

Abstract
Background
Variant ATM heterozygotes have an increased risk of developing cancer, cardiovascular diseases, and diabetes. Costs and time of sequencing and ATM variant complexity make large-scale, general population screenings not cost-effective yet. Recently, we developed a straightforward, rapid, and inexpensive test based on p53 mitotic centrosomal localization (p53-MCL) in peripheral blood mononuclear cells (PBMCs) that diagnoses mutant ATM zygosity and recognizes tumor-associated ATM polymorphisms.
Methods
Fresh PBMCs from 496 cancer patients were analyzed by p53-MCL: 90 cases with familial BRCA1/2-positive and -negative breast and/or ovarian cancer, 337 with sporadic cancers (ovarian, lung, colon, and post-menopausal breast cancers), and 69 with breast/thyroid cancer. Variants were confirmed by ATM sequencing.
Results
A total of seven individuals with ATM variants were identified, 5/65 (7.7 %) in breast cancer cases of familial breast and/or ovarian cancer and 2/69 (2.9 %) in breast/thyroid cancer. No variant ATM carriers were found among the other cancer cases. Excluding a single case in which both BRCA1 and ATM were mutated, no p53-MCL alterations were observed in BRCA1/2-positive cases.
Conclusions
These data validate p53-MCL as reliable and specific test for germline ATM variants, confirm ATM as breast cancer susceptibility gene, and highlight a possible association with breast/thyroid cancers.
Electronic supplementary material
The online version of this article (doi:10.1186/s13046-016-0410-3) contains supplementary material, which is available to authorized users.
Keywords: ATM cancer susceptibility gene, Early-onset breast cancer, BRCA1/2, p53-mitotic centrosomal localization (p53-MCL)
Background
Biallelic mutations in the ATM gene cause Ataxia-telangiectasia (A-T), a rare autosomal recessive multisystemic disorder characterized by progressive cerebellar ataxia, immune defects, insulin-resistant diabetes, radiosensitivity, and high risk for malignancy [1, 2]. The ATM gene spans approximately 160 Kb of genomic DNA containing 66 exons [3] and encodes ATM protein, a serine/threonine kinase mainly involved in DNA damage response pathways following DNA double strand breaks [4]. An enormous number of mutations (more than 600) can occur in the coding and noncoding regions of the ATM gene without hotspots [5]. In A-T patients, the large majority of ATM mutations are protein-truncations or splice-junction variants that can be easily distinguished by the numerous ATM polymorphisms [6, 7].
Heterozygous carriers of variants in the ATM gene (from here on, ATM carriers) are usually asymptomatic and largely considered healthy carriers. However, they have been reported to be more sensitive to ionizing radiation and susceptible to ischemic heart disease, diabetes, and cancer, particularly of the breast, but also digestive tract and lung [2, 8]. Many association studies have been performed on breast cancer susceptibility. Initially, epidemiological studies on relatives of A-T patients revealed a two to fivefold increased in the risk of breast cancer for female obligate ATM carriers [9]. The increased risk of breast cancer in ATM carriers was then confirmed by direct ATM sequencing in breast cancer cases compared to controls [10] and ATM is now considered a moderate-penetrance cancer susceptibility gene in BRCA1/2-negative patients with familial early-onset breast cancer [11]. Along with A-T associated mutations, several ATM screenings in cancer patients identified missense ATM variants, particularly amino acid substitutions that are not expected to be associated with A-T [12]. However, discrimination of these ATM variants from ATM polymorphisms and their contribution to health risks is still controversial. In addition, distinguishing between deleterious and neutral ATM alterations is required to allow the definition of standard-of-care clinical guidelines for the management of ATM carriers and their families [11]. Systematic review and meta-analysis of ATM sequencing data have been conducted to evaluate the health risks for parents and siblings of A-T patients, but similar large-scale screenings for ATM carriers in the general population by direct sequencing are not cost-effective yet [13].
Recently, we have developed a rapid, reliable and non-expensive test based on the ATM-dependent p53-mitotic centrosomal localization (p53-MCL) that clearly discriminates ATM carriers of A-T mutations and at least some of the ATM cancer predisposition variants in lymphoblastoid cell lines (LCLs) and PBMCs. At variance with other diagnostic tests, the p53-MCL assay does not measure a continuous quantitative variation (e.g., radiosensitivity, ATM protein levels, phosphorylation of ATM targets) but a “binary” outcome. Indeed, at the single cell level, p53 does or does not localize at the centrosomes while, at the cell-population level, the number of cells showing one or the other phenotype allows to unambiguously diagnose A-T homozygotes and ATM carriers [14].
In a preliminary set up and validation of the p53-MCL test, we showed that it is highly sensitive, specific, and precise. In particular, we assessed the specificity by analyzing LCLs from monogenic disease carrying mutations in a series of DNA-damage related factors, such as MRE11, NBS1, SMC1A, WRN, ATR, FANC-A, and p53 [14]. In addition, p53-MCL test revealed 7 ATM carriers among 80 sporadic breast cancer patients. By direct ATM sequencing of 3 of these carriers, we identified the cancer-prone intronic c.8786 + 8A > C variant [15] in one patient and the c.2572 T > C (p.F858L) missense mutation [16] in other two patients. No ATM carriers were observed in a comparable cohort of healthy donors [14]. These data support p53-MCL as promising candidate test for cost-effective, large-scale screenings of ATM carriers.
Here, we examined validity and specificity of p53-MCL analyzing 15 LCLs from familial breast and ovarian cancer cases and fresh PBMCs from a total of 496 cases including BRCA1/2-positive and -negative familial breast and ovarian cancer and different sporadic cancers.
Methods
Patients
A total of 496 cancer patients were enrolled from 2010 to 2015 at three different Italian institutes: Policlinico Umberto I (University La Sapienza), Sant’Andrea Hospital (University La Sapienza), and Regina Elena National Cancer Institute - IRCCS. In particular, according to previously described criteria [17], we selected 90 unrelated families affected with breast and/or ovarian cancer after interview at the Hereditary Tumors Counseling Centre of the Policlinico Umberto I. Pre-test counseling was performed by an expert cancer risk counselor and the probands analyzed in this study belonged to different high-risk classes. During the genetic counseling, we calculated the a priori probability of carrying a pathogenic BRCA1/2 germline mutation by the statistical model BRCAPRO that considers: proband health state (un/affected); current age and age at diagnosis of the proband and all family members of four consecutive generations (only first/second degree relatives); typology of the existent tumors (unilateral/bilateral breast cancer, other cancers) [18]. Then, 69 patients with breast and thyroid cancer regardless of the sequence of appearance were recruited from Sant’Andrea Hospital and Regina Elena National Cancer Institute. Others 337 patients unselected cancer patients diagnosed at any age were recruited from Regina Elena National Cancer Institute.
Cells and culture conditions
EBV-immortalized LCLs and freshly isolated PBMCs were cultured in RPMI-1640 GlutaMAX supplemented with 15 % heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Invitrogen, CA, USA). PBMCs were isolated from donors’ heparinized blood samples by Lympholyte-H (Cedarlane, Burlington, USA) density gradient centrifugation. PBMCs were stimulated to proliferate by incubation with 5 μg/ml PHA (Sigma-Aldrich, St. Louis, MO, USA) and incubated at 37 °C in a 5 % CO2 atm for 60 h [14].
p53-MCL test
Proliferating cells (i.e., LCLs or PHA-stimulated PBMCs) were set up for p53-MCL test as previously described [14]. Cells were examined under an Olympus BX53 microscope equipped with epifluorescence. Percentages of p53-MCL were measured by counting 100 cells in metaphase and analyzing two coverslips for each sample. The percentage of p53 mitotic localization is from 75 to 90 % for normal subjects, from 40 to 55 % for ATM carriers, and from 0 to 30 % for A-T patients.
BRCA1/2 gene sequencing
Genomic DNA was extracted from peripheral blood of all probands using commercial kit (Qiamp Blood Kit, Qiagen, Hilden, Germany). The entire coding sequence and each intron/exon boundary of BRCA1 and BRCA2 were screened by direct sequencing. All truncating and/or novel genetic variants were confirmed by sequencing different samples on both DNA strands. Sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit and a 3130xl Genetic Analyzer (Applied Biosystems, CA, USA). Reference sequence for BRCA1 was Genebank NM_007294.3, NG_005905.2 and reference sequence for BRCA2 was Genebank, NM_000059.3, NG_012772.3. BRCA1/2 genomic rearrangements were searched by the Multiple Ligation dependent Probe Amplification (MLPA) methodology. MLPA procedure was carried out according to the manufacturer’s instructions. Variations in peak height were evaluated comparing each sample with a normal control and by a cumulative comparison.
ATM gene sequencing
Genomic DNA was extracted from PBMCs by Quick-gDNA MiniPrep (Zymo Research, CA, USA) according to the manufacturer’s instructions. Sixty-two ATM exons were amplified using AmpliTaq Gold (Applied Biosystems, CA, USA), as described [14], and subjected to direct sequencing at the Genechron Laboratory (Rome, Italy). Reference sequence for ATM was Genebank U82828.1.
In silico analysis
To predict possible impact of amino acid substitutions on the structure and function of human proteins, the PolyPhen-2 (Polymorphism Phenotyping v2) software, that uses straightforward physical and evolutionary comparative considerations [19], was employed.
Efficient splicing of many exons requires splicing-enhancers to promote splicing at unfavorable splice-sites or splicing-silencers to repress more favorable splice-sites nearby. As a consequence, single nucleotide changes in an exon or intron close to these splice-sites may be predicted to disrupt splicing. For in silico prediction of the effects of mutations on normal splicing, the interactive biosoftware Alamut v2.3 [20] was adopted.
Statistic
We determined statistical significance of differences between two groups by 2-tailed Student’s t test. P values less than 0.01 were considered significant.
Results
Beyond the high penetrance BRCA1/2 genes, mutations of several cancer susceptibility genes, including ATM, CHK2 and PALB2, have been shown to associate, with a moderate penetrance, with familial breast and/or ovarian cancers [21, 22]. Thus, we first verified p53-MCL test specificity compared to mutant BRCA1/2 and CHK2 proteins that are also involved in DNA damage response and centrosome amplification and localization [23–25]. LCLs from 15 familial breast and ovarian cancer (Table 1) including four BRCA1-positive, 2 BRCA2-positive, 1 CHK2-positive and 8 BRCA1/2/CHK2/PALB2-negative LCLs were analyzed by the p53-MCL test. As shown in Table 1 and Fig. 1a, the percentage of p53-MCL was compatible with that of wild-type cells in 13 out of 15 LCLs, while two cases showed a p53-MCL reduction typical of ATM carriers, one in a BRCA1-positive case and the other in a BRCA1/2/CHK2/PALB2-negative case. Direct ATM sequencing confirmed the presence of ATM variants (Table 1) in both cases demonstrating p53-MCL specificity for ATM variants also in comparison with mutations in the centrosome related factors BRCA1/2 and CHK2.
Table 1.
Genetic status and p53-MCL rate of LCLs from familial breast and ovarian cancer
| BRCA1 | BRCA2 | CHK2 | PALB2 | ATM | p53-MCL% | |
|---|---|---|---|---|---|---|
| BR36 | wt | mut | nt | nt | nt | 80 |
| BR13 | wt | mut | nt | nt | nt | 76 |
| BR409-3 | mut | wt | nt | nt | nt | 97 |
| BR324-1 | mut | wt | nt | nt | nt | 85 |
| BR404-1 | mut | wt | wt | wt | wt | 76 |
| BR317 | mut | wt | nt | nt | mut | 50 |
| BR377 | wt | wt | wt | wt | mut | 50 |
| BR60-1 | wt | wt | wt | wt | wt | 71 |
| BR107-1 | wt | wt | wt | wt | wt | 90 |
| BR362-1 | wt | wt | wt | wt | wt | 92 |
| BR494 | wt | wt | wt | wt | wt | 86 |
| BR38 | wt | wt | wt | wt | wt | 91 |
| BR48 | wt | wt | wt | wt | wt | 70 |
| BR278-1 | wt | wt | wt | wt | wt | 80 |
| BR501 | wt | wt | mut | wt | wt | 80 |
wt wild type, mut mutated, nt not tested
Fig. 1.
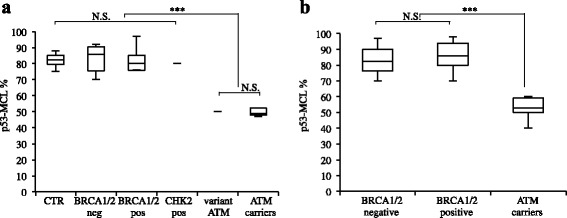
p53-MCL in LCLs and in PBMCs from familial breast and/or ovarian BRCA1/2-positive and –negative patients. (a) Comparison of p53-MCL percentages, among LCLs derived from wild type ATM donors (CTR, n = 11), BRCA1/2-negative (n = 7), BRCA1/2-positive (n = 5), CHK2-positive (n = 1), and ATM variants carriers (n = 2) and ATM carriers (n = 9) previously tested. Not significant differences among the groups including wild type ATM (CTR, BRCA1/2-neg, BRCA1/2-pos and CHK2-pos), but significant differences between these groups and ATM carriers groups (ATM variants and ATM carriers). (b) Comparison of p53-MCL percentages among PBMCs from familial breast and/or ovarian BRCA1/2-positive, BRCA1/2–negative and ATM carrier patients. Not significant differences between BRCA1/2-positive and -negative patients, but significant differences between wild type ATM group (BRCA1/2-positive and -negative) and ATM carriers. ***P < 0.0001; NS = Not Significant; 2-tailed Student’s t test
Next, we analyzed 90 cases of familial breast and/or ovarian cancer (Table 2). All 90 patients were screened for mutations in the BRCA1 and BRCA2 genes and 20 were found to carry pathogenic variants with an overall mutation rate of 22.2 % (Table 2). In particular BRCA1 pathogenic mutation recurred in about 26 % (8/30) of the Hereditary Breast and Ovarian Cancer (HBOC) families and in about 7 % (4/60) of the Hereditary Breast Cancer (HBC) families, while BRCA2 pathogenic mutation occurred in about 13 % (4/30) and 7 % (4/60) of the HBOC and HBC families, respectively. Of the 20 mutation-positive probands, 11 had breast cancer alone (5 BRCA1 and 6 BRCA2), three had ovarian cancer alone (3 BRCA1), three had bilateral breast cancer (2 BRCA1 and 1 BRCA2), two had both breast and ovarian cancer (1 BRCA1 and 1 BRCA2) and one had both bilateral breast and ovarian cancer (BRCA1). When fresh PBMCs from BRCA1/2-positive and -negative patients were analyzed by the p53-MCL test, five out of 90 cases showed aberrant p53-MCL (Fig. 1b). Interestingly p53-MCL positive cases accounted for 6.6 and 5 % of the HBOC and HBC families, respectively (Table 2). Of note, this rate is similar to that of BRCA2 mutations [26].
Table 2.
Characteristics of familial breast and/or ovarian cancer patients
| Variable | Study population n = 90 | BRCA1/2 carriers n = 20/90 (22.2 %) | ATM carriers n = 5/90(5.5 %) |
|---|---|---|---|
| Age-years | |||
| Median | 52.9 | 53.7 | 44.4 |
| Range | 30–82 | 40–75 | 32–51 |
| Familial aggregation | |||
| HBOC | 30 (33.3 %) | BRCA1: 8/30 (26 %) | 2/30 (6.6 %) |
| BRCA2: 4/30 (13 %) | |||
| HBC | 60 (66.7 %) | BRCA1: 4/60 (7 %) | 3/60 (5 %) |
| BRCA2: 4/60 (7 %) | |||
| BRCAPRO | 27.3 % | 50.1 % | 52.2 % |
HBOC hereditary breast and ovarian cancer, HBC hereditary breast cancer
The five individuals with ATM variants identified by the p53-MCL test were all breast cancer patients (5/65, 7.7 %). Four of them were BRCA1/2 negative patients and one was a BRCA1 positive patient (Fig. 2). No ATM carriers were detected by p53-MCL in patients with ovarian cancer, bilateral breast cancer, or multiple cancers (breast cancer and at least one other non-breast cancer) (n = 27) (Table 3).
Fig. 2.
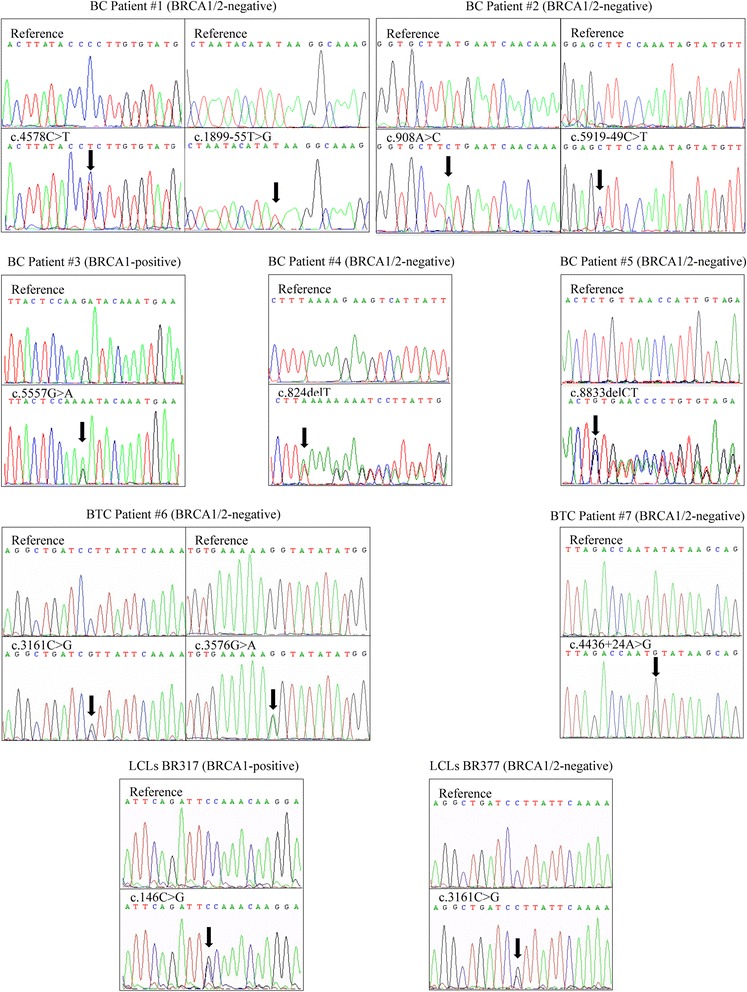
ATM variants in ATM carriers. Electropherograms showing ATM variants identified in PBMCs derived from five familial breast cancer (BC) patients (BC patient#1/#5), two breast-thyorid cancer (BTC) patients (BTC patient#6/#7) and two LCLs (BR317; BR377). All sequences are compared with wild-type reference sequence. Arrows indicates the position of the substitution and/or deletion
Table 3.
Histopathological characteristics of all cancer patients
| Tumor type | Cases n = 496 | BRCA1/2 carriers n = 20 | ATM carriers n = 7 |
|---|---|---|---|
| Familial Cancer cases | 90 | 20 | 5 |
| Breast | 65 (70.6 %) | 11/65 (16.9 %) | 5/65 (7.7 %) |
| Ovarian | 8 (8.7 %) | 3/8 (37.5 %) | 0/8 |
| Bilateral Breast | 10 (10.9 %) | 3/10 (30 %) | 0/10 |
| Breast-Ovarian | 4 (4.3 %) | 2/4 (50 %) | 0/4 |
| Colon-Breast | 1 (1.1 %) | 0/1 | 0/1 |
| Uterin-Breast | 1 (1.1 %) | 0/1 | 0/1 |
| Anal-Bilateral Breast | 1 (1.1 %) | 0/1 | 0/1 |
| Bilateral Breast-Ovarian | 1 (1.1 %) | 1/1 (100 %) | 0/1 |
| Ipsilateral Breast | 1 (1.1 %) | 0/1 | 0/1 |
| Sporadic cancer cases | 406 | - | 2/406 |
| Ovarian | 49 | - | 0/49 |
| Lung | 150 | - | 0/150 |
| Colon | 80 | - | 0/80 |
| Post-menopausal Breast | 58 | - | 0/58 |
| Breast/Thyroid | 69 | - | 2/69 (2.9 %) |
Besides early onset-breast cancer, ATM carriers have been reported to be more susceptible to other types of cancer, such as digestive tract, lung, and thyroid cancers [2, 8]. Thus, we performed the p53-MCL test on fresh PBMCs from 403 patients with sporadic cancers, including ovarian (n = 49), lung (n = 150), colon (n = 80), post-menopausal breast (n = 58), and both breast/thyroid (n = 69) cancer. Two individuals with ATM variants were identified among breast/thyroid cancer cases (2/69, 2.9) (Table 4). No ATM carriers were found in sporadic ovarian cancer (0/49), non-small cell lung cancer (0/150), colorectal cancer (0/80), and post-menopausal breast cancer (0/58).
Table 4.
Clinical and pathological characteristics of breast and thyroid cancer patients
| Variable | Study population (n = 69) | p53-MCL positive (n = 2) |
|---|---|---|
| Age-years | ||
| Average | 60.2 | 58 |
| Range | 30–77 | 58–63 |
| Age onset Breast tumor appearance | ||
| Average | 49.1 | 48 |
| Range | 25–70 | 48–51 |
| Breast tumor Diagnosis | ||
| Ductal | 59 (85.5 %) | 1 (50 %) |
| Lobular | 3 (4.3 %) | 0 |
| Unknown | 7 (10.2 %) | 1 (50 %) |
| ER status | ||
| Negative | 17 (24.6 %) | 0 |
| Positive | 34 (49.3 %) | 2 (100 %) |
| Unknown | 18 (26.1 %) | 0 |
| PgR status | ||
| Negative | 17 (24.6 %) | 0 |
| Positive | 34 (49.3 %) | 2 (100 %) |
| Unknown | 18 (26.1 %) | 0 |
ER estrogen receptor, PgR progesterone receptor
As shown in Fig. 2 and Additional file 1: Table S1, 11 different ATM variants were detected among the 9 p53-MCL positive cases (i.e., 2 LCLs and 7 PBMCs). Specifically, in breast cancer cases, patient #1 presents c.4578C > T and c.1899-55 T > G variants. The c.4578C > T was previously described [27] and corresponds to a synonymous substitution (P1526P), which is predicted to create a new exonic site with increased affinity for the SRp55 splicing factor by an in silico analysis (Fig. 3a). The c.1899-55 T > G is a previously described intronic variant [28] predicted to increase the affinity for the SRp40 splicing factor (Fig. 3b). Patient #2 presents two variants not previously described in the literature nor in the ATM variation database (c.908A > C and c.5919-49C > T). Our in silico analyses predict a higher affinity for the splicing factor SC35, for both variants (Fig. 3c, d). The c.5557G > A (D1853N) variant presents in patient #3 is predicted to be “possibly damaging” by the PolyPhen-2 tool and has been intensively studied with respect to its possible association with breast cancer susceptibility [29]. Patient #4 and patient #5 present two deleterious variants, c.824delT and c.8833delCT, that induce early protein truncation at the level of exon 9 and 63, respectively, which have already been described in ATM families [30, 31]. In the breast and thyroid cancer cases, patient #6 presents two already described variants, c.3161C > G and c.3576G > A. The c.3161C > G (P1054R) is a substitution predicted to be “possibly damaging” by PolyPhen-2 and implicated in breast and prostate cancer risk [32]. The c.3576G > A variant leads to exon 26 skipping and causes A-T syndrome in homozygosis [30]. Patient #7 presents the intronic variant c.4436 + 24A > G that in silico analyses predict a mild higher affinity for the splicing factors SC35 and SRp55 (Fig. 3e). LCLs BR317 presents c.146C > G (S49C) variant, a substitution predicted to be possibly damaging by PolyPhen-2 and reported as breast cancer susceptibility variant [16]. Finally, LCLs BR377 presents the same c.3161C > G (P1054R) variant found in patient #6.
Fig. 3.
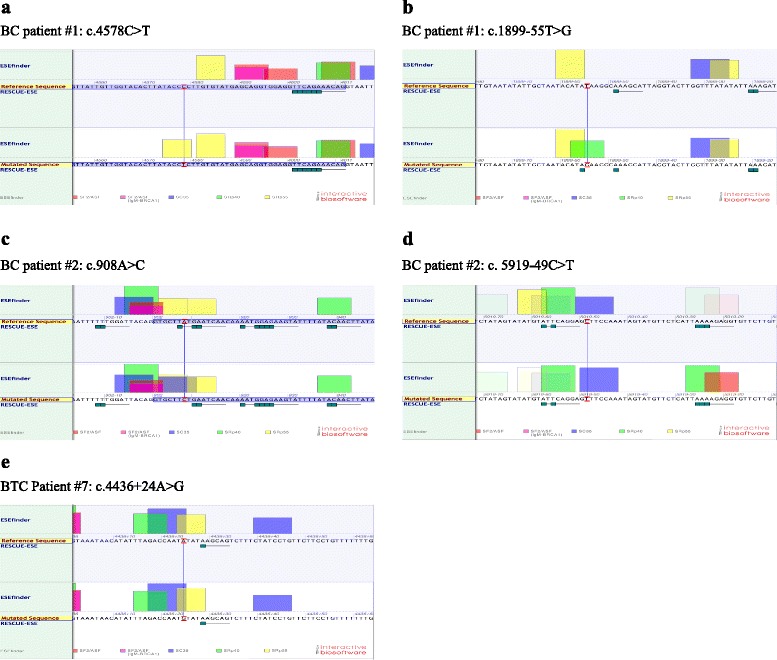
In silico analysis using Alamut software. ATM variants identified in PBMCs derived from two familial breast cancer patients (BC patient#1 and #2) and one breast-thyorid cancer patient (BTC patient#7) lead to an increased affinity and/or appearance of a de novo sites for SR proteins
Discussion
Genetic susceptibility plays an important role in several common chronic diseases including many types of cancer. Genetic testing for large-scale, general population screening can be very expensive and non-cost effective for National Health Services. Concerning the ATM gene, the complex genomic organization, the large number of polymorphisms, the absence of mutation hot-spots, and the frequent occurrence of variants of yet uncharacterized but predicted deleterious functions, make direct gene sequencing not yet a cost-effective approach. This is particularly evident for large-scale surveys of mutant/variant ATM carriers for which gene sequencing is not sufficient to classify the rare hits. All these factors hinder both genetic counseling and clinical guidelines for risk management of ATM carriers and their families. The use of the recently developed p53-MCL functional test to detect ATM carriers might overcome at least some of these limitations. Here, we established the p53-MCL specificity for ATM in respect to the high-risk, BRCA1/2 breast/ovarian cancer susceptibility genes and confirmed p53-MCL as reliable test to detect variant ATM carriers in cancer patients.
BRCA1/2 and ATM proteins share functional activities both in DNA damage response pathways and in centrosome regulation [33]. Thus, it was mandatory to establish whether a functional test, such as the p53-MCL test, would discriminate between ATM and BRCA1/2 variant carriers. Analysis of BRCA1/2-positive LCLs (n = 6) and PBMCs (n = 20), and of 1 CHK2-positive LCL established the specificity of p53-MCL test on ATM in respect to BRCA1/2 and CHK2 mutations (Table 1 and Fig. 1).
A total of seven individuals with ten ATM variants were identified by p53-MCL test in PBMCs: five breast cancer patients in HBC and HBOC families and two breast/thyroid cancer patients. The finding that all cases were among pre-menopausal breast cancer cases but not among patients with post-menopausal breast cancer or ovarian cancer is consistent with previous studies showing prevalence of germline ATM mutations detected by sequencing in patients with familial early-onset breast cancer [34] or by p53-MCL in sporadic breast cancer [14]. Interestingly, the a priori probability of carrying a pathogenic BRCA1/2 germline mutation measured by the BRCAPRO score showed a mean for the entire cohort of 27.3 while the mean for the five ATM carriers was 52.2 % (Table 2). This difference is statistically significant (P =0.01) and supports the presence of a strong genetic component in the ATM carriers with early-onset breast cancer, also considered that the BRCAPRO score for the BRCA positive cases is very similar to ATM positive ones (50.1 % vs. 52.2 %).
Evaluation of the histopathologic features of the breast cancers developed in the seven identified ATM carriers showed the presence of estrogen and progesterone receptors and the absence of HER2 receptor in four out of five cases that have been tested (Tables 4 and 5), confirming our previous results [14]. Thus, the presence of germline ATM variants recognized by p53-MCL appears to identify a subset of tumors with a more favorable biomarker asset, despite their earlier onset. In addition, a possible association between breast and thyroid cancer was highlighted by the identification of two ATM carriers among the sporadic cancer patients with breast/thyroid cancer (2/69, 2.9 %), encouraging for further studies.
Table 5.
Clinical and pathological characteristics of breast cancer patients of HBOC and HBC
| Variable | Study population (n = 65) | p53-MCL positive (n = 5) |
|---|---|---|
| Age-years | ||
| Average | 50.4 | 44.4 |
| Range | 30–82 | 32–51 |
| Age onset tumor appearance | ||
| Average | 42.5 | 38.6 |
| Range | 24–67 | 28–43 |
| Diagnosis | ||
| Ductal | 47 (72.3 %) | 3 (60 %) |
| Lobular | 8 (12.3 %) | 0 |
| Unknown | 8 (12.3 %) | 2 (40 %) |
| Other | 2 (3.1 %) | 0 |
| ER status | ||
| Negative | 15 (23.1 %) | 0 |
| Positive | 36 (55.4 %) | 5 (100 %) |
| Unknown | 14 (21.5 %) | 0 |
| PgR status | ||
| Negative | 19 (29.2 %) | 0 |
| Positive | 32 (49.2 %) | 5 (100 %) |
| Unknown | 14 (21.5 %) | 0 |
| HER2 status | ||
| Negative | 29 (44.6 %) | 4 (80 %) |
| Positive | 16 (24.6 %) | 1 (20 %) |
| Unknown | 20 (30.8 %) | 0 |
| BRCAPRO | 32.3 | 52.2 |
ER estrogen receptor, PgR progesterone receptor, HER2 human epidermal growth factor receptor 2
Familial breast and ovarian cancers are linked to highly penetrant mutations in the BRCA1/2 susceptibility genes that overall account for 20–25 % of hereditary breast cancers and 15 % of ovarian cancers [26]. Gene panel next generation sequencing approaches identified moderate-penetrant mutations in the ATM gene in 2.9 % [11] and 2.3 % [34] of BRCA1/2-negative cases. By the p53-MCL test, in this study we found 5.5 % (5/90) of ATM mutation carriers, which adds up to the 27.7 % (20/90) carrying BRCA1/2 mutations. In particular, three ATM mutations occurred among the 60 HBC cases, with a mutation rate (5 %) very close to that observed for BRCA2 (7 %) in the same subset. These observations suggest that the fast and non-expensive p53-MCL test should precede or be performed in parallel with BRCA1/2 sequencing.
Most of the ATM mutations occurring in A-T patients are frameshift or nonsense mutations leading to protein truncation or splice junction variants [6, 7] The role of A-T-causing mutations in cancer susceptibility (in particular breast cancer) is still debated and some studies have shown that a subset of rare, evolutionarily unlikely missense substitutions are important [15, 35, 36]. Here, the p53-MCL test identified 11 ATM variants in 9 breast and breast/thyroid patients. Three variants (c.824delT; c.8833delCT; c.3576G > A) identified in 3 different patients are known to cause A-T in homozigosity [30, 31]. Other 3 variants (c.5557G > A; c.146C > G; c.3161C > G) identified in 3 different patients have been shown to be associated with an increased cancer risk [16, 29, 32]. The last 3 patients present 5 different variants (c.4578C > T; c.1899-55 T > G; c.908A > C; c.5919-49C > T; c.4436 + 24A > G) that our in silico analysis predicted to increase the affinity for splicing factor and modify alternative splicing activities. Whether these predicted modifications alter ATM function/s and cancer predisposition remain to be evaluated. Since loss of p53 centrosomal localization was the functional readout through which we diagnosed the ATM carriers, we can conclude that these variants are at least able to impair the mitotic localization of p53 at the centrosomes. It will be relevant to study whether all ATM variants induce this p53 defect or whether only functionally relevant variants are able to impair p53-MCL. Whether this impaired p53 localization has a role in tumorigenesis is presently unknown. Studying the mechanistic basis of p53 centrosomal localization will give insights on the contribution that different ATM variants with uncertain significance might have in cancer predisposition. Application of the p53-MCL test to LCLs or PBMCs with a broad spectrum of ATM variants will help to define these issues.
Conclusions
Our results show that p53-MCL test may offer the opportunity for screening of the general population and to identify the differences among deleterious, neutral and beneficial variants helping, in the future, to define the guidelines for ATM carriers not only in the A-T families.
Acknowledgments
We are grateful to all patients and families taking part in this study. We thank Mustapha Haoui for technical assistance.
Availability of data and materials
Please, contact authors for data request.
Authors’ contributions
A Prodosmo designed and performed experiments. He contributed to analyze data and write the manuscript. A Buffone generated sequencing data and molecular analyses. MM performed the p53 MCL-test. A Barnabei and A Persichetti collected blood samples and clinical information of patients related to breast and thyroid cancer cases. ADL collected blood samples related to breast and thyroid cancer cases. MLA coordinated the study related to breast and thyroid cancer cases. AN produced lymphoblastoid cell lines. AC collected blood samples and clinical information of patients related to familial breast and/ovarian cancer cases. S Sciacchitano coordinated the study and collected blood samples and clinical information of patients related to breast and thyroid cancer cases. CG collected blood samples and clinical information of patients related to post menopausal breast cancer cases. PP collected clinical information of patients related to post menopausal breast cancer cases. GS coordinated the study related to post menopausal breast cancer cases. LS performed statistical analyses. GA collected blood samples and clinical information of patients related to lung cancer cases. FF coordinated the study related to lung cancer cases. MC collected blood samples and clinical information of patients related to colon cancer cases. GLG coordinated the study related to colon cancer cases. GC collected blood samples and clinical information of patients related to ovarian cancer cases. EV coordinated the study related to ovarian cancer cases. GG coordinated the study related to familial breast and/or ovarian cancer cases, performed genetic counseling, contributed to design the study and to write the manuscript. S Soddu conceived and designed the experiments. She coordinated the entire study and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The Institutional Ethics Committee approved this study (I.F.O. CE/160/09 and CE/763/13) and all patients studied signed an informed consent for participation.
Financial support
This work was supported by grants from Italian Association for Cancer Research (AIRC) to S.S. (5 per mille #9979) and (IG #14592), and to G.G. (IG#12116 and IG#17734). A. Prodosmo and A. Buffone were recipients of a fellowship from Fondazione Umberto Veronesi “Pink is Good”.
Abbreviations
- A-T
Ataxia telangiectasia
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3 related
- BRCA
Breast cancer susceptibility gene
- CHK2
Checkpoint kinase 2
- EBV
Epstein barr virus
- FANC-A
Fanconi anemia complementation group A
- HBC
Hereditary breast cancer
- HBOC
Hereditary breast and ovarian cancer
- LCLs
Lymphoblastoid cell lines
- MRE11
Meiotic recombination 11
- NBS1
Nijmegen breakage syndrome 1
- p53-MCL
p53-Mitotic centrosomal localization
- PALB2
Partner and localizer of BRCA2
- PBMCs
Peripheral blood mononuclear cells
- PHA
Phytohemagglutinin
- SMC1A
Structural maintenance of chromosomes 1A
- WRN
Werner syndrome
Additional file
Characteristics of ATM variants. (DOCX 75 kb)
Contributor Information
Andrea Prodosmo, Email: prodo77@hotmail.com.
Amelia Buffone, Email: maybuf@yahoo.it.
Manlio Mattioni, Email: manlio.mattioni@ifo.gov.it.
Agnese Barnabei, Email: agnese.bernabei@ifo.gov.it.
Agnese Persichetti, Email: agnese.persichetti@ifo.gov.it.
Aurora De Leo, Email: aurora.dileo@ifo.gov.it.
Marialuisa Appetecchia, Email: marialuisa.appetecchia@ifo.gov.it.
Arianna Nicolussi, Email: arianna.nicolussi@uniroma1.it.
Anna Coppa, Email: anna.coppa@uniroma1.it.
Salvatore Sciacchitano, Email: ssciacchitano@hotmail.it.
Carolina Giordano, Email: carolina.giordano@ifo.gov.it.
Paola Pinnarò, Email: paola.pinnaro@ifo.gov.it.
Giuseppe Sanguineti, Email: giuseppe.sanguineti@ifo.gov.it.
Lidia Strigari, Email: lidia.strigari@ifo.gov.it.
Gabriele Alessandrini, Email: gabriele.alessandrini@ifo.gov.it.
Francesco Facciolo, Email: francesco.facciolo@ifo.gov.it.
Maurizio Cosimelli, Email: maurizio.cosimelli@ifo.gov.it.
Gian Luca Grazi, Email: gianluca.grazi@ifo.gov.it.
Giacomo Corrado, Email: giacomo.corrado@ifo.gov.it.
Enrico Vizza, Email: enrico.vizza@ifo.gov.it.
Giuseppe Giannini, Phone: +3906 49255136, Email: giuseppe.giannini@uniroma1.it.
Silvia Soddu, Phone: +390652662492, Email: silvia.soddu@ifo.gov.it.
References
- 1.Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine (Baltimore) 1991;70:99–117. doi: 10.1097/00005792-199103000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Su Y, Swift M. Mortality rates among carriers of ataxia-telangiectasia mutant alleles. Ann Intern Med. 2000;133:770–8. doi: 10.7326/0003-4819-133-10-200011210-00009. [DOI] [PubMed] [Google Scholar]
- 3.Uziel T, Savitsky K, Platzer M, Ziv Y, Helbitz T, Nehls M, et al. Genomic organization of the ATM gene. Genomics. 1996;33:317–20. doi: 10.1006/geno.1996.0201. [DOI] [PubMed] [Google Scholar]
- 4.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 5.Campbell C, Mitui M, Eng L, Coutinho G, Thorstenson Y, Gatti RA. ATM mutations on distinct SNP and STR haplotypes in ataxia-telangiectasia patients of differing ethnicities reveal ancestral founder effects. Hum Mutat. 2003;21:80–5. doi: 10.1002/humu.10156. [DOI] [PubMed] [Google Scholar]
- 6.Mitui M, Campbell C, Coutinho G, Sun X, Lai CH, Thorstenson Y, et al. Independent mutational events are rare in the ATM gene: haplotype prescreening enhances mutation detection rate. Hum Mutat. 2003;22:43–50. doi: 10.1002/humu.10232. [DOI] [PubMed] [Google Scholar]
- 7.Concannon P, Gatti RA. Diversity of ATM gene mutations detected in patients with ataxia-telangiectasia. Hum Mutat. 1997;10:100–7. doi: 10.1002/(SICI)1098-1004(1997)10:2<100::AID-HUMU2>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 8.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–6. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med. 1987;316:1289–94. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- 10.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 11.Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med. 2015;17:630–8. doi: 10.1038/gim.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gatti RA, Tward A, Concannon P. Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab. 1999;68:419–23. doi: 10.1006/mgme.1999.2942. [DOI] [PubMed] [Google Scholar]
- 13.Shen L, Yin ZH, Wan Y, Zhang Y, Li K, Zhou BS. Association between ATM polymorphisms and cancer risk: a meta-analysis. Mol Biol Rep. 2012;39:5719–25. doi: 10.1007/s11033-011-1381-2. [DOI] [PubMed] [Google Scholar]
- 14.Prodosmo A, De Amicis A, Nisticò C, Gabriele M, Di Rocco G, Monteonofrio L, et al. p53 centrosomal localization diagnoses ataxia-telangiectasia homozygotes and heterozygotes. J Clin Invest. 2013;123:1335–42. doi: 10.1172/JCI67289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meier M, den Boer ML, Hall AG, Irving JA, Passier M, Minto L, et al. Relation between genetic variants of the ataxia telangiectasia-mutated (ATM) gene, drug resistance, clinical outcome and predisposition to childhood T-lineage acute lymphoblastic leukaemia. Leukemia. 2005;19:1887–95. doi: 10.1038/sj.leu.2403943. [DOI] [PubMed] [Google Scholar]
- 16.Stredrick DL, Garcia-Closas M, Pineda MA, Bhatti P, Alexander BH, Doody MM, et al. The ATM missense mutation p.Ser49Cys (c.146C>G) and the risk of breast cancer. Hum Mutat. 2006;27:538–44. doi: 10.1002/humu.20323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannini G, Capalbo C, Ristori E, Ricevuto E, Sidoni T, Buffone A, et al. Novel BRCA1 and BRCA2 germline mutations and assessment of mutation spectrum and prevalence in Italian breast and/or ovarian cancer families. Breast Cancer Res Treat. 2006;100:83–91. doi: 10.1007/s10549-006-9225-9. [DOI] [PubMed] [Google Scholar]
- 18.Berry DA, Iversen ES, Jr, Gudbjartsson DF, Hiller EH, Garber JE, Peshkin BN, et al. BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J Clin Oncol. 2002;20:2701–12. doi: 10.1200/JCO.2002.05.121. [DOI] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biosoftware Alamut v2.3. http://www.interactive-biosoftware.com
- 21.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032–7. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Apostolou P, Fostira F. Hereditary breast cancer: the era of new susceptibility genes. Biomed Res Int. 2013;2013:747318. doi: 10.1155/2013/747318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 24.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–92. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 25.Wu J, Lu LY, Yu X. The role of BRCA1 in DNA damage response. Protein Cell. 2010;1:117–23. doi: 10.1007/s13238-010-0010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palma M, Ristori E, Ricevuto E, Giannini G, Gulino A. BRCA1 and BRCA2: the genetic testing and the current management options for mutation carriers. Crit Rev Oncol Hematol. 2006;57:1–23. doi: 10.1016/j.critrevonc.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Thorstenson YR, Shen P, Tusher VG, Wayne TL, Davis RW, Chu G, et al. Global analysis of ATM polymorphism reveals significant functional constraint. Am J Hum Genet. 2001;69:396–412. doi: 10.1086/321296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Concannon P, Haile RW, Børresen-Dale AL, Rosenstein BS, Gatti RA, Teraoka SN, et al. Variants in the ATM gene associated with a reduced risk of contralateral breast cancer. Cancer Res. 2008;68:6486–91. doi: 10.1158/0008-5472.CAN-08-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mao C, Chung VC, He BF, Luo RC, Tang JL. Association between ATM 5557G > A polymorphism and breast cancer risk: a meta-analysis. Mol Biol Rep. 2012;39:1113–8. doi: 10.1007/s11033-011-0839-6. [DOI] [PubMed] [Google Scholar]
- 30.Sandoval N, Platzer M, Rosenthal A, Dörk T, Bendix R, Skawran B, et al. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum Mol Genet. 1999;8:69–79. doi: 10.1093/hmg/8.1.69. [DOI] [PubMed] [Google Scholar]
- 31.Telatar M, Teraoka S, Wang Z, Chun HH, Liang T, Castellvi-Bel S, et al. Ataxia-telangiectasia: identification and detection of founder-effect mutations in the ATM gene in ethnic populations. Am J Hum Genet. 1998;62:86–97. doi: 10.1086/301673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angèle S, Falconer A, Edwards SM, Dörk T, Bremer M, Moullan N, et al. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer. 2004;91:783–7. doi: 10.1038/sj.bjc.6602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang S, Hemmerich P, Grosse F. Centrosomal localization of DNA damage checkpoint proteins. J Cell Biochem. 2007;101:451–65. doi: 10.1002/jcb.21195. [DOI] [PubMed] [Google Scholar]
- 34.Brunet J, Gutiérrez-Enríquez S, Torres A, Bérez V, Sanjosé S, Galceran J, et al. ATM germline mutations in Spanish early-onset breast cancer patients negative for BRCA1/BRCA2 mutations. Clin Genet. 2008;73:465–73. doi: 10.1111/j.1399-0004.2008.00987.x. [DOI] [PubMed] [Google Scholar]
- 35.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–22. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 36.Tavtigian SV, Oefner PJ, Babikyan D, Hartmann A, Healey S, Le Calvez-Kelm F, et al. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am J Hum Genet. 2009;85:427–46. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Please, contact authors for data request.