and logistic regression models were used to test association between genotype and categorical phenotype. Multivariate analysis was carried out to determine independent predictors of NAFLD.
Results
The clinical, demographic and biochemical characteristics of study subjects (N = 341; BMI =48 ± 9.13, Age = 44 ± 11.2, 41.9 % Non-NASH NAFLD, 30.4 % NASH, 27.5 % controls) are summarized in Table 1.
Table 1.
Clinical and demographic characteristic of Study Cohort
| Demographic/Clinical Variable | Average ± SD or N (%) |
|---|---|
| Age (yrs) | 44 ± 11.2 |
| Males | 88 (25 %) |
| BMI (kg/m2) | 48 ± 9.13 |
| ALT (U/L) | 34.59 ± 25.9 |
| AST (U/L) | 26.37 ± 19.35 |
| Glucose (mg/dl) | 108.8 ± 36.76 |
| Triglycerides (mg/dl) | 157.9 ± 93.45 |
| Total cholesterol (mg/dl) | 187.69 ± 39.3 |
| HDL (mg/dl) | 47.28 ± 12.85 |
| LDL (mg/dl) | 108.2 ± 35.8 |
| Normal liver biopsy | 94 (27.5 %) |
| Non-NASH NAFLD | 143 (41.9 %) |
| Histologic NASH | 104 (30.4 %) |
Continuous variables are represented as mean ± standard deviation (SD), and categorical variables as percentages
Patients with histologic NASH when compared to non-NASH NAFLD had higher levels of serum aminotransferases compared to patients with non-NASH NAFLD (p < 0.001) (Table 2). In contrast, fasting serum HDL and platelets were lower in NASH patients compared to those with non-NASH NAFLD (p < 0.001) (Table 2). Fasting serum glucose, serum aminotransferases, serum triglycerides and total cholesterol was higher, while serum HDL and platelets were lower (p < 0.001) (Table 2) in patients with Non-NASH NAFLD compared to controls with normal liver histology. Among the NASH and non-NASH NAFLD subjects, there was no significant difference in the mean BMI, age, serum triglycerides, serum cholesterol, serum LDL and fasting glucose levels (p > 0.05) (Table 2). Similarly, subjects with normal liver histology (controls) had no significant difference in the mean BMI, WBC, platelets, serum LDL and HDL levels when compared to Non-NASH NAFLD subjects (p > 0.05) (Table 2). Notably, patients with normal liver histology were more likely to be younger and had higher platelets counts when compared to those with NASH (p < 0.001) (Table 2).
Table 2.
Comparisons of Controls (normal liver histology) with Non-NASH NAFLD and NASH
| Variable | Normal liver histology (N = 94) | Non-NASH NAFLD (N = 143) | NASH (N = 104) | P-value* | P-value† | P-value‡ |
|---|---|---|---|---|---|---|
| Age (yrs) | 41.48 ± 10.86 | 44.08 ± 11.39 | 46.42 ± 10.94 | 0.0814 | 0.0017 | 0.1053 |
| BMI (kg/m2) | 46.80 ± 7.98 | 47.80 ± 8.50 | 49.47 ± 10.56 | 0.3733 | 0.0512 | 0.1715 |
| WBC (x 103/uL) | 7.81 ± 2.54 | 8.08 ± 2.21 | 8.82 ± 8.81 | 0.3784 | 0.2855 | 0.3366 |
| Platelets (x 103/uL) | 303.51 ± 69.73 | 291.01 ± 64.23 | 268.69 ± 66.97 | 0.1632 | 0.0005 | 0.0094 |
| Insulin Resistance | 1.92 ± 2.83 | 2.40 ± 1.18 | 8.37 ± 11.00 | 0.4879 | 0.0413 | 0.0154 |
| Glucose (mg/dL) | 94.16 ± 21.74 | 110.47 ± 36.20 | 120.22 ± 43.50 | 0.0002 | <.0001 | 0.0641 |
| ALT (U/L) | 22.20 ± 9.74 | 31.58 ± 16.35 | 49.51 ± 37.25 | <.0001 | <.0001 | <.0001 |
| AST (U/L) | 19.41 ± 5.57 | 23.49 ± 11.04 | 36.43 ± 29.69 | 0.0013 | <.0001 | <.0001 |
| Triglycerides (mg/dL) | 125.84 ± 58.42 | 160.26 ± 87.31 | 182.10 ± 117.04 | 0.0018 | 0.0001 | 0.1152 |
| LDL (mg/dL) | 100.96 ± 35.75 | 110.75 ± 35.40 | 111.71 ± 36.34 | 0.0620 | 0.0646 | 0.8555 |
| Total cholesterol (mg/dL) | 175.84 ± 34.00 | 193.24 ± 39.40 | 190.10 ± 41.96 | 0.0011 | 0.0150 | 0.5683 |
| HDL (mg/dL) | 50.40 ± 12.42 | 48.58 ± 13.63 | 42.68 ± 10.96 | 0.3461 | <.0001 | 0.0014 |
| White: N (%) | 66 (70.2 %) | 112 (78.3 %) | 84 (80.8 %) | 0.1579 | 0.0835 | 0.6389 |
| Diabetes: N (%) | 17 (18.3 %) | 44 (30.8 %) | 46 (44.2 %) | 0.0322 | <.0001 | 0.030 |
| Hyperlipidemia: N (%) | 34 (37.4 %) | 69 (50.0 %) | 59 (59.6 %) | 0.0599 | 0.0022 | 0.1438 |
| Hypertension: N (%) | 40 (44.4 %) | 81 (57.9 %) | 64 (65.3 %) | 0.0468 | 0.0041 | 0.2464 |
| Sleep Apnea: N (%) | 53 (57.6 %) | 100 (71.9 %) | 81 (80.2 %) | 0.0241 | 0.0007 | 0.1425 |
*Pairwise comparison between controls (normal liver histology) and Non-NASH NAFLD cohorts; †Pairwise comparison between controls (normal liver histology) and NASH cohorts; ‡Pairwise comparison between Non-NASH NAFLD and NASH cohorts. Continuous variables are represented as mean ± standard deviation (SD), and categorical variables as percentages. For convenience, all statistically significant p-values are highlighted by bold font
Both NAFLD and NASH are associated with PNPLA3 rs738409 variants
In previous studies, PNPLA3 variant I148M (rs738409) has been associated with both the obese phenotype and NAFLD. In order to assess any potential interaction between PNPLA3 and mitochondrial DNA, we genotyped rs738409 in all of our patients. The prevalence of the homozygous CC allele in study population was 58 %, while GG allele frequency was 7.5 % and GC allele frequency was 33.8 %. For the PNPLA3 SNP rs738409 alleles, observed frequencies were in accordance with Hardy-Weinberg equilibrium [34–36].
In controls, the CC genotype of PNPLA3 rs738409 was more prevalent than in the NASH cohort (72.3 vs 43.3 %, p < 0.001). Similarly, the CC genotype was more common in patients with non-NASH NAFLD as compared to NASH (60.8 vs 43.3 %, p = 0.006), while the prevalence GC genotype was higher in patients with NASH as compared to those with Non-NASH NAFLD (51.9 vs 25.5 %, p < 0.0001) and higher as compared with the controls (51.9 vs 26.6 %, p = 0.0003). Interestingly, the homozygosity for the risk allele (GG genotype) had higher prevalence in Non-NASH NAFLD patients as compared to NASH patients (14 vs 4.8 %, p = 0.018) (Fig. 1).
Fig. 1.
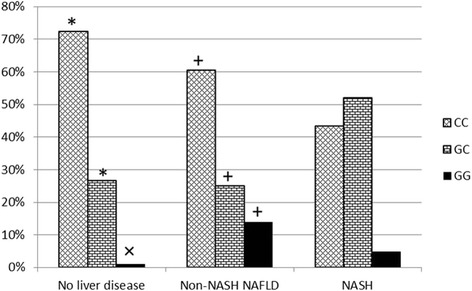
The prevalence of PNPLA3 rs738409 variant in NASH, Non-NASH NAFLD and Normal Liver Histology cohorts (p < 0.05 significant). * Significant pair-wise comparison between controls with normal liver histology and NASH; + Significant pair-wise comparison between Non-NASH NAFLD and NASH; x Significant pair-wise comparison between controls with normal liver histology and Non-NASH NAFLD
L mitochondrial haplogroup and NAFLD
In our study cohort, six common mitochondrial haplogroups (H, L, U, K, J and T) had a prevalence of greater than 5 %. For the purpose of further analysis, all the uncommon (<5 %) haplotypes (A, C, I, W, M, N, X, B and R) were pooled together into one group (“Other”). Among the self-reported ethnic groups, 77 % were White, 14.7 % were African Americans, 3.5 % were Hispanic (Additional file 1: Figure S1).
The overall distribution of haplogroup lineages within the self-identified race/ethnicity cohorts was in agreement with a recent survey of haplogroups in United States [37]. For example, in self-identified population of whites, European lineage haplogroups were the most common haplogroup (35.5 %), with haplogroup H (41 %) being the most common, followed by haplogroups U (17 %), K (12 %), J (11 %) and T (9 %). These data are in agreement with the H frequency reported recently to be 43 % in non-hispanic whites of United States [37]. Among self-identified African Americans, 91 % were confirmed as being of African lineages (Fig. 2) with L2 (40 %) being the most common haplogroup followed by L3 (36 %). Among self-identified Hispanics, 70 % were confirmed as of Asian/American Indian lineage (Fig. 2).
Fig. 2.
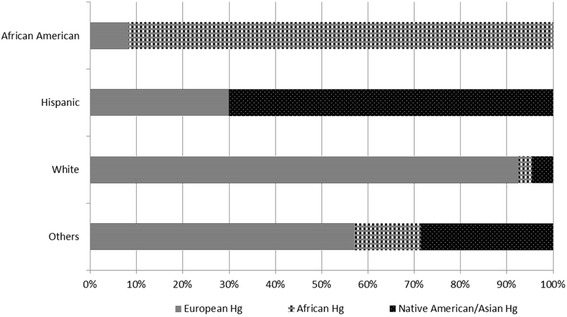
Haplogroup distribution by self-identified ethnicity in our study population. European lineages: H, T, U, V, X, K, N, I, J, HV, W; African Lineages: L, L1, L2, L3, L3; Asian/American Indian: A, B, F, macro-haplogroup M (C, D, E, and G), X
Mitochondrial haplogroup assessment showed that African lineage Haplogroup L (encompasses L0, L1, L2 and L3 subtypes) was less prevalent in Non-NASH NAFLD and NASH cohorts as compared to the cohort with normal liver histology, p < 0.018 and p < 0.0002, respectively. This suggests that L haplogroup may protect against NAFLD (Table 3).
Table 3.
Distribution of mitochondrial haplotypes in study groups
| Variable | Normal liver histology (N = 94) | Non-NASH NAFLD (N = 143) | NASH (N = 104) | P value* | P-value† | P value‡ |
|---|---|---|---|---|---|---|
| Haplotype | ||||||
| H | 28 (29.8 %) | 56 (39.2 %) | 37 (35.6 %) | 0.1400 | 0.3863 | 0.5660 |
| L | 23 (24.5 %) | 22 (15.4 %) | 6 (5.8 %) | 0.0811 | 0.0002 | 0.0186 |
| U | 11 (11.7 %) | 14 (9.8 %) | 17 (16.3 %) | 0.6393 | 0.3490 | 0.1247 |
| K | 8 (8.5 %) | 12 (8.4 %) | 11 (10.6 %) | 0.9743 | 0.6221 | 0.5595 |
| J | 11 (11.7 %) | 11 (7.7 %) | 5 (4.8 %) | 0.2980 | 0.0755 | 0.3631 |
| T | 6 (6.4 %) | 9 (6.3 %) | 6 (5.8 %) | 0.9780 | 0.8566 | 0.8647 |
| Other | 4 (4.3 %) | 13 (9.1 %) | 18 (17.3 %) | 0.1582 | 0.0035 | 0.0543 |
*Pairwise comparison between controls (normal liver histology) and Non-NASH NAFLD cohorts; †Pairwise comparison between controls (normal liver histology) and NASH cohorts; ‡Pairwise comparison between Non-NASH NAFLD and NASH cohorts. For convenience, significant p-values are highlighted by bold font
Nuclear and mitochondrial genotypes as NASH predictors
An initial univariate analysis showed that in population of patients with NAFLD (N = 247), the heterozygosity for PNPLA3 rs738409 variant may serve as independent predictor of having histologic NASH (OR 2.9: 95 % CI 1.67 – 5.05). When similar analyses were performed for mitochondrial haplogroups, only L haplogroup provided protection against histologic NASH (OR 0.34: 95 % CI 0.13 – 0.86) (Table 4).
Table 4.
Univariate-adjusted analysis for NASH vs Non-NASH NAFLD
| Variable Name | Sample Size | P Value | Odds Ratio | Lower OR | Upper OR |
|---|---|---|---|---|---|
| Age (yrs) | 247 | 0.106 | 1.02 | 1.00 | 1.04 |
| BMI (kg/m2) | 244 | 0.173 | 1.02 | 0.99 | 1.05 |
| WBC (x 103/uL) | 246 | 0.396 | 1.02 | 0.97 | 1.08 |
| Platelet (x 103/ul) | 242 | 0.011 | 0.99 | 0.99 | 1.00 |
| IR (HOMA) | 40 | 0.009 | 2.20 | 1.22 | 3.97 |
| Glucose (mg/dL) | 232 | 0.068 | 1.01 | 1.00 | 1.01 |
| ALT (U/L) | 245 | 0.000 | 1.03 | 1.02 | 1.05 |
| AST (U/L) | 245 | 0.000 | 1.05 | 1.03 | 1.08 |
| Triglycerides (mg/dL) | 219 | 0.127 | 1.00 | 1.00 | 1.01 |
| LDL (mg/dL) | 193 | 0.854 | 1.00 | 0.99 | 1.01 |
| Total Cholesterol (mg/dL) | 223 | 0.567 | 1.00 | 0.99 | 1.00 |
| HDL (mg/dL) | 198 | 0.002 | 0.96 | 0.94 | 0.99 |
| Male | 247 | 0.000 | 4.47 | 2.48 | 8.06 |
| White | 247 | 0.639 | 1.16 | 0.62 | 2.18 |
| Diabetes | 247 | 0.031 | 1.78 | 1.06 | 3.02 |
| Hyperlipidemia | 237 | 0.144 | 1.47 | 0.88 | 2.49 |
| Hypertension | 238 | 0.247 | 1.37 | 0.80 | 2.34 |
| Haplotype, | |||||
| H | 247 | 0.566 | 0.86 | 0.51 | 1.45 |
| L | 247 | 0.023 | 0.34 | 0.13 | 0.86 |
| U | 247 | 0.128 | 1.80 | 0.84 | 3.84 |
| Other | 247 | 0.058 | 2.09 | 0.98 | 4.49 |
| K | 247 | 0.560 | 1.29 | 0.55 | 3.05 |
| J | 247 | 0.367 | 0.61 | 0.20 | 1.80 |
| T | 247 | 0.866 | 0.91 | 0.31 | 2.65 |
| PNPLA3 SNP, | |||||
| CC | Reference | ||||
| GC | 247 | 0.000 | 2.90 | 1.67 | 5.05 |
| GG | 247 | 0.172 | 0.48 | 0.17 | 1.37 |
For convenience, significant p-values are highlighted by bold font
In non-L haplotype cohort, further multivariate analysis after adjusting for gender, age and ALT showed that patients with GC genotype of PNPLA3 locus are twice more likely to have NASH as compared to those with genotype CC (OR 2.66: 95 % CI 1.43 – 4.95).
Nuclear and mitochondrial genotypes in pericellular fibrosis
Since fibrosis is an important predictor of clinical outcome, we assessed the association between mitochondrial haplogroups and PNPLA3 genotype with pericellular fibrosis as well as with other histologically features.
NAFLD patients without pericellular fibrosis (N = 247) had higher platelet and serum HDL levels (Table 5) compared to those with pericellular fibrosis (N = 95). Serum aminotransferases were lower in these subjects compared to those with pericellular fibrosis. Notably, NAFLD patients with pericellular fibrosis were enriched for L haplogroup (L0, L1, L2, L3) (15.8 vs 4.2 %, p = 0.005), compared to NAFLD patients without pericellular fibrosis.
Table 5.
Comparisons of NAFLD patients with pericellular fibrosis with NAFLD patients without pericellular fibrosis
| Variable | NAFLD without | NAFLD with | P-value |
|---|---|---|---|
| p. fibrosis (N = 152) | p. fibrosis (N = 95) | ||
| Age (yrs) | 44.03 ± 11.08 | 46.73 ± 11.35 | 0.0609 |
| BMI (kg/m2) | 48.05 ± 8.94 | 49.23 ± 10.19 | 0.1752 |
| WBC (x 103/uL) | 8.16 ± 2.35 | 8.77 ± 9.15 | 0.5668 |
| Platelets (x 103/uL) | 290.32 ± 63.79 | 267.63 ± 67.91 | 0.0094 |
| Insulin Resistance | 2.42 ± 1.16 | 8.69 ± 11.25 | 0.0009 |
| Glucose (mg/dL) | 111.01 ± 36.85 | 120.25 ± 43.34 | 0.2630 |
| ALT (U/L) | 32.38 ± 17.62 | 49.95 ± 37.96 | <.0001 |
| AST (U/L) | 23.97 ± 11.78 | 36.88 ± 30.51 | <.0001 |
| Triglycerides (mg/dL) | 159.48 ± 86.89 | 185.17 ± 119.26 | 0.0760 |
| LDL (mg/dL) | 112.12 ± 36.03 | 109.52 ± 35.33 | 0.7968 |
| Total cholesterol (mg/dL) | 193.96 ± 39.81 | 188.62 ± 41.43 | 0.3486 |
| HDL (mg/dL) | 48.38 ± 13.47 | 42.69 ± 11.20 | 0.0004 |
| Male: N (%) | 26 (17.1 %) | 45 (47.4 %) | <.0001 |
| White: N (%) | 119 (78.3 %) | 77 (81.1 %) | 0.6017 |
| HAPLOTYPE, N (%) | |||
| H | 58 (38.2 %) | 35 (36.8 %) | 0.8355 |
| L | 24 (15.8 %) | 4 (4.2 %) | 0.0052 |
| U | 17 (11.2 %) | 14 (14.7 %) | 0.4123 |
| Other | 13 (8.6 %) | 18 (18.9 %) | 0.0164 |
| K | 12 (7.9 %) | 11 (11.6 %) | 0.3324 |
| J | 11 (7.2 %) | 5 (5.3 %) | 0.5398 |
| T | 10 (6.6 %) | 5 (5.3 %) | 0.6736 |
| PNPLA3_SNP, N (%) | |||
| CC | 94 (61.8 %) | 38 (40.0 %) | 0.0008 |
| GC | 38 (25.0 %) | 52 (54.7 %) | <0.0001 |
| GG | 20 (13.2 %) | 5 (5.3 %) | 0.0454 |
For convenience, significant p-values are highlighted by bold font
Homozygous genotypes for PNPLA3: CC and GG, were more prevalent in NAFLD patients without pericellular fibrosis (61.8 vs 40 % for CC, p = 0.0008 and 13.2 vs 5.3 %, p = 0.04). However, the association of GG genotype with pericellular fibrosis should be considered with caution due to the small sample size of GG genotype. GC genotype, on the other hand was more common in pericellular fibrosis cohort (54.7 vs 25 %, p < 0.001).
Univariate analysis revealed that L haplotype was protective against odds of developing pericellular fibrosis (OR 0.23: 95 % CI 0.08–0.70) and patients with heterozygosity in PNPLA3 locus had a 3 times higher odds of developing pericellular fibrosis as compared to patients CC genotype (OR 3.39: 95 % CI 1.93 – 5.94) (Table 6). After adjusting for age, number of platelets, levels of ALT, AST, triglycerides, HDL and male gender, individuals with PNPLA3 GC genotype remained associated with higher likelihood of having pericellular fibrosis than those with PNPLA3 genotype CC (OR 4.38: 95 % CI 2.17 – 8.85).
Table 6.
Univariate-adjusted analysis for Pericellular fibrosis
| Variable Name | Sample Size | P Value | Odds Ratio | Lower OR | Upper OR |
|---|---|---|---|---|---|
| Age (yrs) | 247 | 0.067 | 1.02 | 1.00 | 1.05 |
| BMI (kg/m2) | 244 | 0.345 | 1.01 | 0.99 | 1.04 |
| WBC (x 103/uL) | 246 | 0.467 | 1.02 | 0.97 | 1.07 |
| Platelet (x 103/ul) | 242 | 0.011 | 0.99 | 0.99 | 1.00 |
| IR (HOMA) | 40 | 0.008 | 2.26 | 1.24 | 4.14 |
| Glucose (mg/dL) | 232 | 0.087 | 1.01 | 1.00 | 1.01 |
| ALT (U/L) | 245 | 0.000 | 1.03 | 1.01 | 1.04 |
| AST (U/L) | 245 | 0.000 | 1.05 | 1.02 | 1.07 |
| Triglycerides (mg/dL) | 219 | 0.080 | 1.00 | 1.00 | 1.01 |
| LDL (mg/dL) | 193 | 0.623 | 1.00 | 0.99 | 1.01 |
| Total Cholesterol (mg/dL) | 223 | 0.338 | 1.00 | 0.99 | 1.00 |
| HDL (mg/dL) | 198 | 0.003 | 0.96 | 0.94 | 0.99 |
| Male | 247 | 0.000 | 4.36 | 2.43 | 7.82 |
| White | 247 | 0.602 | 1.19 | 0.62 | 2.25 |
| Diabetes | 247 | 0.011 | 1.99 | 1.17 | 3.38 |
| Hyperlipidemia | 237 | 0.012 | 1.99 | 1.16 | 3.42 |
| Hypertension | 238 | 0.254 | 1.37 | 0.80 | 2.36 |
| Haplotype | |||||
| H | 247 | 0.836 | 0.95 | 0.56 | 1.61 |
| L | 247 | 0.009 | 0.23 | 0.08 | 0.70 |
| U | 247 | 0.414 | 1.37 | 0.64 | 2.93 |
| Other | 247 | 0.019 | 2.50 | 1.16 | 5.38 |
| K | 247 | 0.335 | 1.53 | 0.65 | 3.62 |
| J | 247 | 0.541 | 0.71 | 0.24 | 2.12 |
| T | 247 | 0.674 | 0.79 | 0.26 | 2.38 |
| PNPLA3_SNP | |||||
| CC | Reference | ||||
| GC | 247 | 0.000 | 3.39 | 1.93 | 5.94 |
| GG | 247 | 0.370 | 0.62 | 0.22 | 1.77 |
For convenience, significant p-values are highlighted by bold font
Discussion
Oxidative stress is an important player in development of progressive NAFLD or NASH. In fact, oxidative stress related cellular damage, reflected as hepatic necrosis or apoptosis, is a mitochondrial dependent factor that potentially influences the progression of NAFLD [38–40]. Given that mitochondria are critical players in maintaining balance of energy input and expenditure [41] as well as the pathogenesis of NASH, it is enticing to explore mitochondrial genome variation as a potential genetic factor that could contribute to the pathogenesis of NASH.
Mitochondria have their own circular DNA that can be categorized into various haplogroups based on variants within its non-coding hypervariable region. Notably, the non-coding region is indispensable for replication and transcription of the mitochondrial genome. Thus variations in this region are expected to influence the ability of mitochondria to produce ATP and ROS. In turn, availability of ATP and the production of ROS are known to affect tissue responses to extrinsic or intrinsic stressors and energy demands. Consequently, mitochondrial DNA variations have been the focus of studies in patients with altered metabolism [42–44]. Additionally, in a recent study, there is evidence that mitochondrial DNA haplogroups may influence expression of nuclear genes that are induced under oxidative stress [45].
NAFLD is a complex disease with genetic and environmental factors interacting to influence the development and progression of the disease. The relative importance of these factors may vary between populations depending on genetic background and lifestyle. This study is unique as we show for the first time the differential prevalence of mitochondrial haplogroups in obese patients (BMI > 35) with varying severity of NAFLD. We also examine the interaction between mitochondrial genome variation and nuclear PNPLA3 genotype in this patient cohort.
In this study, we present evidence that mitochondrial haplogroup L is less prevalent in subjects with NASH and may thus be protective against NASH (Table 3). Haplogroup L has the highest sequence divergence from other haplogroups [46]. A recent study demonstrated that L haplogroup is also functionally different [45]. When compared to the cybrids with H haplogroup, those with L haplogroup demonstrated lower levels of the ROS production but similar levels of ATP generation, indicative of more efficient oxidative phosphorylation with lower electron leakage and higher efficiency of aerobic respiration [45]. These observations support the hypothesis that the patients with L haplogroup may be intrinsically protected from oxidative stress and thus, potential liver damage by ROS.
It is also important to note that the mitochondrial haplogroups are commonly used as a proxy to ethnic origin of individual. According to the population-based studies, NAFLD and NASH are more prevalent in individuals of Hispanic origin [3, 47], while African Americans are somewhat protected against high prevalence of NAFLD and NASH [48]. The high prevalence of L haplogroup in subjects of African origin may serve as a physiological explanation for this phenomenon, although further studies will be needed to determine the causality. In our study cohort, 88 % (N = 44) of the self-reported African Americans had L haplotype and 66 % (N = 33) of them had normal liver biopsy, 22 % (N = 11) had steatosis and 12 % (N = 6) had NASH. Additional studies with a larger African American cohort will be needed to further validate this trend. Notably, the haplotype results of our study are consistent with the haplogroup proportions reported in a recent large population based study in United States [37].
In addition to mitochondrial haplotypes, we also measured the well-described gene PNPLA3 variant rs738409 associated with NAFLD. The rs738409 variant is located in coding region of PNPLA3 gene. This variant corresponds to Isoleucine to Methionine substitution (p.I148M) and has previously been identified as risk factor for steatohepatitis, NAFLD-cirrhosis [14, 49] and hepatocellular carcinoma [15, 20, 50–52]. The 148 M variant has also been linked to elevated aminotransferase levels [53] and liver fibrosis [54]. However, the effect of the I148M substitution on the functioning of PNPLA3 is still enigmatic [15]. The 148 M variation is reported to result in loss of hydrolase activity (loss-of-function model) [22, 24, 55] and thus cause accumulation of triglyceride in the liver. However, studies in knock-out mice show contrary results [26, 56]. These studies show that loss of PNPLA3 hydrolase activity does not alter hepatic triglycerides. PNPLA3 variant has also been shown to have a gain of lysophosphatidic acid transacetylase (LPAAT) activity. The 148 M substitution increases LPAAT activity and results in increased TAG synthesis (gain-of-function model) [15, 23]. Thus, PNPLA3 148 M variant may increase hepatic triglyceride levels by multiple changes in triglyceride metabolism.
Our results with regards to PNPLA3 are in agreement with the previously reported studies. The alleles of PNPLA3 locus showed differential distribution in cohorts with NAFLD, NASH (Table 2) and pericellular fibrosis. Moreover, multivariate analysis showed that heterozygosity in this locus is independently associated with higher risk of having NASH as well as with pericellular fibrosis. Overall, carrying G allele in position rs738409 appears to increase the odds of developing NASH and pericellular fibrosis. Importantly, in the final multivariate model of NAFLD (Table 4), no interaction between the nuclear rs738409 variant and mitochondrial haplogroup was detected. This indicates that the somatic genotype and mitochondrial genotype can have independent and important impact in the pathogenesis of NASH.
Conclusion
In conclusion, our data shows that the presence of haplogroup L exhibits certain degree of protection against the development of NASH and pericellular fibrosis. This suggests that, besides nuclear genome variants and environmental factors, the mitochondrial genetics may play an important role in NAFLD probably by modulation of oxidative stress and the efficiency of oxidative phosphorylation. Further studies will be needed to confirm the causality. Moreover, United States cohorts are substantially admixed, resulting in ambiguity in the questionnaire-based ethnicity assessments [57]. Thus, ethnicity verifying mitochondrial haplogroup data should be coupled with the other biomarkers to serve as a valuable resource for future studies investigating NAFLD and NASH in such populations.
Acknowledgements
This study has been supported by the Beatty Liver and Obesity Fund at Inova Health System and the Liver Disease Outcomes Fund of the Center for Liver Diseases at Inova Fairfax Hospital, Falls Church, VA.
Funding
This study has been supported by the Beatty Liver and Obesity Fund at Inova Health System and the Liver Disease Outcomes Fund of the Center for Liver Diseases at Inova Fairfax Hospital, Falls Church, VA.
Availability of data and material
The data supporting the findings have been provided in the manuscript and as supplementary information where applicable.
Authors’ contribution
RM conceived the study, performed sequence alignment, participated in study design, coordinated all efforts, drafted the manuscript and provided daily supervision for KJ and AK, MS students in training. KJ extracted DNA, performed mitochondrial sequencing and sequence alignment. AK performed SNP profiling and validation. ZO performed statistical analysis. ZG read the liver biopies. AB and ZM participated in the design of the study and finalized the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
NA.
Ethics approval and consent to participate
This study has been approved by Internal Review Board of Inova Fairfax Hospital (Federal Assurance FWA00000573). The informed written consent was obtained for study from participants.
Abbreviations
- ALT
Alanine transaminase
- AST
Aspartate aminotransferase
- ATP
Adenosine triphosphate
- BMI
Body mass index
- DNA
Deoxyribonucleic acids
- HDL
High density lipoproteins
- LDL
Low density lipoproteins
- NAFLD
Non-alcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- PCR
Polymerase chain reaction
- PNPLA3
patatin-like phospholipase domain containing 3
- ROS
Reactive oxygen species
- SNP
Single nucleotide polymorphism
Additional file
Table S1 Forward and reverse primer sequences for mitochondrial control region amplification. Figure S1 Self-reported ethnicity distribution in the study cohort. (DOCX 47 kb)
Contributor Information
Rohini Mehta, Email: mehta.rohini@gmail.com.
Kianoush Jeiran, Email: kiajeiran@gmail.com.
Aaron B. Koenig, Email: koenigab@gmail.com
Munkzhul Otgonsuren, Email: Munkhzul.Otgonsuren@inova.org.
Zachary Goodman, Email: Zachary.Goodman@inova.org.
Ancha Baranova, Email: aancha@gmail.com.
Zobair Younossi, Phone: (703) 776-2540, Email: Zobair.younossi@inova.org.
References
- 1.Erickson SK. Nonalcoholic fatty liver disease. J Lipid Res. 2009;50(Suppl):S412–S416. doi: 10.1194/jlr.R800089-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 3.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 4.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American association for the study of liver diseases, American College of gastroenterology, and the American gastroenterological association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 5.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatol Baltim Md. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 6.Kneeman JM, Misdraji J, Corey KE. Secondary causes of nonalcoholic fatty liver disease. Ther Adv Gastroenterol. 2012;5:199–207. doi: 10.1177/1756283X11430859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishra A, Younossi ZM. Epidemiology and natural history of Non-alcoholic fatty liver disease. J Clin Exp Hepatol. 2012;2:135–144. doi: 10.1016/S0973-6883(12)60102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–649. doi: 10.3109/07853890.2010.518623. [DOI] [PubMed] [Google Scholar]
- 9.Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabet Endocrinol. 2014;2:901-10. [DOI] [PubMed]
- 10.Younossi ZM, Stepanova M, Negro F, Hallaji S, Younossi Y, Lam B, Srishord M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine (Baltimore) 2012;91:319–327. doi: 10.1097/MD.0b013e3182779d49. [DOI] [PubMed] [Google Scholar]
- 11.Veena J, Muragundla A, Sidgiddi S, Subramaniam S. Non-alcoholic fatty liver disease: need for a balanced nutritional source. Br J Nutr. 2014;112:1858–1872. doi: 10.1017/S0007114514002591. [DOI] [PubMed] [Google Scholar]
- 12.Al-Serri A, Anstee QM, Valenti L, Nobili V, Leathart JBS, Dongiovanni P, Patch J, Fracanzani A, Fargion S, Day CP, Daly AK. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case–control and intra-familial allele association studies. J Hepatol. 2012;56:448–454. doi: 10.1016/j.jhep.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Naik A, Košir R, Rozman D. Genomic aspects of NAFLD pathogenesis. Genomics. 2013;102:84–95. doi: 10.1016/j.ygeno.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maglio C, Pirazzi C, Pujia A, Valenti L, Romeo S. The PNPLA3 I148M variant and chronic liver disease: When a genetic mutation meets nutrients. Spec Issue 3rd Foodomics Conf Foodomics New Compr Approach Food Nutr. 2014;63(Part B):239–243. [Google Scholar]
- 16.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatol Baltim Md. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 17.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, NASH CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatol Baltim Md. 2010;52:894–903. doi: 10.1002/hep.23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zain SM, Mohamed R, Mahadeva S, Cheah PL, Rampal S, Basu RC, Mohamed Z. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum Genet. 2012;131:1145–1152. doi: 10.1007/s00439-012-1141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romeo S, Sentinelli F, Dash S, Yeo GSH, Savage DB, Leonetti F, Capoccia D, Incani M, Maglio C, Iacovino M, O’Rahilly S, Baroni MG. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes. 2010;34:190–194. doi: 10.1038/ijo.2009.216. [DOI] [PubMed] [Google Scholar]
- 20.Petit J-M, Guiu B, Masson D, Duvillard L, Jooste V, Buffier P, Terriat B, Bouillet B, Brindisi M-C, Loffroy R, Robin I, Hillon P, Cercueil J-P, Verges B. Specifically PNPLA3-mediated accumulation of liver Fat in obese patients with type 2 diabetes. J Clin Endocrinol Metab. 2010;95:E430–E436. doi: 10.1210/jc.2010-0814. [DOI] [PubMed] [Google Scholar]
- 21.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro G, Vanni E, Bugianesi E, Maggioni M, Fracanzani AL, Fargion S, Day CP. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatol Baltim Md. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Cohen JC, Hobbs HH. Expression and Characterization of a PNPLA3 Protein Isoform (I148M) Associated with Nonalcoholic Fatty Liver Disease. J Biol Chem. 2011;286:37085–37093. doi: 10.1074/jbc.M111.290114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA, Knittelfelder O, Rechberger GN, Birner-Gruenberger R, Eder S, Brown HA, Haemmerle G, Oberer M, Lass A, Kershaw EE, Zimmermann R, Zechner R. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, Montalcini T, Hedfalk K, Romeo S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta. 1841;2014:574–580. doi: 10.1016/j.bbalip.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Ståhlman M, Taskinen M-R, Orho-Melander M, Perman J, Pujia A, Andersson L, Maglio C, Montalcini T, Wiklund O, Borén J, Romeo S. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol. 2012;57:1276–1282. doi: 10.1016/j.jhep.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 26.Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, Yang K, Kumari M, Gross RW, Zechner R, Kershaw EE. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52:318–329. doi: 10.1194/jlr.M011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumashiro N, Yoshimura T, Cantley JL, Majumdar SK, Guebre-Egziabher F, Kursawe R, Vatner DF, Fat I, Kahn M, Erion DM, Zhang X-M, Zhang D, Manchem VP, Bhanot S, Gerhard GS, Petersen KF, Cline GW, Samuel VT, Shulman GI. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatol Baltim Md. 2013;57:1763–1772. doi: 10.1002/hep.26170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santoro N, Caprio S, Giannini C, Kim G, Kursawe R, Pierpont B, Shaw MM, Feldstein AE. Oxidized fatty acids: A potential pathogenic link between fatty liver and type 2 diabetes in obese adolescents? Antioxid Redox Signal. 2014;20:383–389. doi: 10.1089/ars.2013.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, Burza MA, Indiveri C, Ferro Y, Montalcini T, Maglio C, Dongiovanni P, Fargion S, Rametta R, Pujia A, Andersson L, Ghosal S, Levin M, Wiklund O, Iacovino M, Borén J, Romeo S. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23:4077–4085. doi: 10.1093/hmg/ddu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovarova M, Königsrainer I, Königsrainer A, Machicao F, Häring H-U, Schleicher E, Peter A. The genetic variant I148M in PNPLA3 is associated with increased hepatic retinyl-palmitate storage in humans. J Clin Endocrinol Metab. 2015;100:E1568–1574. doi: 10.1210/jc.2015-2978. [DOI] [PubMed] [Google Scholar]
- 31.Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, Goodman Z. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatol Baltim Md. 2011;53:1874–1882. doi: 10.1002/hep.24268. [DOI] [PubMed] [Google Scholar]
- 32.Fan L, Yao Y-G. An update to MitoTool: Using a new scoring system for faster mtDNA haplogroup determination. Mitochondrion. 2013;13:360–363. doi: 10.1016/j.mito.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 33.Falleti E, Fabris C, Cmet S, Cussigh A, Bitetto D, Fontanini E, Fornasiere E, Bignulin S, Fumolo E, Bignulin E, Pirisi M, Toniutto P. PNPLA3 rs738409C/G polymorphism in cirrhosis: relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int Off J Int Assoc Study Livee. 2011;31:1137–1143. doi: 10.1111/j.1478-3231.2011.02534.x. [DOI] [PubMed] [Google Scholar]
- 34.Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for Histologic nonalcoholic fatty liver disease but Not metabolic disease. Hepatol Baltim Md. 2010;52:904–912. doi: 10.1002/hep.23768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ, Crn the N The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology. 2010;52:894–903. doi: 10.1002/hep.23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tai C-M, Huang C-K, Tu H-P, Hwang J-C, Chang C-Y, Yu M-L. PNPLA3 genotype increases susceptibility of nonalcoholic steatohepatitis among obese patients with nonalcoholic fatty liver disease. Surg Obes Relat Dis. 2015;11:888-94. [DOI] [PubMed]
- 37.Mitchell SL, Goodloe R, Brown-Gentry K, Pendergrass SA, Murdock DG, Crawford DC. Characterization of mitochondrial haplogroups in a large population-based sample from the United States. Hum Genet. 2014;133:861–868. doi: 10.1007/s00439-014-1421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albano E, Mottaran E, Occhino G, Reale E, Vidali M. Review article: role of oxidative stress in the progression of non-alcoholic steatosis. Aliment Pharmacol Ther. 2005;22:71–73. doi: 10.1111/j.1365-2036.2005.02601.x. [DOI] [PubMed] [Google Scholar]
- 39.Madan K, Bhardwaj P, Thareja S, Gupta SD, Saraya A. Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD) J Clin Gastroenterol. 2006;40:930–935. doi: 10.1097/01.mcg.0000212608.59090.08. [DOI] [PubMed] [Google Scholar]
- 40.Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in Non-alcoholic steatohepatitis (NASH) Int J Mol Sci. 2013;14:20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng Z, Ristow M. Mitochondria and metabolic homeostasis. Antioxid Redox Signal. 2013;19:240–242. doi: 10.1089/ars.2013.5255. [DOI] [PubMed] [Google Scholar]
- 42.Nardelli C, Labruna G, Liguori R, Mazzaccara C, Ferrigno M, Capobianco V, Pezzuti M, Castaldo G, Farinaro E, Contaldo F, Buono P, Sacchetti L, Pasanisi F. Haplogroup T is an obesity risk factor: mitochondrial DNA haplotyping in a morbid obese population from southern Italy. BioMed Res Int. 2013;2013:e631082. doi: 10.1155/2013/631082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tranah GJ, Manini TM, Lohman KK, Nalls MA, Kritchevsky S, Newman AB, Harris TB, Miljkovic I, Biffi A, Cummings SR, Liu Y. Mitochondrial DNA variation in human metabolic rate and energy expenditure. Mitochondrion. 2011;11:855–861. doi: 10.1016/j.mito.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF. Recent Mitochondrial DNA Mutations Increase the Risk of Developing Common Late-Onset Human Diseases. PLoS Genet. 2014;10:e1004369. doi: 10.1371/journal.pgen.1004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kenney MC, Chwa M, Atilano SR, Falatoonzadeh P, Ramirez C, Malik D, Tarek M, del Carpio JC, Nesburn AB, Boyer DS, Kuppermann BD, Vawter MP, Jazwinski SM, Miceli MV, Wallace DC, Udar N. Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: Implications for population susceptibility to diseases. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2014;1842:208–219. doi: 10.1016/j.bbadis.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238:211–230. doi: 10.1016/S0378-1119(99)00295-4. [DOI] [PubMed] [Google Scholar]
- 47.Pan J-J, Fallon MB. Gender and racial differences in nonalcoholic fatty liver disease. World J Hepatol. 2014;6:274–283. doi: 10.4254/wjh.v6.i5.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 49.Severson TJ, Besur S, Bonkovsky HL. Genetic factors that affect nonalcoholic fatty liver disease: A systematic clinical review. World J Gastroenterol. 2016; 22:6742-56. [DOI] [PMC free article] [PubMed]
- 50.Nischalke HD, Berger C, Luda C, Berg T, Müller T, Grünhage F, Lammert F, Coenen M, Krämer B, Körner C, Vidovic N, Oldenburg J, Nattermann J, Sauerbruch T, Spengler U. The PNPLA3 rs738409 148 M/M genotype is a risk factor for liver cancer in alcoholic cirrhosis but shows No or weak association in hepatitis C cirrhosis. PLoS ONE. 2011;6:e27087. doi: 10.1371/journal.pone.0027087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corradini SG, Burza MA, Molinaro A, Romeo S. Patatin-like phospholipase domain containing 3 sequence variant and hepatocellular carcinoma. Hepatology. 2011;53:1776. doi: 10.1002/hep.24244. [DOI] [PubMed] [Google Scholar]
- 52.Valenti L, Rumi M, Galmozzi E, Aghemo A, Del Menico B, De Nicola S, Dongiovanni P, Maggioni M, Fracanzani AL, Rametta R, Colombo M, Fargion S. Patatin-Like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C. Hepatology. 2011;53:791–799. doi: 10.1002/hep.24123. [DOI] [PubMed] [Google Scholar]
- 53.Kollerits B, Coassin S, Beckmann ND, Teumer A, Kiechl S, Döring A, Kavousi M, Hunt SC, Lamina C, Paulweber B, Kutalik Z, Nauck M, van Duijn CM, Heid IM, Willeit J, Brandstätter A, Adams TD, Mooser V, Aulchenko YS, Völzke H, Kronenberg F. Genetic evidence for a role of adiponutrin in the metabolism of apolipoprotein B-containing lipoproteins. Hum Mol Genet. 2009;18:4669–4676. doi: 10.1093/hmg/ddp424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krawczyk M, Grünhage F, Zimmer V, Lammert F. Variant adiponutrin (PNPLA3) represents a common fibrosis risk gene: Non-invasive elastography-based study in chronic liver disease. J Hepatol. 2011;55:299–306. doi: 10.1016/j.jhep.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 55.He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, Cohen JC, Hobbs HH. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52:1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seldin MF, Pasaniuc B, Price AL. New approaches to disease mapping in admixed populations. Nat Rev Genet. 2011;12:523–528. doi: 10.1038/nrg3002. [DOI] [PMC free article] [PubMed] [Google Scholar]