

Abstract
Introduction
The increased risk of end-stage kidney disease among hypertensive African Americans is partly related to APOL1 allele variants. The initial glomerulosclerosis of hypertension-associated arterionephrosclerosis consists of focal global glomerulosclerosis, but in biopsy studies, focal segmental glomerulosclerosis is found with progression to end-stage kidney disease, particularly in African Americans.
Methods
This is a study of arterionephrosclerosis in successfully APOL1-genotyped autopsy kidney tissue of 159 African Americans and 135 whites aged 18 to 89 years from a general population with no clinical renal disease.
Results
Glomerulosclerosis was nearly exclusively focal global glomerulosclerosis with 3 subjects having focal segmental glomerulosclerosis–like lesions that were unrelated to APOL1 risk status. For both races, in multivariable analysis, the dependent variables of arteriosclerosis and glomerulosclerosis were significantly related to the independent variables of older age (P < 0.001) and hypertension (P < 0.001). A relationship between APOL1 genotype and arteriosclerosis was apparent only after 35 years of age, when, for any level of elevated blood pressure, more severe arteriosclerosis was found in the interlobular arteries of 14 subjects with 2 APOL1 risk alleles compared to African Americans with none (n = 37, P = 0.02) or 1 risk allele (n = 35, P = 0.02).
Discussion
With the limitation of the small number of subjects contributing to the positive results, the findings imply that APOL1 risk alleles recessively augment small-vessel arteriosclerosis in conjunction with age and hypertension. Focal segmental glomerulosclerosis was not a significant finding, indicating that in the early stages of arterionephrosclerosis, the primary pathologic influence of APOL1 genotype is vascular rather than glomerular.
Keywords: aging, APOL1 variants, arteriosclerosis, hypertension, race
The risk of end-stage kidney disease (ESKD) among Americans with nondiabetic kidney disease is 3.5-fold greater for African Americans than for whites, with the greatest burdens falling in the diagnostic categories of hypertension-associated nephrosclerosis, focal segmental glomerulosclerosis (FSGS), and HIV nephropathy.1 In 2010, variants in the apolipoprotein L1 (APOL1) gene were reported by Genovese et al.2 as being a major factor in this risk. Two APOL1 risk variants were identified. G1 consists of 2 nonsynonymous amino acid substitutions, S342G and I384M, and G2 consists of the deletion of 2 amino acid residues, N388 and Y389.2 It is thought that the risk is largely a recessive trait requiring the inheritance of 2 risk alleles that can be either G1 or G2.1, 3
In the United States, hypertension is the attributed cause of 25% of ESKD for whites and 34% for African Americans.4 There is a graded relationship between the level of hypertension and the observed risk of ESKD that is 3.1 for mild, 6.0 for moderate, and 11.2 for severe hypertension compared to reference subjects with optimal blood pressure. This makes high blood pressure, itself, a logical causative factor of ESKD; nevertheless, fewer than 0.5% of persons with hypertension progress to the late stages of chronic kidney disease (CKD).5 The susceptibility for ESKD among hypertensive African Americans with 2 risk alleles is estimated at somewhere between the 4% lifetime risk for FSGS in non–HIV-infected persons and the 50% risk with HIV nephropathy.6 This is a wide range indicative of the uncertain mechanisms underlying the progression of kidney disease attributed to hypertension.1, 6, 7
Arterionephrosclerosis, the pathologic accompaniment of hypertension-associated nephrosclerosis, is characterized by arteriosclerosis, global glomerulosclerosis, and cortical fibrosis with tubular atrophy and loss.7, 8, 9, 10, 11, 12 The arteriosclerosis affects 3 levels of renal arteries. The arcuate arteries and interlobular arteries develop varying degrees of fibrous intimal thickening, and hyaline material accumulates in the walls of afferent arterioles. Arcuate arteries have been referred to as close and interlobular arteries remote according to their proximity to the aorta, and their intimal thickening designated as Itc for arcuate and Itr for interlobular arteries.9, 13, 14, 15 Tracy et al.9, 13 observed that Itc and Itr had somewhat different relationships to age and blood pressure, with Itc reflecting age-related large artery stiffness and Itr being more closely correlated with blood pressure. In those studies, Itc appeared to precede hypertension and to occur before Itr, leading to a proposal that hypertension may be not a primary disorder but secondary to small artery disease.7, 8, 9
Nevertheless, both Itc and Itr correlate strongly with hypertension and glomerulosclerosis,13, 14, 15 while the associations with arteriolar hyalinization are less certain. Hill et al.16, 17 thought that arteriolar hyalinization may be related to a loss of autoregulatory vascular tone and the development of FSGS in conjunction with glomerular hypertrophy and possible hyperfiltration, particularly in African Americans, while Tracy et al.18 found an unpredictable relationship to blood pressure and no relationship to race.
Arterionephrosclerosis is most commonly accompanied by glomerular obsolescence that in the late stages consists of a contracted hyalinized tuft surround by intracapsular fibrosis within a largely intact Bowman’s capsular basement membrane. This pattern of glomerulosclerosis is thought to be the result of ischemia secondary to preglomerular arteriosclerosis and will be hereafter referred to as focal global glomerulosclerosis (FGGS). A contrasting pattern of glomerular loss consists of FSGS that is inconsistently found in arterionephrosclerosis. The late stages of FSGS result in glomerular solidification in which the tuft is expanded by increased mesangial matrix and often contains hyaline insudates. Bowman’s capsule basement membrane is fragmented, and there is an absence of intracapsular fibrosis. In patients with presumed primary hypertension, FSGS and glomerular solidification are described mainly in biopsies of patients with azotemia or proteinuria and, when identified, tend to raise the question of a primary glomerular disease.7, 16, 17 Whether the glomerulosclerosis is global or segmental, the end result is a loss of renal function. This loss of function was evaluated in the Mayo Clinic biopsy series of transplant donors in which lower pre-donation glomerular filtration rate (GFR) was significantly associated with age, hypertension, and increased FGGS.19
Recently, we reported that African Americans with 2 APOL1 risk alleles have an exaggerated glomerular loss in the first 38 years of adult life compared to subjects with no risk alleles or 1 risk allele.20 In the current study, we examine the effects of APOL1 status, age, and hypertension on the structural features of mild to moderate arterionephrosclerosis, and, in particular, arteriosclerosis in the different levels of renal arteries.
Methods
Autopsy Cohort and Collection of Specimens and Clinical Data
The analyzed specimens consist of right kidneys collected at autopsy between 1998 and 2005 at the University of Mississippi Medical Center (Jackson, MS). Race was ascertained from a combination of medical records, reports from next of kin, and physical examinations. Subjects with known renal disease, with kidneys of significantly unequal size, and with grossly scarred or contracted kidneys were specifically excluded. Kidneys were included if they were grossly normal or showed mild to moderate arterionephrosclerosis. Such arterionephrosclerotic kidneys were collected from patients with a clinical diagnosis of diabetes but were excluded if there was microscopically a diabetic nephropathy. Clinical and demographic characteristics were obtained from University of Mississippi Medical Center records. Causes of death were documented and medical records were reviewed for a history of hypertension. Patients were categorized as hypertensive on the basis of clinical history of hypertension, consistently elevated blood pressures (≥140/90 mm Hg), the presence of cardiomegaly, and the severity of intrarenal arteriosclerosis as previously described.12, 14, 15 Mean arterial blood pressure (MAP) was obtained on 113 African Americans and 66 whites and was calculated from at least 3 blood pressure determinations. Blood pressures from terminal admissions were not used unless patients were identified as hypertensive and blood pressures were elevated. Patients were recorded as treated for hypertension if there was any statement of such treatment in their medical record or medical examiner’s report. Approval was obtained from the Institutional Review Board of the University of Mississippi Medical Center and the Human Research Ethics Committee of Monash University, Victoria, Australia, and consent provided by next of kin.
Measurement of Glomerulosclerosis, Cortical Fibrosis, and Arteriosclerosis
Representative kidney blocks from the upper pole and the mid-portion of the kidney not sampled for stereology were paraffin embedded. Sections were cut at 4 μm and stained with periodic acid–Schiff and hematoxylin and with picrosirius red for fibrillar collagen. Measurement of the percentage of obsolete glomeruli and arterial intimal thickening used sections stained with periodic acid–Schiff and hematoxylin. The percentage of sclerotic glomeruli was estimated by counting sclerosed and non-sclerosed glomeruli in non-overlapping 100X microscopic fields moving from the subcapsular surface to inner cortex, and with at least 400 glomeruli being counted per subject. The severity of arteriosclerosis was measured as a percentage of the thickness of the intima to the outer wall diameter at original magnifications of 200X to 400X in interlobular arteries 90 to 250 μm in diameter and arcuate arteries >250 μm in diameter using the linear measurement function of Image-Pro Plus morphometric software (Media Cybernetics, Bethesda, MD). The percentage thickening of interlobular or remote arteries was designated Itr and the percentage thickening of arcuate or close arteries as Itc. Cortical fibrosis was measured in original 200X images as the percentage of cortex staining red with the picrosirius stain using the automated Image-Pro Plus area counting function.
Measurement of Total Glomerular Number and Glomerular Volume
Kidney tissue was sent to Monash University for stereologic estimation of total glomerular number (Nglom). Nglom was estimated using the physical disector/fractionator combination as previously described.12, 21, 22 In brief, perfusion-fixed kidneys were sampled to provide a systematic, uniform random sample of tissue blocks. These blocks were embedded in glycolmethacrylate and then sectioned at 20 μm. Every tenth and eleventh sections (a section pair) were then stained with the periodic acid–Schiff stain, and glomeruli in identical fields in each section pair were counted using the disector principle. Stereologic point counting at the same time on the same sampled fields allowed the calculation of glomerular volume (Vglom).
Genetic Analysis
DNA was isolated from formalin-fixed, paraffin-embedded tissue as previously described.23 APOL1 risk alleles were genotyped using TaqMan assays (ABI, Foster City, CA). The APOL1 G1 allele was defined as the presence of the S342G variant (rs73885319A>G) with or without the I384M (rs60910145T>G) variant, and the G2 allele was defined as the deletion of N388Y389 (rs71785313 TTATAA/-).
Statistical Analysis
Continuous variables were tabulated using mean ± SD and discrete variables as numbers of subjects and percents. Clinical characteristics were grouped by race and by risk genotype status as no risk alleles, 1 risk allele, and 2 risk alleles. The microstructural features of arterionephrosclerosis were grouped by ages of 18 to 34 years, 35 to 54 years, and ≥55 years. Analyses of data were performed with Stata/IC 10.0 (StataCorp Statistical Software, StataCorp LP, College Station, TX) or with SigmaStat 3.5 (Systat Software Inc., Richmond, CA).
Multiple group comparisons of continuous variables by age or risk allele status were tested using a Kruskal–Wallis analysis of variance on ranks with Dunn’s post hoc pairwise tests. Two-way comparisons of continuous variables were performed by Wilcoxon rank sum tests. Discrete variables were evaluated by χ2 tests, and 2-sided Fisher’s exact tests were used to evaluate whether genotype frequencies were in Hardy–Weinberg equilibria. The microstructural features of glomerulosclerosis, cortical fibrosis, Itc, Itr, and arteriolar hyalinization had marked positive skews with zero values and were transformed as log10 (y + 10). Multivariable analyses used fractional polynomial regressions for untransformed data and linear regressions for transformed or untransformed data when normality tests were met or closely approximated normality. Differences in arteriosclerosis by risk genotype are tabulated using transformed and untransformed data. Significant allele group differences in arteriosclerosis were evaluated using recessive (2 > 0 or 1 risk alleles), dominant (2 and 1 > 0 risk alleles), and additive (2 > 1 > 0 risk alleles) models.
Results
Clinical Characteristics and APOL1 Genotypes of the Study Cohort
This is an autopsy cohort study of 159 African Americans and 135 whites ≥18 years old that had successful APOL1 genotyping performed on archived kidney tissue that had been previously analyzed stereologically and morphometrically. The causes of death were coronary artery disease 33%, cerebrovascular disease 9%, pulmonary embolus 8%, neoplastic disease 15%, chronic lung disease 8%, accident 14%, homicide 7%, suicide 1%, and other or unknown 4%. Among African Americans, 30 (19%) had 2 risk alleles (12, 2 G1; 13, G1G2; and 5, 2 G2), 68 (43%) had 1 risk allele (36, G1; 32, G2), and 61 (39%) had no risk alleles. Among whites, 3 (2%) had 1 risk allele (3, G1). The genotype distributions in African Americans were in Hardy–Weinberg equilibrium, and allele frequencies were similar to those in a sample of middle-aged African Americans enrolled in the Atherosclerosis Risk in Community (ARIC) study.24 Females had a slightly, but not significantly, higher proportion of APOL1 risk alleles than expected, and more female than male African Americans had 2 rather than none or 1 risk alleles (P = 0.02).
Table 1 summarizes the clinical and genotypic characteristics of the autopsy subjects, comparing the races with no risk alleles and African Americans by risk genotype. Gender distribution, body mass index (BMI), and rates of obesity were similar between races and between risk genotypes. Overall, whites were slightly older than all African Americans (P = 0.04), but not by race with no risk alleles (P = 0.40). African Americans had higher blood pressures than whites, overall (P = 0.02) and when comparing subjects with no risk alleles (P = 0.03). The average MAP was normotensive for whites and hypertensive for African Americans. Among African Americans, 11% were classified as severely hypertensive (MAP ≥ 133 mm Hg) compared to 1.5% of whites. There was no significant difference in MAP among African Americans by risk genotype. A similar proportion of African Americans and whites were treated for hypertension. The hypertensive medications consisted of β-blocking agents, angiotensin-converting enzyme inhibitors, hydrochlorothiazide, calcium channel blockers, and, for 1 subject, spironolactone. Among African Americans there were no significant differences by risk genotype in frequency of hypertension, treated hypertension, or MAP.
Table 1.
Clinical and genotypic characteristics of autopsy subjects by race and APOL1 risk genotype
| Characteristic | White |
African American |
P |
|||
|---|---|---|---|---|---|---|
| 0 risk alleles n = 132 |
0 risk alleles n = 61 |
1 risk allele n = 68 |
2 risk alleles n = 30 |
Racea | AA risk genotype | |
| Age (yr) | 44.2 ± 14.2 | 42.5 ± 12.7 | 42.0 ± 12.2 | 39.9 ± 15.2 | 0.40 | 0.67 |
| Male | 83 (62.9%) | 36 (59.0%) | 37 (54.4%) | 10 (33.3%) | 0.90 | 0.38 |
| BMI (kg/m2) | 30.2 ± 8.4 | 29.4 ± 8.4 | 31.3 ± 12.0 | 27.7 ± 6.8 | 0.98 | 0.22 |
| BMI ≥30 kg/m2 | 53 (40.2%) | 26 (42.6%) | 25 (36.7%) | 6 (20%) | 0.95 | 0.32 |
| Hypertension | 48 (35.6%) | 32 (52.5%) | 41 (60.3%) | 14 (46.7%) | 0.23 | 0.77 |
| Treated hypertension | 19 (14.4%) | 16 (26.2%) | 18 (26.5%) | 6 (20.0%) | 0.15 | 0.85 |
| MAP (mm Hg) | 102.9 ± 13.0 | 108.9 ± 16.0 | 108.3 ± 17.5 | 107.0 ± 16.3 | 0.03 | 0.90 |
| CAD/CVD death | 45 (32.6%) | 19 (31.1%) | 31 (45.6%) | 10 (33.3%) | 0.67 | 0.49 |
| Age CAD/CVD death | 53.1 ± 13.0 | 49.9 ± 10.1 | 41.8 ± 9.7 | 42.9 ± 6.3 | 0.69 | 0.01 |
| Diabetes | 2 (1.5%) | 3 (4.9%) | 4 (5.9%) | 0 (0%) | 1.00 | DNT |
AA, African American; BMI, body mass index; CAD/CVD, coronary artery disease/cerebrovascular disease; DNT, does not test; MAP, mean arterial blood pressure.
Race is tested between whites and African Americans with no risk alleles by Wilcoxon rank sum tests for continuous variables and χ2 test for discrete variables (Fisher’s exact test for diabetes). For African Americans APOL1 risk genotype is tested by 3-way analysis of variance for continuous variables and χ2 tests for discrete variables.
The proportion of cardio-cerebrovascular disease (coronary artery disease/cerebrovascular disease [CAD/CVD]) deaths was not significantly different between whites and African Americans. The age of CAD/CVD death was similar for African Americans with 1 and 2 risk alleles, and in both groups was significantly younger than among African Americans with no risk alleles (P = 0.01). The difference in the age of death from CAD/CVD is not significantly different between whites and African Americans with no risk alleles (P = 0.69).
Seven African Americans were diabetic. All were male and hypertensive, and 3 died of CAD/CVD. Four African American diabetics had 1 APOL1 risk allele, 3 no risk alleles, and none 2 APOL1 risk alleles. Two whites were diabetic; 1 was hypertensive and dying of CAD/CVD. Four African American diabetics and 1 white were treated for hypertension.
There were 3 African Americans who died after age 70. All were hypertensive, and all had 1 or 2 APOL1 risk alleles: a 74-year-old woman (G1,G2) with 5.4% glomerulosclerosis, and cause of death uterine cancer; a 79-year-old man (G1) with 4.6% glomerulosclerosis and cause of death fulminant hepatic failure; and an 83-year-old woman (G1,G1) with 6.7% glomerulosclerosis and cause of death bleeding gastric ulcer.
Microstructural Characteristics and Vascular Pathology and Relationships to Age, Race, and Risk Genotype
The kidneys of the entire cohort ranged from normal to having varying degrees of arterionephrosclerosis. Three subjects with severe hypertension having MAP ≥133 mm Hg (1 white with no risk alleles, 1 African American with no risk alleles, and 1 African American with 1 risk allele) had FSGS-like lesions involving some glomeruli but with a predominant pattern (>50%) of FGGS. In all other kidneys, the glomerulosclerosis was FGGS.
Table 2 compares the microanatomy and MAP of whites and African Americans with no risk alleles by the ages of 18 to 34 (group 1), 35 to 54 (group 2), and ≥55 years old (group 3), and Table 3 compares African Americans by risk genotype in the same age groups. Before age 35, the structural features of arterionephrosclerosis were absent or very mild, and no racial or significant risk genotype differences were apparent. After 35 years of age, for both races and all 3 APOL1 risk allele categories, there was increased blood pressure and a marked increase in all microstructural aspects of arterionephrosclerosis that generally followed a graded relationship with group 3 ≥ group 2 > group 1 (P ≤ 0.001). Among subjects with no risk alleles, African Americans had more severe Itc, Itr, and glomerulosclerosis than whites that became significant after 54 years old. Among African Americans in age groups 2 and 3, subjects with 2 risk alleles tended to have more severe Itc, Itr, and arteriolar hyalinization than subjects with none or 1 risk allele, but differences were not significant.
Table 2.
Microstructural kidney features and mean arterial blood pressure compared by age categories and race in subjects with no risk alleles
| White (no APOL1 risk alleles) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Group 1, 18–34 yrs |
Group 2, 35–54 yrs |
Group 3, ≥55 yrs |
P age groups | Pairwise P < 0.05 |
||||
| n = 34 | P race | n = 70 | P race | n = 28 | P race | |||
| MAP | 99.0 ± 13.0 | 0.28 | 103.4 ± 11.6 | 0.08 | 107.2 ± 17.8 | 0.10 | 0.07 | NS |
| Itc | 5.2 ± 5.2 | 0.29 | 11.7 ± 6.7 | 0.07 | 15.9 ± 6.3 | 0.02 | <0.001 | 3>2>1 |
| Itr | 2.2 ± 3.1 | 0.29 | 6.6 ± 6.8 | 0.05 | 9.6 ± 7.0 | 0.01 | <0.001 | 3≈2>1 |
| Hyaline | 5.1 ± 12.1 | 0.63 | 13.0 ± 16.6 | 0.91 | 9.9 ± 11.4 | 0.01 | 0.001 | 3≈2>1 |
| GS | 1.3 ± 2.0 | 0.03 | 2.5 ± 3.1 | 0.56 | 6.1 ± 6.6 | 0.01 | <0.001 | 3>2>1 |
| Cort fib | 2.4 ± 3.0 | 0.70 | 4.1 ± 3.6 | 0.26 | 7.2 ± 7.0 | 0.17 | <0.001 | 3≈2>1 |
| Nglom | 957,268 ± 315,660 | 0.45 | 939,161 ± 311,394 | 0.73 | 908,179 ± 339,201 | 0.59 | 0.87 | NS |
| Vglom | 5.83 ± 1.92 | 0.01 | 7.35 ± 2.56 | 0.21 | 7.07 ± 2.52 | 0.50 | <0.01 | 2>3≈1 |
| African Americans (no APOL1 risk alleles) | ||||||||
| n = 15 | n = 33 | - | n = 13 | - | ||||
| MAP | 94.3 ± 8.9 | - | 110.3 ± 14.9 | - | 116.9 ± 17.0 | - | <0.01 | 3≈2>1 |
| Itc | 3.2 ± 2.8 | - | 14.5 ± 7.3 | - | 20.4 ± 8.1 | - | <0.001 | 3≈2>1 |
| Itr | 2.0 ± 3.9 | - | 9.0 ± 7.3 | - | 16.6 ± 8.2 | - | <0.001 | 3>2>1 |
| Hyaline | 3.1 ± 7.1 | - | 11.4 ± 12.2 | - | 27.8 ± 25.3 | - | <0.001 | 3>2≈1 |
| GS | 0.8 ± 1.4 | - | 3.0 ± 5.4 | - | 11.2 ± 8.4 | - | <0.001 | 3>2>1 |
| Cort fib | 2.3 ± 3.0 | - | 5.7 ± 5.9 | - | 10.1 ± 7.9 | - | <0.001 | 3≈2>1 |
| Nglom | 902,597 ± 281,603 | - | 952,724 ± 290,787 | - | 820,489 ± 262,534 | - | 0.36 | NS |
| Vglom | 8.01 ± 2.98 | - | 7.99 ± 2.39 | - | 7.86 ± 3.43 | - | 0.74 | NS |
Age categories tested by Kruskal–Wallis 1-way analysis of variance on ranks with Dunn’s post hoc pairwise method. Race tested by Wilcoxon rank sum method. Arteriolar hyalinization, glomerulosclerosis (GS), and cortical fibrosis (Cort fib) are percentages of the affected structures (i.e., percent of arterioles hyalinized, percent of glomeruli that are globally sclerotic, and percent of cortex that is fibrotic). Itc and Itr represent the intimal thickening of arcuate (Itc) and interlobular (Itr) arteries as a percentage of the outer diameter of the artery wall.
MAP, mean arterial blood pressure (mm Hg); Nglom, total estimated glomerular number of right kidney; NS, not significant; Vglom, average glomerular volume (μm3 × 106).
Table 3.
African American microstructural kidney features and mean arterial blood pressure compared by age categories and risk alleles
| African Americans (no APOL1 risk alleles) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Group 1, 18–34 yrs |
Group 2, 35–54 yrs |
Group 3, ≥55 yrs |
P age groups | PW age groups | ||||
| n = 15 | P alleles | n = 33 | P alleles | n = 13 | P alleles | |||
| MAP | 94.3 ± 8.9 | 0.67 | 110.3 ± 14.9 | 0.86 | 116.9 ± 17.0 | 0.93 | <0.01 | 3≈2>1 |
| Itc | 3.2 ± 2.8 | 0.05 | 14.5 ± 7.3 | 0.45 | 20.4 ± 8.1 | 0.65 | <0.001 | 3≈2>1 |
| Itr | 2.0 ± 3.9 | 0.04 | 9.0 ± 7.3 | 0.15 | 16.6 ± 8.2 | 0.51 | <0.001 | 3>2>1 |
| Hyaline | 3.1 ± 7.1 | 0.93 | 11.4 ± 12.2 | 0.86 | 27.8 ± 25.3 | 0.56 | <0.001 | 3>2≈1 |
| GS | 0.8 ± 1.4 | 0.50 | 3.0 ± 5.4 | 0.18 | 11.2 ± 8.4 | 0.51 | <0.001 | 3>2>1 |
| Cort fib | 2.3 ± 3.0 | 0.97 | 5.7 ± 5.9 | 0.69 | 10.1 ± 7.9 | 0.80 | <0.001 | 3≈2>1 |
| Nglom | 902,597 ± 281,603 | 0.25 | 952,724 ± 290,787 | 0.59 | 820,489 ± 262,534 | 0.29 | 0.36 | NS |
| Vglom | 8.01 ± 2.98 | 0.21 | 7.99 ± 2.39 | 0.23 | 7.86 ± 3.43 | 0.18 | 0.74 | NS |
| African Americans (1 APOL1 risk allele) | ||||||||
| n = 18 | n = 41 | n = 9 | ||||||
| MAP | 100.7 ± 16.7 | 110.1 ± 18.4 | 114.0 ± 7.5 | 0.24 | NS | |||
| Itc | 6.8 ± 5.2 | 14.2 ± 7.3 | 21.2 ± 8.3 | <0.001 | 3>2>1 | |||
| Itr | 3.8 ± 4.5 | 10.2 ± 7.8 | 14.9 ± 6.9 | <0.001 | 3≈2>1 | |||
| Hyaline | 6.1 ± 11.8 | 12.9 ± 15.7 | 20.1 ± 22.1 | 0.02 | 3>2≈1 | |||
| GS | 1.1 ± 1.6 | 4.8 ± 5.3 | 7.3 ± 4.2 | <0.001 | 3≈2>1 | |||
| Cort fib | 3.1 ± 4.8 | 5.8 ± 5.4 | 9.9 ± 3.0 | <0.001 | 3>2>1 | |||
| Nglom | 1,036,861 ± 366,869 | 894,230 ± 308,028 | 874,517 ± 260,299 | 0.25 | NS | |||
| Vglom | 6.97 ± 2.31 | 9.01 ± 3.03 | 8.68 ± 2.74 | 0.04 | 3≈2>1 | |||
| African Americans (2 APOL1 risk alleles) | ||||||||
| n = 12 | PW alleles | n = 15 | PW alleles | n = 3 | PW alleles | |||
| MAP | 94.5 ± 9.9 | NS | 113.8 ± 16.3 | NS | 116.0 ± 7.1 | NS | 0.02 | 3≈2>1 |
| Itc | 4.8 ± 3.3 | NS | 16.8 ± 8.8 | NS | 26.0 ± 16.4 | NS | <0.001 | 3≈2>1 |
| Itr | 0.9 ± 1.4 | NS | 14.8 ± 13.3 | NS | 21.6 ± 14.5 | NS | <0.001 | 3≈2>1 |
| Hyaline | 2.8 ± 5.7 | NS | 14.6 ± 18.3 | NS | 16.2 ± 6.8 | NS | <0.01 | 3≈2>1 |
| GS | 2.3 ± 4.8 | NS | 3.9 ± 5.6 | NS | 6.0 ± 0.7 | NS | 0.01 | 3>2≈1 |
| Cort fib | 2.4 ± 3.7 | NS | 7.3 ± 10.3 | NS | 7.5 ± 3.1 | NS | <0.01 | 3≈2>1 |
| Nglom | 1,051,442 ± 286,088 | NS | 871,486 ± 254,699 | NS | 1,118,975 ± 487,746a | NS | 0.08a | NS |
| Vglom | 6.33 ± 2.00 | NS | 8.08 ± 2.84 | NS | 6.29 ± 1.62 | NS | 0.31 | NS |
Age and APOL1 risk allele categories tested by Kruskal–Wallis 1-way analysis of variance with Dunn’s post hoc pairwise method.
Cort fib, cortical fibrosis; GS, glomerulosclerosis; Itc, intimal thickening of arcuate arteries as a percentage of the outer diameter of the artery wall; Itr, intimal thickening of interlobular arteries as a percentage of the outer diameter of the artery wall; MAP, mean arterial blood pressure (mm Hg); Nglom, total estimated glomerular number of right kidney; NS, not significant; PW, pairwise comparisons, P < 0.05; Vglom, average glomerular volume (μm3 × 106).
Includes an 83-year-old woman having 2 G1 alleles and 1,678,280 glomeruli. This is greater than the 95th percentile of Nglom (1,416,713) for subjects ≥40 years old and could be considered an outlier. For age ≤70 years, the loss of glomeruli for subjects with 2 APOL1 risk alleles is significant by linear regression (P = 0.04, coefficient = –9311 glomeruli per year).
There were no significant differences in Nglom, overall or by age categories between African Americans and whites. With similar Nglom, African Americans had significantly larger Vglom than whites in the age groups 18 to 34 among subjects with no risk alleles (P = 0.01). There was a trend for Vglom to increase with increasing arterionephrosclerosis that was significant for whites (P < 0.01) and for African Americans with 1 risk allele but not with none or 2 risk alleles.
Relationships Between Microstructural Characteristics and Clinical Features
In multivariable analysis (Table 4), model 1 compares African Americans and whites having no risk alleles. The microstructural features of Itc, Itr, arteriolar hyalinization, glomerulosclerosis, and cortical fibrosis are dependent variables, and race, age, sex, BMI, hypertension, treatment of hypertension, CAD/CVD, and diabetes are independent variables. In this model, age and hypertension are strongly related to Itc, Itr, glomerulosclerosis, and cortical fibrosis (all, P ≤ 0.001). African American race is significantly related only to Itr (P = 0.01) and glomerulosclerosis (P < 0.05). Arteriolar hyalinization is related to hypertension (P < 0.001), CAD/CVD (P = 0.04), and diabetes (P = 0.001) but not to race (P = 0.89).
Table 4.
Multivariable analysis of microstructural features of arterionephrosclerosis and clinical characteristics
| Model 1: African Americans and whites (both with no risk alleles) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Feature | AA race | Age (yr) | Male sex | BMI | HTN | RxHTN | CAD/CVD | Diabetes |
| Itc | 1.45 | 0.30 | 0.08 | 0.04 | 5.37 | 0.45 | 2.42 | 0.26 |
| –0.40 to 3.31 | 0.21 to 0.39 | –1.68 to 1.84 | –0.14 to 0.05 | 3.11 to 7.63 | –2.13 to 3.02 | 0.37 to 4.47 | –5.02 to 5.53 | |
| 0.12 | <0.001 | 0.93 | 0.40 | <0.001 | 0.73 | 0.02 | 0.92 | |
| Itr | 2.54 | 3.69 | 1.00 | –0.06 | 4.91 | –0.15 | 3.24 | –2.79 |
| 0.67 to 4.43 | 1.99 to 5.39 | –0.77 to 2.79 | –0.17 to 0.05 | 2.63 to 7.19 | –2.78 to 2.48 | 1.14 to 5.35 | –8.13 to 2.54 | |
| 0.01 | <0.001 | 0.26 | 0.26 | <0.001 | 0.91 | 0.01 | 0.30 | |
| Hyaline | 0.29 | 0.08 | –0.27 | –0.21 | 12.8 | –2.09 | 4.98 | 16.5 |
| –4.10 to 4.69 | –0.11 to 0.22 | –4.45 to 3.90 | –0.45 to 0.03 | 7.5 to 18.2 | –8.20 to 4.01 | 0.14 to 9.82 | 4.0 to 28.9 | |
| 0.89 | 0.51 | 0.90 | 0.09 | <0.001 | 0.50 | 0.04 | 0.001 | |
| GS | 1.36 | 0.03 | –0.47 | –0.04 | 2.38 | –0.76 | 0.92 | 2.56 |
| 0.01 to 2.71 | 0.02 to 0.04 | –1.75 to 0.81 | –0.12 to 0.03 | 0.95 to 4.02 | –2.14 to 1.12 | –0.57 to 2.40 | –1.27 to 6.38 | |
| 0.048 | <0.001 | 0.47 | 0.19 | <0.01 | 0.42 | 0.22 | 0.19 | |
| Cort fib | 1.24 | 0.02 | 0.01 | –0.01 | 2.91 | –0.01 | –0.27 | 3.48 |
| –0.25 to 2.74 | 0.01 to 0.03 | –1.40 to 1.42 | –0.09 to 0.07 | 1.11 to 4.72 | –2.09 to 2.08 | –1.92 to 1.38 | –0.75 to 7.72 | |
| 0.10 | <0.001 | 0.99 | 0.82 | 0.01 | 0.99 | 0.75 | 0.11 | |
| Model 2: African Americans | ||||||||
|---|---|---|---|---|---|---|---|---|
| Feature | APOL1 | Age (yr) | Male sex | BMI | MAP | RxHTN | CAD/CVD | Diabetes |
| Itc | 0.58 | 0.31 | –1.45 | –0.12 | 0.23 | –1.46 | 1.07 | NT |
| –0.91 to 2.08 | 0.21 to 0.41 | –3.76 to 0.86 | –0.23 to –0.01 | 0.15 to 0.32 | –4.1 to 1.23 | –1.4 to 3.5 | ||
| 0.44 | <0.001 | 0.22 | 0.03 | <0.001 | 0.39 | 0.39 | ||
| Itr | 1.44 | 0.27 | 0.95 | –0.13 | 0.27 | –2.91 | 1.90 | NT |
| –0.24 to 3.12 | 0.16 to 0.390 | –1.65 to 3.56 | –0.25 to 0.007 | 0.18 to 0.37 | –5.95 to 0.13 | –1.07 to 4.5 | ||
| 0.09 | <0.001 | 0.47 | 0.04 | <0.001 | 0.06 | 0.23 | ||
| Hyaline | –1.12 | 0.10 | –0.68 | –0.31 | 0.48 | –3.74 | 3.37 | NT |
| –4.3 to 2.0 | –0.11 to 0.31 | –5.5 to 4.2 | –0.53 to –0.07 | 0.31 to 0.66 | –9.4 to 2.0 | –1.9 to 8.6 | ||
| 0.48 | 0.37 | 0.78 | 0.01 | <0.001 | 0.20 | 0.20 | ||
| GS | 0.16 | 0.15 | –0.37 | –0.04 | 0.11 | –2.23 | –0.64 | NT |
| –0.98 to 1.3 | 0.07 to 0.22 | –2.1 to 1.4 | –0.13 to 0.04 | 0.04 to 0.17 | –4.29 to –0.17 | –2.5 to 1.2 | ||
| 0.78 | <0.001 | 0.68 | 0.29 | 0.001 | 0.03 | 0.50 | ||
| Cort fib | 0.46 | 0.10 | –0.52 | –0.09 | 0.19 | –2.38 | –2.05 | NT |
| –0.99 to 1.91 | 0.004 to 0.19 | –2.7 to 1.7 | –0.20 to 0.01 | 0.11 to 0.27 | –4.99 to 0.24 | –4.45 to 0.35 | ||
| 0.53 | 0.04 | 0.65 | 0.09 | <0.001 | 0.08 | 0.09 | ||
Correlation coefficients derived from fractional polynomials of untransformed data. Within the cells, the upper values are correlation coefficients with 95% confidence intervals and the lower values are P values. Significant values are in bold.
AA, African American; APOL1, genotype designated as 0, 1, and 2 risk alleles; BMI, body mass index (kg/m2); CAD/CVD, coronary artery disease/cerebrovascular disease; Cort fib, cortical fibrosis; GS, glomerulosclerosis; HTN, hypertension; Itc, intimal thickening of arcuate arteries as a percentage of the outer diameter of the artery wall; Itr, intimal thickening of interlobular arteries as a percentage of the outer diameter of the artery wall; MAP, mean arterial pressure (mm Hg); NT, not tested, no African Americans with diabetes and 2 risk alleles; RxHTN, treated hypertension.
In model 2, which is applied only to African Americans, APOL1 genotype replaces race as an independent variable, the continuous variable of MAP is substituted for the dichotomous variable of hypertension, and diabetes is dropped because of an absence of diabetics among subjects with 2 risk alleles. In this model, increased age and MAP were significantly associated with Itc, Itr, glomerulosclerosis, and cortical fibrosis (all, P ≤ 0.001), but APOL1 genotype was not significantly associated with any of the features of arterionephrosclerosis and approached significance only for Itr (P = 0.09). In model 2, all of the microscopic features of arterionephrosclerosis were negatively associated with treatment of hypertension. The reduced glomerulosclerosis was significant (P = 0.03), and a reduction in Itr tended toward significance (P = 0.06). There was a curious negative association between BMI and the severity of arterionephrosclerosis among African Americans (Figure 1) that was significant for Itc (P = 0.03), Itr (p = 0.04), and arteriolar hyalinization (P = 0.01). This was possibly because of a not-significant trend toward earlier death among overweight and obese African Americans but not whites (age vs. BMI: African Americans, r = –0.102, coefficient = –0.167, P = 0.11; whites, r = 0.070, coefficient 0.113, P = 0.41) that suggested that arterionephrosclerosis in African Americans may not have had time to more fully develop.
Figure 1.

Arteriolar hyalinization versus body mass index among African Americans. Shown is an inverse relationship by polynomial regression between the percent hyalinized arterioles and body mass index (BMI). The 2-way relationship is not significant (r = –0.73, P = 0.21, coefficient = –0.12), but the relationship becomes significant in multivariable regression with age and hypertension (age: coefficient = 0.21, P = 0.04; BMI: coefficient = –0.25, P = 0.04; hypertension: coefficient = 11.83, P < 0.001; α = 0.05:1.000).
Vascular Pathology: Relationships to Age, Blood Pressure, and APOL1 Genotype
Increased Itc and Itr began to be seen in late adolescence (Figure 2a). Under 35 years old, Itc averaged 2.3-fold Itr, with the difference diminishing to 1.3-fold Itr at 55 years of age and older. When Itc is plotted on the y-axis against Itr on the x-axis, the regression line is fitted down toward the x-axis and shifted slightly to the right, indicating an earlier, more pronounced pathology of Itc compared to Itr (Figure 2b). Among African Americans, the relationships between vascular changes and age by APOL1 genotype for Itr, Itc, and arteriolar hyalinization are shown in Figure 3, Figure 4, Figure 5. Two-way correlations between age and Itr and between age and Itc showed a strong direct relationship for all 3 risk genotypes (P < 0.001) with the slope of the regressions for none, 1, and 2 APOL1 risk alleles, respectively, being r = 0.68, r = 0.68, and r = 0.73 for Itc; and r = 0.55, r = 0.52, and r = 0.67 for Itr. The differences in the regressions between 2 and none or 1 risk allele were not significant for either level of blood vessel (see figure legends). Nevertheless, the plots demonstrated a greater elevation of Itr and to a lesser extent Itc in subjects with 2 risk alleles beginning at about 35 years of age, but with no such separation being seen for arteriolar hyalinization.
Figure 2.

Intimal thickening of arcuate and interlobular arteries. (a) Arterial intimal thickening for arcuate (Itc) and interlobular (Itr) arteries. Linear regression showing the relationships between age and Itc (r = 0.58, P < 0.001, coefficient = 0.961) and age and Itr (r = 0.48, P < 0.001, coefficient = 0.791). The coefficients represent the percent increase in intimal thickening per year after 18 years old. All subjects are included. The slopes of the regressions are not significantly different (P = 0.98). (b) Intimal thickening of arcuate (Itc) versus interlobular arteries (Itr). Linear regression between Itc and Itr. The relationship is shifted to the right and downward along the x-axis, indicating that Itc begins somewhat before and is more pronounced than Itr in the early development of arteriosclerosis.
Figure 3.

Arcuate artery intimal thickening (Itc) versus age by APOL1 risk allele status for African Americans. Shown are relationships by polynomial regression between age and Itc by APOL1 genotype (0 risk alleles, r = 0.68, P < 0.001, coefficient = 0.46; 1 risk allele, r = 0.68, P < 0.001, coefficient = 0.45; 2 risk alleles, r = 0.73, P > 0.001, coefficient = 0.51). There is no significant difference between the alleles (0 vs. 1, P = 0.75; 0 vs. 2, P = 0.40; 1 vs. 2, P = 0.66).
Figure 4.

Interlobular artery (Itr) intimal thickening versus age by APOL1 risk allele status for African Americans. Shown are relationships by polynomial regression between age and Itr by APOL1 genotype (0 risk alleles, r = 0.55, P < 0.001, coefficient = 0.36; 1 risk allele, r = 0.52, P < 0.001, coefficient = 0.33; 2 risk alleles, r = 0.67, P > 0.001, coefficient = 0.56). There is no significant difference between the alleles (0 vs. 1, P = 0.77; 0 vs. 2, P = 0.22; 1 vs. 2, P = 0.36), but Itr for 2 risk alleles begins to separate from 0 and 1 risk allele at about 35 years of age.
Figure 5.
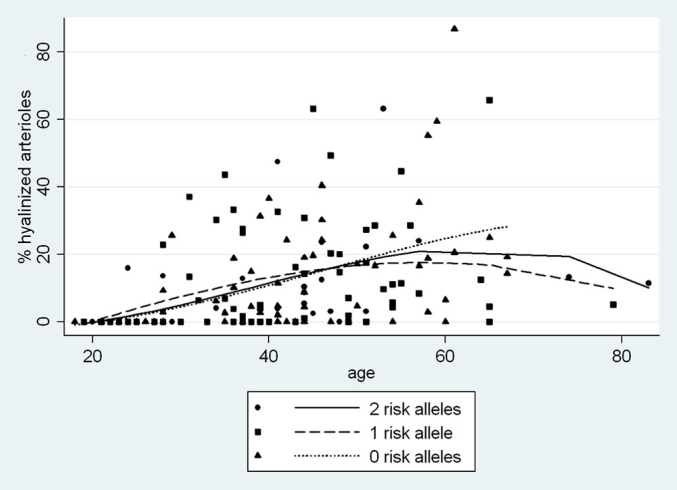
Arteriolar hyalinization versus age by APOL1 risk allele status for African Americans. Shown are relationships by polynomial regression between age and percent hyalinized arterioles by APOL1 genotype (0 risk alleles, r = 0.48, P < 0.001, coefficient = 0.64; 1 risk allele, r = 0.26, P = 0.04, coefficient = 0.34; 2 risk alleles, r = 0.37, P = 0.04, coefficient = 0.36). There is no significant difference between the alleles (0 vs. 1, P = 0.89; 0 vs. 2, P = 0.66; 1 vs. 2, P = 0.65).
Table 5 shows the effects of blood pressure, race, and APOL1 genotype on arteriosclerosis in multivariable analysis for all age groups. With no risk alleles, Itc, Itr, and arteriolar hyalinization were determined by MAP (P < 0.001) without a significant contribution from race. Among African Americans, all aspects of arteriosclerosis were strongly determined by MAP for all risk genotypes (P < 0.001 for all) with the correlation coefficients indicating a more pronounced severity of Itc and Itr for subjects with 2 APOL1 risk alleles compared to none or 1 risk allele. Table 6 illustrates the effect of age on the relationships between APOL1 risk genotype and arteriosclerosis among African Americans by comparing all age groups with subjects ≥35 years old. When the influence of subjects in the 18-to-34-year-old age group who have little or no hypertension or arteriosclerosis are included, the differences in the effects of MAP on Itr with 2 risk alleles compared to none or 1 risk allele approach but do not achieve significance (P = 0.08). The differences, however, become significant when only subjects ≥35 years old are analyzed (P = 0.02).
Table 5.
Multivariable analyses of arteriosclerosis versus mean arterial blood pressure comparing race and APOL1 risk genotype
| African American and white | ||||
|---|---|---|---|---|
| Risk genotype | Coeff. (95% CI) | P | Coeff. (95% CI) | P |
| 0 risk alleles | Itc | Itc transformed | ||
| MAP | 0.34 (0.25 to 0.42) | <0.001 | 0.007 (0.005 to 0.008) | <0.001 |
| African American | –0.11 (–2.62 to 2.40) | 0.93 | –0.008 (–0.059 to 0.043) | 0.74 |
| 0 risk alleles | Itr | Itr transformed | ||
| MAP | 0.31 (0.23 to 0.39) | <0.001 | 0.007 (0.005 to 0.009) | <0.001 |
| African American | 0.64 (–1.75 to 3.04) | 0.60 | 0.009 (–0.043 to 0.062) | 0.71 |
| 0 risk alleles | Hyaline | Hyaline transformed | ||
| MAP | 0.65 (0.45 to 0.84) | <0.001 | 0.011 (0.008 to 0.014) | <0.001 |
| African American | –4.37 (–10.06 to 1.31) | 0.13 | –0.64 (–0.151 to 0.023) | 0.15 |
| African Americans | ||||
| Itc vs. MAP | Itc transformed vs. MAP | |||
| 0 risk alleles | 0.34 (0.25 to 0.42) | <0.001 | 0.007 (0.005 to 0.008) | <0.001 |
| 1 risk allele | 0.20 (0.10 to 0.30) | <0.001 | 0.004 (0.002 to 0.006) | <0.001 |
| 2 risk alleles | 0.45 (0.22 to 0.69) | 0.001 | 0.007 (0.003 to 0.011) | 0.001 |
| Itr vs. MAP | Itr transformed vs. MAP | |||
| 0 risk alleles | 0.32 (0.24 to 0.40) | <0.001 | 0.007 (0.004 to 0.009) | <0.001 |
| 1 risk allele | 0.22 (0.11 to 0.33) | <0.001 | 0.004 (0.002 to 0.007) | 0.001 |
| 2 risk alleles | 0.56 (0.28 to 0.85) | <0.001 | 0.010 (0.005 to 0.014) | <0.001 |
| Hyaline vs. MAP | Hyaline transformed | |||
| 0 risk alleles | 0.62 (0.43 to 0.81) | <0.001 | 0.010 (0.008 to 0.014) | <0.001 |
| 1 risk allele | 0.41 (0.18 to 0.64) | 0.001 | 0.007 (0.003 to 0.012) | 0.001 |
| 2 risk alleles | 0.14 (0.03 to 0.24) | 0.013 | 0.007 (0.001 to 0.012) | 0.017 |
Correlation coefficients derived from fractional polynomial regressions for untransformed and linear regression for transformed data. Number of subjects with mean arterial blood pressure: 0 risk alleles: white, n = 65; African American, n = 45. 1 risk allele: African American, 46; 2 risk alleles: African American, 22.
CI, confidence interval; Itc, intimal thickening of arcuate arteries as a percentage of the outer diameter of the artery wall; Itr, intimal thickening of interlobular arteries as a percentage of the outer diameter of the artery wall; MAP, mean arterial blood pressure (mm Hg).
Table 6.
Multivariable analyses of arteriosclerosis versus mean arterial blood pressure among African Americans by APOL1 risk genotype and by age
| African Americans, all age groups | ||||
|---|---|---|---|---|
| Risk genotype | Coeff. (95% CI) | P | Coeff. (95% CI) | P |
| Itc | Itc transformed | |||
| MAP (mm Hg) | 0.39 (0.28 to 0.51) | <0.001 | 0.004 (0.003 to 0.007) | <0.001 |
| 0 vs. 2 risk alleles | 1.16 (–0.76 to 3.07) | 0.23 | 0.02 (–0.01 to 0.05) | 0.25 |
| 0 vs. 1 risk allele | –0.31 (–3.04 to 2.42) | 0.82 | 0.005 (–0.05 to 0.06) | 0.82 |
| 1 vs. 2 risk alleles | 2.25 (–1.43 to 5.93) | 0.23 | 0.03 (–0.04 to 0.09) | 0.45 |
| Itr | Itr transformed | |||
| MAP (mm Hg) | 0.40 (0.28 to 0.52) | <0.001 | 0.006 (0.004 to 0.008) | <0.001 |
| 0 vs. 2 risk alleles | 1.90 (–0.21 to 4.0) | 0.08 | 0.03 (–0.01 to 0.07) | 0.14 |
| 0 vs. 1 risk allele | –0.17 (–2.93 to 2.59) | 0.90 | 0.004 (–0.06 to 0.07) | 0.88 |
| 1 vs. 2 risk alleles | 3.72 (–0.51 to 7.96) | 0.08 | 0.05 (–0.03 to 0.13) | 0.23 |
| Hyaline | Hyaline transformed | |||
| MAP (mm Hg) | 0.61 (0.36 to 0.85) | <0.001 | 0.009 (0.006 to 0.014) | <0.001 |
| 0 vs. 2 risk alleles | –0.53 (–3.68 to 2.61) | 0.73 | –0.001 (–0.05 to 0.05) | 0.96 |
| 0 vs. 1 risk allele | –0.56 (–6.0 to 4.85) | 0.84 | –0.02 (0.11 to 0.08) | 0.74 |
| 1 vs. 2 risk alleles | –0.77 (–7.53 to 5.98) | 0.82 | 0.01 (–0.11 to 0.12) | 0.90 |
| African Americans ≥35 years old | ||||
| Itc | Itc transformed | |||
| MAP (mm Hg) | 0.24 (0.17 to 0.45) | <0.001 | 0.004 (0.003 to 0.007) | <0.001 |
| 0 vs. 2 risk alleles | 1.73 (–0.65 to 4.1) | 0.15 | 0.03 (–0.01 to 0.06) | 0.17 |
| 0 vs. 1 risk allele | –0.59 (–3.6 to 2.4) | 0.70 | –0.004 (–0.05 to 0.05) | 0.89 |
| 1 vs. 2 risk alleles | 4.26 (–0.29 to 8.8) | 0.07 | 0.06 (–0.15 to 0.13) | 0.10 |
| Itr | Itr transformed | |||
| MAP (mm Hg) | 0.33 (0.17 to 0.49) | <0.001 | 0.005 (0.003 to 0.007) | <0.001 |
| 0 vs. 2 risk alleles | 3.25 (0.52 to 6.0) | 0.021 | 0.06 (0.01 to 0.10) | 0.019 |
| 0 vs. 1 risk allele | –0.19 (–3.5 to 3.1) | 0.91 | 0.001 (–0.07 to 0.01) | 0.99 |
| 1 vs. 2 risk alleles | 6.79 (1.3 to 12.3) | 0.017 | 0.12 (0.02 to 0.21) | 0.016 |
| Hyaline | Hyaline transformed | |||
| MAP (mm Hg) | 0.53 (0.34 to 0.84) | <0.001 | 0.009 (0.006 to 0.06) | <0.001 |
| 0 vs. 2 risk alleles | –1.34 (–5.5 to 2.9) | 0.53 | –0.01 (–0.08 to 0.05) | 0.66 |
| 0 vs. 1 risk allele | –1.25 (–7.6 to 5.1) | 0.69 | –0.03 (–0.13 to 0.08) | 0.58 |
| 1 vs. 2 risk alleles | –1.19 (–9.9 to 7.5) | 0.78 | 0.01 (–0.14 to 0.14) | 0.94 |
Correlation coefficients derived from fractional polynomial regressions for untransformed and linear regression for transformed data. Risk allele regressions are 2-way comparisons between 0 and 2, 0 and 1, and 1 and 2 risk alleles. Transformed values = log10 (y + 10). Significant values are in bold. Risk alleles: number of subjects ≥35 years old with MAP (n); power of tests. 0: African American (n = 37); by race, α = 0.05 (Itc, Itr, hyaline, ≥: 0.997). 1: African American (n = 35); 0 vs. 1, α = 0.05 (Itc: 0.993, Itr: 0.992, hyaline: 0.999). 2: African American (n = 14); 0 vs. 2, α = 0.05 (Itc: 0.993, Itr: 0.995, hyaline: 0.994). 1 vs. 2, α = 0.05 (Itc: 0.963, Itr: 0.987, hyaline: 0.994).
CI, confidence interval; Itc, intimal thickening of arcuate arteries as a percentage of the outer diameter of the artery wall; Itr, intimal thickening of interlobular arteries as a percentage of the outer diameter of the artery wall; MAP, mean arterial blood pressure.
Figure 6 demonstrates that despite similar levels of blood pressure, African Americans ≥35 years old with 2 APOL1 risk alleles demonstrated increased Itr for any increase in MAP over subjects with no (P = 0.02) or 1 risk allele (P = 0.02). As shown in Table 7, the greater difference in Itr was observed despite the treatment of hypertension producing a significant reduction in Itr in subjects with 2 risk alleles (coefficient = –14.3, P = 0.04) but not in subjects with none (coefficient = –0.17, P = 0.95) or 1 (coefficient = –0.81, P = 0.77) risk allele. A phenotype analysis of Itr and the response of Itr to blood pressure (Itr/MAP) demonstrates that after age 34, subjects with 2 risk alleles had a greater degree of interlobular artery intimal thickening in relationship to blood pressure than subjects with none or 1 risk allele, with the relationship to blood pressure being significant (P = 0.02). These are findings consistent with a recessive rather than a dominant or additive trait (Table 8).
Figure 6.

African Americans ≥35 years old. Interlobular artery intimal thickening (transformed) versus MAP by APOL1 risk allele status. Shown are relationships by linear regression between the percent interlobular artery intimal thickening (Itr) transformed as log10 (Itr + 10) and MAP by APOL1 genotype. In multivariable regression, there is a significant difference between the alleles (0 vs. 1, P = 0.99, normality = 0.72, constant variance = 0.23, α = 0.05:0.986; 0 vs. 2, P = 0.019, normality = 0.24, constant variance = 0.58, α = 0.05:0.996; 1 vs. 2, P = 0.016, normality = 0.65, constant variance = 0.08, α = 0.05:0.982).
Table 7.
Multivariable analyses of Itr versus mean arterial blood pressure and treatment of hypertension by APOL1 risk genotype in African Americans ≥35 years old
| Characteristic | Itr | Itr transformed | ||
|---|---|---|---|---|
| 2 risk alleles, n = 14 | Coeff. (95% CI) | P | Coeff. (95% CI) | P |
| MAP (mm Hg) | 0.59 (0.16 to 1.01) | 0.012 | 0.007 (0.002 to 0.013) | 0.016 |
| RxHTN, n = 4 | –14.30 (–29.0 to –0.55) | 0.043 | –0.19 (–0.37 to 0.01) | 0.041 |
| α = 0.05:854 normality = 0.81 |
α = 0.05:832 normality = 0.66 |
|||
| 1 risk allele, n = 35 | ||||
| MAP (mm Hg) | 0.21 (0.05 to 0.36) | 0.012 | 0.004 (0.001 to 0.007) | 0.017 |
| RxHTN, n = 13 | –0.81 (–6.4 to 4.7) | 0.77 | –0.033 (–0.15 to 0.08) | 0.57 |
| α = 0.05:791 normality = 0.13 |
α = 0.05:713 normality = 0.42 |
|||
| 0 risk alleles, n = 37 | ||||
| MAP (mm Hg) | 0.27 (0.11 to 0.44) | <0.01 | 0.005 (0.002 to 0.009) | <0.01 |
| RxHTN, n = 11 | –0.17 (–5.6 to 5.3) | 0.95 | 0.007 (–0.10 to 0.12) | 0.89 |
| α = 0.05:914 normality = 0.04 |
α = 0.05:920 normality = 0.31 |
|||
Linear regressions are shown with α and normality tests. Significant P values for treatment effect are in bold.
Itr, % intimal thickening of interlobular arteries; Itr transformed, log10 (Itr + 10); MAP, mean arterial blood pressure; RxHTN, treatment of hypertension.
Table 8.
African Americans ≥35 years old
| Risk genotype | Itr | Itr/MAP |
|---|---|---|
| 0 | 11.0 ± 8.2 | 0.09 ± 0.07 |
| 1 | 11.0 ± 7.8 | 0.09 ± 0.06 |
| 2 | 15.9 ± 13.3 | 0.15 ± 0.10 |
| P, 0 vs. 1 | 0.88 | 0.80 |
| P, 0 vs. 2 | 0.15 | 0.02 |
| P, 1 vs. 2 | 0.19 | 0.03 |
Comparisons by Wilcoxon rank sum tests between APOL1 risk allotypes for Itr and Itr divided by MAP. The Itr (% intimal thickening of interlobular arteries) phenotype as a response to mean arterial blood pressure (MAP) is recessive, that is, risk genotype 2 > risk genotype 0 or 1.
Discussion
This work represents a cross-sectional study of subjects selected by autopsy from the general population of Jackson, Mississippi. As such, it evaluates the microstructural kidney changes in adult subjects among whom whites serve as age-, gender-, and BMI-matched case controls for African Americans with no risk alleles, and African Americans with no risk alleles serve as case controls for those with 1 or 2 risk alleles. The collection criteria excluded clinical renal disease including diabetic nephropathy. While diabetes and cancer were underrepresented and sudden death by misadventure overrepresented, the sample contained the high frequency of CAD/CVD deaths, hypertension, and obesity that is characteristic of the general Mississippi population.25, 26
Jackson is 1 of the 4 communities participating in the ARIC study, which has recently published results of a more than 20-year follow-up of age-related estimated GFR (eGFR) decline among African Americans and whites ≥45 years old at baseline among whom white participants served as the reference population.24, 27 African Americans with none or 1 APOL1 risk alleles were defined as low risk and those with 2 risk alleles as high risk. Both low- and high-risk APOL1 genotypes had higher rates of new-onset hypertension and diabetes and more rapid eGFR decline than whites. APOL1 high-risk subjects had a more rapid eGFR decline and higher rates of ESKD despite having rates of hypertension and diabetes similar to those of low-risk African Americans. We recently reported that African Americans carrying 2 APOL1 risk alleles have an exaggerated glomerular loss during the first 30 to 40 years of adult life compared to subjects with none or 1 risk allele.20 This corresponds to the results of the ARIC study with glomerulosclerosis and nephron loss being the microstructural counterpart of declining eGFR.
In the present study, we show that older age and hypertension were associated with increased glomerulosclerosis and arteriosclerosis beginning at about age 35 in both African Americans and whites. Aging African Americans had more severe manifestations of arterionephrosclerosis than whites that might be attributed to their more severe hypertension, but with subjects with 2 APOL1 risk alleles exhibiting, in a recessive model, a more pronounced degree of fibrous intimal thickening in the interlobular arteries for any level of increased blood pressure. We also show that there were no racial or APOL1 risk allele differences in arteriosclerosis before age 35, implying that interlobular artery differences were acquired and not a preceding feature of race or APOL1 risk status. It is notable that the more pronounced intimal thickening after age 35 was found despite antihypertensive treatment having a negative effect on interlobular artery intimal thickening that was significant for 2 risk alleles but not none or 1 risk alleles. This suggests that without treatment the differences might be even more pronounced.
The data that we present on arterial intimal thickening and the differences between close or arcuate arteries (Itc) and remote or interlobular arteries (Itr) are very similar to those reported by Tracy et al.13, 18 in their autopsy studies on the African American population of New Orleans. Our findings support their concept that the anatomic levels of renal blood vessels respond differently to age and hypertension, and further indicate a more enhanced effect of blood pressure in the presence of 2 APOL1 risk variants. The effect of APOL1 status was found primarily in the distal, interlobular arteries and was present but not as notably in proximal, arcuate arteries. Arteriolar hyalinization had no relationship to APOL1 genotype, a finding congruent with the observation by Tracy et al.18 that arteriolar hyalinization had little relationship to race.
The glomerulosclerosis occurring in these kidneys was nearly entirely FGGS, with FSGS being found in only 3 subjects, all with severe hypertension and with only 1 of the subjects having a single APOL1 risk allele. While the individual features of FGGS, arteriosclerosis, and cortical fibrosis correlate closely with each other, it is not at all clear how arterionephrosclerosis advances to ESKD. The intimal thickening of small renal arteries might in time produce enough FGGS that GFR would diminish to a level considered late-stage CKD. This is suggested by Elsherbiny et al.19 in 6-month postoperative evaluations of Mayo Clinic kidney donors. Rather than showing a compensatory increase, the (single-kidney) GFR of 33% of the donors was <60 ml/min per 1.73 m2 with lower function being predicted by FGGS in pre-donation biopsies.
Nevertheless, other biopsy studies indicate that at least part of the progression of arterionephrosclerosis to the later stages of CKD is not simply an accrual of FGGS.16, 17, 28, 29, 30 Biopsies of hypertension-associated nephropathy from African Americans, including those from African American Study of Kidney Disease and Hypertension (AASK) patients, demonstrate proportionately less FGGS and more FSGS and solidified glomeruli than those from whites.28, 29 More recently Larsen et al.30 examined the effect of APOL1 variants on biopsies of arterionephrosclerosis and found an increased frequency of FSGS and solidified glomeruli in subjects with 2 APOL1 risk alleles. These biopsies were performed for reduced renal function with an average serum creatinine of 2.9 mg/dl in AASK patients and 3.6 to 4.3 mg/dl in the APOL1 study and imply that the structural alterations of arterionephrosclerosis might change in the later stages of CKD.10, 28, 29, 30
The possibility of fundamental differences in the mechanisms involved in early- and late-stage hypertension-attributed renal disease and their relationship to APOL1 status might be inferred from the study of Lipkowitz et al.,31 who found that AASK patients with APOL1 risk variants were more likely to have progressive disease than a control group of African Americans and that progression was unrelated to blood pressure control. The possible transition may be related to a more exaggerated decompensation of renal function in African Americans with 2 APOL1 risk alleles in which the initiating changes are related to FGGS and are superseded by FSGS with progressive disease.16, 19, 31
FGGS and FSGS appear to have different underlying pathogenetic mechanisms that in the context of arterionephrosclerosis may involve variations in glomerular volume. FSGS, particularly adaptive FSGS, has been related to larger glomerular volume and a depletion of podocytes covering the expanded glomerular capillary surfaces.32, 33, 34
The estimation of Vglom represents an average glomerular volume for the entire kidney and provides no information about the distribution of individual glomerular sizes. We have analyzed individual glomerular volume (IGV) in our autopsy series and have found increased IGV variability in older, hypertensive subjects that is most marked in African Americans.35 The variability is skewed toward larger size with some glomeruli becoming very large. The skew is possibly secondary to glomerular loss owing to FGGS and may identify a pool of glomeruli susceptible to segmental sclerosis in the later stages of CKD.
The current study is limited by the small number of subjects with 2 risk alleles. The P = 0.02 for Itr leaves an approximately 2% to 3% risk for false positive results. Nevertheless, this is the largest study of its kind and includes detailed morphometric and stereologic analyses that will be difficult to repeat. As an autopsy study, it is restricted to subjects selected by death due to causes unrelated to clinical renal disease and provides little or no indication of structural factors other than the general features of arterionephrosclerosis that might indicate a risk for ESKD. This is reflected by our presentation of 3 African Americans aged 74, 79, and 83 years old whose deaths were neither vascular nor renal. One had a single and 2 had 2 APOL1 risk alleles. All were hypertensive and had from 4.5% to 6.7% glomerulosclerosis, findings that emphasize that subjects with APOL1 risk variants can live to old age with a degree of arterionephrosclerosis that probably represents a continuum of normal for the elderly. In keeping with a vascular mechanism, we demonstrate that subjects with 2 APOL1 risk alleles died of arteriosclerotic cardiac and cerebrovascular diseases at an average of 6.7 years younger than African Americans with no risk alleles, possibly corresponding to the greater cardiovascular death rate for risk allele variants reported by Ito et al.36
While the current findings indicate a primary role for the vascular phenotype in the initial stages of APOL1-associated arterionephrosclerosis, there is a considerable gap between autopsy and biopsy studies in renal function and structure that are important to our understanding of the effects of aging and hypertension on the kidney. Many of our concepts about hypertensive renal disease are derived from experimental models that have an inflammatory component and produce glomerular injury characterized by FSGS.37, 38 The role of inflammation in human hypertension is becoming more appreciated. APOL1 protein may, itself, be an inflammatory mediator through mechanisms involving apoptosis and autophagy with the potential signaling pathways offering opportunities for investigating influences on the structural attributes of arteries and glomeruli.7, 39, 40
Disclosure
All the authors declared no competing interests.
Acknowledgments
The study has been supported by a project grant from the National Health and Medical Research Council (NHMRC) of Australia (#194276), by an untied grant from the Colonial Foundation of Australia (to WEH, 2001–2011), by an NHMRC Australian Fellowship (#511081), by the National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; R01-DK065970-01), and by a grant from the American Heart Association, Southeastern Affiliate (2001–2003). This work was supported in part by the Intramural Research Program of the NIDDK, NIH, and by federal funds from the National Cancer Institute, NIH, under contract HHSN26120080001.
References
- 1.Freedman B.I., Skoreki K. Gene-gene and gene-environment interactions in apolipoprotein L1 gene-associated nephropathy. Clin J Am Soc Nephrol. 2014;9:2006–2013. doi: 10.2215/CJN.01330214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genovese G., Friedman D.J., Ross M.D. Association of trypanolytic APOL1 variants of kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freedman B.I., Kopp J.B., Langefeld C.D. The apolipoprotein L1 (APOL1) gene and non-diabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21:1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.US Renal Data System. USRDS 2004 Annual Data Report. Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. 2004. Bethesda, MD, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
- 5.Klag M.J., Whelton P.K., Randall B.L. Blood pressure and end-stage renal disease in men. N Engl J Med. 1996;334:13–18. doi: 10.1056/NEJM199601043340103. [DOI] [PubMed] [Google Scholar]
- 6.Kopp J.B., Nelson G.W., Sampath K. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kopp J.B. Rethinking hypertensive kidney disease: arterionephrosclerosis as a genetic, metabolic, and inflammatory disorder. Curr Opin Nephrol Hypertens. 2013;22:266–272. doi: 10.1097/MNH.0b013e3283600f8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson R.J., Feig D.I., Nakagawa T. Pathogenesis of essential hypertension: historical paradigms and modern insights. J Hypertension. 2008;26:381–391. doi: 10.1097/HJH.0b013e3282f29876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tracy R.E. Renal vasculature in essential hypertension: a review of some contrarian evidence. Contrib Nephrol. 2011;169:327–336. doi: 10.1159/000314908. [DOI] [PubMed] [Google Scholar]
- 10.Marcantoni C., Fogo A.B. A perspective on arterionephrosclerosis: from pathology to potential pathogenesis. J Nephrol. 2007;20:518–524. [PubMed] [Google Scholar]
- 11.Glassock R.J., Rule A.D. The implications of anatomical and functional changes in the aging kidney: with emphasis on glomeruli. Kidney Int. 2012;82:270–277. doi: 10.1038/ki.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughson M.D., Puelles V.G., Hoy W.E. Hypertension, glomerular hypertrophy and nephrosclerosis: the effect of race. Nephrol Dial Transplant. 2014;29:1399–1409. doi: 10.1093/ndt/gft480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tracy R.E., Velez-Duran M., Heigle T., Oalmann M.C. Two variants of nephrosclerosis separately related to age and blood pressure. Am J Pathol. 1988;131:270–282. [PMC free article] [PubMed] [Google Scholar]
- 14.Hughson M.D., Douglas-Denton R., Bertram J.F., Hoy W.E. Hypertension, glomerular number, and birth weight in African Americans and whites in the Southeastern United States. Kidney Int. 2006;69:671–678. doi: 10.1038/sj.ki.5000041. [DOI] [PubMed] [Google Scholar]
- 15.Hughson M.D., Gobe G., Douglas-Denton R.N. Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am J Kidney Dis. 2008;52:18–28. doi: 10.1053/j.ajkd.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 16.Hill G.S., Heudes D., Jacquot C. Morphometric evidence for impairment of renal autoregulation in advanced essential hypertension. Kidney Int. 2006;69:823–831. doi: 10.1038/sj.ki.5000163. [DOI] [PubMed] [Google Scholar]
- 17.Hill G.S. Hypertensive nephrosclerosis. Curr Opin Nephrol Hypertens. 2008;17:266–270. doi: 10.1097/MNH.0b013e3282f88a1f. [DOI] [PubMed] [Google Scholar]
- 18.Tracy R.E., Bhandaru S.Y., Oalmann M.C. Blood pressure and nephrosclerosis in black and white men and women aged 25-54. Mod Pathol. 1991;4:602–609. [PubMed] [Google Scholar]
- 19.Elsherbiny H.E., Alexander M.P., Kremers W.K. Nephron hypertrophy and glomerulosclerosis and their association with kidney function and risk factors among living kidney donors. Clin J Am Soc Nephrol. 2014;9:1892–1902. doi: 10.2215/CJN.02560314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoy W.E., Hughson M.D., Kopp J.B. APOL1 risk alleles are associated with exaggerated age-related changes in glomerular number and volume in African-American adults: an autopsy study. J Am Soc Nephrol. 2015;26:3179–3189. doi: 10.1681/ASN.2014080768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertram J.F. Counting in the kidney. Kidney Int. 2001;59:792–796. doi: 10.1046/j.1523-1755.2001.059002792.x. [DOI] [PubMed] [Google Scholar]
- 22.Bertram J.F. Analyzing renal glomeruli with the new stereology. Int Rev Cytol. 1995;161:111–172. doi: 10.1016/s0074-7696(08)62497-3. [DOI] [PubMed] [Google Scholar]
- 23.Atta M.G., Estrella M.M., Kuperman M. HIV-associated nephropathy patients with and without apolipoprotein L1 gene variants have similar clinical and pathological characteristics. Kidney Int. 2012;82:338–343. doi: 10.1038/ki.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster M.C., Coresh J., Formage M. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol. 2013;24:1484–1491. doi: 10.1681/ASN.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The State of Obesity: Better Policies for a Healthier America. Mississippi state obesity data, rates and trends. Available at: http://stateofobesity.org/states/ms.
- 26.Akil L., Ahmad H.A. Relationships between obesity and cardiovascular diseases in four Southern States and Colorado. J Health Care Poor Underserved. 2011;22:61–72. doi: 10.1353/hpu.2011.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grams M.E., Rebholz C.M., Chen Y. Race, APOL1 risk, and eGFR decline in the general population. J Am Soc Nephrol. 10 March 2016 doi: 10.1681/ASN.2015070763. [e-pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogo A., Breyer J.A., Smith M.C. Accuracy of the diagnosis of hypertensive nephrosclerosis in African Americans: a report from the African American study of kidney disease (AASK) trail. AASK Pilot Study Investigators. Kidney Int. 1997;51:244–252. doi: 10.1038/ki.1997.29. [DOI] [PubMed] [Google Scholar]
- 29.Marcantoni C., Ma L.-J., Federspiel C., Fogo A.B. Hypertensive nephrosclerosis in African Americans versus Caucasians. Kidney Int. 2002;62:172–180. doi: 10.1046/j.1523-1755.2002.00420.x. [DOI] [PubMed] [Google Scholar]
- 30.Larsen C.P., Beggs M.L., Saeed M. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod Pathol. 2015;28:95–102. doi: 10.1038/modpathol.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipkowitz M.S., Freedman B.I., Langefeld C.D., SK investigators Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans. Kidney Int. 2013;83:114–120. doi: 10.1038/ki.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wanner N., Hartleben B., Herbach N. Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol. 2014;25:707–716. doi: 10.1681/ASN.2013050452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puelles V.G., Douglas-Denton R.N., Cullen-McEwen L.A. Podocyte number in children and adults: associations with glomerular size and numbers of other glomerular resident cells. J Am Soc Nephrol. 2015;26:2277–2288. doi: 10.1681/ASN.2014070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukuda A., Chowdhury M.A., Ventkatareddy M.P. Growth dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol. 2012;23:1351–1363. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoy W.E., Hughson M.D., Diouf B. Distribution of volumes of individual glomeruli in kidneys at autopsy: association with physical and clinical characteristics and with ethnic group. Am J Nephrol. 2011;33:15–20. doi: 10.1159/000327044. [DOI] [PubMed] [Google Scholar]
- 36.Ito K., Bick A.G., Flannick J. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circ Res. 2014;114:845–850. doi: 10.1161/CIRCRESAHA.114.302347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian N., Thrasher K.D., Gundy P.D. Antioxidant treatment prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Hypertension. 2005;45:934–939. doi: 10.1161/01.HYP.0000160404.08866.5a. [DOI] [PubMed] [Google Scholar]
- 38.Tian N., Moore R.S., Phillips W.F. NAPD oxidase contributes to renal damage and dysfunction in Dahl salt-sensitive hypertension. Am J Physiol Regul Integr Physiol. 2008;295:R1858–R1865. doi: 10.1152/ajpregu.90650.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman D.J., Pollak M.R. Genetics of kidney failure and the evolving story of APOL1. J Clin Invest. 2011;201:3367–3374. doi: 10.1172/JCI46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhaorigetu S., Wan G., Kaini R. APOL1, a BH3-only lipid binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–1082. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]