

Abstract
DNA methylation, through 5-methyl- and 5-hydroxymethylcytosine (5mC and 5hmC), is considered to be one of the principal interfaces between the genome and our environment, and it helps explain phenotypic variations in human populations. Initial reports of large differences in methylation level in genomic regulatory regions, coupled with clear gene expression data in both imprinted genes and malignant diseases, provided easily dissected molecular mechanisms for switching genes on or off. However, a more subtle process is becoming evident, where small (<10 %) changes to intermediate methylation levels are associated with complex disease phenotypes. This has resulted in two clear methylation paradigms. The latter “subtle change” paradigm is rapidly becoming the epigenetic hallmark of complex disease phenotypes, although we are currently hampered by a lack of data addressing the true biological significance and meaning of these small differences.
Our initial expectation of rapidly identifying mechanisms linking environmental exposure to a disease phenotype led to numerous observational/association studies being performed. Although this expectation remains unmet, there is now a growing body of literature on specific genes, suggesting wide ranging transcriptional and translational consequences of such subtle methylation changes. Data from the glucocorticoid receptor (NR3C1) has shown that a complex interplay between DNA methylation, extensive 5′UTR splicing, and microvariability gives rise to the overall level and relative distribution of total and N-terminal protein isoforms generated. Additionally, the presence of multiple AUG translation initiation codons throughout the complete, processed mRNA enables translation variability, hereby enhancing the translational isoforms and the resulting protein isoform diversity, providing a clear link between small changes in DNA methylation and significant changes in protein isoforms and cellular locations. Methylation changes in the NR3C1 CpG island alters the NR3C1 transcription and eventually protein isoforms in the tissues, resulting in subtle but visible physiological variability.
This review addresses the current pathophysiological and clinical associations of such characteristically small DNA methylation changes, the ever-growing roles of DNA methylation and the evidence available, particularly from the glucocorticoid receptor of the cascade of events initiated by such subtle methylation changes, as well as addressing the underlying question as to what represents a genuine biologically significant difference in methylation.
Keywords: DNA methylation, EWAS, Association studies, Biomarker, Transcriptional microvariability, Gene-environment interactions
Background
Two clear methylation paradigms
DNA methylation and hydroxyl-methylation are amongst the more intensely studied epigenetic mechanisms. These two modifications consist of either a methyl or a hydroxymethyl group being covalently bound to the 5 position of cytosine in palindromic cytosine-phosphate-guanine (CpG) dinucleotides, abbreviated to 5mC and 5hmC, respectively [1–4]. CpG dinucleotides occur infrequently, and 98 % of the mammalian genome is CpG-deficient. The remaining ~2 % contains short high-frequency stretches of CpGs called CpG islands (CGIs) that are mainly associated with gene promoter and regulatory regions. Single CpGs are often found in repetitive DNA elements and centromeric regions [1, 2, 4, 5].
There are two concurrent paradigms for DNA methylation: the first paradigm is a clear mechanism for switching genes on/off through complete methylation or demethylation of genomic regulatory regions. DNA methylation has long been considered a marker of permanent gene silencing (imprinting) or reactivation [6]. In malignant diseases, this simple on/off switch is often observed activating or silencing oncogenes and tumour suppressor genes, respectively [7], e.g. O6-methylguanine-DNA-methyltransferase (MGMT) methylation levels vary from 0 to >60 %. Although it is not the focus of this review, and has been extensively reviewed and meta-reviewed elsewhere, the principal diagnostic epigenetic cancer biomarkers available such as VIM, SEPT9, SHOX2, GST1, APC, and RASSF1A share this clear pattern of no or little methylation, and clear (>60 %) hypermethylation, with almost nothing in-between [8]. However, this simple paradigm has been challenged, and a second paradigm is emerging. In this second paradigm, intermediary DNA methylation levels are fine-tuned, often influenced by the external environment, and are becoming the epigenetic hallmark of many complex non-malignant disorders. In this case, the association of DNA methylation with an observed phenotype occurs through small differences in the methylation level of <10 % and often only 1–5 %, at single CpGs or over very limited genomic regions [3, 9]. Such limited differences in DNA methylation are known to be set during periods of epigenetic sensitivity [10]. Additionally, they have been shown to play a role in creating a large diversity in phenotypes linked to the onset of many complex non-malignant diseases, such as type 2 diabetes, major depression, schizophrenia, hypertension, and cardiovascular diseases [9, 11]. Epigenetic phenotypes are not necessarily restricted to an exposed individual. Some epigenetic marks are transgenerational, hereby transmitting the phenotypic trait and possibly the linked disease to the offspring [6, 11–13].
This split into two paradigms has been accompanied by the expansion of the roles of 5mC and 5hmC. Both are now considered important factors assuring the quantitative, spatial, and temporal regulation of gene expression as well as normal development and differentiation [3, 5, 6]. By targeting promoter CpGs and CGIs, DNA methylation was mainly thought to interfere with the transcription initiation and consequently gene silencing or reactivating [1, 6, 14]. Genome-wide analysis techniques showed that DNA methylation influences many other mechanisms, such as alternative splicing, alteration of enhancer, insulator, and regulatory element function, hence altering gene expression [9, 14–16]. For both tissue-specific regulation and non-malignant disorders, changes in gene expression are frequently caused by small changes in methylation levels, often at single CpG dinucleotides or over a limited genomic region. Such small differences have a big impact on the phenotype diversity that is linked to the onset of non-malignant diseases [15, 17]. Plasticity in methylation levels allows environmental adaptations, transient changes, and long-term alterations of the cell’s transcriptomic profile, hereby contributing to the diversity of characteristics, both biochemical and physiological, and hence the phenotypic variations observed in human populations [2, 3, 6]. These mechanisms have been associated with the onset and maintenance of pathogenesis [2, 18, 19], and methylation has increasingly been associated with the aetiology and onset of multiple, non-malignant, complex disorders [15, 18–21].
DNA methylation can be summarised as either discrete hyper- and hypomethylation coupled with clear gene silencing, and easily dissected molecular mechanisms, or a more subtle complex process where small (<10 %) methylation changes are associated with disease phenotypes and many transcriptional processes. This leads us to the fundamental question of the biological significance of such small changes and how they give rise to the final disease phenotype. There is currently doubt over the true biological relevance of such small changes, if they are genuinely meaningful, what mechanisms link such limited changes in methylation to the phenotype, and how this affects our view of what a gene is. In this review, we summarise the pathophysiological and clinical associations that have been made to small, subtle methylation changes; the ever-growing roles of DNA methylation; and the evidence available, particularly from the glucocorticoid receptor of the cascade of events initiated by such subtle methylation changes, and conclude that such small changes may reflect genuine biological differences.
Environmental influence on phenotype diversity: a role for small epigenetic changes?
Environmental influence on DNA methylation, gene expression, phenotype, and disease onset have been extensively studied. In the framework of the Developmental Origins of Health and Disease (DOHaD) paradigm, in utero or early-life conditions programme lifelong health trajectories. This paradigm focusses on organisms’ biological plasticity to adjust their phenotype to their environment over the short and long term in which epigenetic processes such as DNA methylation are thought to be involved. Mismatches between the pre-/post-natally anticipated and the actual mature environment predisposes organisms to disease (Fig. 1) [22–24].
Fig. 1.
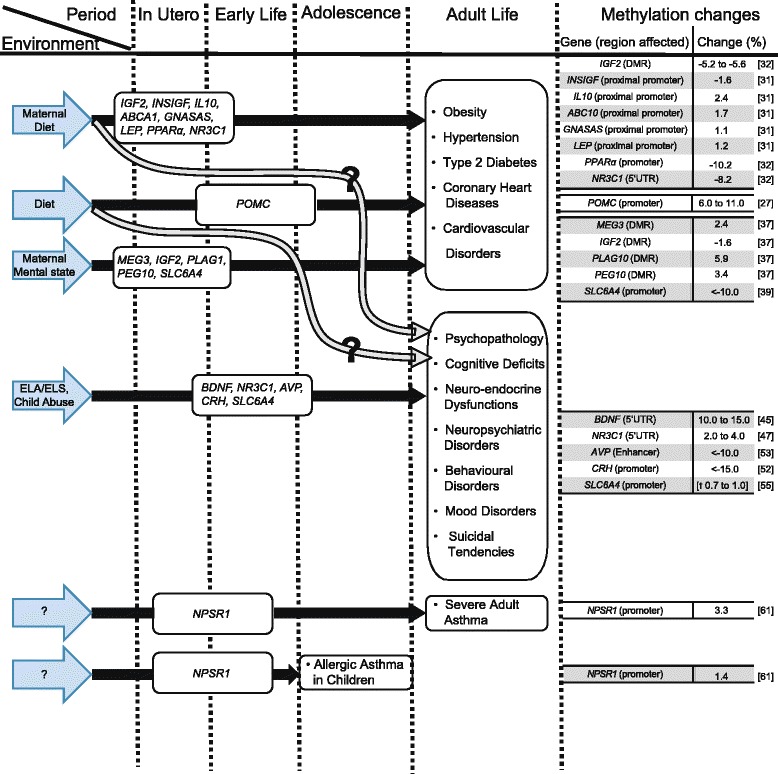
Methylation changes have been associated with adult pathology and environmental factors in association studies. Environmental factors during certain periods of life have been linked to genes or disorders. The changes in methylation are listed next to the phenotype in adult life. ELA early-life adversity, ELS early-life stress, dagger data are infinium β values
Obesity, hypertension, cardiovascular diseases, diabetes
The prevalence of obesity, hypertension and the accompanying cardiovascular disorders, and diabetes have been associated with early-life environmental factors, such as diet, parental diet, and maternal mood during pregnancy (Fig. 1) [25, 26]. In the ‘small litter’ neonatal overfeeding model, appetite was dysregulated via hypermethylation of the POMC promoter at the NF-kB and Sp1 binding sites necessary for inducing POMC expression by leptin and insulin [27]. Consequently, POMC expression will be reduced despite insulin or leptin presence [25, 26]. Parental diet strongly influenced their offspring’s methylation profile and phenotype (Fig. 1) [12, 25]. Gestational high fat diets increased the offspring’s probability of developing obesity, metabolic syndrome, insulin insensitivity, and diabetes in both humans and animal models (Fig. 1) [25, 28, 29]. Conversely, a low-protein maternal diet peri-conceptually or during gestation was associated with lower birth weight, schizophrenia, an increased risk of the offspring developing cardiovascular diseases, hypertension, dyslipidaemia, and obesity (Fig. 1) [12, 25, 30–32]. A well-known natural experiment for transgenerational nutri-epigenomics was the ‘Dutch Hunger Winter’. Dutch individuals exposed in utero to malnutrition and their direct descendants [30] had higher rates of obesity (BMI raise of 7.4 % in women [33]), hypertension (odds ratio (OR) 1.44 [34]), an increased risk for cardiovascular disorders (coronary heart disease: OR 3.0) and impaired glucose homeostasis later on in life (glucose tolerance index: prenatally −21 %; late gestation −4 %; midgestation −24 %; early gestation −37 %) [35, 36]. This was accompanied by hypomethylation in IGF2 (−5.2 to −5.6 %) and INSIGF (−1.6 %), and hypermethylation of IL10 (2.4 %), ABCA1 (1.7 %), GNASAS (1.1 %) and LEP (1.2 %) (Fig. 1) [31, 32]. Late gestational exposure appeared to be a less sensitive period, as it only affected the methylation profile of GNASAS (−1.1 %) from the limited number of target genes investigated [31]. An equally important factor affecting the offspring’s methylation profile and phenotype was maternal mental state during pregnancy (Fig. 1). Gestational depression during pregnancy associated with a lower birth weight (OR 3.6, 95 % CI 1.1–11.4), obesity, as well as cardiovascular disorders and diabetes in later life (Fig. 1) [37, 38]. This was accompanied by higher MEG3 methylation levels (2.4 %) and decreased methylation of IGF2 (−1.6 %) compared to children with normal birth weight [37]. Offspring with a higher birth weight than normal showed a hypermethylation of PLAG1 and PEG10 (5.9 and 3.4 %, respectively), genes that have previously been linked to the regulation of placental and foetal growth and development, growth in general, and diabetes [37]. Maternal depression during the second trimester of pregnancy on the other hand was linked with hypomethylation of the SLC6A4 promoter region for both mother and child (Fig. 1) [37, 39]. Overall, it seems that individuals subjected to poor diets in utero or early life or born out of mothers suffering from severe depression during gestation develop phenotypes with a higher prevalence of obesity, hypertension and the accompanying cardiovascular disorders and diabetes. It remains unclear, however, whether the methylation changes are part of the mechanism increasing disorder prevalence or rather an additional consequence.
Psychopathology and behaviour
The risk of developing psychopathologies, cognitive, behavioural, anxiety and mood disorders or suicidal tendencies later in life have been related to stressful/traumatic experiences during early development and early life (Fig. 1). These early life periods profoundly affect development of the central nervous system, the limbic structures or hypothalamus-pituitary-adrenal (HPA) axis regulation. Although the underlying mechanisms are unknown, the detection of methylome and gene expression changes between phenotypes highlights the importance of DNA methylation [3, 40–44]. BDNF, a gene involved in neurodevelopment, neuroplasticity, the onset of psychiatric disorders and suicidal behaviour, has been associated to early-life adversity (ELA) (Fig. 1). Rat and mouse models for ELA and depression showed that the epigenetic processes controlling BDNF transcription were stress sensitive. The BDNF promoter region was hypermethylated (10 % to 15 % per CpG on average), with the ensuing lower expression levels [41, 45]. Similar BDNF methylation patterns were observed in post-mortem adult brains from suicide completers [41, 42]. HPA axis and stress response dysregulation have been amongst the most consistent biological findings in major depression and psychopathology [46]. NR3C1, coding the central HPA axis regulating glucocorticoid receptor, was frequently investigated as part of the mechanism linking ELA and the predisposition towards psychopathology or suicide risk (Fig. 1) [41, 47, 48]. In rat models, ELA caused a hypomethylation of the hippocampal NR3C1 promoter (2 to 4 %), which significantly altered the gene expression and HPA-axis responsivity [1, 47, 49, 50]. Post-mortem brain analyses and clinical studies observed similar trends [41, 47, 48, 50]. Suicide completers with a history of ELA had increased hippocampal NR3C1 promoter methylation and decreased NR3C1 expression (Fig. 1) [41, 47, 50, 51]. Similarly, higher hippocampal and leukocyte NR3C1 methylation levels were observed for healthy individuals previously exposed to ELA. As such, the evidence is now strong that NR3C1 methylation is part of the panoply of changes linking ELA events to later life psychopathology, although there is no definite evidence as to whether it is a direct mechanism or an additional independent event [48]. HPA axis changes were not limited to NR3C1; ELA and early-life stress (ELS) also induced a sustainable hypomethylation of AVP (<10 % per CpG position) and CRH (<15 % per CpG position [3, 52, 53], as well as psychopathology-associated genes such as SLC6A4 [54, 55]. The methylation status of the SLC6A4 promoter was shown to be affected by abuse as well as genotype [54]. Although DNA methylation appears to explain the link between ELA and psychopathology through HPA axis regulation, robust proof of principle remains, however, to be provided as connecting methylome profile alterations and gene expression robustly failed [54]. The link between epigenetic alterations and neuropsychiatric disorders remains unproven.
Asthma and allergic pathologies
Genetic makeup has been seen in many studies to be one of the strongest risk factors for eventually developing allergic symptoms [56, 57] consistent with epidemiological evidence of an increased allergic rhinitis (AR) concordance in twin studies [58]. Although many candidate genes have been suggested, genome-wide association studies (GWAS) have not, so far, identified “overlapping and consistent genetic components” [59, 60], and epigenetic mechanisms have been proposed to play an equally important role. For example, the promoter methylation level of NPSR1 showed small but significant differences for persons suffering from severe adult or allergic asthma in children (Fig. 1). NPSR1, normally highly methylated (>75 %), was hypomethylated by −3.29 and −1.40 % for severe adult asthma and allergic asthma in children, respectively [61]. DNA methylation levels have also been associated with factors such as the current smoking behaviour, parental smoking during infancy, and the month in which the sample was taken [61], which are thought to be implicated in the onset of both asthma and allergic diseases.
Associations and hypotheses, not mechanisms
The increased number of association studies has given us a better insight of the environmental impact on phenotype development (Fig. 1). Yet, as the majority of these observational studies did not address the underlying mechanisms, we are left with associations and hypotheses. In order to enhance our understanding, future research should address the underlying process and try to provide robust evidence for the exact cascade of events linking environment and phenotype differences. A good example of such a clear link is the viable yellow Agouti (Avy) mouse model, where the offspring’s coat colour shifts between yellow and brown due to incomplete erasure of the maternal epiallele during embryogenesis. The Agouti gene has a methylation-sensitive intracisternal-A particle retrotransposon inserted at the 5′ end that functions as a transcription start site. Large changes in the methylation of the A locus from ~70 to ~25 % result in a yellow rather than the natural brown coat. The offspring phenotype and methylation level appeared to be heavily influenced by those of the mother. Oocyte transfer to surrogate mothers of a different epigenetic background, however, was necessary to demonstrate that the offspring epigenotype depended on the incomplete erasure of the maternal methylation during embryogenesis, rather than the uterine environment [62]. For the studies above, such detailed mechanistic studies are unfortunately absent.
Currently, epigenome-wide association studies (EWAS) data such as those highlighted above are a perfect storm of visibly low methylation levels, of which the biological meaning is uncertain and a large variety of confounding factors influencing their methylation state [63]. There is a void, with limited information or guidelines on how to design and conduct meaningful EWAS. Adopting a set of guidelines or rules for best practices, in a similar manner to GWAS, would benefit EWAS interpretation and increase their relevance.
Highlight: improving and interpreting EWAS studies
Sample variability
To reduce variability, purified single cell types must be the sample of choice. If this is not possible, heterogeneity should be measured, e.g. by flow cytometry and controlled for statistically. For whole blood, the post hoc Houseman treatment may be used. Care needs to be taken in the choice of sampling surrogate or target cells.
Data format
In order to enable direct comparisons between relative methylation values and absolute methylation levels, it is suggested to report absolute methylation values or β values. When possible, relative methylation levels should be confirmed by a direct quantitative technique such as pyrosequencing.
Confounding variables
Genetic, demographical, clinical, and environmental factors contribute to the overall phenotype. Comprehensive metadata collection is important. Minimum data includes age, sex, smoking habits and BMI, in addition to technical data to account for batch effects and technical variability. Whenever possible, sequencing-based techniques should be preferred, allowing access to the underlying genomic sequence.
Study design
Successful interpretation of small methylation changes calls for a well-planned study design. It is essential to estimate the minimally required sample size to reach the necessary statistical power, taking into account the cohort type (control-case, monozygotic twins) and potential confounding co-factors, which will need to be sampled and modelled for correct analyses and interpretation.
What is a biologically meaningful change in methylation level?
As we [38] and others [64] have previously noted, there is doubt over the true biological relevance of small changes in absolute methylation levels, and it has been suggested that authors may have increased confidence in the biological significance of methylation differences >10 %, and conversely, must treat differences of <5 % with extreme caution [65].
Reducing sample variability
Different cell types have specific epigenetic profiles [66], and measuring aggregate levels over a large populations is a major source of variability. Since methylation is essentially binary, i.e. in any given cell, a specific CpG is either methylated, unmethylated, or potentially hemi-methylated (asymmetric methylation of two alleles), the methylation levels measured simply reflect the proportion of methylated cells in the original sample [38, 67–69]. Consequently, minor changes in methylation may actually represent small changes in the cellular composition of the original sample rather than a genuine difference due to the disease or paradigm studied. As an aside the most widely used sample, blood is unfortunately one of the most variable, although there is now a well-established procedure that adequately corrects for this variability [70, 71].
The impact of the data format
Teasing out the biologically relevant changes in methylation levels is further complicated by the current trend towards reporting fold changes rather than absolute methylation values. The appropriate data to report is naturally specific for the analysis method employed. For example, MedIP-Seq and Infinium arrays (Illumina) give M and β values that may correlate to the percentage methylation, they are relative values, and they may be considerably different from the direct measurement (e.g. by pyrosequencing) of the absolute methylation levels. Although there is no direct comparison available, it has been suggested that ‘a β-value of 0.8 might correspond to a level of 30 % methylation’ [64], however, as highlighted above, when methylation levels are low, as in the case of NR3C1, a relatively small change in the absolute methylation level will be represented as a wildly exaggerated fold change or percentage increase. In the current situation, where small differences in methylation or low methylation values are being reported, there are additional technical concerns with data analysis and reporting. Illumina β values are predominantly reported as they can be considered an approximation to the percentage methylation present in the original sample. However, this is only valid for values in the ‘middle methylation range’ [72], with severe heteroscedasticity for low and high methylation values. This has lead authors to suggest that statistical analyses are performed with M values, but to report β values [72].
Confounding variables
Interpretation of small methylation changes is further complicated by the numerous sources of epigenetic variability that are currently poorly defined. There is significant evidence that many genetic, demographical, clinical, and environmental factors are strong cofounding variables [64]. However, these underappreciated confounding variables all contribute to the overall measured phenotype. This was highlighted by the low intra-individual, but high inter-individual, difference in methylation levels we observed throughout the human brain [73]. Population-wide, 5-mC levels are both reduced and redistributed with age [74] and are generally higher genome-wide in males than females [75, 76]. Locus-specific differential hyper- or hypomethylation has, however, been reported for both men and women [77–80]. Equally, the underlying genomic sequence heavily influences DNA methylation levels. Although there are numerous other examples [81–85], the best estimate is that approximately 2 % of the investigated CpGs that cover up to 9.5 % of genes represent methylation quantitative trait loci (mQTLs) and may operate over distances up to 5 kb [86]. Our NR3C1 data demonstrated that methylation of the NR3C1 promoter 1H was associated with a complete haplotype (haplotype 2), rather than a specific SNP, operating over approximately 3 kbp. The effect of the underlying genome sequence is also highlighted by pervasive asymmetric methylation in diploid genomes (i.e. difference between the two alleles), particularly outside imprinted regions [83, 87–89]. This asymmetry is known to be regulated by underlying heterozygotic genetic variants. In transgenerational epigenetic inheritance, there is now convincing evidence that it is the genomic sequence, rather than the parental DNA methylation levels that determines 5mC levels during embryogenesis [90]. Furthermore, allele-specific methylation events are found in unrelated individuals with the same haplotype/genotype as well as in multiple inter-individual tissues [83]. Although the evidence for these confounding factors is growing, there are still no population-epigenetics principles available to guide study design, analysis, and interpretation. However, we suggest that moving towards sequencing-based techniques (whole genome bisulphite sequencing, reduced representation bisulphite sequencing, MeDIP-Seq, etc.) will allow access to the genomic variants that is not available in array-based techniques.
The importance of study design and best practice guideline
As for GWAS, a study design adapted to the chosen research hypothesis is important [91, 92]. Frequently adopted designs are case-control and monozygotic twin designs, each of them having different sample size and statistical power requirements. Current best estimates suggest that to observe a ~13 % difference in methylation, there is little difference in the power of discordant twin or case control designs, however, smaller differences especially below 10 %, required 178 pairs of monozygotic twins or 211 control and 211 cases to detect 7 % methylation differences genome-wide significance with a statistical power of 80 % (Table 1) [91, 93]. These estimations have only been performed for array-based EWAS and useful information or guidelines on sequencing-based EWAS are unfortunately absent. The surge of sequencing-based technologies and the possibility of greater in-depth examination genome-wide of methylation differences requires guidelines for best practices. Not solely guidelines concerning study design, sample size, and power but also guidelines concerning interpretation and importance of the small methylation differences that are regularly observed together with co-variables and confounding factors such as gender, age, and smoking that will significantly affect the methylation.
Table 1.
Estimated cohort sizes for 80 % power at individual loci and genome wide in twin and case-control EWAS designs (from [93], under CC BY 4.0 licence)
| Difference (%) | Twin | Case-control | ||||||
|---|---|---|---|---|---|---|---|---|
| P < 0.05 | P < 1 × 10−6 | P < 0.05 | P < 1 × 10−6 | |||||
| t testa | Wilcoxb | t testa | Wilcoxb | t testc | Wilcoxd | t testc | Wilcoxd | |
| 7 | 30 | 30 | 178 | 178 | 37 | 37 | 211 | 211 |
| 8 | 25 | 25 | 145 | 149 | 30 | 30 | 169 | 169 |
| 9 | 20 | 20 | 117 | 117 | 24 | 24 | 137 | 137 |
| 10 | 17 | 18 | 98 | 102 | 20 | 21 | 112 | 110 |
| 11 | 15 | 15 | 81 | 83 | 17 | 18 | 96 | 95 |
| 12 | 13 | 13 | 71 | 71 | 15 | 16 | 80 | 80 |
| 13 | 11 | 12 | 63 | 69 | 13 | 13 | 70 | 70 |
| 14 | 10 | 11 | 55 | 62 | 11 | 13 | 61 | 63 |
| 15 | 9 | 10 | 50 | 57 | 10 | 11 | 54 | 57 |
a t test, paired t test
bWilcox, Wilcoxon signed-rank test
c t test, two-sample t test
dWilcox, Wilcoxon rank-sum test
Study purpose
The current interest in DNA methylation is primarily to exploit its potential as a biomarker. In both malignant and complex non-malignant diseases, work has centred on associating methylation changes with the external environment, particularly to exploit the latency between exposure and disease development. In both the DOHaD and ‘foetal origins’ models, early-life events induce epigenetic changes that are maintained lifelong. Similarly, many environmental factors, e.g. chemical, biological (e.g. toxins, allergens), or heavy metal exposure alter the epigenome, ultimately increasing the risk of developing cancer [94, 95], for example, asbestos exposure leads to DNMT overexpression, highly specific methylation patterns, and eventually malignant pleural mesothelioma [96, 97]. In both cases, there is a considerable period of latency from the exposure to clinically discernible disease ranging from a few years (autism, obesity) to many decades (cardiovascular disease, mesothelioma). During this latent period, the epigenetic marks are, however, present. If the interest in DNA methylation is solely as a biomarker, then the question of the origin and biological relevance of these changes is somewhat irrelevant. If the observed changes can be robustly validated and replicated, then their simple representation of a change in the sampled cell population may be adequate for their exploitation as a biomarker [8, 98]. However, when changes are observed in purified cell populations, such subtle changes in methylation may give significant insight into underlying pathophysiological mechanisms. If we consider post-partum depression (PPD), pre-symptom onset epigenetic markers have been identified, potentially allowing the identification of susceptible women [99]. Although the epigenetic markers had a >80 % predictive accuracy and have significant potential as PPD biomarkers, they also provide significant mechanistic insight into the pathophysiology of PPD. It has long been postulated that PPD is linked to the significant fluctuation in hormonal levels during pregnancy and, indeed, the epigenetic marks have all been linked to 17β oestradiol (E2). Although PPD has a range of previously identified biological and environmental risk factors, it is unlikely to have a single underlying cause, and the methylation changes identified may represent a ‘final common pathway’ [100] integrating many potential pathways. However, this highlights that differentially methylated regions are not exclusively biomarkers as they are often reported, but may provide significant insight into the underlying mechanisms.
Overall, we are forced to conclude that there is currently no accurate estimate of what represents a genuine, biologically relevant, change in methylation and what may be ascribed to any of a multitude of external factors, although it should be emphasised that all these outside factors contribute to measurable differences in the observed phenotype, and that small changes may represent a genuine biological difference.
Methylation: single CpG or clusters?
It is becoming clear that despite numerous reports of single CpGs associating with disease phenotypes, methylation levels are regulated in clusters. This has brought into question the functional effect of limited changes to the methylation level of single CpG dinucleotides [38, 101]. Using the NR3C1 as an example, that methylation over a region of ~45 consecutive CpGs within one of the many promoter regions efficiently silenced the associated transcripts [102]. However, methylation of smaller regions of ~125 bp (~12CpGs) reduced promoter activity by 75 % [47]. There is currently no evidence for the NR3C1 that single CpG methylation has functional effects on gene expression [38]. Both individual [67] and promoter-wide [103] CpG methylation increases have been associated with clinical post-traumatic stress disorder (PTSD) symptoms. Our NR3C1 methylation data concords with the latter observation, where a strong distance-dependent correlation throughout the NR3C1 promoter was observed both in man [73, 101] and rat [104], suggesting that for the NR3C1, methylation occurs in clusters over ~80 bp. Similar results have been observed at the whole epigenome level as well as the population level [105, 106]. Importantly, at the population level, methylation clusters appeared to behave in a manner similar to genetic variants with multiple clusters of methylation in ‘linkage-disequilibrium’ covering distances up to 300 kbp [107].
Towards a mechanism linking subtle methylation changes to phenotypes?
The glucocorticoid receptor as a model
The glucocorticoid receptor (NR3C1, GR) has well-characterised transcriptional and translational variability. The association of receptor levels and variants with disease [108–110] has made it a particularly useful model to explore both the functional relevance and the effects of small methylation changes [102, 111–114] and the association between methylation and pathology at the single gene level [51, 73, 101, 115]. The NR3C1 5′ structure, containing multiple alternative non-coding first exons (1A to 1J) with a multitude of transcription factor binding sides (Fig. 2a), was initially reported by to be responsible for the quantitative, spatial, and temporal expression of the NR3C1 [108–110]. Recently, however, NR3C1’s transcription was shown to be exceptionally permissive rather than being initiated at fixed positions (Fig. 2). We observed a total of 358 statistically significant transcription start sites (TSS) located in 38 contiguous loci in the absence of any particular stimuli, with a further 185 stimuli specific [111]. For instance, demethylation with 5-AZA-2′-deoxycytidine (AZA) had a profound influence on the TSSs used, with 127 stimuli-specific TSSs induced by demethylation. This permissivity, covering a large 3-kbp region, is called transcriptional microvariability (Fig. 2) [111]. Although such microvariability appears to be stochastic, in the case of the NR3C1, it has a significant effect on translation. Small differences in TSS location (<10 nt) within any given locus redirected ribosomes to initiate translation from internal (downstream) ATG codons, altering the balance of the translational GR isoforms produced (Fig. 2) [25]. A shift in TSS location results in an altered mRNA secondary structure and half-life and influences the overall translational efficiency in a ‘length-dependent, but sequence-independent manner’ [111, 113]. For the NR3C1, this microvariability vastly inflates the associated proteome. The GR is classically cytosolic; however, we have demonstrated that the membrane bound form of the receptor (mGR [31]) is derived from the classical NR3C1 gene [114], and further refined its molecular origin to the epigenetically regulated alternative first exon, 1D [113]. As such, microvariability influences not only the final protein form but also the final cellular distribution of the GR proteins. Our data lead us to conclude that physiological differences in glucocorticoid secretion and response are the result of DNA methylation altering TSS/first exon usage, with the consequentially proteomic difference [49, 73, 102, 108, 116–118]. Importantly, our data suggest that, at least for the NR3C1, neither single nor clusters of CpGs that are methylated switch off transcription of any particular splice variant, rather, they orchestrate the final proteomic landscape, and potentially alter the splicing internally or at the 3′ end [113, 119].
Fig. 2.
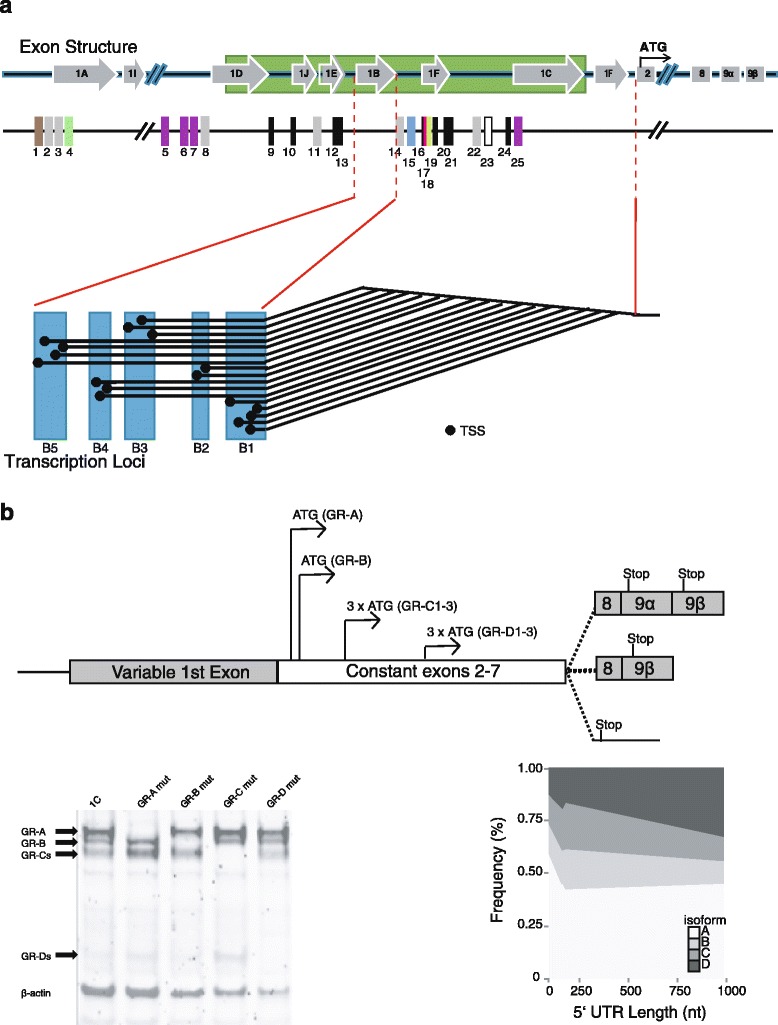
a A schematic representation of the NR3C1 5′ UTR structure, showing the alternative first exons (1A–1J, CpG island:  ), transcription factor binding sites (1–25), transcriptional loci (B1–B5), and microvariable transcription start sites (•). Transcription factor binding sites: (
), transcription factor binding sites (1–25), transcriptional loci (B1–B5), and microvariable transcription start sites (•). Transcription factor binding sites: ( ) IRF-1 and IRF-2 (position 1); (
) IRF-1 and IRF-2 (position 1); ( ) glucocorticoid response elements (GRE, positions 2, 3, 8, 11, 14, and 22); (
) glucocorticoid response elements (GRE, positions 2, 3, 8, 11, 14, and 22); ( ) c-Myb, c-Ets1/2 and PU1 (position 4); (
) c-Myb, c-Ets1/2 and PU1 (position 4); ( ) Ying Y and 1 (positions 5, 6, 7, and 25); (■) Sp1 binding sites (positions 9, 10, 12, 13, 16, 19, 20, 21, and 24); (
) Ying Y and 1 (positions 5, 6, 7, and 25); (■) Sp1 binding sites (positions 9, 10, 12, 13, 16, 19, 20, 21, and 24); ( ) Ap-1 (position 15); (
) Ap-1 (position 15); ( ) NGFI-A binding site (position 17); (
) NGFI-A binding site (position 17); ( ) glucocorticoid response factor-1 (GRF-1, position 18); (□) Ap-2 (position 23). b Structure of the GR mRNA with the internal ATG translation initation codons, a western blot demonstrating the different transcriptional isoforms (from [113] with permission), and the frequency of the different protein isoforms with increasing 5′ UTR length (adapted from [111] with permission)
) glucocorticoid response factor-1 (GRF-1, position 18); (□) Ap-2 (position 23). b Structure of the GR mRNA with the internal ATG translation initation codons, a western blot demonstrating the different transcriptional isoforms (from [113] with permission), and the frequency of the different protein isoforms with increasing 5′ UTR length (adapted from [111] with permission)
Expanding the mechanism from the NR3C1 to the complete transcriptome and proteome?
It has been recognised for many years that both the complexity and phenotypic diversity increase as the relative size of non-coding genomic regions and the regulatory elements and variability within them increase throughout evolution [120, 121]. Features like alternative first exons, transcriptional microvariability, and alternative in-frame downstream ATG initiation codons are found genome-wide; ~60 % of all genes are thought to possess a highly variable 5′ structure with many alternative first exons [122], and this transcription variability has been reported in many cases to be responsible for spatio-temporal gene expression patterns [123]. Similarly, multiple alternative in-frame ATG translation initiation codons within mature mRNA are found ubiquitously through evolution, occurring in many plant, invertebrate, and vertebrate species [124–126].
In light of data on the origin and evolution of new TSSs and exons in different species [127–130], transcriptional microvariability is not unexpected, and now, several reports have observed transcription starting over small multiple small loci genome-wide [122] and in model organisms [131]. Multiple alternative in-frame ATG translation initiation codons have been observed in a wide range of genes. Although no systematic review of their occurrence has been performed, they are thought to be ubiquitous and cover both leaky ribosome scanning and internal ribosome entry [132]. These observations and the ubiquity of the features made researchers suggest that the 5′ UTR, together with intergenic regions and the 75 % of the human and mouse genomes that are transcribed are the key to understanding the vastly inflated proteome [111, 120, 133]. It therefore seems logical that the mechanisms outlined for NR3C1 above should be expandable to the complete transcriptome and proteome. Irrespective of whether 5′ variability comes from the mRNA structure, the TSS location, transcriptional microvariability, or alternative mRNA splicing, this variability will give rise “to high complex and diverse transcriptomes and proteomes” [111] (Fig. 3).
Fig. 3.
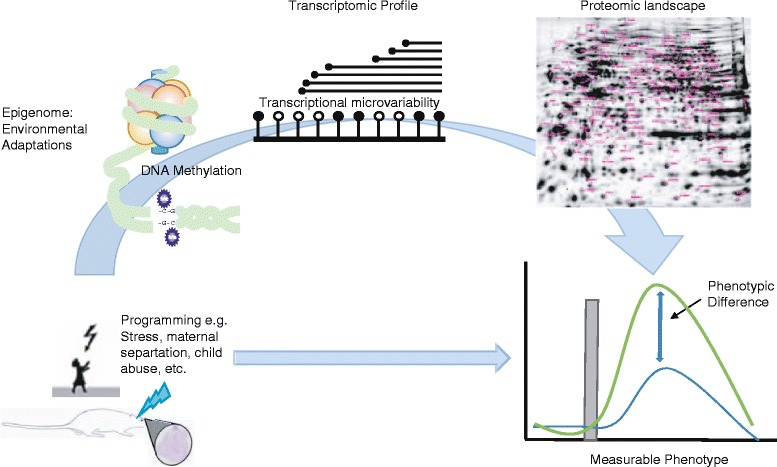
Proposed mechanism for the creation of phenotype diversity by environmental factors. Epigenetic marks, such as DNA methylation, are proposed to influence transcriptional variability and hence the proteomic landscape, resulting in a phenotype diversity
Re-defining a ‘gene’?
The significant increase in transcriptional and translational complexity observed for the NR3C1 concords with the recent movement towards re-defining a ‘gene’. While the definition of ‘gene’ has changed considerably over the last century, the current definition used worldwide for genome annotation is ‘a DNA segment that contributes to phenotype/function. In the absence of demonstrated function a gene may be characterized by sequence, transcription or homology’ [134]. This definition has come under scrutiny over the last decade [135, 136]. Large-scale sequencing projects such as ENCODE/GENCODE have identified several phenomena that are changing our perception of what a gene is, including universal alternative splicing, pervasive and intergenic transcription, and dispersed patterns of transcription regulation [137–139]. Gerstein et al. metaphorically described the classical definition of a gene as ‘subroutines in the genomic operating system’ [135]. This analogy was further broken down into the genome being a complete human ‘operating system’ and with gene being a clear, well-defined ‘subroutines’ where a genomic region is assembled as in a homologous manner to computer code, with transcription and translation considered the homologues of calling and running a subroutine. In this analogy, gene elements (5′, 3′ UTR, intron, exon, etc.) were considered as the syntax. GENCODE and subsequent data have called this neat definition into question. The vastly inflated transcriptome and proteome suggest that the process is rather ‘higgledy-piggledy’ or stochastic, with the gene ‘subroutine’ very poorly defined with many starting points. Post GENCODE, the definition of a gene was simplified taking into account this variability as ‘a gene is a genomic sequence (DNA or RNA) directly encoding functional product molecules, either RNA or protein’ [135]. The two definitions can be compared to strict Boolean or fuzzy logic. This definition is amenable to the integration of data, such as ours, from the epigenetic regulation of the NR3C1, as it would appear that a combination of genetic and epigenetic variants underpin and orchestrate the higgledy-piggledy or fuzzy processes into a concerted, specific response to the external environment.
Conclusions
It has become clear that DNA methylation occurs either as discrete hyper- and hypomethylation coupled with a clear on/off switch of genes as often observed in oncogenes, and easily dissected molecular mechanisms, or in a second paradigm as a more subtle complex process where small (<10 %) methylation changes have been associated with divers phenotypes and epigenetic programming events. Despite the observational association studies’ aim to increase our understanding of the environmental impact on phenotype development, the underlying mechanisms linking subtle methylation changes to an eventual phenotype remained unaddressed (Fig. 3). Consequently, hampering our interpretation of the associations with subtle changes in methylation due to a lack of data addressing the true biological relevance and function of such small differences.
We are now starting to gain insight into the function and relevance of such small changes in methylation from genes, such as NR3C1. These data suggest that the 5′ UTR is the key to controlling gene expression. Small changes in methylation throughout this region impact mechanisms such as alternative splicing and transcriptional microvariability, altering enhancer and insulator use, and the function of regulatory elements. Methylation of single CGs affect the TSS usage within a gene promotor region, i.e. silence a specific location, whereas methylation of multiple closely related CG’s will rather silence a transcription loci, i.e. a whole site of adjacent TSSs. Recent studies demonstrate that small changes in methylation levels seem to be regulated in clusters rather than single CpGs. But whether they act as single CpGs or in clusters, these small changes do not function as an on/off switch, rather redistributing the transcriptional landscape, affect translational isoform production, and orchestrating the final proteomic landscape.
Technologies such as next-generation sequencing (NGS) have enabled researchers to study these subtle methylation changes in greater detail genome-wide. The emerging approach, combining both NGS and single cell technology, will allow a far more in depth analysis of this phenomenon and its importance overall as well as on the single-cell level. The recognition and appreciation of the functional significance of such small differences in methylation highlights the importance of unaddressed EWAS questions, particularly in identifying the correct confounding variables. Being able to control for different sources of variability has become more important in order to ensure the small changes observed are genuine biological differences and to be able to subsequently interpret them.
Acknowledgments
Funding
The authors are currently funded by the Fonds National de Recherche, Luxembourg [C12/BM/3985792] and the Ministry of Higher Education and Research (Luxembourg), and work performed by us and reviewed here through the Fonds National de Recherche [PHD-80-053; TR-PHD-BFR07/127; TRPHD-BFR-07/043], the Deutsche Forschungsgemeinschaft (GRK 1389/1) and Ministry of Higher Education and Research (Luxembourg). The funding bodies played no role in the writing of this manuscript, nor in the design, data collection, analysis, and interpretation of our previously published data reviewed here.
Availability of data and materials
Please refer to the individual publications cited in this review for the availability of materials and data within them.
Authors’ contributions
This review was conceived by JDT, written by FL and JDT, and revised into its final format by FL, CPM, and JDT. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 5hmC
5-Hydroxymethylcytosine
- 5mC
5-Methylcytosine
- AR
Allergic rhinitis
- AZA
5-AZA-2′-deoxycytidine
- CGI
CpG island
- CpG
Cytosine-phosphate-guanine
- DOHaD
Developmental Origins of Health and Disease
- ELA
Early-life adversity
- ELS
Early-life stress
- EWAS
Epigenome-wide association studies
- GR
Glucocorticoid receptor
- GWAS
Genome-wide association studies
- HPA
Hypothalamus-pituitary-adrenal
- MGMT
Methylguanine-DNA-methyltransferase
- mQTLs
Methylation quantitative trait loci
- NGS
Next-generation sequencing
- OR
Odds ratio
- PPD
Post-partum depression
- PTSD
Post-traumatic stress disorder
- TSS
Transcription start site
Contributor Information
Fleur A. D. Leenen, Email: fleur.leenen@lih.lu
Claude P. Muller, Email: claude.muller@lih.lu
Jonathan D. Turner, Phone: +352 26970629, Email: jonathan.turner@lih.lu
References
- 1.Garcia-Carpizo V, Ruiz-Llorente L, Fraga M, Aranda A. The growing role of gene methylation on endocrine function. J Mol Endocrinol. 2011;47(2):R75–89. doi: 10.1530/JME-11-0059. [DOI] [PubMed] [Google Scholar]
- 2.Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6(1):e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neidhart M. DNA methylation and complex human diseases. 1. Amsterdam: Academic Press; 2016. [Google Scholar]
- 4.Serre D, Lee BH, Ting AH. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38(2):391–9. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 6.Mazzio EA, Soliman KF. Basic concepts of epigenetics: impact of environmental signals on gene expression. Epigenetics. 2012;7(2):119–30. doi: 10.4161/epi.7.2.18764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Smet C, Lurquin C, Lethe B, Martelange V, Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Mol Cell Biol. 1999;19(11):7327–35. doi: 10.1128/MCB.19.11.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes (Basel) 2014;5(3):821–64. doi: 10.3390/genes5030821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levenson VV. DNA methylation as a universal biomarker. Expert Rev Mol Diagn. 2010;10(4):481–8. doi: 10.1586/erm.10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner JD, Kirschner S, Molitor AM, Evdokimov K, Muller CP. Epigenetics. 2nd Edition ed. International Encyclopedia of Behavioural Sciences. Oxford: Elsevier; 2015. [Google Scholar]
- 11.Guerrero-Bosagna C, Weeks S, Skinner MK. Identification of genomic features in environmentally induced epigenetic transgenerational inherited sperm epimutations. PLoS One. 2014;9(6):e100194. doi: 10.1371/journal.pone.0100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, de Rooij SR. Hungry in the womb: what are the consequences? Lessons from the Dutch famine. Maturitas. 2011;70(2):141–5. doi: 10.1016/j.maturitas.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 13.Guerrero-Bosagna C, Settles M, Lucker B, Skinner MK. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One. 2010;5(9). doi:10.1371/journal.pone.0013100. [DOI] [PMC free article] [PubMed]
- 14.Yagi S, Hirabayashi K, Sato S, Li W, Takahashi Y, Hirakawa T, et al. DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression. Genome Res. 2008;18(12):1969–78. doi: 10.1101/gr.074070.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liyanage VR, Jarmasz JS, Murugeshan N, Del Bigio MR, Rastegar M, Davie JR. DNA modifications: function and applications in normal and disease states. Biology (Basel) 2014;3(4):670–723. doi: 10.3390/biology3040670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carrio E, Suelves M. DNA methylation dynamics in muscle development and disease. Front Aging Neurosci. 2015;7:19. doi: 10.3389/fnagi.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levenson VV, Melnikov AA. DNA methylation as clinically useful biomarkers—light at the end of the tunnel. Pharmaceuticals (Basel) 2012;5(1):94–113. doi: 10.3390/ph5010094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Zeng C. Role of DNA methylation in cardiovascular diseases. Clin Exp Hypertens. 2016;38(3):261–7. doi: 10.3109/10641963.2015.1107087. [DOI] [PubMed] [Google Scholar]
- 19.Moarii M, Boeva V, Vert JP, Reyal F. Changes in correlation between promoter methylation and gene expression in cancer. BMC Genomics. 2015;16(1):873. doi: 10.1186/s12864-015-1994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulahannan N, Greally JM. Genome-wide assays that identify and quantify modified cytosines in human disease studies. Epigenetics Chromatin. 2015;8:5. doi: 10.1186/1756-8935-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laker RC, Ryall JG. DNA methylation in skeletal muscle stem cell specification, proliferation, and differentiation. Stem Cells Int. 2016;2016:5725927. doi: 10.1155/2016/5725927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gluckman PD, Hanson MA, Mitchell MD. Developmental origins of health and disease: reducing the burden of chronic disease in the next generation. Genome Med. 2010;2(2):14. doi: 10.1186/gm135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gluckman PD, Hanson MA, Buklijas T. A conceptual framework for the developmental origins of health and disease. J Dev Orig Health Dis. 2010;1(1):6–18. doi: 10.1017/S2040174409990171. [DOI] [PubMed] [Google Scholar]
- 24.Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61(5 Pt 2):5R–10R. doi: 10.1203/pdr.0b013e318045bedb. [DOI] [PubMed] [Google Scholar]
- 25.Lillycrop KA. Effect of maternal diet on the epigenome: implications for human metabolic disease. Proc Nutr Soc. 2011;70(1):64–72. doi: 10.1017/S0029665110004027. [DOI] [PubMed] [Google Scholar]
- 26.Lillycrop KA, Burdge GC. The effect of nutrition during early life on the epigenetic regulation of transcription and implications for human diseases. J Nutrigenet Nutrigenomics. 2011;4(5):248–60. doi: 10.1159/000334857. [DOI] [PubMed] [Google Scholar]
- 27.Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol. 2009;587(Pt 20):4963–76. doi: 10.1113/jphysiol.2009.176156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunn GA, Bale TL. Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology. 2011;152(6):2228–36. doi: 10.1210/en.2010-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunn GA, Bale TL. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology. 2009;150(11):4999–5009. doi: 10.1210/en.2009-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bygren LO. Intergenerational health responses to adverse and enriched environments. Annu Rev Public Health. 2013;34:49–60. doi: 10.1146/annurev-publhealth-031912-114419. [DOI] [PubMed] [Google Scholar]
- 31.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18(21):4046–53. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105(44):17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999;70(5):811–6. doi: 10.1093/ajcn/70.5.811. [DOI] [PubMed] [Google Scholar]
- 34.Stein AD, Zybert PA, van der Pal-de Bruin K, Lumey LH. Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: evidence from the Dutch Famine. Eur J Epidemiol. 2006;21(10):759–65. doi: 10.1007/s10654-006-9065-2. [DOI] [PubMed] [Google Scholar]
- 35.de Rooij SR, Painter RC, Phillips DI, Osmond C, Michels RP, Godsland IF, et al. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care. 2006;29(8):1897–901. doi: 10.2337/dc06-0460. [DOI] [PubMed] [Google Scholar]
- 36.de Rooij SR, Painter RC, Roseboom TJ, Phillips DI, Osmond C, Barker DJ, et al. Glucose tolerance at age 58 and the decline of glucose tolerance in comparison with age 50 in people prenatally exposed to the Dutch famine. Diabetologia. 2006;49(4):637–43. doi: 10.1007/s00125-005-0136-9. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Murphy SK, Murtha AP, Fuemmeler BF, Schildkraut J, Huang Z, et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics. 2012;7(7):735–46. doi: 10.4161/epi.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vinkers CH, Kalafateli AL, Rutten BP, Kas MJ, Kaminsky Z, Turner JD, et al. Traumatic stress and human DNA methylation: a critical review. Epigenomics. 2015;7(4):593–608. doi: 10.2217/epi.15.11. [DOI] [PubMed] [Google Scholar]
- 39.Devlin AM, Brain U, Austin J, Oberlander TF. Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS One. 2010;5(8):e12201. doi: 10.1371/journal.pone.0012201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kundakovic M, Gudsnuk K, Herbstman JB, Tang D, Perera FP, Champagne FA. DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci U S A. 2015;112(22):6807–13. doi: 10.1073/pnas.1408355111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labonte B, Turecki G. Epigenetic effects of childhood adversity in the brain and suicide risk. In: Dwivedi Y, editor. The Neurobiological Basis of Suicide. Frontiers in Neuroscience. Boca Raton (FL): CRC Press/Taylor & Francis; 2012. [PubMed] [Google Scholar]
- 42.Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, et al. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch Gen Psychiatry. 2010;67:258–67. doi: 10.1001/archgenpsychiatry.2010.9. [DOI] [PubMed] [Google Scholar]
- 43.Stenz L, Zewdie S, Laforge-Escarra T, Prados J, La Harpe R, Dayer A, et al. BDNF promoter I methylation correlates between post-mortem human peripheral and brain tissues. Neurosci Res. 2015;91:1–7. doi: 10.1016/j.neures.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 44.Li S, Papale LA, Zhang Q, Madrid A, Chen L, Chopra P, et al. Genome-wide alterations in hippocampal 5-hydroxymethylcytosine links plasticity genes to acute stress. Neurobiol Dis. 2016;86:99–108. doi: 10.1016/j.nbd.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65(9):760–9. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31(9):464–8. doi: 10.1016/j.tins.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 47.McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342–8. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PLoS One. 2012;7(1):e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 50.Suderman M, McGowan PO, Sasaki A, Huang TC, Hallett MT, Meaney MJ, et al. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc Natl Acad Sci U S A. 2012;109(Suppl 2):17266–72. doi: 10.1073/pnas.1121260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alt SR, Turner JD, Klok MD, Meijer OC, Lakke EA, Derijk RH, et al. Differential expression of glucocorticoid receptor transcripts in major depressive disorder is not epigenetically programmed. Psychoneuroendocrinology. 2010;35(4):544–56. doi: 10.1016/j.psyneuen.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Elliott E, Ezra-Nevo G, Regev L, Neufeld-Cohen A, Chen A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat Neurosci. 2010;13(11):1351–3. doi: 10.1038/nn.2642. [DOI] [PubMed] [Google Scholar]
- 53.Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12(12):1559–66. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- 54.Vijayendran M, Beach SR, Plume JM, Brody GH, Philibert RA. Effects of genotype and child abuse on DNA methylation and gene expression at the serotonin transporter. Front Psychiatry. 2012;3:55. doi: 10.3389/fpsyt.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beach SR, Brody GH, Todorov AA, Gunter TD, Philibert RA. Methylation at SLC6A4 is linked to family history of child abuse: an examination of the Iowa Adoptee sample. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(2):710–3. doi: 10.1002/ajmg.b.31028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi SH, Yoo Y, Yu J, Rhee CS, Min YG, Koh YY. Bronchial hyperresponsiveness in young children with allergic rhinitis and its risk factors. Allergy. 2007;62(9):1051–6. doi: 10.1111/j.1398-9995.2007.01403.x. [DOI] [PubMed] [Google Scholar]
- 57.Blumenthal MN. The role of genetics in the development of asthma and atopy. Curr Opin Allergy Clin Immunol. 2005;5(2):141–5. doi: 10.1097/01.all.0000162306.12728.c2. [DOI] [PubMed] [Google Scholar]
- 58.Kim JS, Ouyang F, Pongracic JA, Fang Y, Wang B, Liu X, et al. Dissociation between the prevalence of atopy and allergic disease in rural China among children and adults. J Allergy Clin Immunol. 2008;122(5):929–35 e4. doi: 10.1016/j.jaci.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J, Zhang Y, Lin X, Yu R, Wang Y, Wang C, et al. Association between DNA hypomethylation at IL13 gene and allergic rhinitis in house dust mite-sensitized subjects. Clin Exp Allergy. 2015. doi:10.1111/cea.12647. [DOI] [PubMed]
- 60.Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol. 2010;126(3):453–65. doi: 10.1016/j.jaci.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reinius LE, Gref A, Saaf A, Acevedo N, Joerink M, Kupczyk M, et al. DNA methylation in the neuropeptide S receptor 1 (NPSR1) promoter in relation to asthma and environmental factors. PLoS One. 2013;8(1):e53877. doi: 10.1371/journal.pone.0053877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23(3):314–8. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 63.Birney E, Smith GD, Greally JM. Epigenome-wide association studies and the interpretation of disease -omics. PLoS Genet. 2016;12(6):e1006105. doi: 10.1371/journal.pgen.1006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hogg K, Price EM, Robinson WP. Improved reporting of DNA methylation data derived from studies of the human placenta. Epigenetics. 2014;9(3):333–7. doi: 10.4161/epi.27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10(10):949–55. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 66.Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013;23(9):1522–40. doi: 10.1101/gr.156539.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Labonte B, Azoulay N, Yerko V, Turecki G, Brunet A. Epigenetic modulation of glucocorticoid receptors in posttraumatic stress disorder. Transl Psychiatry. 2014;4:e368. doi: 10.1038/tp.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, et al. Differential glucocorticoid receptor exon 1(b), 1(c), and 1(h) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry. 2012;72(1):41–8. doi: 10.1016/j.biopsych.2012.01.034. [DOI] [PubMed] [Google Scholar]
- 69.McGowan PO, Suderman M, Sasaki A, Huang TC, Hallett M, Meaney MJ, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One. 2011;6(2):e14739. doi: 10.1371/journal.pone.0014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30(10):1431–9. doi: 10.1093/bioinformatics/btu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao-Lei L, Suwansirikul S, Jutavijittum P, Meriaux SB, Turner JD, Muller CP. Glucocorticoid receptor gene expression and promoter CpG modifications throughout the human brain. J Psychiatr Res. 2013;47(11):1597–607. doi: 10.1016/j.jpsychires.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 74.Liu L, van Groen T, Kadish I, Li Y, Wang D, James SR, et al. Insufficient DNA methylation affects healthy aging and promotes age-related health problems. Clin Epigenetics. 2011;2(2):349–60. doi: 10.1007/s13148-011-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuke C, Shimabukuro M, Petronis A, Sugimoto J, Oda T, Miura K, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet. 2004;68(Pt 3):196–204. doi: 10.1046/j.1529-8817.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 76.Shimabukuro M, Sasaki T, Imamura A, Tsujita T, Fuke C, Umekage T, et al. Global hypomethylation of peripheral leukocyte DNA in male patients with schizophrenia: a potential link between epigenetics and schizophrenia. J Psychiatr Res. 2007;41(12):1042–6. doi: 10.1016/j.jpsychires.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 77.Sandovici I, Kassovska-Bratinova S, Loredo-Osti JC, Leppert M, Suarez A, Stewart R, et al. Interindividual variability and parent of origin DNA methylation differences at specific human Alu elements. Hum Mol Genet. 2005;14(15):2135–43. doi: 10.1093/hmg/ddi218. [DOI] [PubMed] [Google Scholar]
- 78.Sarter B, Long TI, Tsong WH, Koh WP, Yu MC, Laird PW. Sex differential in methylation patterns of selected genes in Singapore Chinese. Hum Genet. 2005;117(4):402–3. doi: 10.1007/s00439-005-1317-9. [DOI] [PubMed] [Google Scholar]
- 79.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38(12):1378–85. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.El-Maarri O, Becker T, Junen J, Manzoor SS, Diaz-Lacava A, Schwaab R, et al. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet. 2007;122(5):505–14. doi: 10.1007/s00439-007-0430-3. [DOI] [PubMed] [Google Scholar]
- 81.Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299(24):2877–83. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bock C, Walter J, Paulsen M, Lengauer T. Inter-individual variation of DNA methylation and its implications for large-scale epigenome mapping. Nucleic Acids Res. 2008;36(10):e55. doi: 10.1093/nar/gkn122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7(8):e1002228. doi: 10.1371/journal.pgen.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heijmans BT, Kremer D, Tobi EW, Boomsma DI, Slagboom PE. Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Hum Mol Genet. 2007;16(5):547–54. doi: 10.1093/hmg/ddm010. [DOI] [PubMed] [Google Scholar]
- 85.Xie H, Wang M, de Andrade A, Bonaldo Mde F, Galat V, Arndt K, et al. Genome-wide quantitative assessment of variation in DNA methylation patterns. Nucleic Acids Res. 2011;39(10):4099–108. doi: 10.1093/nar/gkr017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15(2):R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Choufani S, Shapiro JS, Susiarjo M, Butcher DT, Grafodatskaya D, Lou Y, et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 2011;21(3):465–76. doi: 10.1101/gr.111922.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40(7):904–8. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 89.Schalkwyk LC, Meaburn EL, Smith R, Dempster EL, Jeffries AR, Davies MN, et al. Allelic skewing of DNA methylation is widespread across the genome. Am J Hum Genet. 2010;86(2):196–212. doi: 10.1016/j.ajhg.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang A, Huang Y, Li Z, Wan S, Mou L, Yin G, et al. Analysis of a four generation family reveals the widespread sequence-dependent maintenance of allelic DNA methylation in somatic and germ cells. Sci Rep. 2016;6:19260. doi: 10.1038/srep19260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12(8):529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chadwick LH, Sawa A, Yang IV, Baccarelli A, Breakefield XO, Deng H-W, et al. New insights and updated guidelines for epigenome-wide association studies. Neuroepigenetics. 2015;1:14–9. doi: 10.1016/j.nepig.2014.10.004. [DOI] [Google Scholar]
- 93.Tsai PC, Bell JT. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. International journal of epidemiology. 2015. doi:10.1093/ije/dyv041. [DOI] [PMC free article] [PubMed]
- 94.Choi JD, Lee JS. Interplay between epigenetics and genetics in cancer. Genomics Inform. 2013;11(4):164–73. doi: 10.5808/GI.2013.11.4.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang S. Genetic and non-genetic instability in tumor progression: link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013;32(3-4):423–48. doi: 10.1007/s10555-013-9435-7. [DOI] [PubMed] [Google Scholar]
- 96.Christensen BC, Houseman EA, Godleski JJ, Marsit CJ, Longacker JL, Roelofs CR, et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res. 2009;69(1):227–34. doi: 10.1158/0008-5472.CAN-08-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kohno H, Amatya VJ, Takeshima Y, Kushitani K, Hattori N, Kohno N, et al. Aberrant promoter methylation of WIF-1 and SFRP1, 2, 4 genes in mesothelioma. Oncol Rep. 2010;24(2):423–31. doi: 10.3892/or_00000875. [DOI] [PubMed] [Google Scholar]
- 98.Robles AI, Harris CC. Integration of multiple “OMIC” biomarkers: a precision medicine strategy for lung cancer. Lung Cancer. 2016. doi:10.1016/j.lungcan.2016.06.003. [DOI] [PMC free article] [PubMed]
- 99.Guintivano J, Arad M, Gould TD, Payne JL, Kaminsky ZA. Antenatal prediction of postpartum depression with blood DNA methylation biomarkers. Mol Psychiatry. 2014;19(5):560–7. doi: 10.1038/mp.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kimmel M, Kaminsky Z, Payne JL. Biomarker or pathophysiology? The role of DNA methylation in postpartum depression. Epigenomics. 2013;5(5):473–5. doi: 10.2217/epi.13.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li-Tempel T, Larra MF, Sandt E, Meriaux SB, Schote AB, Schachinger H, et al. The cardiovascular and hypothalamus-pituitary-adrenal axis response to stress is controlled by glucocorticoid receptor sequence variants and promoter methylation. Clin Epigenetics. 2016;8:12. doi: 10.1186/s13148-016-0180-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cao-Lei L, Leija SC, Kumsta R, Wust S, Meyer J, Turner JD, et al. Transcriptional control of the human glucocorticoid receptor: identification and analysis of alternative promoter regions. Hum Genet. 2011;129(5):533–43. doi: 10.1007/s00439-011-0949-1. [DOI] [PubMed] [Google Scholar]
- 103.Yehuda R, Flory JD, Bierer LM, Henn-Haase C, Lehrner A, Desarnaud F, et al. Lower methylation of glucocorticoid receptor gene promoter 1 F in peripheral blood of veterans with posttraumatic stress disorder. Biol Psychiatry. 2015;77(4):356–64. doi: 10.1016/j.biopsych.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 104.Breivik T, Gundersen Y, Murison R, Turner JD, Muller CP, Gjermo P, et al. Maternal deprivation of Lewis rat pups increases the severity of experimental periodontitis in adulthood. Open Dent J. 2015;9:65–78. doi: 10.2174/1874210601509010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27(4):361–8. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12(1):R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu Y, Li X, Aryee MJ, Ekstrom TJ, Padyukov L, Klareskog L, et al. GeMes, clusters of DNA methylation under genetic control, can inform genetic and epigenetic analysis of disease. Am J Hum Genet. 2014;94(4):485–95. doi: 10.1016/j.ajhg.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turner JD, Alt SR, Cao L, Vernocchi S, Trifonova S, Battello N, et al. Transcriptional control of the glucocorticoid receptor: CpG islands, epigenetics and more. Biochem Pharmacol. 2010;80(12):1860–8. doi: 10.1016/j.bcp.2010.06.037. [DOI] [PubMed] [Google Scholar]
- 109.Turner JD, Muller CP. Structure of the glucocorticoid receptor (NR3C1) gene 5′ untranslated region: identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol. 2005;35(2):283–92. doi: 10.1677/jme.1.01822. [DOI] [PubMed] [Google Scholar]
- 110.Turner JD, Schote AB, Macedo JA, Pelascini LP, Muller CP. Tissue specific glucocorticoid receptor expression, a role for alternative first exon usage? Biochem Pharmacol. 2006;72(11):1529–37. doi: 10.1016/j.bcp.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 111.Leenen FA, Vernocchi S, Hunewald OE, Schmitz S, Molitor AM, Muller CP, et al. Where does transcription start? 5′-RACE adapted to next-generation sequencing. Nucleic Acids Res. 2016;44(6):2628–45. doi: 10.1093/nar/gkv1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Turner JD, Pelascini LP, Macedo JA, Muller CP. Highly individual methylation patterns of alternative glucocorticoid receptor promoters suggest individualized epigenetic regulatory mechanisms. Nucleic Acids Res. 2008;36:7207–18. doi: 10.1093/nar/gkn897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Turner JD, Vernocchi S, Schmitz S, Muller CP. Role of the 5′-untranslated regions in post-transcriptional regulation of the human glucocorticoid receptor. Biochim Biophys Acta. 2014;1839(11):1051–61. doi: 10.1016/j.bbagrm.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 114.Vernocchi S, Battello N, Schmitz S, Revets D, Billing AM, Turner JD, et al. Membrane glucocorticoid receptor activation induces proteomic changes aligning with classical glucocorticoid effects. Mol Cell Proteomics. 2013;12(7):1764–79. doi: 10.1074/mcp.M112.022947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Elwenspoek MM, Hengesch X, Schachinger H, Muller CP, Turner JD. Increased methylation of the GR 1F promoter in post-institutionalized adults. Psychoneuroendocrinology. 2015;61:64. doi: 10.1016/j.psyneuen.2015.07.564. [DOI] [Google Scholar]
- 116.Buttgereit F, Scheffold A. Rapid glucocorticoid effects on immune cells. Steroids. 2002;67(6):529–34. doi: 10.1016/S0039-128X(01)00171-4. [DOI] [PubMed] [Google Scholar]
- 117.Moser D, Molitor A, Kumsta R, Tatschner T, Riederer P, Meyer J. The glucocorticoid receptor gene exon 1-F promoter is not methylated at the NGFI-A binding site in human hippocampus. World J Biol Psychiatry. 2007;8(4):262–8. doi: 10.1080/15622970701429862. [DOI] [PubMed] [Google Scholar]
- 118.Witzmann SR, Turner JD, Meriaux SB, Meijer OC, Muller CP. Epigenetic regulation of the glucocorticoid receptor promoter 1(7) in adult rats. Epigenetics. 2012;7(11):1290–301. doi: 10.4161/epi.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Turner JD, Schote AB, Keipes M, Muller CP. A new transcript splice variant of the human glucocorticoid receptor: identification and tissue distribution of hGRDelta313-338, an alternative exon 2 transactivation domain isoform. Ann N Y Acad Sci. 2007;1095:334–41. doi: 10.1196/annals.1397.037. [DOI] [PubMed] [Google Scholar]
- 120.Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69(21):3613–34. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424(6945):147–51. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 122.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38(6):626–35. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 123.Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, et al. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res. 2011;21(8):1260–72. doi: 10.1101/gr.120535.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bohrer AS, Yoshimoto N, Sekiguchi A, Rykulski N, Saito K, Takahashi H. Alternative translational initiation of ATP sulfurylase underlying dual localization of sulfate assimilation pathways in plastids and cytosol in Arabidopsis thaliana. Front Plant Sci. 2014;5:750. doi: 10.3389/fpls.2014.00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wamboldt Y, Mohammed S, Elowsky C, Wittgren C, de Paula WB, Mackenzie SA. Participation of leaky ribosome scanning in protein dual targeting by alternative translation initiation in higher plants. Plant Cell. 2009;21(1):157–67. doi: 10.1105/tpc.108.063644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tournillon AS, Lopez I, Malbert-Colas L, Naski N, Olivares-Illana V, Fahraeus R. The alternative translated MDMX(p60) isoform regulates MDM2 activity. Cell Cycle. 2015;14(3):449–58. doi: 10.4161/15384101.2014.977081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Merkin JJ, Chen P, Alexis MS, Hautaniemi SK, Burge CB. Origins and impacts of new mammalian exons. Cell Rep. 2015;10(12):1992–2005. doi: 10.1016/j.celrep.2015.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shabalina SA, Ogurtsov AY, Spiridonov NA, Koonin EV. Evolution at protein ends: major contribution of alternative transcription initiation and termination to the transcriptome and proteome diversity in mammals. Nucleic Acids Res. 2014;42(11):7132–44. doi: 10.1093/nar/gku342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shabalina SA, Spiridonov AN, Spiridonov NA, Koonin EV. Connections between alternative transcription and alternative splicing in mammals. Genome Biol Evol. 2010;2:791–9. doi: 10.1093/gbe/evq058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Irimia M, Rukov JL, Penny D, Roy SW. Functional and evolutionary analysis of alternatively spliced genes is consistent with an early eukaryotic origin of alternative splicing. BMC Evol Biol. 2007;7:188. doi: 10.1186/1471-2148-7-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matteau D, Rodrigue S. Precise identification of genome-wide transcription start sites in bacteria by 5′-rapid amplification of cDNA ends (5′-RACE) Methods Mol Biol. 2015;1334:143–59. doi: 10.1007/978-1-4939-2877-4_9. [DOI] [PubMed] [Google Scholar]
- 132.Ghesquiere B, Jonckheere V, Colaert N, Van Durme J, Timmerman E, Goethals M, et al. Redox proteomics of protein-bound methionine oxidation. Mol Cell Proteomics. 2011;10(5):M110 006866. doi: 10.1074/mcp.M110.006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–8. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wain HM, Bruford EA, Lovering RC, Lush MJ, Wright MW, Povey S. Guidelines for human gene nomenclature. Genomics. 2002;79(4):464–70. doi: 10.1006/geno.2002.6748. [DOI] [PubMed] [Google Scholar]
- 135.Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17(6):669–81. doi: 10.1101/gr.6339607. [DOI] [PubMed] [Google Scholar]
- 136.Laubichler MD, Stadler PF, Prohaska SJ, Nowick K. The relativity of biological function. Theory Biosci. 2015;134(3-4):143–7. doi: 10.1007/s12064-015-0215-5. [DOI] [PubMed] [Google Scholar]
- 137.Shafik A, Schumann U, Evers M, Sibbritt T, Preiss T. The emerging epitranscriptomics of long noncoding RNAs. Biochim Biophys Acta. 2016;1859(1):59–70. doi: 10.1016/j.bbagrm.2015.10.019. [DOI] [PubMed] [Google Scholar]
- 138.de Andres-Pablo A, Morillon A, Wery M. LncRNAs, lost in translation or licence to regulate? Curr Genet. 2016. doi:10.1007/s00294-016-0615-1. [DOI] [PubMed]
- 139.Berretta J, Morillon A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009;10(9):973–82. doi: 10.1038/embor.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Please refer to the individual publications cited in this review for the availability of materials and data within them.