

SUMMARY
The UbiB protein kinase-like (PKL) family is widespread—comprising one-quarter of microbial PKLs and five human homologs—yet its biochemical activities remain obscure. COQ8A (ADCK3) is a mammalian UbiB protein associated with ubiquinone (CoQ) biosynthesis and an ataxia (ARCA2) through unclear means. We show that mice lacking COQ8A develop a slowly progressive cerebellar ataxia linked to Purkinje cell dysfunction and mild exercise intolerance, recapitulating ARCA2. Interspecies biochemical analyses show that COQ8A and yeast Coq8p specifically stabilize a CoQ biosynthesis complex through unorthodox PKL functions. While COQ8 was predicted to be a protein kinase, we demonstrate that it lacks canonical protein kinase activity in trans. Instead, COQ8 has ATPase activity and interacts with lipid CoQ intermediates—functions that are likely conserved across all domains of life. Collectively, our results lend insight into the molecular activities of the ancient UbiB family and elucidate the biochemical underpinnings of a human disease.
eTOC Blurb
The ancient UbiB protein kinase-like (PKL) family has 5 human members (ADCK1–5), which are largely uncharacterized. Stefely et al. demonstrate that COQ8A (ADCK3) possesses conserved unorthodox protein kinase-like (uPKL) functionalities essential for maintaining a CoQ biosynthesis complex. Loss of COQ8A disrupts this complex, causing CoQ deficiency and cerebellar ataxia.
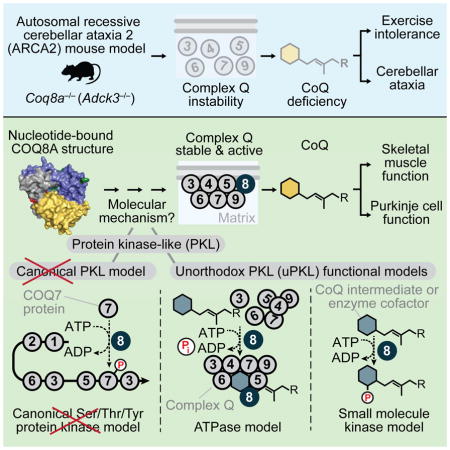
INTRODUCTION
Coenzyme Q (CoQ, ubiquinone) is a redox-active lipid found in all domains of life and virtually all cellular membranes (Crane et al., 1957; Lester and Crane, 1959; Morton, 1958; Sastry et al., 1961). CoQ functions as an essential component of the mitochondrial electron transport chain to enable oxidative phosphorylation, acts as a lipophilic antioxidant, and contributes to membrane structure. Yet, our understanding of CoQ biosynthesis remains incomplete. In the yeast Saccharomyces cerevisiae, at least 11 genes (coq1–coq9, yah1, arh1) are required for CoQ biosynthesis (Pierrel et al., 2010; Tran and Clarke, 2007), but the biochemical functions of Coq4p, Coq8p, and Coq9p remain unclear.
In humans, mutations in 9 putative CoQ biosynthesis genes have been linked to primary CoQ deficiencies, which affect diverse tissues and range from severe infantile multisystemic disease to adult-onset isolated myopathy (Laredj et al., 2014; Quinzii and Hirano, 2010). Tissue specificities likely reflect cell type-specific differences in CoQ production, uptake, and demand. Differences in clinical severity may be influenced by whether the mutated gene encodes a CoQ biosynthesis enzyme or a regulatory protein. Yet, no clear explanation exists for this extreme clinical heterogeneity, in part due to the absence of suitable animal models.
The most frequent form of hereditary CoQ deficiency, Autosomal Recessive Cerebellar Ataxia 2 (ARCA2, SCAR9), is a rare ataxia due to mutations in ADCK3, an ortholog of yeast coq8 (Lagier-Tourenne et al., 2008; Mollet et al., 2008). ARCA2 is characterized by slowly progressive gait abnormality, cerebellar atrophy, and moderate CoQ deficiency in skeletal muscle. More variable features include exercise intolerance, epilepsy, and intellectual deficiency. The molecular mechanisms underlying ARCA2 are obscure, largely because ADCK3 lacks a clear biochemical function.
ADCK3 belongs to the ancient UbiB family, which comprises one-quarter of all microbial protein kinase-like (PKL) genes (Kannan et al., 2007; Leonard et al., 1998; Oruganty et al., 2016) and has diverse biological functions in cell migration (Simpson et al., 2008), tumor cell viability (Brough et al., 2011; Wiedemeyer et al., 2010), and lipid metabolism (Lundquist et al., 2013; Martinis et al., 2013; Tan et al., 2013). The UbiB family has five mammalian members (ADCK1–5), which are classified as atypical kinases (Manning et al., 2002). ADCK4 is the closest homolog of ADCK3, and ADCK4 mutations cause a nephrotic syndrome associated with CoQ deficiency (Ashraf et al., 2013). Based on phylogenetic analyses defining ADCK3 and ADCK4 as co-orthologs of yeast Coq8p and functional studies presented in this manuscript, we propose to rename ADCK3 and ADCK4 as COQ8A and COQ8B, respectively.
While Coq8p and COQ8A have been linked to CoQ through genetic studies, whether these proteins act exclusively in CoQ biosynthesis is unknown. Moreover, the mild CoQ deficiency of ARCA2 has raised questions about the role of COQ8A in CoQ production. Coq8p and COQ8A localize to the matrix face of the inner mitochondrial membrane (Do et al., 2001; Pagliarini et al., 2008; Rhee et al., 2013; Tauche et al., 2008), where CoQ is produced. Additionally, using affinity enrichment mass spectrometry (AE-MS), we recently demonstrated that COQ8A and COQ8B interact with CoQ biosynthesis enzymes in a complex we term “complex Q” (Floyd et al., this issue of Mol Cell), consistent with reports that yeast proteins form a similar complex (He et al., 2014). In yeast, lack of coq8 perturbs post-translational modifications (PTMs) of Coq3p, Coq5p, and Coq7p (Xie et al., 2011), but whether this effect is direct or indirect—or whether these PTMs are important for complex Q function—is unknown.
The putative biochemical function of Coq8p, COQ8A, and the larger UbiB family is protein phosphorylation, but this idea remains largely untested and is based primarily on PKL sequence homology. Unorthodox PKL functions, including lipid kinase activity (Walker et al., 1999) and nucleotide-regulated conformational changes (Kung and Jura, 2016), are increasingly recognized. Moreover, 10% of the human kinome consists of pseudokinases with non-catalytic functions (Zeqiraj and van Aalten, 2010). Differentiating these diverse modes of action remains a central challenge in studying the kinome. For COQ8A in particular, unique structural features are positioned to inhibit protein kinase activity, raising the possibility of atypical functionality (Stefely et al., 2015).
Here, we show that Coq8a knockout (Coq8a−/−) mice recapitulate the primary features of ARCA2. We trace the main pathophysiology of this disease to dysfunctional cerebellar Purkinje cells, defective skeletal muscle, and disruption of complex Q. Leveraging interspecies biochemical investigations, we provide evidence for unorthodox PKL COQ8A activity and present a new model for how COQ8A supports CoQ biosynthesis.
RESULTS
Coq8a−/− Mice Develop Ataxia and Seizures
To investigate COQ8A function in vivo, we generated a constitutive knockout (Coq8a−/−) mouse model (Figure S1A) that showed normal Mendelian inheritance, growth, and life-span, despite the complete absence of COQ8A in all tissues tested (Figures 1A and S1B). No overt clinical phenotypes were observed. Evaluation of standard blood parameters revealed hyperlipidemia (Figure S1C), suggesting a defect in lipid metabolism.
Figure 1. Coq8a−/− Mice Develop Cerebellar Ataxia and Mild Exercise Intolerance.

(A) Immunoblot analysis of COQ8A abundance in 30-week-old mice.
(B) Accelerating rotarod retention times of 10-week-old mice (mean ± SEM, n = 8–12). ANOVA test P<0.05.
(C) Left, representative footprints of 10-week-old mice. Right, “linearity coefficient”, which is proportional to non-linear movement (mean ± SEM, n = 8–12), *P<0.05, **P<0.01.
(D) Number of hind limb coordination errors (mistakes) by mice on beam test (mean ± SEM, n = 8–12), *P<0.05, **P<0.01, ***P<0.01.
(E) Hematoxylin and eosin (H&E) (upper) and Calbindin (lower) staining of cerebellar sections of 15- and 40-week-old mice, respectively. *Shrunken Purkinje neurons; scale bar, 20 μm.
(F) Electron microscopy of 30-week-old cerebella. G, Golgi apparatus; *dilated Golgi; scale bars, 5 μm upper, 1 μm lower.
(G) Electrophysiological recordings of mouse Purkinje cells. ISI and CV2 (mean ± SEM) (n = 3, > 40 cells/mouse); ** Mann-Whitney U test P<0.01.
(H) Maximum speed (upper) and duration (lower) on treadmill test by 10-month-old mice (mean ± SEM, n = 8), *P<0.05.
(I) H&E stained skeletal muscle sections of 7-month-old mice. Scale bar, 50 μm.
(J) Electron microscopy of quadriceps showing broken mitochondria with collapsing cristae (arrows). Upper: 7-month-old mice, scale bar, 2 μm. Lower: 15-month-old mice, scale bar, 1 μm.
See also Figure S1.
To assess the neurological consequences of COQ8A depletion, we conducted behavioral analyses. By 10 weeks of age, Coq8a−/− mice showed decreased performance on accelerating rotarod (Figure 1B), an increase in nonlinear movement in footprint analysis (Figure 1C), and a decrease in hindlimb coordination on the beam test that worsened with age (Figure 1D). Thus, Coq8a−/− mice develop a slowly progressive loss of coordination after birth. Epilepsy and seizures are also observed in ARCA2 patients. Consistently, Coq8a−/− mice developed occasional seizures during daily manipulation and showed increased seizure susceptibility after pentylenetetrazole (PTZ) injection (Figure S1D). Some ARCA2 patients also show moderate intellectual disability, although no clear genotype-phenotype correlation exists. Coq8a−/− mice show only a slight delay in spatial memory in a Morris water maze test (Figures S1E and S1F). Collectively, these results show that the neurological phenotypes of Coq8a−/− mice recapitulate those seen in patients, and thus constitute a good model to explore ARCA2 pathophysiology.
Coq8a−/− Purkinje Cells are Dysfunctional
Histological analysis of the central nervous system (CNS) revealed neither cerebellar atrophy nor gross CNS defects in Coq8a−/− mice throughout their life span. However, Coq8a−/− mice showed a specific defect in the cerebellar Purkinje cell (PC) layer, with the presence of dark shrunken neurons and patches of gaps in calbindin staining, suggesting neuronal degeneration (Figure 1E). Ultrastructural analysis demonstrated that ~10% of Coq8a−/− PCs were dark and shrunken, with dysmorphic nuclei and abnormal membrane structures (Figure 1F). Interestingly, most Coq8a−/− PCs also displayed a dilated and fragmented Golgi apparatus and dilated cisternae of ribosome-rich endoplasmic reticulum (Figures 1F and S1G). Consistent with an important function for COQ8A in PCs, Coq8a mRNA is abundant in the cerebellar PC layer (Figure S1H). Yet, PC mitochondria appear structurally normal (Figure S1G). Coq8a−/− cerebella also showed normal levels of PC-specific molecular markers and lacked apoptotic cells (Figures S1I and S1J), suggesting no massive PC loss, even at advanced stages of the disease.
The electrophysiological function of PCs, which normally exhibit regular pacemaking action potentials (Hausser and Clark, 1997), was assessed by measuring spontaneous firing in acute cerebellar slices (Figure 1G). PCs from 3-month-old Coq8a−/− mice exhibited a significant increase in interspike interval (ISI), while the coefficient of variation between adjacent spikes (CV2) was normal (Figure 1G). A distribution analysis demonstrated that a significant proportion of Coq8a−/− PCs have high ISI values (>40) (Figure S1K). Interestingly, 8-month-old Coq8a−/− PCs exhibited a significant increase in CV2, while the ISI was normal (Figure 1G). Thus, Coq8a−/− PCs have altered pacemaker activity, with a progressive phenotype. Together, these results show that specific PC dysfunction underlies the Coq8a−/− ataxia.
Coq8a−/− Mice Develop Exercise Intolerance
In addition to neurological symptoms, exercise intolerance is a common feature of ARCA2. On an accelerating treadmill, the maximum speed reached by Coq8a−/− mice was significantly decreased, and a trend toward lower endurance was observed (Figure 1H). No difference in muscle strength was observed by grip analysis (Figure S1L). Together, these data demonstrate mild exercise intolerance in Coq8a−/− mice. Consistent with an important role for COQ8A in muscle function, COQ8A mRNA is highly expressed in skeletal muscle (Figures S1M and S1N). Histological analyses of 7-month-old Coq8a−/− mouse skeletal muscle revealed no gross defects (Figure 1I), but ultrastructural analyses revealed abnormal mitochondrial morphology with broken or collapsed cristae and mild fiber degeneration (Figures 1J). Despite these perturbations, mitochondrial DNA levels (Figure S1O), cellular respiration (Figure S1P), and metabolites of central carbon metabolism (Figure S1Q) were found to be within WT ranges, further showing that the Coq8a−/− muscle phenotypes are mild, as observed in ARCA2.
Collectively, this work establishes Coq8a−/− mice as a model system that recapitulates the key features of ARCA2, deepens our understanding of its pathogenesis, and provides a foundation for molecular studies. Here, we conduct parallel studies of mammalian COQ8A and yeast Coq8p to test their functional homology and to help elucidate the biochemistry of ARCA2.
Coq8a−/− Mice Develop Specific CoQ Deficiency
ARCA2 patients have mild skeletal muscle CoQ10 deficiency, which could cause exercise intolerance. 7-month-old Coq8a−/− mice show significant CoQ deficiencies in kidney, liver, and skeletal muscle (Figure 2A). However, CoQ levels were normal in younger mice (Figure S2A). Because the CoQ deficiency was mild in skeletal muscle and absent from whole cerebella, we used mass spectrometry (MS) lipidomics to more broadly explore lipid metabolism. These analyses demonstrated that CoQ is significantly and specifically deficient in Coq8a−/− skeletal muscle (Figure 2B and Table S1). Similarly, yeast lacking coq8 (Δcoq8) show specific CoQ deficiency (Figure S2B and Table S2). However, normal CoQ levels were observed in whole Coq8a−/− cerebella (Figure 2B), providing further evidence that Purkinje cells are specifically affected by loss of COQ8A. No significant difference in serum CoQ was observed (Figure 2B). Together, these results show that lack of COQ8A causes mild, tissue-specific CoQ deficiency.
Figure 2. Loss of COQ8A or Coq8p Causes Deficiency of Complex Q and CoQ.

(A) CoQ9 content in 7-month-old mice (mean ± SD, n = 6). *P<0.05, **P<0.01, ***P<0.001.
(B) Fold changes in mouse lipid abundances (log2(Coq8a−/−/WT), n = 4) versus statistical significance (−log10(p-value)) as quantified by LC-MS for quadriceps, cerebellum, and serum.
(C) Fold changes in mouse protein abundances (log2(Coq8a−/−/WT), n = 4) versus statistical significance as quantified by LC-MS/MS for cerebellum, quadriceps, heart, and kidney.
(D) Expression of COQ7 in muscles of 10-week-old mice
(E) Expression of COQ7 in myoblasts from 7–9-day-old pups
(F) Fold changes in yeast protein abundances (log2(Δcoq8/WT), n = 3) across the diauxic shift (Figure S2E) versus statistical significance as quantified by LC-MS/MS
See also Figure S2
COQ8A Specifically Maintains CoQ Biosynthesis Proteins
Toward understanding the molecular basis for the CoQ deficiency of Coq8a−/− mice, we quantified protein abundance changes by MS-based proteomics (Figure 2C and Table S3). Proteins of the recently defined mammalian complex Q (COQ3–9) (Floyd et al., this issue) were significantly and specifically deficient across multiple Coq8a−/− tissues, including the affected cerebellum and skeletal muscle, but also unaffected tissues to varying degrees (Figures 2C–2E, S2C, and S2D). Together with the co-purification studies by Floyd et al., these Coq8a−/− proteomics results provide further, complementary evidence to support the existence of a mammalian complex Q and its physiological significance. Immunoblot analyses of Coq8a−/− tissues from young mice (Figures 2D and S2C) and myoblasts derived from pups (Figure 2E) showed COQ7 deficiency, suggesting that complex Q dysregulation is an early feature of ARCA2. In yeast, loss of Coq8p caused a similarly specific deficiency of homologous complex Q proteins during the diauxic shift, a metabolic transition from fermentation to respiration (Figures 2F, S2E–S2H, and Table S4). Together, these results show that Coq8p and COQ8A specifically maintain complex Q in yeast and mammals, respectively.
COQ8A Interacts Dynamically with Complex Q
The observed COQ8A-dependent stability of complex Q could be driven by COQ8A-complex Q protein-protein interactions. In pairwise co-expression studies in COS cells, COQ8A-FLAG robustly co-purified with COQ5-HA, but not with COQ3-HA or COQ9-HA (Figure 3A). To further explore the interaction network of COQ8A, we performed AE-MS analyses of human COQ8A-FLAG expressed in HEK293 cells. Compared to MLS-GFP-FLAG, COQ8A-FLAG robustly co-purified with endogenous COQ5, COQ7, COQ6, COQ4, and COQ3 (Figures 3B, 3C, and S3A). The enrichment of COQ3 was relatively weak and could be indirectly mediated by complex Q interactions. To test whether perturbation of the COQ8A active site alters these interactions, we conducted COQ8A-FLAG AE-MS experiments with two COQ8A point mutants: D507N, which disrupts Mg-nucleotide binding, and K276H, a point mutation of the UbiB family-specific KxGQ motif (Figures 3D and 3E). Both mutations altered complex Q, suggesting that the activity of COQ8A modulates the interactions between CoQ proteins. Consistently, we recently observed dynamic, metabolic state-dependent COQ8A/B-complex Q interactions (Floyd et al., this issue).
Figure 3. COQ8A Interacts with Complex Q but Lacks Protein Kinase Activity in trans.

(A) Immunoblot (IB) analysis of interactions between COQ8A-FLAG and COQ5-HA, COQ3-HA, or COQ9-HA transfected into COS cells and immunoprecipitated (IP’d) using anti-FLAG beads.
(B) Heatmap showing the top 25 endogenous proteins most enriched by COQ8A-FLAG IP’d from HEK293 cells compared to various control IPs (mean, n = 4).
(C) Relative abundances of endogenous proteins co-purifying with COQ8A-FLAG compared to MLS-GFP-FLAG (mean, n = 4) IP’d from HEK293 cells as assessed by LC-MS/MS.
(D) Cartoon of COQ8 active site residues (based on PDB 4PED).
(E) Relative abundances of endogenous COQ proteins co-purifying with COQ8A-FLAG (log2(mutant/WT)) IP’d from HEK293 cells as assessed by LC-MS/MS.
(F) SDS-PAGE analysis of in vitro Mg[γ-32P]ATP autophosphorylation reactions with Coq8NΔ41 variants or PKA. BSA, bovine serum albumin (reaction buffer component).
(G) Divalent cation dependence of Coq8NΔ41 autophosphorylation.
(H) Time course of Coq8NΔ41 autophosphorylation.
(I) Coq8NΔ41 autophosphorylation sites identified by LC-MS/MS mapped onto a homology model of Coq8p (based on COQ8A structure, PDB 4PED).
(J) SDS-PAGE analysis of in vitro Mg[γ-32P]ATP autophosphorylation reactions with combinations of Coq8NΔ41 variants. MBP, maltose binding protein tag.
(K) SDS-PAGE analysis of in vitro Mg[γ-32P]ATP kinase reactions with PKA or Coq8NΔ41 and potential substrate proteins.
See also Figure S3.
COQ8A Lacks Protein Kinase Activity in trans
The observed COQ8A protein interactions could fit with the prevailing model that COQ8A is a protein kinase that phosphorylates COQ proteins. To test the protein kinase hypothesis, we examined Coq8a−/− mouse phosphoproteome changes. We quantified >7,500 cerebellar phosphopeptides and >6,000 skeletal muscle phosphopeptides, but we did not detect phosphopeptides from COQ3, COQ5, or COQ7 (Figure S2I and Table S5), the hypothesized COQ8A substrates (Xie et al., 2011), despite detecting representative peptides for each protein (Figure 2C). In skeletal muscle, COQ9 pS81 and pY88 were observed, but were not significantly altered when normalized to protein abundance changes. Similarly, COQ9 pS81 was moderately decreased in Coq8a−/− cerebellums (Coq8a−/−/WT = 0.6), likely due to the observed decrease in COQ9 abundance (Figure 2C). Likewise, in yeast, we quantified >6,000 phosphopeptides across the diauxic shift (Figures S2J and S2K and Table S6), but no phosphopeptides from Coq proteins were observed, despite detecting peptides from Coq3p–Coq9p (Figure 2F). These results suggest that complex Q proteins are not abundantly phosphorylated in vivo and led us to test the protein kinase hypothesis directly in vitro.
Mutants of a truncated COQ8A construct (COQ8ANΔ250) can autophosphorylate in vitro (Stefely et al., 2015), suggesting that COQ8A might act as a protein kinase in vivo. Here, we purified the full mitochondrial form of Coq8p (Coq8NΔ41), which maintains its mature N-terminus (Figure S3B) and thus eliminates caveats of the COQ8A truncation. Like COQ8ANΔ250 (Stefely et al., 2015), Coq8NΔ41 preferentially binds adenine nucleotides, with selectivity for ADP over ATP that can be reversed by an A-rich loop A-to-G mutation (Figures S3C and S3D). Mutating the A-rich loop or the invariant KxGQ motif enhances Coq8NΔ41 autophosphorylation activity, which is dependent on active site residue D365, Mg2+, and time (Figures 3F–3H, S3E, and S3F). Thus, the basic biochemical properties of COQ8A and Coq8p are conserved.
LC-MS/MS analyses showed that Coq8NΔ41 autophosphorylates residues of the KxGQ domain (Figures 3I and S3G), which occludes the canonical peptide substrate pocket. The KxGQ domain is accessible in cis or in trans, but reactions with combinations of active (WT or A197G,K134H) and inactive (D365N) Coq8NΔ41 showed that these Coq8p variants autophosphorylate exclusively in cis (Figures 3J and S3H). Similar results were observed for COQ8ANΔ250 (Figure S3I). Importantly, mutating Coq8NΔ41 autophosphorylation sites did not inhibit respiratory yeast growth (Figure S3J), showing that the autophosphorylation observed in vitro is dispensable for function in vivo.
Screening potential protein substrates for COQ8A and Coq8p further demonstrated their lack of trans protein kinase activity. While protein kinase A (PKA) exhibited robust protein kinase activity in trans, neither WT nor A197G,K134H Coq8NΔ41 catalyzed phosphorylation of myelin basic protein or mixtures of COQ proteins in trans (Figure 3K). Similar results were obtained for COQ8ANΔ250, which also does not catalyze phosphorylation of general protein kinase substrates (Figures S3K and S3L). Collectively, these results demonstrate that the enzymatic activities of mammalian COQ8A and yeast Coq8p are highly similar, and that neither catalyzes canonical protein kinase activity. These findings argue against the protein kinase hypothesis and demand a new model for the “unorthodox protein kinase-like” (uPKL) functionality of COQ8.
Coq8p Catalyzes ATPase Activity with its KxGQ Motif
To begin testing potential uPKL functions, we examined Coq8NΔ41 for ATPase activity, which can be considered as small molecule “water kinase” activity. WT Coq8NΔ41 has ATPase activity and an A197G mutation, which enhances MgATP binding in vitro, increased the ATPase activity (Figures 4A and S4A–S4C). In contrast, K134H and A197G,K134H mutations decreased ATPase activity to a level near that of a mutant that does not bind nucleotides, D365N. Thus, Coq8p ATPase activity is enhanced by the UbiB-specific KxGQ motif. The inhibition of ATPase activity seen with the K134H mutants is in stark contrast to their dramatically enhanced autophosphorylation activity. Importantly, the K134H mutation eliminates CoQ production in vivo (Stefely et al., 2015), suggesting that, contrary to autophosphorylation, the ATPase activity is likely related to the endogenous activity of Coq8p.
Figure 4. COQ8A and Coq8p Exhibit Unorthodox PKL Activities.

(A) kcat and kcat/Km values for the ATPase activity of Coq8NΔ41 variants measured by observing production of phosphate (mean ± SD, n = 3), *P<0.05.
(B) Relative protein abundances in Δcoq8 yeast transformed with the indicated coq8 (Coq8p) variants as quantified by label free quantitation LC-MS/MS analysis (mean ± SD, n = 4).
(C) Immunoblot analysis of COQ8A localization in submitochondrial fractions. Mitochondrial markers: COQ7 (inner membrane, peripheral), CYC1 (intermembrane space), NFU1 (matrix), Prohibitin (inner membrane, integral), and TOM20 (outer membrane, integral). Supe., supernatant.
(D) Heatmap showing enrichment of lipids co-purifying with MBP-Coq8NΔ41 variants compared to the bacterial lysate from which the proteins were purified as assessed by LC-MS (mean, n = 6).
(E) Relative abundances (mutant/WT) of CoQ biosynthesis intermediates (R, polyisoprenyl tail) co-purifying with MBP-Coq8NΔ41 variants as assessed by LC-MS (mean ± SD, n = 6).
See also Figure S4.
To test the impact of these differential Coq8p mutations on complex Q stability in vivo, we examined Δcoq8 yeast rescued with coq8 variants by MS proteomics (Figures 4B and S4D). While WT Coq8p rescued the abundance of complex Q proteins in vivo, the K134H mutant did not. The essentially equal need for D365 (MgATP binding) and K134 (ATPase, uPKL activity) suggests that uPKL activity stabilizes complex Q. The observation that the A197G mutation enhanced ATPase activity in vitro but decreased function in vivo could suggest that Coq8p does not act as an ATPase in vivo, but this A-rich loop perturbation likely has multifactorial effects because this loop is known to impact substrate binding and protein structure in other PKLs (Bossemeyer, 1994; Hemmer et al., 1997). While the A-to-G mutation is useful for boosting MgATP binding in vitro, this alanine residue is highly conserved, and our results show that it does indeed support Coq8p function in vivo. More generally, these results do not necessarily demonstrate that COQ8 ATPase activity per se stabilizes complex Q in vivo because water could mimic the hydroxyl group of another small molecule substrate. The COQ8 ATPase activity could be more broadly indicative of small molecule kinase activity or lipid kinase activity.
Coq8p Binds Lipid CoQ Intermediates
An interaction between COQ8A and lipids could be a feature of either lipid kinase activity or interactions with the membrane-associated complex Q. Sub-fractionation and Na2CO3 treatment of mouse mitochondria showed that endogenous COQ8A is partially buried in the inner mitochondrial membrane with its C-terminus facing the matrix (Figure 4C). Consistently, COQ8ANΔ250 and Coq8NΔ41 bind to liposomes (Figure S4E). Thus, COQ8A interacts physically with lipid membranes.
To examine specific lipids that bind to Coq8p, we conducted LC-MS/MS analyses of lipids that co-purify with recombinant Coq8NΔ41 expressed in E. coli, which possesses a CoQ biosynthesis pathway similar to that found in eukaryotes. The E. coli CoQ biosynthesis intermediates octaprenylhydroxybenzoate (OHB) and octaprenylphenol (OPP) co-purified with Coq8NΔ41, but not with PKA, and Coq8p active site mutations decreased binding of OHB and OPP (Figure S4F). An independent set of experiments showed enrichment of OHB and OPP in samples of isolated Coq8NΔ41 compared to their abundance in the bacterial lysate from which they were purified (Figure 4D). Furthermore, binding of OHB, OPP, and heptaprenylphenol (HPP) was decreased by Coq8p active site mutations D365N and K134H (Figures 4D and 4E). Interestingly, Coq8p did not enrich for CoQ (detected in the oxidized form in this experiment), and Coq8p active site mutations did not perturb co-purification of CoQ, suggesting that Coq8p preferentially binds CoQ biosynthesis intermediates.
Nucleotide Binding Activates COQ8A and Opens Small Molecule Pockets
Our previous structure of apo COQ8ANΔ254 showed that its UbiB family specific KxGQ domain occludes the typical substrate binding pocket (Stefely et al., 2015), and no obvious hydrophobic binding pockets that could bind a lipid were present. However, nucleotide binding could open a COQ8A substrate binding pocket. To test this idea, we crystallized COQ8ANΔ254 R611K with adenosine 5′-(β,γ-imido)triphosphate (AMPPNP) and solved an X-ray structure at a resolution of 2.3 Å (Figure 5A and Table 1). The R611K mutant was used because we were unable to obtain WT-nucleotide co-crystals.
Figure 5. Structure and Dynamics of Nucleotide-Bound COQ8A.

(A) Surface representations of COQ8ANΔ254 R611K bound to AMPPNP (PDB 5I35), colored by domains (see Figure 3I). Two hydrophobic pockets (KxGQ pockets 1 and 2) are highlighted.
(B) Surface representation of apo COQ8ANΔ254 (PDB 4PED).
(C) Surface representation of PKA (PDB 1ATP) (Knighton et al., 1991).
(D) Representative snapshots of MD analyses of COQ8A showing interactions of D488 with either R611 or the KxGQ motif. Left, apo; Middle and right, MgATP-bound.
See also Figure S5.
Table 1.
X-ray Data Collection and Refinement Statistics
| Data collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c (Å) | 150.05, 59.12, 97.69 |
| α, β, γ (°) | 90, 97.69, 90 |
| Resolution (Å) | 39.42–2.30 (2.38–2.30)a |
| Rsym | 0.088 (1.068) |
| I/σI | 15.6 (1.9) |
| Completeness (%) | 99 (100) |
| Redundancy | 7.4 (7.6) |
| Refinement | |
| Resolution (Å) | 39.42–2.30 |
| No. reflections | 19830 (1940) |
| Rwork/Rfree | 0.184/0.259 |
| No. atoms | |
| Protein | 3105 |
| Ligand/ion | 31 |
| Water | 26 |
| B-factors | |
| Protein | 72.4 |
| Ligand/ion | 74.8 |
| Water | 68.8 |
| Root-mean-square deviation | |
| Bond lengths (Å) | 0.024 |
| Bond angles (°) | 2.2 |
| Ramachandran plot (%) | |
| Favored | 94 |
| Allowed | 5 |
| Outliers | 1 |
The highest-resolution shell is shown in parentheses.
Overall, the apo and nucleotide-bound COQ8A structures are largely similar (Figures 5A, 5B, and S5A). Even with a nucleotide bound, the KxGQ motif is positioned to occlude the typical peptide substrate binding site and preclude in trans protein phosphorylation. Nucleotide-bound COQ8A is also structurally related to Rio family enzymes (Figures S5A and S5B)—PKLs that function as ATPases (Ferreira-Cerca et al., 2012). Detailed comparisons of apo and nucleotide-bound COQ8A show that most residues deviate by only 2–3 Å, but notably larger 7–12 Å deviations of two loops in the KxGQ domain were observed: the GQα2–GQα3 loop and the GQα5–GQα6 loop (“Q switch 1” and “Q switch 2”, respectively). The movement of these two COQ8A “Q switches” due to nucleotide binding opens two hydrophobic pockets in the KxGQ domain (KxGQ pockets 1 and 2, respectively), which are not present in the apo COQ8A structure (Figures 5A and S5D). KxGQ pocket 1 is near the putative phosphoryl acceptor substrate binding region, and it could potentially bind the kinase’s substrate. In contrast, KxGQ pocket 2, which is partially formed by the “x” of the KxGQ motif, has the potential to bind an allosteric effector molecule that could communicate with the active site through the KxGQ motif.
KxGQ pocket 1 appears to be open sufficiently to accept a small molecule kinase substrate, but other structural features also play important roles in kinase activation. While COQ8A has intact “spine”-like structures (Figure S5E), an important feature of PKL enzyme activation (Kornev et al., 2006), the putative catalytic base D488 is locked in a salt bridge in the apo COQ8A structure (Figure S5F). To test the idea that nucleotide binding might allow D488 to move into an active conformation, we conducted 200 ns molecular dynamics (MD) simulations of apo, MgADP- and MgATP- bound COQ8A. Kullback–Leibler (KL) divergence between simulations revealed clusters of residues with differential dynamics in the nucleotide pocket and the KxGQ domain (Figure S5G). In apo COQ8A, D488 remains locked inside the protein core (Figure S5H). However, in the nucleotide bound form, because of slight repacking of the GQα2 and GQα3 helices, D488 displays large side chain flexibility. Surprisingly, in the MgATP simulation, D488 flips from inside the hydrophobic core into a catalytically-competent, solvent-exposed posture (Figure 5D), as indicated in the X1 angle plot, which falls into two distinct ensembles (Figure S5H). This D488 flip is facilitated by hydrogen bonding with the KxGQ glutamine (Figure 5D). In addition, during our simulation, K276 can also form an ionic interaction with the γ-phosphate of ATP (Figure 5D), suggesting that the KxGQ motif may participate directly in catalyzing phosphoryl transfer. Together, these results demonstrate that COQ8A can adopt a conformation poised to catalyze phosphoryl transfer, not only in spite of the KxGQ domain’s position in the typical peptide substrate pocket, but likely facilitated by this very position of the KxGQ motif. Collectively, these structural observations provide further evidence for the uPKL functionality of COQ8A.
DISCUSSION
Insight into CoQ Biosynthesis
Two overarching problems continue to confound studies of CoQ biosynthesis in human health and disease. First, primary CoQ deficiencies have an extraordinary range of tissue specificities and clinical severities for unknown reasons (Laredj et al., 2014). Variable expression of COQ8A and COQ8B likely influence this clinical heterogeneity. Second, CoQ biosynthesis has many biochemical knowledge gaps, including proteins of unknown molecular function. These CoQ-associated proteins could support the pathway directly and specifically or, alternatively, indirectly as part of a broader biological function. The biochemical function of COQ8A is particularly difficult to define because, as we demonstrated here, it has complex functionalities outside of the canonical protein kinase model (Figure 6A). Completely defining the biochemistry of CoQ biosynthesis could lead to new strategies for treating CoQ deficiency.
Figure 6. A Model for the Molecular Basis of COQ8 Biology.
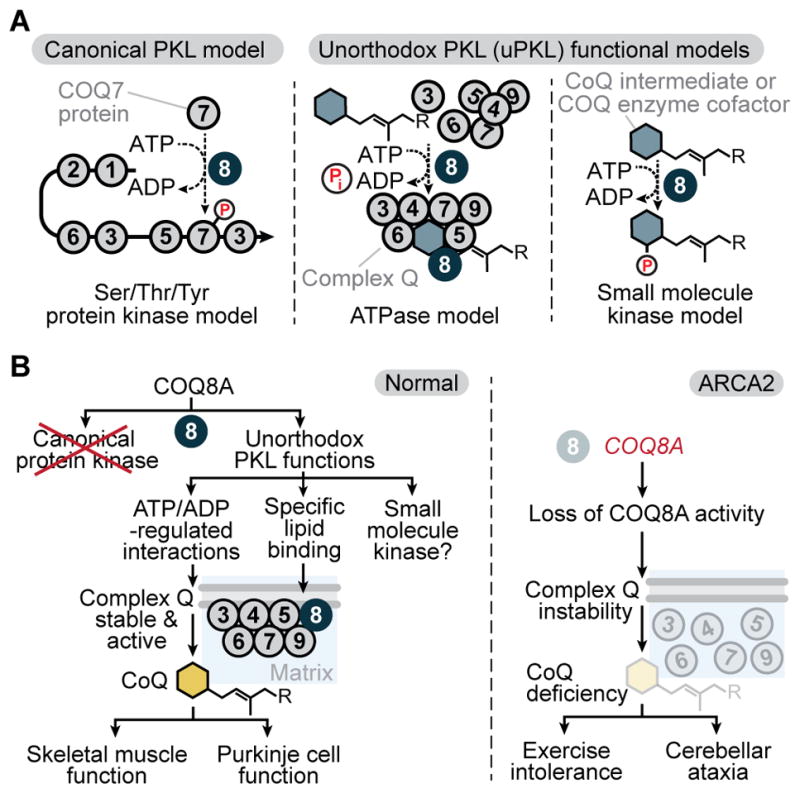
(A) Models for the molecular activity of COQ8 (COQ8A/B and Coq8p).
(B) Models for COQ8A biology and its disruption in ARCA2.
Our results provide biological and biochemical evidence that mammalian COQ8A and yeast Coq8p function similarly by directly and specifically supporting CoQ biosynthesis through maintenance of complex Q. These results, in combination with phylogenetic analyses, led us to rename ADCK3 and ADCK4 as COQ8A and COQ8B, respectively. This nomenclature provides more functionally descriptive names and properly designates them as co-orthologs of Coq8p.
A Mouse Model of Moderate CoQ Deficiency
Generating animal models for studying CoQ deficiency is difficult because Coq gene knockouts are often embryonic lethal (Licitra and Puccio, 2014). Here, in contrast, we demonstrated that lack of COQ8A in mice results in a mild phenotype with progressive cerebellar ataxia, mild exercise intolerance, and moderate CoQ deficiency (Figure 6B), recapitulating the more frequent features of ARCA2 (Aure et al., 2004; Barca et al., 2016; Gerards et al., 2010; Horvath et al., 2012; Lagier-Tourenne et al., 2008; Mignot et al., 2013; Mollet et al., 2008). Thus, this work establishes Coq8a−/− mice as a valid model system for studying ARCA2 and testing potential treatments.
The pathophysiologies of cerebellar ataxias are diverse (Wolf and Koenig, 2013). Similar to other ataxia models (Custer et al., 2006; Maltecca et al., 2009; Stankewich et al., 2010), Coq8a−/− mice displayed dark degenerating Purkinje cells (PCs), while other cerebellar cells were spared. Our biochemical studies showed a deficit of complex Q proteins, but normal cerebellar CoQ levels. PCs account for a small fraction (<0.1%) of cerebellar cells (Lange, 1975), and a PC-specific CoQ deficiency could be masked by other cells. Unexpectedly, Coq8a−/− PCs displayed Golgi morphology defects, but normal mitochondria. Golgi pathology is a hallmark of neurodegenerative diseases including Parkinson’s disease and amyotrophic lateral sclerosis (Sundaramoorthy et al., 2015). The Coq8a−/− Golgi defects could be secondary to mitochondrial CoQ deficiency or, alternatively, due to defective Golgi CoQ biosynthesis, which is biologically important (Mugoni et al., 2013). Golgi defects could in turn disrupt intracellular protein trafficking and, thus, PC electrophysiology. Our Coq8a−/− mice provide a model system to study CoQ production across different organelles, cells, and tissues.
Organism-wide, loss of COQ8A has complex, tissue-specific effects. Coq8a−/− skeletal muscle is significantly deficient in complex Q and in CoQ, and Coq8a−/− mice have low exercise fitness. However, CoQ and complex Q were also affected across Coq8a−/− tissues that do not have obvious biological phenotypes. Tissue-specific variability in COQ8A and COQ8B expression could contribute to these differential effects. Indeed, the two tissues most affected by loss of COQ8A—cerebellum and skeletal muscle—show low expression of COQ8B (Figures S1H, S1M, and S1N). In contrast, the kidney, which is relatively unaffected by loss of COQ8A (Figure S2D), shows high COQ8B expression (Figures S1M and S1N). More distantly related COQ8A/B homologs (ADCK1, ADCK2, and ADCK5) could also play a role. Cell type-specific differences in CoQ transport and uptake (Anderson et al., 2015), could further complicate the relationship between complex Q abundance and CoQ abundance. Our proteomics and phosphoproteomics results provide a resource for continued investigation of the tissue-specific effects of COQ8A deficiency.
Unorthodox Activity of UbiB Family Kinases
Initially, COQ8A was predicted to be a protein kinase with a non-essential regulatory role in CoQ biosynthesis. However, our results argue against the canonical protein kinase model and instead support an unorthodox protein kinase-like (uPKL) functionality for COQ8 (referring to both mammalian COQ8A/B and yeast Coq8p, which we propose to function through the same fundamental mechanisms). Thus, our work adds COQ8 to an expanding list of PKL superfamily members with non-canonical activities that are central to their biological functions (Kung and Jura, 2016).
The precise molecular mechanism by which COQ8 enhances complex Q activity to support CoQ biosynthesis is not yet fully resolved. However, our past (Stefely et al., 2015) and present work shows that COQ8’s endogenous function requires adenine nucleotide binding, a conserved active site that catalyzes phosphoryl transfer, and the UbiB-specific KxGQ motif. Our results support at least two potential uPKL activities that incorporate these features (Figure 6A): (1) ATPase activity that enhances complex Q interactions or supports complex Q assembly and (2) small molecule kinase activity that phosphorylates a complex Q component (e.g. a CoQ intermediate or cofactor), which would support both the stability and the activity of complex Q.
Rigorously defining which uPKL functional model operates in vivo will require extensive work, but our results establish biochemical uPKL activities of COQ8 that are more likely important for its endogenous function than its in vitro cis autophosphorylation activity. Mutating the lysine of the UbiB family-specific KxGQ motif provides a particularly informative perturbation, because it enhances cis autophosphorylation but inhibits other COQ8 activities, including support of in vivo respiratory metabolism, CoQ production, complex Q stability, ATPase activity (which could be indicative of small molecule kinase activity), and lipid CoQ intermediate binding.
The ability of Coq8p to bind CoQ intermediates provides an important addition to its known molecular functions. Interestingly, OPP is the intermediate that accumulates in ΔubiB E. coli (Cox et al., 1969; Poon et al., 2000), which lack the prokaryotic coq8 homolog, and hexaprenylhydroxybenzoate (an OHB analog) is the intermediate that accumulates in Δcoq8 yeast. Although we have not yet observed COQ8-dependent phosphorylation of OPP or OHB, potentially due to product lability, a lipid CoQ intermediate could bind to KxGQ pocket 1 as a kinase substrate. Phosphorylation of a CoQ intermediate could potentially make it more accessible to enzymes peripherally associated with the lipid membrane or create a protecting group to maintain the intermediate in a reduced or energetically-charged state for a particular biosynthetic reaction (e.g. the C2 methylation catalyzed by COQ5). Alternatively, or in addition, an isoprenoid lipid could bind to KxGQ pocket 2 as an allosteric regulator of COQ8 activity.
Together, our work supports a model wherein the uPKL functionality of COQ8—which involves interactions with both CoQ intermediates and the protein COQ5—enhances complex Q stability and activity to support CoQ biosynthesis and various in vivo functions (e.g. skeletal muscle function and Purkinje cell function in the case of COQ8A) (Figure 6B). More broadly, our work on COQ8 provides a molecular foundation for studying the larger UbiB family, which has diverse functions in lipid metabolism (Tan et al., 2013), cell migration (Simpson et al., 2008), and tumor cell viability (Brough et al., 2011; Wiedemeyer et al., 2010). Because all UbiB family members have a KxGQ motif and a predicted KxGQ domain—which we have shown to play a central role in the uPKL functions of COQ8—we predict uPKL functionality to be conserved throughout this ancient protein family.
Collectively, this work establishes a robust mammalian model system for studying neurodegeneration, muscle disorders, potential treatments, and the complex effects of CoQ deficiency on health and disease. Our studies also show that the molecular basis for the inherited disease ARCA2 involves deficiency of complex Q and CoQ due to loss of unorthodox protein kinase-like activity, which is likely conserved in homologs across all three superkingdoms of life.
EXPERIMENTAL PROCEDURES
Further details for all procedures can be found in the Supplemental Experimental Procedures.
Mice and Cell Culture
Coq8a−/− (Adck3−/−) mice deleted for exons 9–14 were generated by homologous recombination. Motor coordination was assessed by accelerating rotating rod, footprint analysis, and beam-walking tests. Memory and learning capacity were assessed with Y-Maze, object recognition, and Morris water maze tests. Physical fitness was assessed by treadmill and grip tests. Sex- and age- matched mice were used in experiments in accordance with national ethical rules. Primary myoblasts were generated from hind limb muscle of 7–9-day-old mice.
Microscopy and PC Electrophysiology
For immunohistochemistry (IHC) and transmission electron microscopy (TEM), mice were perfused with 4% paraformaldehyde. For IHC, tissues were either frozen or paraffin embedded. For TEM, tissues were fixed in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, and embedded in Epon. For PC electrophysiology, mice were decapitated under isoflurane anesthesia, acute 330 μm-thick cerebellar slices were prepared, and extracellular recordings of individual PCs were performed using 15–30 MΩ pipettes.
CoQ Quantitation and Lipidomics
Lipids were extracted from tissue homogenates with organic solvents. For targeted CoQ measurements, lipids were separated by liquid chromatography (LC) and quantified by electrochemical detection using CoQ8 as an internal standard. For unbiased lipidomics, lipids were separated, identified, and quantified by LC-MS/MS performed on a C18 column coupled to a Q Exactive Focus mass spectrometer by a heated ESI source.
Proteomics and Phosphoproteomics
Proteins from lysates were digested into peptides, labeled with isobaric tags, and fractionated by strong cation exchange. Phosphopeptides were enriched by immobilized metal affinity chromatography (IMAC). LC-MS/MS was performed on a C18 column coupled to an Orbitrap Elite mass spectrometer by a nanoESI source.
In Vitro Kinase and ATPase Assays
8His-MBP-[TEV]-Coq8NΔ41 was expressed in E. coli and purified using cobalt IMAC resin, tobacco etch virus (TEV) cleavage, and a second subtractive IMAC purification to remove 8His-MBP and isolate Coq8NΔ41. For kinase assays, unless otherwise indicated, Coq8NΔ41 (4 μM), MBP-Coq8NΔ41 (4 μM), or PKA (4 μM) was mixed with [γ-32P]ATP (0.25 μCi/μL, 100 μM [ATP]total), MgCl2 (20 mM), and substrate proteins and incubated (30 °C, 60 min) (final concentrations). Reactions were quenched with 4×LDS buffer, [γ-32P]ATP was separated from Coq8NΔ41 by SDS-PAGE, and the gel was stained, dried, and imaged. For ATPase assays, Coq8NΔ41 (2 μM) was mixed with ATP (0–2 mM) and phosphate detection reagents and incubated (22 °C).
Identification of Lipids Bound to Coq8p
8His-MBP-Coq8NΔ41 was expressed in E. coli and isolated by cobalt IMAC. Lipids were extracted with CHCl3/MeOH (1:1, v/v) and analyzed by unbiased LC-MS/MS.
Protein Co-Immunoprecipitation
COQ8A-FLAG was expressed in HEK293 cells and immunoprecipitated with anti-FLAG M2 beads. Co-purifying proteins were digested into peptides with trypsin and analyzed by label-free quantitation LC-MS/MS.
Protein Crystallization
Crystals of selenomethionine-labeled COQ8ANΔ254 R611K were obtained by hanging drop vapor diffusion using 2 μL of 0.2 mM COQ8A, 2 mM AMPPNP, 4 mM MgCl2 mixed with 2 μL of reservoir solution, 26% sodium acrylate 5100, 20 mM MgCl2, and 100 mM NaHEPES pH 7.5. Crystals were cryopreserved by increasing the sodium acrylate 5100 concentration to 30%.
Supplementary Material
HIGHLIGHTS.
Coq8a−/− mice recapitulate the most frequent form of human genetic CoQ deficiency
COQ8A maintains a mammalian coenzyme Q (ubiquinone) biosynthesis complex
COQ8 stabilizes complex Q through unorthodox kinase-like mechanisms
COQ8 binds lipid CoQ intermediates with a conserved structural feature (KxGQ domain)
Acknowledgments
We thank Nadia Messadeq, Hamid Meziane, Marie-France Champy and the ICS facility for mouse phenotyping support, Carol Mercer for support, Bob Smith for crystallomics assistance, and Michael Becker for beamline assistance. This work was supported by Searle Scholars and Vilas Associates Awards and by NIH grants U01GM94622, R01DK098672, R01GM112057, and R01GM115591 (to D.J.P.); the UK ataxia association, FRM Physiopathologie Mitochondriale DPM20121125555, and ERC 206634/ISCATAXIA (to H.P.); NIH Ruth L. Kirschstein NRSA F30AG043282 (to J.A.S.); NIH T32GM008505 (to A.G.R.); NIH 5T32GM08349 and NSF DGE-1256259 (to D.C.L.); NIH T32DK007665 (to B.J.F.); NIH R35GM118110 (to J.J.C.); EPFL and the Swiss NSF (200021_157217) (to M.D.P.); ANR grant pABACoQ (to F.P.); IGBMC IPP Fellowships (to F.L. and T.J.) (LabEx INRT funds, ANR-10-LABX-0030-INRT, Investissements d’Avenir ANR-10-IDEX-0002-02); FRM DEQ20140329514 and ANR-13-SAMA-0010-01 (to P.I.); U01GM098248 and U54GM094584 (C.A.B.); the US DOE (DE-AC02-06CH11357) (Advanced Photon Source use); the Michigan Economic Development Corporation (085P1000817) (LS-CAT); and NIH NCI (Y1-CO-1020) and NIGMS (Y1-GM-1104) (GM/CA@APS).
Footnotes
ACCESSION NUMBERS
The coordinates and structure factors have been deposited in the Protein Data Bank with accession code 5I35.
Supplemental Information includes Supplemental Experimental Procedures, 5 figures, and 6 tables.
AUTHOR CONTRIBUTIONS
J.A.S., F.L., L.L., H.P., and D.J.P. conceived of the project and its design and wrote the manuscript. All authors performed experiments and data analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson CM, Kazantzis M, Wang J, Venkatraman S, Goncalves RL, Quinlan CL, Ng R, Jastroch M, Benjamin DI, Nie B, et al. Dependence of brown adipose tissue function on CD36-mediated coenzyme Q uptake. Cell Rep. 2015;10:505–515. doi: 10.1016/j.celrep.2014.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombes A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–729. doi: 10.1212/01.wnl.0000134607.76780.b2. [DOI] [PubMed] [Google Scholar]
- Barca E, Musumeci O, Montagnese F, Marino S, Granata F, Nunnari D, Peverelli L, DiMauro S, Quinzii CM, Toscano A. Cerebellar ataxia and severe muscle CoQ deficiency in a patient with a novel mutation in ADCK3. Clin Genet. 2016 doi: 10.1111/cge.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossemeyer D. The glycine-rich sequence of protein kinases: a multifunctional element. Trends Biochem Sci. 1994;19:201–205. doi: 10.1016/0968-0004(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Brough R, Frankum JR, Sims D, Mackay A, Mendes-Pereira AM, Bajrami I, Costa-Cabral S, Rafiq R, Ahmad AS, Cerone MA, et al. Functional viability profiles of breast cancer. Cancer Discov. 2011;1:260–273. doi: 10.1158/2159-8290.CD-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox GB, Young IG, McCann LM, Gibson F. Biosynthesis of ubiquinone in Escherichia coli K-12: location of genes affecting the metabolism of 3-octaprenyl-4-hydroxybenzoic acid and 2-octaprenylphenol. J Bacteriol. 1969;99:450–458. doi: 10.1128/jb.99.2.450-458.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane FL, Hatefi Y, Lester RL, Widmer C. Isolation of a quinone from beef heart mitochondria. Biochim Biophys Acta. 1957;25:220–221. doi: 10.1016/0006-3002(57)90457-2. [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- Do TQ, Hsu AY, Jonassen T, Lee PT, Clarke CF. A defect in coenzyme Q biosynthesis is responsible for the respiratory deficiency in Saccharomyces cerevisiae abc1 mutants. J Biol Chem. 2001;276:18161–18168. doi: 10.1074/jbc.M100952200. [DOI] [PubMed] [Google Scholar]
- Ferreira-Cerca S, Sagar V, Schafer T, Diop M, Wesseling AM, Lu H, Chai E, Hurt E, LaRonde-LeBlanc N. ATPase-dependent role of the atypical kinase Rio2 on the evolving pre-40S ribosomal subunit. Nat Struct Mol Biol. 2012;19:1316–1323. doi: 10.1038/nsmb.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerards M, van den Bosch B, Calis C, Schoonderwoerd K, van Engelen K, Tijssen M, de Coo R, van der Kooi A, Smeets H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion. 2010;10:510–515. doi: 10.1016/j.mito.2010.05.008. [DOI] [PubMed] [Google Scholar]
- Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- He CH, Xie LX, Allan CM, Tran UC, Clarke CF. Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim Biophys Acta. 2014;1841:630–644. doi: 10.1016/j.bbalip.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- Horvath R, Czermin B, Gulati S, Demuth S, Houge G, Pyle A, Dineiger C, Blakely EL, Hassani A, Foley C, et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry. 2012;83:174–178. doi: 10.1136/jnnp-2011-301258. [DOI] [PubMed] [Google Scholar]
- Kannan N, Taylor SS, Zhai Y, Venter JC, Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007;5:e17. doi: 10.1371/journal.pbio.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA. 2006;103:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JE, Jura N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure. 2016;24:7–24. doi: 10.1016/j.str.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Tazir M, Lopez LC, Quinzii CM, Assoum M, Drouot N, Busso C, Makri S, Ali-Pacha L, Benhassine T, et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am J Hum Genet. 2008;82:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange W. Cell number and cell density in the cerebellar cortex of man and some other mammals. Cell Tissue Res. 1975;157:115–124. doi: 10.1007/BF00223234. [DOI] [PubMed] [Google Scholar]
- Laredj LN, Licitra F, Puccio HM. The molecular genetics of coenzyme Q biosynthesis in health and disease. Biochimie. 2014;100:78–87. doi: 10.1016/j.biochi.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Leonard CJ, Aravind L, Koonin EV. Novel families of putative protein kinases in bacteria and archaea: evolution of the “eukaryotic” protein kinase superfamily. Genome Res. 1998;8:1038–1047. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- Lester RL, Crane FL. The natural occurrence of coenzyme Q and related compounds. J Biol Chem. 1959;234:2169–2175. [PubMed] [Google Scholar]
- Licitra F, Puccio H. An overview of current mouse models recapitulating coenzyme q10 deficiency syndrome. Mol Syndromol. 2014;5:180–186. doi: 10.1159/000362942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist PK, Poliakov A, Giacomelli L, Friso G, Appel M, McQuinn RP, Krasnoff SB, Rowland E, Ponnala L, Sun Q, et al. Loss of plastoglobule kinases ABC1K1 and ABC1K3 causes conditional degreening, modified prenyl-lipids, and recruitment of the jasmonic acid pathway. Plant Cell. 2013;25:1818–1839. doi: 10.1105/tpc.113.111120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltecca F, Magnoni R, Cerri F, Cox GA, Quattrini A, Casari G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci. 2009;29:9244–9254. doi: 10.1523/JNEUROSCI.1532-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Martinis J, Glauser G, Valimareanu S, Kessler F. A chloroplast ABC1-like kinase regulates vitamin E metabolism in Arabidopsis. Plant Physiol. 2013;162:652–662. doi: 10.1104/pp.113.218644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Apartis E, Durr A, Marques Lourenco C, Charles P, Devos D, Moreau C, de Lonlay P, Drouot N, Burglen L, et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J Rare Dis. 2013;8:173. doi: 10.1186/1750-1172-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollet J, Delahodde A, Serre V, Chretien D, Schlemmer D, Lombes A, Boddaert N, Desguerre I, de Lonlay P, de Baulny HO, et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am J Hum Genet. 2008;82:623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton RA. Ubiquinone. Nature. 1958;182:1764–1767. doi: 10.1038/1821764a0. [DOI] [PubMed] [Google Scholar]
- Mugoni V, Postel R, Catanzaro V, De Luca E, Turco E, Digilio G, Silengo L, Murphy MP, Medana C, Stainier DY, et al. Ubiad1 is an antioxidant enzyme that regulates eNOS activity by CoQ10 synthesis. Cell. 2013;152:504–518. doi: 10.1016/j.cell.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oruganty K, Talevich EE, Neuwald AF, Kannan N. Identification and classification of small molecule kinases: insights into substrate recognition and specificity. BMC Evol Biol. 2016;16:7. doi: 10.1186/s12862-015-0576-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrel F, Hamelin O, Douki T, Kieffer-Jaquinod S, Muhlenhoff U, Ozeir M, Lill R, Fontecave M. Involvement of mitochondrial ferredoxin and para-aminobenzoic acid in yeast coenzyme Q biosynthesis. Chem Biol. 2010;17:449–459. doi: 10.1016/j.chembiol.2010.03.014. [DOI] [PubMed] [Google Scholar]
- Poon WW, Davis DE, Ha HT, Jonassen T, Rather PN, Clarke CF. Identification of Escherichia coli ubiB, a gene required for the first monooxygenase step in ubiquinone biosynthesis. J Bacteriol. 2000;182:5139–5146. doi: 10.1128/jb.182.18.5139-5146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev. 2010;16:183–188. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry SP, Jayaraman J, Ramasarma T. Distribution of coenzyme Q in rat liver cell fractions. Nature. 1961;189:577. doi: 10.1038/189577a0. [DOI] [PubMed] [Google Scholar]
- Simpson KJ, Selfors LM, Bui J, Reynolds A, Leake D, Khvorova A, Brugge JS. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol. 2008;10:1027–1038. doi: 10.1038/ncb1762. [DOI] [PubMed] [Google Scholar]
- Stankewich MC, Gwynn B, Ardito T, Ji L, Kim J, Robledo RF, Lux SE, Peters LL, Morrow JS. Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc Natl Acad Sci USA. 2010;107:6022–6027. doi: 10.1073/pnas.1001522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefely JA, Reidenbach AG, Ulbrich A, Oruganty K, Floyd BJ, Jochem A, Saunders JM, Johnson IE, Minogue CE, Wrobel RL, et al. Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol Cell. 2015;57:83–94. doi: 10.1016/j.molcel.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy V, Sultana JM, Atkin JD. Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front Neurosci. 2015;9:400. doi: 10.3389/fnins.2015.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T, Ozbalci C, Brugger B, Rapaport D, Dimmer KS. Mcp1 and Mcp2, two novel proteins involved in mitochondrial lipid homeostasis. J Cell Sci. 2013;126:3563–3574. doi: 10.1242/jcs.121244. [DOI] [PubMed] [Google Scholar]
- Tauche A, Krause-Buchholz U, Rodel G. Ubiquinone biosynthesis in Saccharomyces cerevisiae: the molecular organization of O-methylase Coq3p depends on Abc1p/Coq8p. FEMS Yeast Res. 2008;8:1263–1275. doi: 10.1111/j.1567-1364.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- Tran UC, Clarke CF. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion. 2007;7(Suppl):S62–71. doi: 10.1016/j.mito.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Wiedemeyer WR, Dunn IF, Quayle SN, Zhang J, Chheda MG, Dunn GP, Zhuang L, Rosenbluh J, Chen S, Xiao Y, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci USA. 2010;107:11501–11506. doi: 10.1073/pnas.1001613107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf NI, Koenig M. Progressive cerebellar atrophy: hereditary ataxias and disorders with spinocerebellar degeneration. Handb Clin Neurol. 2013;113:1869–1878. doi: 10.1016/B978-0-444-59565-2.00057-5. [DOI] [PubMed] [Google Scholar]
- Xie LX, Hsieh EJ, Watanabe S, Allan CM, Chen JY, Tran UC, Clarke CF. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim Biophys Acta. 2011;1811:348–360. doi: 10.1016/j.bbalip.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E, van Aalten DM. Pseudokinases-remnants of evolution or key allosteric regulators? Curr Opin Struct Biol. 2010;20:772–781. doi: 10.1016/j.sbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.