

SUMMARY
Recurrent mutations in the splicing factor U2AF35 are found in several cancers and myelodysplastic syndrome (MDS). How oncogenic U2AF35 mutants promote transformation remains to be determined. Here we derive cell lines transformed by the oncogenic U2AF35(S34F) mutant, and identify aberrantly processed pre-mRNAs by deep sequencing. We find that in U2AF35(S34F)-transformed cells the autophagy-related factor 7 (Atg7) pre-mRNA is abnormally processed, which unexpectedly is not due to altered splicing but rather selection of a distal cleavage and polyadenylation (CP) site. This longer Atg7 mRNA is translated inefficiently, leading to decreased ATG7 levels and an autophagy defect that predisposes cells to secondary mutations, resulting in transformation. MDS and acute myeloid leukemia patient samples harboring U2AF35(S34F) have a similar increased use of the ATG7 distal CP site, and previous studies have shown that mice with hematopoietic cells lacking Atg7 develop an MDS-like syndrome. Collectively, our results reveal a basis for U2AF35(S34F) oncogenic activity.
eTOC Blurb
Mutants of the splicing factor U2AF35 are oncogenic for unknown reasons. Park et al. show that the U2AF35(S34F) mutant unexpectedly promotes usage of a distal poly(A) site in Atg7 mRNA, generating a longer, inefficiently translated transcript. Decreased ATG7 impairs autophagy, predisposing cells to secondary mutations and transformation.
INTRODUCTION
One of the most unexpected findings that emerged from the sequencing of cancer genomes is the discovery of recurrent somatic mutations in general pre-mRNA splicing factors in a variety of hematological and solid malignancies (Harbour et al., 2013; Imielinski et al., 2012; Mansouri et al., 2013; Network, 2012; Network, 2013; Yoshida et al., 2011). For example, recurrent mutations in the splicing factor U2AF35 (also called U2AF1) have been found in several hematopoietic malignancies, lung cancer, and myelodysplastic syndrome (MDS) (Imielinski et al., 2012; Network, 2013; Visconte et al., 2012; Yoshida et al., 2011). U2AF35 is a component of the essential pre-mRNA splicing factor U2AF, a heterodimer composed of a large (65 kDa; U2AF65, also called U2AF2) and a small (35 kDa) subunit (Zamore and Green, 1989). U2AF plays a critical role in 3’ splice site selection and functions by promoting the first step in spliceosome assembly. In addition to its role in splicing, U2AF has also been shown to regulate mRNA 3’ end formation through interactions with components of the cleavage and polyadenylation machinery (de Vries et al., 2000; Millevoi et al., 2006; Vagner et al., 2000), which catalyzes endonucleotyic cleavage of the nascent RNA and synthesis of a poly(A) tail.
The most common U2AF35 mutations that have been found in cancers are at the highly conserved serine at amino acid position 34 (S34F/Y) (Yoshida et al., 2011). In most cases, the mutation is present in only one of the two U2AF35 alleles, and thus both wild-type and mutant U2AF35 are expressed (Yoshida et al., 2011). The specific basis by which oncogenic U2AF35 mutants promote transformation has been controversial. One study found that overexpression of U2AF35(S34F) led to loss of splicing, resulting in intron retention (Yoshida et al., 2011). Another study reported that ectopic expression of U2AF(S34F) resulted in increased exon exclusion and increased use of cryptic splice sites (Graubert et al., 2012). More recently, studies analyzing acute myeloid leukemia (AML) transcriptomes reported exon inclusion in samples harboring U2AF35 mutations (Brooks et al., 2014; Prasad et al., 1999). Oncogenic U2AF35 mutations have been proposed to cause both gain of function (Graubert et al., 2012) and loss of function (Makishima et al., 2012; Yoshida et al., 2011). Most importantly, in none of these previous studies has it been shown that an alternatively spliced mRNA was functionally linked to the transformed phenotype.
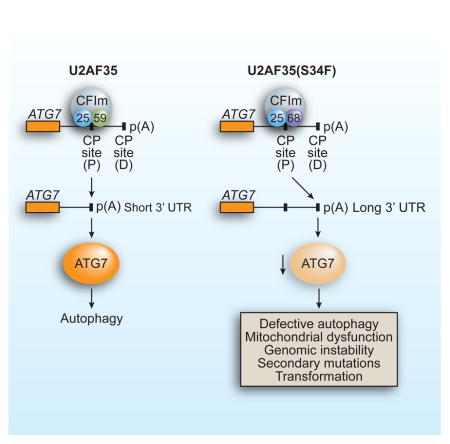
To understand how U2AF35 mutants promote transformation, here we derive cell lines that are transformed by the oncogenic splicing mutant U2AF35(S34F). The derivation of U2AF35(S34F)-transformed cell lines enabled us to perform functional experiments to determine whether altered RNA processing events are responsible for transformation. Unexpectedly, we find that in addition to aberrant splicing, a frequently altered RNA processing event in U2AF35(S34F)-transformed cells is a change in mRNA 3’ end formation, resulting from increased use of a distal cleavage and polyadenylation (CP) site. We go on to show that increased distal CP site use of a specific pre-mRNA, autophagy-related factor 7 (Atg7), which encodes an essential autophagy factor, results in an autophagy defect that leads to transformation.
RESULTS
Derivation of Cell Lines Transformed by Oncogenic U2AF35(S34F)
To derive cell lines that are transformed by the oncogenic U2AF35(S34F) mutant, we used as a model system Ba/F3 cells, an interleukin-3 (IL-3)-dependent immortalized murine bone marrow-derived pro-B cell line that can be transformed by a number of oncogenes, such as BCR-ABL (Daley and Baltimore, 1988), enabling IL-3-independent growth. We stably expressed in Ba/F3 cells either wild-type U2AF35 (Ba/F3-U2AF35 cells), U2AF35(S34F) (Ba/F3-U2AF35(S34F) cells) (Figure S1A) or, as controls, BCR-ABL (Ba/F3-BCR-ABL cells) or empty vector (Ba/F3-V cells) followed by selection in medium lacking IL-3. As expected, Ba/F3-BCR-ABL cells that were able to proliferate in the absence IL-3 emerged rapidly (Figures 1A and S1B). IL-3-independent proliferating Ba/F3-U2AF35(S34F) cells could be detected after 8 days, whereas IL-3-independent Ba/F3-U2AF35 cells did not arise. Notably, the established Ba/F3-U2AF35(S34F) and Ba/F3-BCR-ABL cell lines had comparable growth rates (Figure 1B). Ba/F3-U2AF35(S34F) cells formed tumors in immunocompromised mice, confirming their transformed phenotype (Figure 1C). Tumors could also be formed using Ba/F3-U2AF35(S34F) cells that had not been pre-selected for IL-3-dependent proliferation in culture (Figure S1C).
Figure 1. Derivation of U2AF35(S34F)-transformed Cell Lines.

(A) Proliferation of parental Ba/F3 cells cultured in the presence (+) or absence (−) of IL-3, and Ba/F3-V, Ba/F3-U2AF35, Ba/F3-U2AF35(S34F) or Ba/F3-BCR-ABL cells cultured in the absence of IL-3. A representative graph from n=9 biological replicates is shown. Two other replicates are shown in Figure S1B.
(B) Proliferation of parental Ba/F3 cells, and established Ba/F3-V, Ba/F3-U2AF35, Ba/F3-U2AF35(S34F) or Ba/F3-BCR-ABL cells.
(C) Tumor formation in mice (n=4 mice per group) injected with Ba/F3-V or Ba/F3-U2AF35(S34F) cells. Error bars indicate SD. *P<0.05; **P<0.01. See also Figure S1.
Atg7 Pre-mRNA Undergoes Aberrant CP Site Selection in U2AF35(S34F)-transformed Ba/F3 Cells
To identify pre-mRNAs that were processed abnormally in Ba/F3-U2AF35(S34F) cells, we performed transcriptome profiling (RNA-Seq) experiments. Because U2AF has been shown to affect both pre-mRNA splicing and mRNA 3' end formation (de Vries et al., 2000; Millevoi et al., 2006; Vagner et al., 2000), we analyzed the RNA-Seq data using both Cufflinks, which tests for alternative use of splice sites and untranslated regions (UTRs) (Trapnell et al., 2010), and a modified DaPars algorithm, which specifically tests for alternative use of CP sites (Masamha et al., 2014). Collectively, we identified 184 processing events, corresponding to 127 pre-mRNAs, that were significantly altered in Ba/F3-U2AF35(S34F) cells compared to parental Ba/F3 cells (Tables S1 and S2). The RNA-Seq results are summarized in Figure 2A and reveal, unexpectedly, that the most frequently altered RNA processing event in Ba/F3-U2AF35(S34F) cells was increased use of a distal CP site, which comprised 40.8% of total altered RNA processing events.
Figure 2. Atg7 Pre-mRNA Undergoes Aberrant CP Site Selection in U2AF35(S34F)-transformed Ba/F3 Cells.

(A) Summary of RNA-Seq analysis.
(B) Schematic of the Atg7 pre-mRNA 3’ UTR showing the positions of the proximal (P) and distal (D) CP sites.
(C) qRT-PCR monitoring the relative use of the Atg7 distal versus proximal CP site in Ba/F3-V, Ba/F3-U2AF35(S34F) or Ba/F3-BCR-ABL cells.
(D) Top, northern blot analysis of Atg7 pre-mRNA in Ba/F3-V or Ba/F3-U2AF35(S34F) cells. Gapdh was monitored as a loading control. Bottom, quantification.
(E) qRT-PCR monitoring the relative Atg7 CP site use in Ba/F3-V or Ba/F3-U2AF35(S34F)I cells following doxycycline withdrawal. Error bars indicate SD. *P<0.05; **P<0.01. See also Figure S2.
Of the various pre-mRNAs whose processing was altered, we thought the most potentially relevant to U2AF35(S34F) oncogenic activity was Atg7, which encodes an essential autophagy factor (Komatsu et al., 2005). A variety of studies have shown that autophagy is a context-dependent cancer prevention mechanism (Mathew and White, 2007), and loss of essential autophagy factors have been shown to contribute to tumorigenesis (Liu et al., 2013; Mathew et al., 2007). Most importantly, mice in which Atg7 has been conditionally depleted in hematopoietic cells have an autophagy defect and develop an MDS-like syndrome (Komatsu et al., 2005; Mortensen et al., 2011). The RNA-Seq results indicated that the change in Atg7 pre-mRNA processing was not the result of altered splicing but rather selection of an alternative distal CP site (Figure 2B), which was confirmed by quantitative real-time RT-PCR (qRT-PCR) (Figure 2C) and northern blotting (Figure 2D).
The modified DaPars algorithm also identified an additional 74 pre-mRNAs which, like Atg7, had increased distal CP site use in Ba/F3-U2AF35(S34F) cells compared to parental Ba/F3 cells (Figure S2A and Table S2). The elevated use of a distal CP site in a representative subset of these pre-mRNAs was confirmed by qRT-PCR (Figure S2B).
To determine whether continuous expression of U2AF35(S34F) was required for maintenance of altered Atg7 pre-mRNA processing, we derived a transformed Ba/F3 cell line in which U2AF35(S34F) expression was doxycycline inducible (Ba/F3-U2AF35(S34F)I cells). Following doxycycline withdrawal, loss of U2AF35(S34F) (Figure S2C) resulted in decreased use of the Atg7 distal CP site (Figure 2E).
The U2AF35(S34F) Mutant Results in Decreased Recruitment of CFIm59 to the Atg7 Proximal CP Site, Leading to Increased Use of the Distal CP Site
We next performed experiments to determine the basis by which U2AF35(S34F) increased use of the Atg7 distal CP site. Alternative CP site selection is regulated by the cleavage factor Im (CFIm) complex, which functions by binding to the proximal CP site (Gruber et al., 2012). CFIm is a heterotetramer composed of two CFIm25 (also known as CPSF5 or NUDT21) subunits and either CFIm59 or CFIm68 (also known as CPSF7 or CPSF6, respectively). CFIm59 increases use of proximal CP sites, whereas CFIm68 represses use of proximal CP sites. Figure 3A shows, as expected, that RNAi-mediated knockdown of CFIm59 in Ba/F3 cells (Figure S3A) resulted in decreased use of the Atg7 proximal CP site and, conversely, knockdown of CFIm68 (Figure S3B) resulted in increased use of the Atg7 proximal CP site.
Figure 3. The U2AF35(S34F) Mutant Results in Decreased Recruitment of CFIm59 to the Atg7 Proximal CP Site, Leading to Increased use of the Distal CP Site.

(A) qRT-PCR monitoring relative Atg7 CP site use in Ba/F3 cells expressing a control, CFIm59 or CFIm68 siRNA.
(B) Co-immunoprecipitation analysis. FLAG-tagged CFIm59-bound beads were added to extracts from Ba/F3 cells expressing MYC-U2AF35 or MYC-U2AF35(S34F) in the presence of RNase. The beads were collected and washed, and the bound proteins were analyzed by immunoblotting for U2AF65, MYC-U2AF35 or FLAG-CFIm59. β-actin (ACTB) was monitored as a control. (C-E) RIP assay monitoring binding of CFIm59, CFIm68 or CFIm25 to the Atg7 proximal CP site in Ba/F3-V or Ba/F3-U2AF35(S34F) cells
(C), in Ba/F3 cells expressing a NS or CFIm59 shRNA
(D), or in Ba/F3 cells expressing a NS or CFIm68 shRNA
(E). Binding was normalized to IgG, which was set to 1. *P<0.05; **P<0.01. See also Figure S3.
It has been previously reported that U2AF can interact with the CFIm complex (de Vries et al., 2000; Millevoi et al., 2006). Co-immunoprecipitation experiments showed, as expected, that the U2AF65/U2AF35 heterodimer (U2AF) interacted with CFIm59 (Figure 3B) and CFIm68 (Figure S3C). The co-immunoprecipitations were performed in the presence of RNase, indicating the U2AF–CFIm interaction was not RNA dependent. Notably, the U2AF35(S34F) mutant decreased the U2AF–CFIm59 interaction (Figures 3B and S3D) but did not affect the U2AF–CFIm68 interaction (Figure S3C).
Next, we performed RNA immunoprecipitation (RIP) experiments to measure binding of CFIm subunits to the Atg7 proximal CP site. Figure 3C shows that the level of Atg7 pre-mRNA-bound CFIm59 was substantially lower in Ba/F3-U2AF35(S34F) cells than in Ba/F3-V cells. Conversely, the level of Atg7 pre-mRNA-bound CFIm68 was substantially higher in Ba/F3-U2AF35(S34F) cells than in Ba/F3-V cells. The level of Atg7 pre-mRNA-bound CFIm25 was comparable in the two cell lines. Knockdown of CFIm59 in Ba/F3 cells led to increased levels of Atg7 pre-mRNA-bound CFIm68 (Figures 3D and S3E) and, conversely, knockdown of CFIm68 led to increased levels of Atg7 pre-mRNA-bound CFIm59 (Figures 3E and S3F). Thus, consistent with previous studies (de Vries et al., 2000), binding of CFIm59 or CFIm68 to Atg7 pre-mRNA was mutually exclusive and competitive. Collectively, these results show that the U2AF35(S34F) mutant leads to decreased recruitment of CFIm59 and enhanced association of CFIm68 to the Atg7 proximal CP site, resulting in increased use of the distal CP site. Similar results were obtained with Sfxn3 and Smpd3, two other pre-mRNAs confirmed to have increased use of the distal CP site in Ba/F3-U2AF35(S34F) cells (see Figures S2B and S3G-S3J).
Consistent with the idea that U2AF35 contributes to recruitment of CFIm59 to the proximal site, knockdown of U2AF35 in Ba/F3 cells (Figure S3K) led to increased use of the Atg7 distal CP site (Figure S3L). Notably, a previous study reported increased use of the distal CP site of some pre-mRNAs following U2AF35 knockdown (Kralovicova et al., 2015). By contrast to what we found in parental Ba/F3 cells (see Figure 3A), knockdown of CFIm68 in Ba/F3-U2AF35(S34F) cells (Figure S3M) did not result in decreased use of the Atg7 distal CP site (Figure S3N).
Use of the Atg7 Distal CP Site Results in Decreased ATG7 Proteins Levels
Increased use of the Atg7 distal CP site in Ba/F3-U2AF35(S34F) cells results in a longer 3’ UTR, which is often subject to translational repression (Barrett et al., 2012). Indeed, ATG7 protein levels were markedly reduced in Ba/F3-U2AF35(S34F) cells compared to control Ba/F3-V and Ba/F3-BCR-ABL cells (Figures 4A and S4A). Moreover, depletion of U2AF35(S34F) from Ba/F3-U2AF35(S34F)I cells resulted in increased levels of ATG7 protein (Figures 4B and S4B). Consistent with the results of Figure 3A, knockdown of CFIm59 led to reduced levels of ATG7 protein and, conversely, knockdown of CFIm68 led to increased levels of ATG7 protein (Figure 4C).
Figure 4. Use of the Atg7 Distal CP Site Results in Decreased ATG7 Protein Levels, which can Transform Ba/F3 Cells.

(A) Immunoblot monitoring ATG7 in Ba/F3-V, Ba/F3-U2AF35(S34F) or Ba/F3-BCR-ABL cells. α-tubulin (TUBA) was monitored as a loading control.
(B) Immunoblot monitoring ATG7 in Ba/F3-U2AF35(S34F)I cells following doxycycline (dox) withdrawal.
(C) (Left) Immunoblot monitoring ATG7 and CFIm59 in Ba/F3 cells expressing a control or CFIm59 siRNA. (Right) Immunoblot monitoring ATG7 and CFIm68 in Ba/F3 cells expressing a control or CFIm68 siRNA.
(D) Luciferase reporter assay in HEK293T cells expressing empty vector or a region corresponding to the short or long 3’ UTR of Atg7.
(E) Proliferation of parental Ba/F3, Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells. A representative graph from n=5 biological replicates is shown. Two other replicates are shown in Figure S1B.
(F) Proliferation of parental Ba/F3 cells, and established Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells.
(G) Tumor formation in mice (n=3 mice per group) injected with Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells. Error bars indicate SD. *P<0.05; **P<0.01. See also Figure S4.
To confirm that the Atg7 long 3’ UTR repressed translational activity, we inserted it downstream of a luciferase reporter gene and found that luciferase activity was substantially decreased (Figure 4D). Deletion analysis revealed that the translational repressive element was located between nucleotides 1–350 within the Atg7 long 3’ UTR (Figure S4C), a region that is highly conserved between human and mouse (Figure S4D). Whether this region of the Atg7 long 3’ UTR represses translation through interaction with a miRNA or protein, or by some other mechanism, remains to be determined.
We performed several experiments whose results demonstrated that the decreased level of ATG7 protein was the basis of U2AF35(S34F) oncogenic activity. First, constitutive expression of an Atg7 derivative lacking the 3’ UTR, and thus not subject to translational repression, prevented transformation by U2AF35(S34F) (Figure S4E). Second, knockdown of Atg7 (Figure S4F) could transform Ba/F3 cells, as evidenced by IL-3-independent proliferation (Figures 4E, 4F, and S4G) and tumor formation in immunocompromised mice (Figure 4G). Similar to the results with ATG7, knockdown of two other essential autophagy factors, ATG5 and BECN1 (Watson et al., 2011), also transformed Ba/F3 cells (Figure S4H).
U2AF35(S34F)-transformed Ba/F3 Cells have an Autophagy Defect, Mitochondrial Dysfunction and an Increased Spontaneous Mutation Frequency
We performed several experiments to investigate the basis of U2AF35(S34F)-mediated transformation. First, we sought to confirm that U2AF35(S34F)-transformed cells had an autophagy defect. To measure autophagy, we monitored the levels of two well-characterized markers of autophagic flux: LC3B, whose cytosolic form (LC3B-I) is conjugated to phosphatidylethanolamine to form LC3B-II during autophagy, and p62 (also called STQM1), a protein that is degraded by autophagy (Klionsky et al., 2008; Mizushima et al., 2010). Inhibition of autophagic flux blocks conversion of LC3B-I to LC3B-II (resulting in a decreased LC3B-II/LC3B-I ratio) and degradation of p62 (resulting in increased p62) (Bjorkoy et al., 2009; Mizushima and Yoshimori, 2007). Ba/F3-U2AF35(S34F) cells and, as expected, Ba/F3-Atg7shRNA cells contained a lower LC3B-II/LC3B-I ratio and higher levels of p62 than control Ba/F3-V cells or Ba/F3 cells expressing a non-silencing (NS) shRNA (Ba/F3-NSshRNA cells), indicative of reduced autophagic flux (Figure 5A and Figure S5A). Moreover, PP242, an mTOR inhibitor (Feldman et al., 2009) that activates formation of autophagosome vesicles (Fleming et al., 2011), induced autophagy in control Ba/F3-V and Ba/F3-NSshRNA cells, as expected, but not in Ba/F3-U2AF35(S34F) or Ba/F3-Atg7shRNA cells (Figure 5B).
Figure 5. Clinical Relevance and Demonstration that U2AF35(S34F)-transformed Ba/F3 Cells have an Autophagy Defect, Mitochondrial Dysfunction and an Increased Spontaneous Mutation Frequency.

(A) Immunoblot monitoring LC3B-I, LC3B-II and p62 in Ba/F3-V, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells.
(B) Fluorescent Cyto-ID assay. Ba/F3-V, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells were treated with or without PP242 and stained with Cyto-ID, a fluorescent dye that selectively labels autophagic vacuoles.
(C) Proliferation of Ba/F3-V cells (cultured in the presence of IL-3) or Ba/F3-U2AF35(S34F) cells (cultured in the absence of IL-3) cultured in the presence or absence of doxycycline. (D and E) FACS analysis of Ba/F3-V, Ba/F3-U23AF35, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells cultured in the presence of IL-3 and double-stained with MitoTracker Green and MitoTracker Red
(D) or stained with MitoSOX
(E). The MitoTracker Green-positive, MitoTracker Red-negative population is encircled; quantification of this population is provided in the lower left-hand corner of the plot.
(F) Spontaneous mutation frequency in Ba/F3-V, Ba/F3-U2AF35, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA or Ba/F3-Atg7shRNA control (water) or ascorbate-treated cells, as measured by resistance to 6-thioguanine (6-TG; left) or ouabain (right). The results were normalized to that obtained in control Ba/F3-V or Ba/F3-NSshRNA cells, which was set to 1.
(G) Proliferation of Ba/F3-V, Ba/F3-U2AF35, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells treated with etoposide or, as a positive control, the autophagy inhibitor 3-methyladenine (3-MA), or both.
(H) Proliferation of Ba/F3-V, Ba/F3-U2AF35, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA or Ba/F3-Atg7shRNA cells treated with x-ray irradiation, or x-ray irradiation and 3-MA. Error bars indicate SD. *P<0.05; **P<0.01. See also Figure S5.
We next tested whether U2AF35(S34F) was required for maintenance of the transformed phenotype. We found that following depletion of U2AF35(S34F), Ba/F3-U2AF35(S34F)I cells continued to proliferate in the absence of IL-3 (Figure 5C) despite restoration of normal ATG7 levels (see Figure 4B). Likewise, transient depletion of Atg7 promoted transformation of Ba/F3 cells (Figure S5B). Thus, U2AF35(S34F) is required for the establishment but not maintenance of transformation.
The observations that U2AF35(S34F)-transformed cells emerged relatively slowly (Figure 1A), and that transformation was maintained following depletion of U2AF35(S34F) (Figure 5C), suggested that transformation was the result of a secondary event, such as a mutation. Indeed, previous studies have shown that defective autophagy leads to genomic instability, which is due at least in part to mitochondrial dysfunction resulting in increased levels of reactive oxygen species (ROS) (Mathew et al., 2007; Mathew and White, 2007). Moreover, loss of ATG7 has been found to cause mitochondrial dysfunction, which likely results from impaired clearance of damaged mitochondria (Zhang et al., 2009).
To test Ba/F3-U2AF35(S34F) cells for mitochondrial dysfunction, Ba/F3-V, Ba/F3-U2AF35, Ba/F3-U2AF35(S34F), Ba/F3-NSshRNA, and Ba/F3-Atg7shRNA cells were double-stained with MitoTracker Green (an indicator of mitochondrial mass, or total mitochondria) and MitoTracker Red (an indicator of mitochondrial membrane potential, or respiring mitochondria), or single-stained with MitoSOX (an indicator of mitochondrial superoxide levels, or ROS-generating mitochondria) followed by FACS analysis. Ba/F3-U2AF35(S34F) cells and Ba/F3-Atg7shRNA cells accumulated dysfunctional, non-respiring mitochondria (indicated by MitoTracker Green-positive, Mitotracker Red-negative staining; Zhou et al., 2011) (Figure 5D) and increased mitochondrial ROS production (Figure 5E) compared to control Ba/F3-V and Ba/F3-NSshRNA cells, respectively.
Because ROS is mutagenic (Klaunig et al., 2010), we measured spontaneous mutation frequency using a well established assay in which cells harboring mutations in an essential gene encoding hypoxanthine-guanine phosphoribosyltransferase (HPRT) or Na,K-ATPase are detected by positive selection using 6-thioguanine (6-TG) or ouabain, respectively. Figure 5F shows, consistent with the increased ROS, that Ba/F3-U2AF35(S34F) and Ba/F3-Atg7shRNA cells also had a higher spontaneous mutation frequency, as evidenced by increased frequency of 6-TG- or ouabain-resistant mutants, than Ba/F3-V, Ba/F3-U2AF35 and Ba/F3-NSshRNA cells. Notably, the increased spontaneous mutation frequency was suppressed by treatment of cells with the anti-oxidant ascorbate (Figure 5F), indicative of a causal role for elevated ROS. Furthermore, ascorbate suppressed the ability of U2AF35(S34F) to promote transformation (Figure S5C).
Impaired autophagy can render cells highly sensitive to cytotoxic agents (Gewirtz, 2014). We therefore examined whether the proliferation of Ba/F3-U2AF35(S34F) and Ba/F3-Atg7shRNA cells was preferentially impaired by treatment with cytotoxic agents compared to parental Ba/F3 cells. Figure 5G shows, as expected, that treatment of control Ba/F3-V cells or Ba/F3-NSshRNA cells with the DNA damaging agent etoposide in combination with the autophagy inhibitor 3-methyladenine resulted in reduced proliferation compared to either treatment alone. Notably, proliferation of Ba/F3-U2AF35(S34F) and Ba/F3-Atg7shRNA cells was greatly reduced upon etoposide treatment compared to etoposide-treated Ba/F3-V, Ba/F3-U2AF35 or Ba/F3-NSshRNA cells. Similar results were obtained when cells were treated with two other chemotherapeutic agents, 5-azacytidine or topotecan (Figures S5D and S5E), and when x-ray irradiation was used rather than a drug (Figure 5H).
Aberrant ATG7 CP Site Selection and Decreased ATG7 Protein Levels in U2AF(S34F)-transformed Human Bronchial Epithelial Cells
Previous studies have shown that U2AF35 is also mutated in lung cancer (Imielinski et al., 2012). Therefore, to investigate the generality of our results we analyzed the effect of U2AF35(S34F) expression in small airway (SA) cells, an immortalized but non-transformed human bronchial epithelial cell line (Lundberg et al., 2002). We found that stable expression of U2AF35(S34F) in SA cells (SA-U2AF35(S34F) cells; Figure 6A) resulted in transformation as evidenced by growth in soft agar (Figure 6B) and tumor formation in immunocompromised mice (Figure 6C). Moreover, in SA-U2AF35(S34F) cells there was increased use of the ATG7 distal CP site (Figure 6D), and ATG7 protein levels were reduced relative to those in SA cells expressing empty vector (SA-V cells; Figures 6E, S6A and S6B). As expected, SA-U2AF35(S34F) cells and SA cells expressing an ATG7 shRNA (SA-ATG7shRNA cells) contained a lower LC3B-II/LC3B-I ratio and higher levels of p62 than control SA-V cells or SA-NS shRNA cells, indicative of an autophagy defect (Figures 6F and S6C). Finally, proliferation of SA-U2AF35(S34F) and SA-ATG7shRNA cells was greatly reduced upon etoposide treatment compared to etoposide-treated control SA-V cells or SA-NSshRNA cells (Figure 6G).
Figure 6. Aberrant ATG7 CP Site Selection and Decreased ATG7 Protein Levels in U2AF(S34F)-transformed Human Bronchial Epithelial Cells.

(A) Immunoblot analysis monitoring MYC-U2AF35 and U2AF35 levels in SA cells expressing vector, U2AF35 or U2AF35(S34F). α-tubulin (TUBA) was monitored as a loading control.
(B) Soft agar assay with SA cells expressing vector, U2AF35 or U2AF35(S34F).
(C) Tumor formation in mice (n=3 mice per group) injected with SA cells expressing vector or U2AF35 (S34F).
(D) qRT-PCR analysis monitoring the relative CP site use in the ATG7 pre-mRNA in SA cells expressing vector or U2AF35(S34F).
(E) Immunoblot analysis monitoring ATG7 levels in SA cells expressing vector or U2AF35(S34F).
(F) Immunoblot monitoring LC3B-I, LC3B-II and p62 in SA-V, SA-U2AF35(S34F), SA-NSshRNA or SA-ATG7shRNA cells.
(G) Proliferation of SA-V, SA-U2AF35(S34F), SA-NSshRNA or SA-Atg7shRNA cells treated with etoposide, 3-MA, or both. Error bars indicate SD. *P<0.05; **P<0.01. See also Figure S6.
Increased Selection of the ATG7 Distal CP site in MDS and Acute Myeloid Leukemia Patient Samples
Finally, to investigate the clinical relevance of our results, we analyzed ATG7 mRNA levels in MDS patient bone marrow samples that contained either wild-type U2AF35 or U2AF35(S34F) (Table S3). Similar to our tissue culture results, we found that in MDS patient samples harboring U2AF35(S34F), there was increased use of the ATG7 distal CP site compared to that found in MDS patient samples with wild-type U2AF35 (Figure 7A).
Figure 7. Increased Selection of the ATG7 Distal CP site in MDS and Acute Myeloid Leukemia Patient Samples.

(A) (Left) qRT-PCR monitoring ATG7 pre-mRNA CP site use in MDS patient samples containing wild-type U2AF35 or U2AF35(S34F). The first wild-type U2AF35 sample was set to 1. (Right) Average values for patient samples containing wild-type U2AF35 or U2AF35(S34F). Error bars indicate SD.
(B) Bioinformatic analysis of ATG7 pre-mRNA CP site use in AML samples containing wild-type U2AF35 or U2AF35(S34F). PDUI, percentage distal polyadenylation site usage index. Mean PDUI values are shown; error bars indicate SEM. P<0.05; **P<0.01.
(C) Schematic model. See also Figure S7.
Approximately 30% of patients diagnosed with MDS will ultimately develop acute myeloid leukemia (AML). We were therefore interested in determining whether there might be similar changes in ATG7 CP site selection in AML. Notably, bioinformatic analysis of The Cancer Genome Atlas acute myeloid leukemia (TCGA AML) dataset (Network, 2013) revealed a statistically significant increased use of the ATG7 distal CP site in AML samples containing U2AF35(S34F) (Figure 7B).
DISCUSSION
Based upon our collective results, we propose a model by which the oncogenic U2AF35(S34F) mutant promotes transformation (Figure 7C). The U2AF35(S34F) mutation alters interaction with CFIm59, leading to increased use of a distal CP site in the ATG7 pre-mRNA, decreased levels of ATG7 protein and defective autophagy. The autophagy defect leads to mitochondrial dysfunction, resulting in genomic instability that predisposes cells to secondary oncogenic mutations, ultimately leading to transformation. Consistent with this conclusion, we show that U2AF35(S34F) is required for establishment but not maintenance of transformation. Our results fit well with previous studies showing that loss of essential autophagy factors such as ATG5 and BECN1 contribute to tumorigenesis (Liu et al., 2013; Mathew et al., 2007).
We demonstrate the generality of our results by showing that U2AF35(S34F) can transform both immortalized hematopoietic cells and immortalized epithelial cells with similar effects on both ATG7 pre-mRNA CP site selection and ATG7 protein levels. In addition, several observations indicate that our findings are directly relevant to MDS. First, as described above, mice in which Atg7 has been conditionally depleted in hematopoietic cells develop an MDS-like syndrome (Komatsu et al., 2005; Mortensen et al., 2011). Second, we confirm the clinical relevance of our results by showing that MDS as well as AML patient samples bearing the U2AF35(S34F) mutant have a similar increased use of the ATG7 distal CP site.
Although the phenomenon of alternative CP site use has been known for decades, only recently has the extent of this alternative RNA processing event been fully recognized. The majority of human genes contain at least two CP sites, and widespread alternative CP site use has been found in multiple organisms and systems (Shi, 2012). In general, proliferative cells, such as induced pluripotent stem cells and cancer cells, have a global shortening of 3’ UTRs compared to their less proliferative counterparts (Fu et al., 2011; Ji et al., 2009; Mayr and Bartel, 2009). Consistent with this idea, in cancer cells, which are characterized by uncontrolled proliferation, there is widespread increase in the use of proximal CP sites (Elkon et al., 2013). Here we show how another type of aberrant CP site selection, a switch to a distal CP site, can also lead to transformation.
Although we have shown that decreased ATG7 protein levels are sufficient to transform Ba/F3 cells, the U2AF35(S34F) mutant affects CP site selection in other pre-mRNAs (Figure S2A), some of which may also contribute to transformation. Similar to our results, previous studies have identified mRNAs with altered splicing patterns in cells (Przychodzen et al., 2013; Shao et al., 2014; Yoshida et al., 2011) or tumors (Brooks et al., 2014) containing U2AF35(S34F). Although in no case has an alternatively spliced mRNA been functionally linked to the transformed phenotype, we do not discount the possibility that U2AF35(S34F)-mediated alterations in splicing may also contribute to transformation.
We focused on the U2AF35(S34F) mutant because it is by far the most frequent U2AF35 mutant in MDS and human cancers (Yoshida et al., 2011). However, we also found that MDS patient samples containing U2AF35(S34Y) or U2AF35(Q157P) have a similar increased use of the ATG7 distal CP site (Figure S7). Whether oncogenic U2AF35 mutations in patient samples from other cancer types, such as lung cancer, have a similar effect on ATG7 CP site selection remains to be determined.
In addition to U2AF35, recurrent mutations in other splicing factors such as SF3B1, SRSF2, and ZRSR2 have been identified in various cancer types (Cazzola et al., 2013; Ellis et al., 2012; Furney et al., 2013; Harbour et al., 2013; Papaemmanuil et al., 2011; Quesada et al., 2012; Thol et al., 2012; Wang et al., 2011; Yoshida et al., 2011). Similar to studies with U2AF35(S34F), these other oncogenic splicing factor mutants have been reported to promote aberrant splicing of some pre-mRNAs (Furney et al., 2013; Kim et al., 2015). Whether these other oncogenic splicing factors mutants also directly or indirectly promote alternative CP site selection that is relevant to the transformed phenotype remains to be determined.
As mentioned above, impaired autophagy can render cells highly sensitive to cytotoxic agents (Gewirtz, 2014). Consistent with this general concept, we found that following treatment with DNA damaging agents, the proliferation of Ba/F3 or SA cells expressing U2AF35(S34F) or an Atg7 shRNA was greatly reduced compared to their non-transformed counterparts (see Figures 5G, 5H, 6G, S5D and S5E). Thus, the autophagy defect of U2AF35(S34F)-containing cells may be a vulnerability that can be therapeutically exploited to treat patients with MDS and other malignancies that contain U2AF35(S34F).
EXPERIMENTAL PROCEDURES
Cell Proliferation Assays
Cells (1×105) were seeded in a 6-well plate, and cell viability was determined every 24 h for 4 days using 0.1% HyClone Trypan blue solution (Thermo Fisher Scientific). Viable cells were counted using a Countess Automated Cell Counter (Life Technologies).
Tumor Formation Assays
All animal protocols were approved by the Institution Animal Care and Use Committee at UMMS (A-2247). Ba/F3 or SA cells (6×106) expressing either vector, U2AF35(S34F), NS shRNA or Atg7/ATG7 shRNA were suspended in 100 μl Matrigel (BD Biosciences) and PBS mix (1:1) and injected subcutaneously into the right flank of 5–6 week old athymic BALB/c (nu/nu) male mice (Taconic Farms). Tumor dimensions were measured every 4 days for ~3 weeks and tumor volume was calculated using the formula π/6 × (length) × (width)2.
RNA-Seq
Total RNA from parental Ba/F3 or Ba/F3-U2AF35(S34F) cells was isolated using TriPure Isolation Reagent (Roche), followed by mRNA purification, cDNA synthesis, and amplification. Libraries were sequenced as 100-bp paired ends using Illumina HiSeq 2000. All reads were mapped to the mouse genome (mm10) using TopHat, followed by running Cufflinks to assemble and quantify the transcriptome (Trapnell et al., 2012). Alternative splicing events were first identified using Cuffdiff2 and further analyzed as described in Supplemental Experimental Procedures to select isoforms for further validation (Table S1). Alternative CP site use was evaluated using Identification of novel alternative polyadenylation sites (InPAS, version 0.0.9; http://www.bioconductor.org/packages/release/bioc/html/InPAS.html), the details of which will be published separately. The RNA-Seq data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE63404 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=gdgnakkijbydfsl&acc=GSE63404.
Northern Blot Analysis
Poly(A) RNA was extracted from Ba/F3-V or Ba/F3-U2AF35(S34F) cells using PolyATtract mRNA Isolation System IV (Promega) and analyzed by northern blot analysis using a 32P-labeled probe specific for mouse Atg7 short 3’ UTR or Gapdh (see Supplemental Experimental Procedures for further details). Signals were visualized by phosphorimager and quantified.
RNA Interference
Ba/F3 cells were transfected with 100 nM of the CFIm59 or CFIm68 siRNA (see Supplemental Experimental Procedures for sequences) or control siRNA (Sigma-Aldrich) using a Cell Line Nucleofector kit V (Lonza). For shRNA-mediated knockdown, lentiviral plasmids containing either mouse CFIm59, CFIm68 or Atg7 shRNA (Open Biosystems/GE Dharmacon; clone IDs listed in Supplemental Experimental Procedures) were transfected into HEK293T cells using Effectene (QIAGEN). Viral supernatants were collected 48 h later and used to infect Ba/F3 cells followed by puromycin selection.
Co-immunoprecipitation Assays
HEK293T cells were transfected with a construct expressing Flag-CFIm59 or Flag-CFIm68, and 56 h later cells were harvested, lysed and briefly sonicated. Extracts were cleared by centrifugation and incubated with anti-FLAG M2 affinity gel (Sigma-Aldrich). Beads were washed three times, treated with 10 μg/ml RNase A, and washed again. Flag-CFIm59/68-bound beads were incubated with Ba/F3-U2AF35 or Ba/F3-U2AF35(S34F) nuclear extracts prepared as described in Supplemental Experimental Procedures. Proteins were eluted and analyzed by immunoblotting with the following antibodies: U2AF65 (Santa Cruz Biotechnology, MC3), MYC (Roche, 11-667-149-001), β-actin (Sigma, AC74), FLAG M2 (Sigma, F1804).
RNA Immunoprecipitation Assay
RNA immunoprecipitation was performed as described in the Supplemental Experimental Procedures, using the following antibodies: CFIm25 (Santa Cruz, sc-81109), CFIm59 (Bethyl Laboratories, A301-360A) and CFIm68 (Abcam, ab175237) were used for RNA immunoprecipitation and mouse IgG (Abcam, ab18413) or rabbit IgG (Abcam, ab46540) as a negative control. 1% of the DNase-treated extract was taken for an input fraction. The qRT-PCR levels of the IP samples were normalized against the input of the same samples and calibrated to an IgG control followed by normalization to an irrelevant region in the 18S rRNA. Fold enrichment was calculated by setting the IgG control IP sample to a value of 1.
Immunoblot Analysis
Protein extracts were prepared by lysis in Laemmli buffer [62.5 mM Tris-HCl (pH 6.8), 5% 2-mercaptoethanol, 10% glycerol, 2% SDS, 0.002% bromophenol blue]. Blots were probed with the following antibodies: U2AF65 (Santa Cruz Biotechnology, MC3), MYC (Roche, 11-667-149-001), β-actin (Sigma, AC74), FLAG M2 (Sigma, F1804), ATG7 (ProSci, 3615), CFIm59 (Bethyl Laboratories, A301-359A), CFIm68 (Abcam, ab175237), LC3B (Cell Signaling Technology, #3868), p62 (Santa Cruz Biotechnology, sc-28359), U2AF35 (Proteintech, 10334-1-AP) and α-tubulin (Sigma, B5-1-2). Immunoblots were visualized by a ChemiDoc™ MP System (Bio-Rad).
Luciferase Reporter Assay
Regions of the mouse Atg7 3’UTR cDNA were PCR amplified and cloned downstream of the Renilla luciferase stop codon in psiCHECK-2 (Promega), which also contains firefly luciferase. Constructs were transfected into HEK293T cells using Effectene. Luciferase activity was measured 48–60 h later using the Dual-Luciferase Reporter Assay System (Promega). Transfection efficiency was normalized by firefly luciferase activity.
Cyto-ID Fluorescence Spectrophotometric Assay
Cells were treated with 10 μM PP242 (Sigma-Aldrich) or DMSO for 4 h and then washed with PBS supplemented with 5% FBS. Cells (6x104) were then divided: one half was used for Cyto-ID fluorescence measurement, and the other half for an MTS assay. Cyto-ID (Enzo Life Sciences) staining was performed with minor modifications (see Supplemental Experimental Procedures). Cyto-ID fluorescence was read using a SpectraMax M5 microplate reader (Molecular Devices; excitation 480 nm, emission 530 nm) and was normalized to the cell density among each sample, determined using a Cell-Titer 96 Aqueous One solution cell proliferation assay (MTS) (Promega).
MitoTracker Staining
Cells (5×105) were pelleted and resuspended in pre-warmed staining solution. Mitochondrial mass was measured by staining cells with 100 nM MitoTracker Green FM and MitoTracker Red FM (Molecular Probes/Invitrogen) for 20 min at 37°C. Mitochondrial ROS levels were measured by staining cells with 5 μM MitoSOX (Molecular Probes/Invitrogen) for 20 min at 37°C. Cells were then washed with PBS and resuspended in a PBS solution containing 1% FBS for FACS analysis. Fluorescence emission was analyzed using a BD LSR II flow cytometer (BD Biosciences).
Spontaneous Mutation Frequency Assays
Cells from replicate cultures were counted and resuspended in RPMI medium supplemented with 10% FBS, 0.9% methylcellulose (StemCell Technologies) and 5 ng/ml IL-3. Cells (2.5×105) were plated in selective medium containing 2 mM ouabain (Sigma-Aldrich) or 20 μM 6-TG (Sigma-Aldrich) and incubated at 37°C for 1 week to determine the number of the Na-K-ATPase or HPRT mutant colonies, respectively. Colonies larger than 50 cells were counted under a microscope and mutant frequencies were calculated after correcting for plating efficiency. For ascorbate treatment, (+)-sodium L-ascorbate (Sigma Aldrich) was added to cells (250 μM final concentration) and replenished every three days for one month prior to addition of oubain or 6-TG.
MDS Patient Samples
Bone marrow aspirates from MDS patients (Table S3) were collected using standard procedures at New York Presbyterian/Columbia University Medical Center. Informed consent for sample collection was obtained according to protocols approved by the Columbia University's institutional review board (#AAAL5907). Bone marrow mononuclear cells (BMMNC) were isolated from bone marrow aspirate using Ficoll density gradient centrifugation. BMMNC pellets were re-suspended in TRIzol and stored at −80°C.
The U2AF35 mutation status determination of the length of the human ATG7 3’UTR were performed as described in Supplemental Experimental Procedures. To determine if there was a significant difference in ATG7 CP site use between normal and U2AF35(S34F)-containing samples, the average values for each group were determined.
Soft Agar Anchorage-independent Growth Assay
SA cells (2.5×104) expressing vector, MYC-U2AF35 or MYC-U2AF35(S34F) were suspended in a top layer of BEGM medium (Lonza) and 0.3% Noble agar (Invitrogen) and plated on a bottom layer of growth media and 0.7% Nobel agar in 6-well plates. Colonies were counted 8 weeks after plating.
Acute Myeloid Leukemia Bioinformatic Analysis
RNA-Seq data were obtained for the acute myeloid leukemia (LAML) dataset (which includes 165 samples harboring wild-type U2AF35 and four samples harboring U2AF35(S34F); Table S4) from The Cancer Genome Atlas (TCGA) Data Portal. The percentage distal polyadenylation site usage index (PDUI) values were extracted for ATG7 and filtered by first 100 nt 3' UTR coverage greater than 10.
Cytotoxicity Assays
Ba/F3 cells were plated (2,500 cells per well) in the presence of 1 μM etoposide (Sigma-Aldrich), 1 mM 3-methyladenine (Sigma-Aldrich), or both. SA cells were plated (4,000 cells per well) in the presence of 5 μM etoposide, 5 mM 3-methyladenine (Sigma-Aldrich), or both. For the radiosensitivity test, 2 h following drug treatment, Ba/F3 cells were irradiated with x-rays (4 Gy) at a dose rate of 220 cGy/min. Cytotoxic effects were measured using an MTS assay. The percentage of growth inhibition was calculated as follows: growth inhibition (%) = (1-A of experiment well/A of negative control well)×100% (where A is absorbance, and the negative control is non-treated cells).
Statistics
All quantitative data were collected from experiments performed at least three times with technical replicates; statistically significant results were obtained in independent biological replicates. In general, differences between groups were analyzed using paired two-tailed Student’s t-test using Microsoft Excel, with the exception of Figure 7A, which was analyzed using Student’s unpaired t-test using GraphPad Prism software. Significant differences were considered when P<0.05; *P=0.05, and **P=0.01. Error bars indicate the standard deviation for the technical replicates or standard error of the mean for TCGA data (Figure 7B), as indicated in the figure legend.
Supplementary Material
Highlights.
U2AF35(S34F) transforms immortalized cells by aberrantly processing Atg7 pre-mRNA
The altered Atg7 pre-mRNA has a longer 3’ UTR, resulting in decreased ATG7 levels
Decreased ATG7 results in an autophagy defect that leads to secondary mutations
Altered ATG7 pre-mRNA is found in U2AF35(S34F)-positive MDS and AML patient samples
Acknowledgments
We thank Scott Randell for providing reagents; the UMMS RNAi Core Facility for providing shRNAs and MGC cDNAs; Rod Hardy for providing the pMSCV-IRES-Blasticidin construct; Hyoung-Soo Cho for assistance with FACS analysis; Eric Baehrecke for providing comments on the manuscript; and Sara Deibler for providing editorial assistance. This work was supported by NIH grant R01GM035490 to M.R.G. M.R.G. is an investigator of the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
S.M.P. and M.R.G conceived and designed the experiments. S.M.P. performed the majority of the experiments with the assistance of L.C. for RNA purification and qRT-PCR, T.M.S. for FACS analysis, C-M.V. for virus production, and H.Y. for luciferase assays. J.O. and L.J.Z. performed biostatistical analysis. A.A., S.M. and A.R. provided the MDS patient samples, and discussed the experimental design and interpretation of the results. S.M.P. and M.R.G. analyzed and interpreted the data and wrote the paper. All authors reviewed the paper and provided comments.
Supplemental Information includes Supplemental Experimental Procedures, four figures and three tables and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69:3613–3634. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Rossi M, Malcovati L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood. 2013;121:260–269. doi: 10.1182/blood-2012-09-399725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci USA. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries H, Ruegsegger U, Hubner W, Friedlein A, Langen H, Keller W. Human pre-mRNA cleavage factor II(m) contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J. 2000;19:5895–5904. doi: 10.1093/emboj/19.21.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, Xu A. Differential genome-wide profiling of tandem 3' UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011;21:741–747. doi: 10.1101/gr.115295.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122–1129. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz DA. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem Pharmacol. 2014;90:208–211. doi: 10.1016/j.bcp.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Martin G, Keller W, Zavolan M. Cleavage factor Im is a key regulator of 3' UTR length. RNA Biol. 2012;9:1405–1412. doi: 10.4161/rna.22570. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45:133–135. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, Tian B. Progressive lengthening of 3' untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci U S A. 2009;106:7028–7033. doi: 10.1073/pnas.0900028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovicova J, Knut M, Cross NC, Vorechovsky I. Identification of U2AF(35)-dependent exons by RNA-Seq reveals a link between 3' splice-site organization and activity of U2AF-related proteins. Nucleic Acids Res. 2015;43:3747–3763. doi: 10.1093/nar/gkv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, He Z, von Rutte T, Yousefi S, Hunger RE, Simon HU. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. 2013;5:202ra123. doi: 10.1126/scitranslmed.3005864. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, Randell SH, Stewart SA, Elenbaas B, Hartwell KA, Brooks MW, Fleming MD, Olsen JC, Miller SW, Weinberg RA, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21:4577–4586. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri L, Grabowski P, Degerman S, Svenson U, Gunnarsson R, Cahill N, Smedby KE, Geisler C, Juliusson G, Roos G, et al. Short telomere length is associated with NOTCH1/SF3B1/TP53 aberrations and poor outcome in newly diagnosed chronic lymphocytic leukemia patients. Am J Hematol. 2013;88:647–651. doi: 10.1002/ajh.23466. [DOI] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510:412–416. doi: 10.1038/nature13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, White E. Why sick cells produce tumors: the protective role of autophagy. Autophagy. 2007;3:502–505. doi: 10.4161/auto.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Loulergue C, Dettwiler S, Karaa SZ, Keller W, Antoniou M, Vagner S. An interaction between U2AF 65 and CF I(m) links the splicing and 3' end processing machineries. EMBO J. 2006;25:4854–4864. doi: 10.1038/sj.emboj.7601331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SE, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGAR. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Ramsay AJ, Lopez-Otin C. Chronic lymphocytic leukemia with SF3B1 mutation. N Engl J Med. 2012;366:2530. doi: 10.1056/NEJMc1204033. [DOI] [PubMed] [Google Scholar]
- Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, Zhou J, Qiu J, Jiang L, Li H, et al. Mechanisms for U2AF to define 3' splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol. 2014;21:997–1005. doi: 10.1038/nsmb.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Alternative polyadenylation: new insights from global analyses. Rna. 2012;18:2105–2117. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–3584. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagner S, Vagner C, Mattaj IW. The carboxyl terminus of vertebrate poly(A) polymerase interacts with U2AF 65 to couple 3'-end processing and splicing. Genes Dev. 2000;14:403–413. [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012;26:2447–2454. doi: 10.1038/leu.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AS, Mortensen M, Simon AK. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle. 2011;10:1719–1725. doi: 10.4161/cc.10.11.15673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Green MR. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci USA. 1989;86:9243–9247. doi: 10.1073/pnas.86.23.9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, Ney PA. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood. 2009;114:157–164. doi: 10.1182/blood-2008-04-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.