

Atherosclerosis is a lipid-driven inflammatory disease that underpins myocardial infarction and stroke1, and is the leading cause of death worldwide2. It is not unusual to think of cardiovascular disease as a product of lifestyle3, and indeed, as countries become wealthier and their populations become more sedentary, cardiovascular disease incidence increases. Over the years, epidemiological and experimental research has identified many factors that influence an individual’s cardiovascular risk. Though some of these are inevitable consequences of genetics and the passage of time – sex and age, for example, since men are at higher risk and everyone’s risk increases with age – many others are inextricably linked to lifestyle. Consuming fatty foods, lack of exercise, smoking, excessive psychosocial stress, and poor sleep contribute to measurable risk factors such as high LDL, obesity, hypertension, and diabetes, all of which increase the risk of myocardial infarction and stroke. These connections sometimes lead to the popular misunderstanding that cardiovascular disease can be prevented by making healthy lifestyle choices. Unfortunately, the causes of myocardial infarction and stroke are not exclusively behavioral. An article by Tall, Wang and colleagues4 in this issue of Circulation Research is an excellent illustration of how inheritance crucially shapes our responsiveness to environmental stimuli.
We have all heard it: the diet-conscious runner who collapses from a heart attack at 40 and the obese life-long drinker and smoker who dies at 90. Though anecdotal, do these examples nevertheless tell us something meaningful about cardiovascular disease? Over the last 15 years, genome wide association studies (GWAS) have revealed that certain mutations can either increase the risk of or protect against myocardial infarction5, 6. In other words, inheritance matters. Certain people are more naturally resistant to having a heart attack. As expected, many of these mutations affect genes that participate in cholesterol metabolism. After all, overwhelming evidence indicts high low density lipoprotein (LDL) levels as a major risk factor. Those levels depend on not only what we eat but also on how our bodies process our food. Individuals with mutations in genes that process cholesterol more effectively may find themselves with lower blood cholesterol levels than individuals who lack the mutation, even when those individuals eat the same meal. Identifying mutations that influence the risk of coronary heart disease has been a true GWAS success story that has reaffirmed the importance of lipoprotein metabolism in cardiovascular disease. Surprisingly, however, the vast majority of mutations that associate with coronary artery disease have no connections to cholesterol metabolism. What else is going on?
Tall, Wang, and colleagues focus on a genetic locus, found in the LNK/SH2B3 gene, that is associated with coronary heart disease. Expressed by hematopoietic cells, LNK is an adaptor protein that antagonizes cell proliferation and cytokine signaling by suppressing thrombopoietin signaling via the MPL receptor (see references within). In humans, the genetic locus rs3184504 causes a missense mutation and associates with increased risk of heart disease, elevated platelet counts, and leukocytosis. In mice, deleting LNK leads to more myelopoiesis and megakaryopoiesis, which gives rise to leukocytosis and thrombocytosis, respectively. All of this points to the possibility that LNK confers risk via its effects on hematopoiesis and thrombosis. But does it? And if so, under what conditions?
The authors first ask whether LNK’s effects on hematopoiesis influence the growth of atherosclerotic lesions. To this end, the authors lethally irradiate Ldlr−/− mice, which are frequently used as an atherosclerosis model, reconstitute the mice with bone marrow cells from Lnk−/− donors, and then feed them a diet high in fat and cholesterol. After 10 weeks of feeding, atherosclerotic lesions grow larger in mice whose bone marrow-derived cells cannot produce LNK. The lesions have larger necrotic cores, more macrophages, and more neutrophils. The authors observe that in the context of hypercholesterolemia, LNK-deficient hematopoietic cells expand more dramatically than controls. The marked increase in monocytosis and neutrophilia, the argument goes, may be the result of higher IL-3 and GM-CSF signaling, because Lnk−/− cells stimulated with IL-3 or GM-CSF augment Erk1/2 phosphorylation. In other words, the absence of LNK aggravates atherosclerosis likely due to heightened production of myeloid cells that infiltrate atherosclerotic lesions (Figure 1). This observation is consistent with human studies showing that leukocytosis predicts cardiovascular events7, 8 and mirrors mouse studies showing that atherosclerosis develops via continuous monocyte recruitment9 and hypercholesterolemia-associated monocytosis10, which are driven by heightened myelopoiesis in the bone marrow and spleen11–13. Although Tall and Wang do not definitively test whether LNK limits atherosclerotic lesion growth because of its inibitory effect on hematopoiesis, the data are nevertheless compelling and significantly add to our growing appreciation of hematopoiesis’ role in the development of atherosclerosis.
Figure 1. Proposed mechanism for how LNK/SH2B3 attenuates cardiovascular disease.
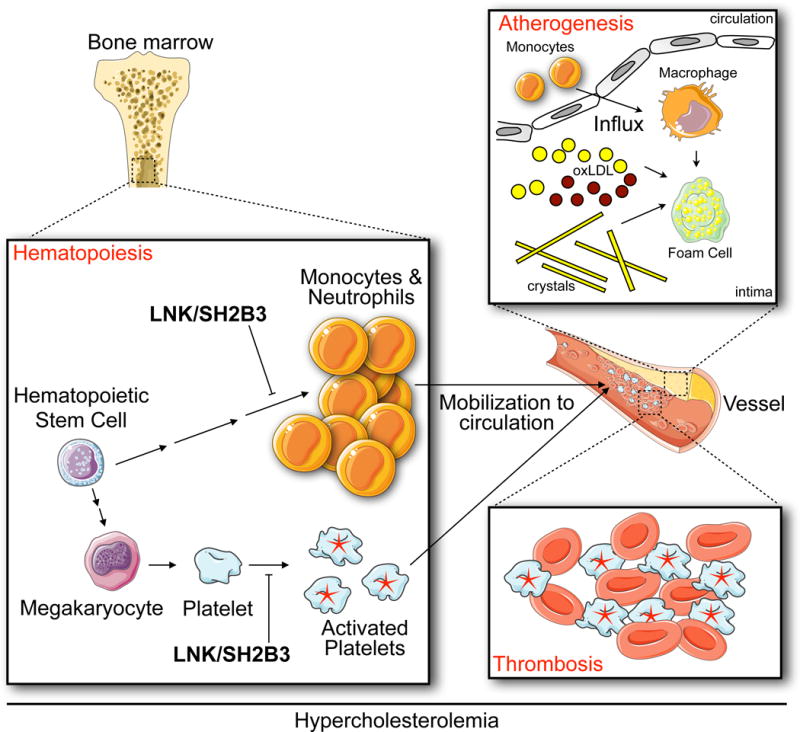
The authors show that, in the context of hypercholesterolemia, LNK/SH2B3 limits myelopoisis and platelet activation. Consequently, reduced LNK/SH2B3 might confer risk of coronary artery disease via both increased production of myeloid cells, which then accumulate in growing atherosclerotic lesions, and overabundantactivated platelets, which promote thrombosis.
The second mechanism the authors investigate is more extensively explored but it comes down to this: in the absence of LNK, hypercholesterolemia activates platelets. The study provides substantial molecular detail regarding how platelet LNK deficiency during hypercholesterolemia leads to lowered platelet LYN kinase activity and decreased SHIP-1 phosphorylation that, together with increased TPO/MPL signaling, leads to elevated AKT phosphorylation and consequent platelet activation. The central message is that LNK negatively regulates platelet activation under hypercholesterolemic conditions, thus predisposing to thrombosis (Figure 1).
Together, these insights suggest that LNK protects against coronary heart disease by influencing two distinct but connected processes. On the one hand, by limiting myelopoiesis, LNK may restrict the size of atherosclerotic lesions, which grow via lipid accumulation and continuous monocyte influx. On the other hand, by keeping platelets relatively quiescent, even in the context of hypercholesterolemia, LNK mitigates the thrombotic events that ultimately block vessels upon lesion rupture. Humans who produce insufficient LNK as a result of a missense mutation might develop larger lesions due to effected hematopoiesis and might have platelets more susceptible to clotting. It is a dangerous combination that warrants further inquiry into possible treatments.
Beyond identifying a potential therapeutic target for treating cardiovascular disease, the paper is notable for several conceptual reasons. Atherosclerosis is frequently described as an inflammatory disease, and indeed, much research focuses on the inflammation that drives atherosclerosis and its complications. Nevertheless, evidence for how inflammatory processes influence disease in humans remains relatively scant and predominantly associative. This paper suggests that monocytes and neutrophils, which are bone marrow-derived innate leukocytes, are causal rather than merely reactive. By focusing on a particular target identified by GWAS, this study reminds us that cardiovascular disease is driven by more than cholesterol or lifestyle. Moreover, by successfully exploring a human SNP in mice with atherosclerosis, the research demonstrates the value of preclinical, fundamental work to understanding human biology. Others in the field should consider such approaches when attempting to decipher the functions of the still-unknown genes identified by GWAS.
Finally, the study represents another nail in the coffin for the idea that atherosclerosis is a local disease. To be sure, there are many locally-occurring processes relevant to lesion growth and stability. For example, monocytes arriving from the blood differentiate to macrophages which then proliferate locally14; “don’t eat me” signals, such as CD47, impair efferocytosis, presumably in the lesion15; and smooth muscle cells from the media migrate to the intima to acquire macrophage-like characteristics16. Yet growing evidence is mounting in favor of the concept that distal processes – and not just those related to cholesterol metabolism – are consequential to atherosclerosis and its complications. The paper by Tall and Wang adds to this growing body of work by suggesting that hematopoiesis in the bone marrow is a crucial decision node that shapes not only the growth of atherosclerotic lesions but also the thrombosis that occurs when lesions rupture. We are likely just scratching the surface.
Acknowledgments
Sources of Funding
This work was supported by NIH grants HL096576, HL095629, NS084863, HL128264, HL095612, HL128264, HL095612 and the MGH Research Scholar.
Footnotes
Disclosure Section
The authors declare no conflict of interest.
References
- 1.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloom DE, Cafiero ET, Jane-Llopis E, Abrahams-Gessel S, Bloom LR, Fathima S, Feigl AB, Gaziano T, Mowafi M, Pandya A, Prettner K, Rosenberg L, Seligman B, Stein AZ, Weinstein C. The Global Economic Burden of Noncommunicable Diseases. Geneva: World Economic Forum; 2011. [Google Scholar]
- 3.Nahrendorf M, Swirski FK. Lifestyle effects on hematopoiesis and atherosclerosis. Circ Res. 2015;116:884–894. doi: 10.1161/CIRCRESAHA.116.303550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang W, Tang Y, Wang Y, Tascau L, Balcerek J, Tong W, Levine R, Welch CL, Tall AR, Wang N. LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis. Circ Res. 2016 doi: 10.1161/CIRCRESAHA.116.308955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148:1242–1257. doi: 10.1016/j.cell.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kessler T, Vilne B, Schunkert H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol Med. 2016;8:688–701. doi: 10.15252/emmm.201506174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berg KE, Ljungcrantz I, Andersson L, Bryngelsson C, Hedblad B, Fredrikson GN, Nilsson J, Bjorkbacka H. Elevated CD14++CD16- monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5:122–131. doi: 10.1161/CIRCGENETICS.111.960385. [DOI] [PubMed] [Google Scholar]
- 8.Coller BS. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol. 2005;25:658–670. doi: 10.1161/01.ATV.0000156877.94472.a5. [DOI] [PubMed] [Google Scholar]
- 9.Swirski FK, Pittet MJ, Kircher MF, Aikawa E, Jaffer FA, Libby P, Weissleder R. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, Tall AR. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, Mulligan-Kehoe MJ, Libby P, Nahrendorf M, Pittet MJ, Weissleder R, Swirski FK. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang M, Subramanian M, Abramowicz S, Murphy AJ, Gonen A, Witztum J, Welch C, Tabas I, Westerterp M, Tall AR. Interleukin-3/granulocyte macrophage colony-stimulating factor receptor promotes stem cell expansion, monocytosis, and atheroma macrophage burden in mice with hematopoietic ApoE deficiency. Arterioscler Thromb Vasc Biol. 2014;34:976–984. doi: 10.1161/ATVBAHA.113.303097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, Leeper NJ. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016 doi: 10.1038/nature18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]