

Abstract
Overexpression of B4GALNT2 (previously GALGT2) inhibits the development of muscle pathology in mouse models of Duchenne muscular dystrophy, congenital muscular dystrophy 1A, and limb girdle muscular dystrophy 2D. In these models, muscle GALGT2 overexpression induces the glycosylation of α dystroglycan with the cytotoxic T cell glycan and increases the overexpression of dystrophin and laminin α2 surrogates known to inhibit disease. Here, we show that GALGT2 gene therapy significantly reduces muscle pathology in FKRP P448Lneo− mice, a model for limb girdle muscular dystrophy 2I. rAAVrh74.MCK.GALGT2-treated FKRP P448Lneo− muscles showed reduced levels of centrally nucleated myofibers, reduced variance, increased size of myofiber diameters, reduced myofiber immunoglobulin G uptake, and reduced muscle wasting at 3 and 6 months after treatment. GALGT2 overexpression in FKRP P448Lneo− muscles did not cause substantial glycosylation of α dystroglycan with the cytotoxic T cell glycan or increased expression of dystrophin and laminin α2 surrogates in mature skeletal myofibers, but it increased the number of embryonic myosin-positive regenerating myofibers. These data demonstrate that GALGT2 overexpression can reduce the extent of muscle pathology in FKRP mutant muscles, but that it may do so via a mechanism that differs from its ability to induce surrogate gene expression.
A variety of forms of muscular dystrophy (MD), ranging from severe congenital forms such as Walker-Warburg syndrome (WWS), Fukuyama congenital MD, muscle eye brain disease, and congenital MD 1C and 1D to adult-onset limb girdle MDs (LGMDs), including LGMD2I, K, L, M, N, O, P, T, and U, arise from mutations in genes known to affect the glycosylation of α dystroglycan with O-mannose-phosphate–linked glycans that are required for laminin binding to the α dystroglycan protein.1, 2, 3, 4, 5 Because all of these disorders share improper post-translational glycosylation of α dystroglycan, they are collectively termed dystroglycanopathies. Dystroglycan is a central component of the dystrophin-associated glycoprotein (DAG) complex in striated muscles.6, 7 Dystroglycan is post-translationally processed into two proteins, α and β dystroglycan, that bind tightly but noncovalently to one another.6, 8 β Dystroglycan is a transmembrane protein that resides within the muscle sarcolemmal membrane, whereas α dystroglycan is a peripheral membrane–associated protein. Extracellular matrix (ECM) proteins, including laminin 211, agrin, and perlecan, bind to α dystroglycan, whereas the intracellular domain of β dystroglycan binds to cytoskeletal-associated proteins, including dystrophin utrophin and plectin 1, which in turn bind to filamentous actin and/or microtubules.7, 9 As such, dystroglycan serves as a vital link within the membrane to connect the ECM, particularly laminin 211 that is a principal component of the basal lamina that surrounds every skeletal myofiber in the adult,10, 11 through the membrane to the intracellular filamentous actin cytoskeleton. ECM proteins bind α dystroglycan through the O-mannose-phosphate–linked oligosaccharide structures present in its mucin-like domain,2 and mutations in well more than a dozen genes that control α dystroglycan glycosylation give rise to congenital or limb girdle forms of MD.3, 4 In their most severe forms, including WWS, Fukuyama congenital MD, and muscle eye brain disease, dystroglycanopathies not only affect skeletal muscle but also eye and brain development, leading to lissencephalic changes in the cerebral cortex due to defects in the glial limitans-pial basement membrane and to ocular malformations that can include retinal detachment.12, 13, 14
One gene that when mutated can give rise to dystroglycanopathies is fukutin-related protein, which is encoded by the FKRP gene. FKRP mutations have been reported to cause WWS, congenital MD 1C, and LGMD2I, with the more severe clinical phenotypes typically being associated with mutations that further affect the glycosylation of α dystroglycan and laminin binding to α dystroglycan.15, 16, 17, 18, 19 Mutations in FKRP and fukutin (FKTN), the gene with which FKRP shares homology and from which FKRP got its name,20, 21 both can give rise to forms of either congenital MD or LGMD.22, 23 Recently, both FKRP and fukutin have been shown to possess ribitol 5-phosphate transferase activity, and a tandem ribitol 5-phosphate moiety was shown to be present on α dystroglycan on which laminin binding glycans could be synthesized and which are themselves absent in FKRP-, fukutin-, and isoprenoid synthase domain containing–deficient cells.24
Lu and colleagues25 have designed a series of mouse knockin models of disease-relevant human FKRP mutations that give rise to varying degrees of muscle disease pathology and dysfunction. Introduction of the FKRP P448L mutation, in the presence of a neomycin gene cassette, gives rise to severe muscle pathology consistent with WWS,26 whereas deletion of the neo cassette from the P448L knockin allele gives rise to a milder disease spectrum more analogous to LGMD2I, with adult onset muscle necrosis and evidence of muscle damage and regeneration, coupled with inflammation, fibrosis, and muscle wasting.25 This mouse model, like human LGMD2I, shows reduced functional glycosylation of α dystroglycan in both heart and skeletal muscles, whereas dystroglycan protein is expressed at or near normal levels.19 Here, we have used the neo-deleted version of the FKRP P448L mouse model (FKRP P448Lneo−) to assess the value of a particular gene therapy for patients with LGMD2I.19, 25
Although defects in α dystroglycan glycosylation can give rise to forms of MD, other enzymes that glycosylate α dystroglycan, when overexpressed, can significantly increase muscle protection and prevent muscle pathology. One such approach involves overexpression of B4GALNT2 (formerly GALGT2). The GALGT2 gene encodes a β1 to 4 N-acetylgalactosaminyl transferase that is confined in expression in adult skeletal muscle to two highly specialized membrane domains, the neuromuscular junction and the myotendinous junction, where it generates the cytotoxic T cell (CT) glycan (GalNAcβ1-4-[Neu5Ac/Gcα2-,3]-Galβ1-4-GlcNAcβ-).27, 28, 29, 30, 31 The CT glycan, as the name implies, was originally identified in CD8+ T cells,32, 33 but this glycan is actually expressed in a number of human organs, with highest expression found in the colon.34, 35 Overexpression of GALGT2 in skeletal muscles, either using transgenic mice or via gene therapy methods using adeno-associated virus (AAV), inhibits the development of muscle pathology associated with MD in the mdx mouse model for Duchenne MD, the dyW mouse model for congenital MD 1A, and the Sgca−/− mouse model for LGMD2D.36, 37, 38, 39, 40
GALGT2 overexpression induces the glycosylation of α dystroglycan with the normally synaptic CT glycan and induces the ectopic overexpression of dystrophin and laminin α2 surrogates that normally bind dystroglycan at the neuromuscular junction and myotendinous junction, including utrophin, plectin 1, laminin α5, and agrin, both in mice and in macaques.29, 36, 37, 39, 41, 42, 43 Overexpression of such proteins, in particular utrophin and agrin, is known to provide therapeutic benefit in mdx and dyW mice, respectively.44, 45, 46, 47, 48 GALGT2 overexpression has a profound impact in preventing muscle injury, including injury caused by eccentric contractions, and can inhibit the development of muscle pathology in mdx mice for almost the entire life span of the animal.38, 40 Recently, we showed that transgenic overexpression of GALGT2 only had a transient effect on muscle pathology in mice where specific muscles had been targeted to delete the gene encoding dystroglycan.49 These data suggest that GALGT2 glycosylation of α dystroglycan is central to its therapeutic mechanism. Here, we tested whether GALGT2 overexpression affects disease when dystroglycan is not deleted but when its impaired glycosylation with O-mannose-phosphate–linked structures causes LGMD2I-like disease.
Materials and Methods
Materials
Agarose-bound lectins (Wisteria floribunda agglutinin, WFA; and wheat germ agglutinin, WGA) were purchased from EY Laboratories (San Mateo, CA). Fluorescein isothiocyanate (FITC)-conjugated WFA was obtained from Vector Laboratories (Burlingame, CA). Both rAAVrh74.MCK (muscle creatine kinase).GALGT2 (human) and rAAV1.CMV (cytomegalovirus). Galgt2 (mouse) were made by the Viral Vector Core at Nationwide Children's Hospital (Columbus, OH). Monoclonal antibodies to dystrophin (Dy4/6D3), utrophin (DRP3/20C5), and embryonic myosin heavy chain (RNMy2/902) were obtained from Novocastra (Newcastle Upon Tyne, UK). A mouse monoclonal antibody to Plectin 1 (10F6) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A monoclonal antibody to the α2 chain of laminin (4H8-2) and polyclonal antibody to glyceraldehyde-3-phosphate dehydrogenase (G9545) were obtained from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal antibodies to α-dystroglycan (IIH6) and agrin (MAB5204) were obtained from EMD Millipore (Temecula, CA). A polyclonal antibody to α-dystroglycan (AF6868) and agrin (AF550) were obtained from R&D Systems (Minneapolis, MN). A polyclonal antibody to embryonic myosin heavy chain (MHY3) was obtained from Proteintech (Chicago, IL). A polyclonal antibody to β-1,4 N-acetylgalactosaminyltransferase 2 (GALGT2 or B4GALNT2) was obtained from Abnova (Taipei City, Taiwan). A rabbit polyclonal antibody to integrin α7B was a gift from Eva Engvall (Burnham Institute, La Jolla, CA). Rabbit polyclonal antibodies to laminin α4 (1100) and laminin α5 (8948) were gifts from Jeffery Miner (Washington University, St. Louis, MO). CT1 and CT2, mouse monoclonal antibodies that recognize the CT glycan, were produced in our laboratory from hybridomas originally generated by Leo Lefrancois (University of Connecticut, Storrs, CT). MANDAG2, a monoclonal anti–β-dystroglycan antibody, was also produced in our laboratory from a hybridoma cell line (Developmental Studies Hybridoma Bank, Iowa City, IA). Horseradish peroxidase- and FITC-conjugated goat anti-mouse immunoglobulin G (IgG)1 and IgG2A secondary antibodies were obtained from Bethyl Laboratories (Montgomery, TX). Fab fragment donkey anti-mouse IgG and all other horseradish peroxidase-, FITC-, cyanine 3- or rhodamine-conjugated secondary antibodies were purchased from Jackson Immunochemicals (Seattle, WA). CT2, polyclonal antibody to embryonic myosin heavy chain, glyceraldehyde-3-phosphate dehydrogenase, B4GALNT2, and monoclonal antibody to agrin antibodies were used only for immunoblotting, whereas embryonic myosin heavy chain, α2 chain of laminin, and polyclonal antibody to agrin antibodies were used only for immunostaining. All other primary antibodies were used for both methods.
Mice
All mouse experiments were performed using protocols approved by the Institutional Animal Care and Use Committee at The Research Institute at Nationwide Children's Hospital. FKRP P448L (neo−) mice were a generous gift of Qi Long Lu (University of North Carolina Charlotte Health Science Center, Charlotte, NC).25 For these experiments, homozygous FKRP P448Lneo− mice were generated and were maintained on a C57Bl/6 background. All untreated control animals were age-matched to treated littermates. C57BL/6 mice were used as age-matched wild-type (WT) controls. To investigate the role of dystroglycan (Dag1) in GALGT2 gene therapy for MD, we used conditional Dag1 knockout mice where gene deletion was mediated by Cre-lox recombination with Cre expression being controlled by Pax3 promoter (P3Pro-Cre) in a transgenic strain in which Cre was only expressed in caudal muscles but not in diaphragm or rostral muscles due to the integration site of the P3Pro-Cre transgene.50 Mice bearing a floxed allele of Dag1 (Dag1lox/lox) and the Pax3-Cre transgene (P3Pro-Cre) were kindly provided by Jeffery Miner (Washington University Medical School). Dag1lox/lox mice were originally generated by Campbell and colleagues,51 and P3Pro-Cre mice were originally generated by Epstein and colleagues.52 For breeding, female Dag1lox/lox mice were bred with male P3Pro-Cre transgenic mice to produce P3Pro-CreDag1lox/+ mice. Male P3Pro-CreTgDag1lox/+ mice were then bred with female Dag1lox/lox mice to obtain P3Pro-CreDag1lox/lox mice in which Dag1 was deleted (Dag1−/−) from caudal skeletal muscles. Dag1lox/lox littermates and P3ProCre mice were used as Dag1+/+ controls, with no difference seen between these two strains and WT mice for the assays performed.
Intramuscular Injection
The gastrocnemius (gastroc), quadriceps (quad), and tibialis anterior (TA) muscle on the left side of 2- to 4-week-old FKRP P448Lneo− mice were injected with 5 × 1011, 5 × 1011, and 1 × 1011 vector genomes (vgs), respectively, of rAAVrh74.MCK.GALGT2 (human gene). At 1, 3, or 6 months after injection, mice were sacrificed and muscles were dissected. One 3-month-old male FKRP P448Lneo− mouse was also injected as described earlier in this paragraph and sacrificed at 6 months after injection. For P3Pro-CreDag1lox/lox mice, the gastroc, quad, TA, biceps, and triceps muscles on the left side of 5-day-old mice were injected once (TA, biceps, triceps) or once daily for 5 days (gastroc, quad) with 1 × 1010 vg of AAV1-CMV-Galgt2 (mouse gene). For all mice, gastroc and quad muscles were injected with AAV vector in a volume of 0.05 mL sterile phosphate-buffered saline (PBS), whereas TA, biceps, and triceps muscles were injected with a volume of 0.025 mL. Muscles on the contralateral (right) side of the mice were mock-injected with an identical volume of sterile PBS. Injections were performed using a 0.3-mL insulin syringe near the midpoint of the belly of the muscle.
Histology and Immunohistochemistry
Muscles were snap-frozen in liquid nitrogen–cooled isopentane and sectioned at 10 μm on a cryostat. Cut cross-sections were either stained with hematoxylin and eosin as described previously53 or immunostained with antibodies IIH6; embryonic myosin heavy chain; integrin α7B; laminin α2, α4, or α5; plectin; or utrophin. Muscle cross-sections were blocked with 10% goat serum and 0.05% Triton X-100 in Tris-buffered saline (TBST) for 1 hour at room temperature. For immunostaining with CT2, sections were blocked in PBS with 3 mg/mL bovine serum albumin for 1 hour at room temperature. For polyclonal antibody to α-dystroglycan immunostaining sections were blocked with 10% donkey serum in TBST for 1 hour at room temperature, and for polyclonal antibody to agrin immunostaining sections were not blocked before incubation with primary antibody. Immunostaining for dystrophin was performed as described by Mundegar et al.54 For staining with mouse monoclonal antibodies (except CT2), sections were incubated with 0.1 mg/mL Fab fragment donkey anti-mouse IgG in TBS for 2 hours at room temperature and washed in TBS before blocking. For plectin and utrophin immunostaining, sections were first fixed in 0.5% paraformaldehyde in TBS and 5% Triton X-100 for 5 minutes at room temperature. For lipid extraction, sections were incubated in −20°C methanol and then in 25°C 1:1 chloroform:methanol for 5 minutes each before blocking. Primary antibodies were diluted in blocking solution, and sections were incubated overnight at 4°C. Sections were washed in TBS or PBS before incubation with appropriate fluorophore-conjugated secondary antibody and for 1 hour at room temperature. Sections were sometimes counterstained with WFA and Hoechst 33342, washed, and mounted using ProLong Gold anti-Fade Reagent (Life Technologies, Grand Island, NY). Staining was visualized on a Zeiss Axiophot epifluorescence microscope (Carl Zeiss, Thornwood, NY) with appropriate filters and images were captured using Zeiss AxioVision LE imaging software version 4.1 (Carl Zeiss). For comparisons of GALGT2-treated and untreated muscles, all comparisons from each antibody stain were time-matched for exposure, and the same concentration of antibody was used for staining.
Quantification of CT Glycan Overexpression, Myofiber Diameter, and IgG+ Muscle Area
To quantify CT glycan overexpression and changes in myofiber diameter, laminin α2 and WFA-FITC–stained cross-sections of injected and contralateral muscles were photographed at 10× magnification. For gastroc, quad, and TA muscles, 7, 8, and 6 representative images, respectively, were taken per side and per animal for analysis. The number of fibers with sarcolemmal WFA staining was expressed as a percentage of all fibers stained with laminin α2. For non-myofiber area, hematoxylin and eosinstained cross-sections of WT and injected and contralateral FKRP P448Lneo− mouse muscles were photographed at 20× magnification. Five characteristic images from each muscle were used for quantification. For quantification of IgG+ muscle area, anti-mouse IgG– and WFA-FITC–stained injected and contralateral FKRP P448Lneo− mouse muscles were photographed at 10× magnification. Five characteristic images per muscle were used for quantification. Fiber counting and semiautomated fiber diameter, nonmyofiber area, and mouse IgG staining quantification were performed with ImageJ software version 1.46r (NIH, Bethesda, MD; http://imagej.nih.gov/ij).
Lectin Precipitation
Gastroc muscles were dissected, minced, and solubilized in NP-40 lysis buffer (Tris-buffered saline pH 7.4, 1% Nonidet-P40, 1 mmol/L EDTA, 1:50 proteinase inhibitor cocktail; Roche Diagnostics, Indianapolis, IN) with shaking overnight at 4°C. The concentration of total protein was determined using a BCA Protein Assay Kit (Life Technologies). Differing amounts of extracted proteins (3.0, 1.0, 0.5, and 0.15 mg) were precipitated with WFA- or WGA-conjugated agarose at 4°C overnight. Precipitated proteins were collected by centrifuging the lectin-conjugated agarose at 1000 × g for 3 minutes, washing three times with NP-40 lysis buffer. The pellet was then boiled in NuPAGE LDS Sample Buffer (Life Technologies) with 100 mmol/L β-mercaptoethanol. Solubilized precipitated material was then separated by SDS-PAGE on 4% to 12% gradient gels and transferred to nitrocellulose. For blotting with CT1 or CT2, blots were blocked in TBS with 0.05% Tween 20 (TBST20) and 1% nonfat dry milk. For all other blotting, TBST20 with 5% nonfat dry milk was used. After blocking, blots were incubated with primary antibody, washed, bound to the appropriate peroxidase-conjugated secondary antibody, washed, and developed using enhanced chemiluminescence (ECL) Ultra (Lumigen, Southfield, MI).
Dystroglycan Immunoprecipitation
Quad muscles were dissected, minced, and solubilized in NP-40 lysis buffer with shaking overnight at 4°C. One or 3 mg of solubilized protein for each sample was then subjected to immunoprecipitation with the anti–β-dystroglycan antibody, MANDAG2,55 essentially as previously described except for the stronger detergent conditions.41 Solubilized precipitated material was then separated by SDS-PAGE on 4% to 12% gradient gels and transferred to nitrocellulose. For blotting with CT1, blots were blocked in TBST20 and 1% nonfat dry milk. For all other blotting, TBST20 with 5% nonfat dry milk was used. After blocking, blots were incubated with primary antibody, washed, bound to the appropriate peroxidase-conjugated secondary antibody, washed, and developed using ECL Ultra (Lumigen).
Immunoblotting
Gastroc muscles were dissected, minced, and solubilized in NP-40 lysis buffer with shaking overnight at 4°C. The concentration of total protein was determined by BCA Protein Assay Kit, after which time 40 μg of protein was denatured by boiling in LDS Sample Buffer with 100 mmol/L β-mercaptoethanol. Samples were then separated by SDS-PAGE on 4% to 12% gradient gels and transferred to nitrocellulose. Blots were blocked in TBST20 with 5% nonfat dry milk, incubated with primary antibody, washed, and bound to the appropriate peroxidase-conjugated secondary antibody, then washed and developed using ECL Ultra (Lumigen).
Real-Time Quantitative PCR of Viral Genomes
DNA was extracted from approximately 10 mg of tissue from treated and contralateral (untreated) gastroc, quad, and TA muscles using the DNeasy Blood & Tissue Kit (Qiagen, Germantown, MD). DNA amount and purity was assessed using a NanoDrop 2000c spectrophotometer (Thermo Scientific, Waltham, MA), and only samples with at least 1.7 for 230/260 nm and 260/280 nm absorbance ratios were used. Samples were diluted to 20 ng DNA/μL with sterile water, and 5 μL of sample was added per PCR reaction. Real-time quantitative PCR was performed with TaqMan Gene Expression Master Mix and a vector-specific primer set (forward, 5′-CCTCAGTGGATGTTGCCTTTA-3′; probe, FAM/5′-AAAGCTGCG/ZEN/GAATTGTACCCGC-3′/3IABkFQ; and reverse, 5′-ATCTTGAGGAGCCACAGAAATC-3′) using the TaqMan ABI 7500 sequence detection system (Applied Biosystems, Life Technologies, Grand Island, NY) as previously described.43 The primer set used did not amplify the endogenous mouse Galgt2 gene or endogenous MCK promoter elements. The pAAV.MCK.GALGT2 plasmid used to calibrate signals was linearized with ClaI, and a standard curve was generated using between 5 × 101 and 5 × 106 DNA copies in increasing logarithmic increments for each assay. Each standard curve had Pearson correlation coefficients of at least 0.98 and did not yield any signal without plasmid present. Untreated WT or FKRP P448Lneo− control sample amplification signals were also undetectable. Each sample and standard reaction were run in triplicate.
Semiquantitative RT-PCR
Approximately 10 mg of tissue from WT, treated, and contralateral (untreated) FKRP P448Lneo− gastroc, quad, and TA muscles were homogenized in TRIzol reagent (Invitrogen, Life Technologies, Grand Island, NY) via high speed shaking using the TissueLyser II (Qiagen). Samples were centrifuged for 5 minutes at 12,000 × g, and RNA was isolated from the supernatant fluid using a Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA). RNA integrity and quantity were measured using a 6000 Nano LabChip kit on a Bioanalyzer 2100 (Agilent, Santa Clara, CA) and a NanoDrop 2000c spectrophotometer, respectively. Only samples of high purity with no evidence of degradation were used. A high-capacity cDNA archive kit (Applied Biosystems) was used to reverse transcribe 600 ng of RNA per the supplied protocol. Synthesized cDNA was diluted 1:100 with nuclease-free water and subjected to RT-PCR in duplicate using TaqMan Gene Expression Master Mix and a TaqMan ABI 7500 sequence detection system with 18S as internal reference (product no. 4319413E; Life Technologies) as described previously.37 Primer sets obtained from Integrated DNA Technologies (Coralville, IA) are as follows: Mus musculus Galgt2: 5′-GCCCACGGAGAGTTCTTTATT-3′ (forward), FAM/5′-ATAATTACA/ZEN/CCGGGGCAA-3′/3IABkFQ (probe), 5′-TCCTTTGGTGGTGTTCTTACTT-3′ (reverse); Integrin α7: 5′-CATAAGACCCTTCCCAGAATCG-3′ (forward), FAM/5′-CCAGCCGGA/ZEN/GACTTGACCTTGAAT-3′/3IABkFQ (probe), 5′-ACCAACATTGATAGCTCAGACC-3′ (reverse); Laminin α2: 5′-CTCACAGAAGTCACCAGCAG-3′ (forward), FAM/5′-AGACCTCAG/ZEN/TATGCCAGAATTGTCAGC-3′/3IABkFQ (probe), 5′-GGACCAACCTTAGGAACCTG-3′ (reverse); Laminin α4: 5′-TTCCTTCTCAACCAGCATACC-3′ (forward), FAM/5′-TGACTTCCC/ZEN/TTCTCATGGAGTCCCT-3′/3IABkFQ (probe), 5′-AACAACTCGGAGAACACACTG-3′ (reverse); and Plectin 1: 5′-GTAAGCGTCCATGTCTTCACT-3′ (forward), FAM/5′-TGCTACATT/ZEN/CACAGTGCCTCCCAA-3′/3IABkFQ (probe), 5′-CAGTCCCAAGTGCCATGAG-3′ (reverse). Primer sets obtained from Applied Biosystems are as follows: agrin (Mm01545840_m1); dystroglycan (Mm00802400_m1), dystrophin (Mm01216926_m1), embryonic myosin heavy chain 3 (Mm01332463_m1), laminin α5 (Mm01222011_m1); and utrophin (Mm01168861_m1). Gene expression was determined as relative changes by the 2−ΔΔCt method,56 and the data are presented as fold difference normalized to 18S ribosomal RNA.
Statistical Analysis
Determinations of significance were estimated using two-way analysis of variance; P < 0.05 was considered significant.
Results
rAAVrh74.MCK.GALGT2 Treatment of FKRP P448Lneo− Muscles Results in High Expression of the CT Glycan
We began by treating specific hindlimb muscles the gastroc, quad, and TA in FKRP P448Lneo− mice with rAAVrh74.MCK.GALGT2 gene therapy. Because the TA muscle is significantly smaller than the quad and gastroc muscles, we used only 1 × 1011 vg to inject this muscle, whereas we used 5 × 1011 vg to inject the gastroc and quad muscles, much as previously done in mdx, dyW, and Sgca−/− mice.37, 39, 40 Mice were injected between 2 and 4 weeks of age, before the development of significant muscle pathology, and were followed at 1 month after treatment, when viral vector expression should become maximal,37, 57 and at 3 and 6 months after treatment, when muscle pathology is evident.25
First, we quantified the transduction of DNA vgs from the AAV vector into the injected muscles using real-time quantitative PCR (Figure 1A). As expected for the doses given, rAAVrh74.MCK.GALGT2 treatment led to high levels of vgs, on the order of 400,000 to 700,000 vg/μg of genomic DNA 1 month after treatment, or roughly three to four vector copies per diploid genome. These levels were reduced at 3 and 6 months after treatment compared with the 1-month after treatment, such that by 6 months titers were at 10,000 to 75,000 vg/μg, with the quad muscles having overall lower levels of transduction than the gastroc and TA muscles. Because the rAAVrh74 serotype can cross the vascular barrier, there was a small amount of leakage of vector out of injected muscles and into the blood stream that carried vector to the contralateral muscle as well, but these levels were several logs lower than the vector amounts measured in the injected muscles.
Figure 1.

GALGT2 and CT glycan expression after intramuscular injection of rAAVrh74.MCK.GALGT2 in FKRP P448Lneo− mice. The gastroc, quad, and TA muscles of P448L FKRP mutant mice were injected with rAAVrh74.MCK.GALGT2 (treated) or PBS (untreated) and compared at 1, 3, and 6 months after injection. A: rAAV vgs delivered were quantified per microgram of genomic DNA. B: Relative levels of AAV-introduced human GALGT2 mRNA transcripts were quantified, relative to endogenous mouse Galgt2 expression, and normalized to 18S mRNA. C: Quantification of the percentage of total myofibers with membrane overexpression of the CT glycan. D: Number of vgs per transduced muscle is plotted against the percentage of myofibers overexpressing CT glycan for muscles used for this study. Error bars represent SEM. n = 9 (A), 6 (B), or 18 to 24 (C) measurements from three muscles per group. *P < 0.05, **P < 0.01, and ***P < 0.001. AAV, adeno-associated virus; CT, cytotoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; quad, quadriceps; rAAV, recombinant adeno-associated virus; TA, tibialis anterior; vg, vector genome.
We extracted mRNA and analyzed, by quantitative RT-PCR, the relative increase in human GALGT2 transcripts compared with endogenous mouse Galgt2 transcripts, normalized to 18S RNA (Figure 1B). We observed a very significant increase in GALGT2 gene expression in all treated muscles. As for vg measures, this expression peaked at 1 month after treatment and then declined at 3 and 6 months after treatment. Even so, at 6 months, we observed a significant amount of human GALGT2 gene overexpression relative to endogenous mouse Galgt2. It is important to note here that endogenous mouse Galgt2 is expressed at very low levels in skeletal muscle, making the denominator quite small for such comparisons. In addition, AAV-GALGT2 treatment never altered endogenous mouse Galgt2 expression, and mouse Galgt2 expression remained equivalent between treated and contralateral untreated muscles.
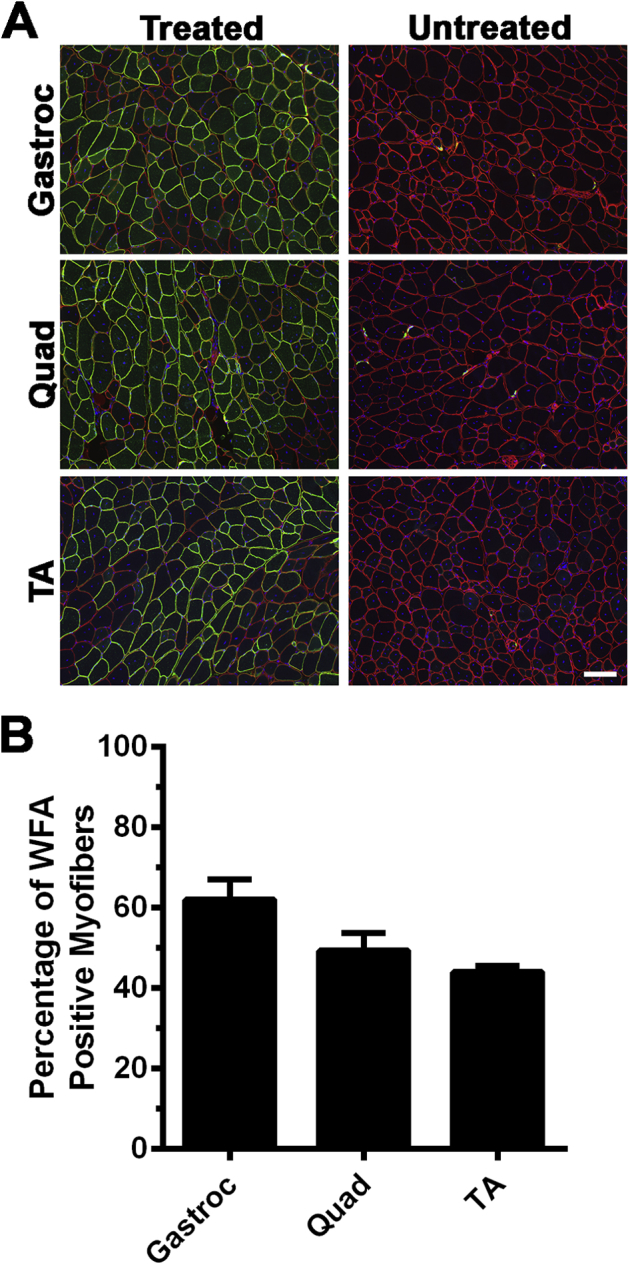
We next stained rAAVrh74.MCK.GALGT2-treated and untreated FKRP P448Lneo− muscles with WFA, a β-GalNAc–binding lectin that can identify GALGT2-overexpressing myofibers (Figure 1C and Figure 2A). WFA staining is analogous to staining with the CT2 anti-CT glycan antibody, which also recognizes increased GALGT2-dependent muscle glycosylation.27 For all three treated muscles (gastroc, quad, and TA), we observed that most myofibers overexpressed GALGT2 activity. Here again, this percentage declined at 3 and 6 months after treatment, with only 20% to 35% of myofibers maintaining CT glycan overexpression at 6 months after treatment. For rAAVrh74.MCK.GALGT2-treated muscles, WFA and CT2 costaining was also performed to confirm that WFA staining recognized overexpressed CT glycan (Figure 2B). The loss of expression may have something to do with the early time of treatment, because injecting a 3-month-old FKRP P448Lneo− mouse with the same doses of rAAVrh74.MCK.GALGT2 in the TA, gastroc, and quad muscles led to CT glycan overexpression in 45% to 60% of myofibers when analyzed 6 months after treatment (Supplemental Figure S1).
Figure 2.

CT glycan staining in rAAVrh74.MCK.GALGT2-treated FKRP P448Lneo− muscle. A: The gastroc, quad, and TA muscles of FKRP P448Lneo− mice were injected with rAAVrh74.MCK.GALGT2 (treated) and analyzed at 1, 3, and 6 months after injection compared with contralateral PBS-injected controls (untreated). Cross-sections of muscle were stained with WFA (green) to label CT glycan overexpression and with an antibody to laminin α2 (red) to label the muscle sarcolemma. Nuclei were stained with DAPI (blue). B: Costaining with WFA lectin (green) and CT2 (red) to show WFA staining of CT glycan recognized by an anti-CT glycan antibody in rAAVrh74.MCK.GALGT2-treated muscle at 3 months after treatment. Overlap is shown in merged image in yellow. Scale bars: 100 μm (A); 50 μm (B). Ab, antibody; CT, cytotoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; quad, quadriceps; rAAV, recombinant adeno-associated virus; TA, tibialis anterior; WFA, Wisteria floribunda agglutinin.
By comparing the vg of rAAVrh74.MCK.GALGT2 for all injected muscles with the percentage of myofibers overexpressing CT glycan, we generated a correlation curve for CT glycan overexpression that showed that the vgs needed to achieve relative percentages of muscle transduction in FKRP P448Lneo− muscles (Figure 1D). These data show that about 20,000 vg/μg genomic DNA is needed to transduce 20% of myofibers, an amount previously shown to be needed to achieve a physiological improvement in mdx skeletal muscle,38 whereas 80,000 vg/μg genomic DNA is needed for 50% transduction and about 500,000 vg/μg genomic DNA, or roughly 3 AAV vgs per diploid mouse genome, for 80% transduction in FKRP P448Lneo− muscle (Figure 1D). These observations are similar to data we previously generated in macaque muscle, where 500,000 vg/μg genomic reflected transduction of 75% of myofibers in the gastroc after vascular delivery of rAAVrh74.MCK.GALGT2.43
Costaining with WFA and a laminin α2 antibody confirmed that most myofibers were transduced in the TA, gastroc, and quad muscles at 1 month after treatment with rAAV74.MCK.GALGT2 and that the expression was maintained at 3 months after treatment (Figure 2A). A significant fraction of myofibers, however, lost expression at 6 months after treatment (Figure 2A). Increased intracellular WFA staining was also evident in some muscles, perhaps reflecting background binding of the lectin to immaturely glycosylated O-linked GalNAc structures or to GalNAcβ1,3GlcNAcβ1,4Man(6P)-O-linked structures on α dystroglycan that would normally be glycosylated with ribitol 5-phosphate by FKRP.24 Merged images of WFA staining and laminin α2 are shown in Figure 2 to show regions of coincident staining. Cumulatively, these data show that rAAVrh74.MCK.GALGT2 treatment of FKRP P448Lneo− muscles led to increased expression of vgs, GALGT2 mRNA, and functional GALGT2 production of the CT glycan in all treated muscles; however, some expression was lost for all three measures as time after treatment increased. In addition, these studies show that one needs about 500,000 vg/μg of genomic DNA in a treated FKRP P448Lneo− muscle to transduce most skeletal myofibers.
rAAVrh74.MCK.GALGT2 Treatment of FKRP P448Lneo− Mice Reduces Skeletal Muscle Pathology
We next assessed the extent of muscle pathology in GALGT2-treated and -untreated FKRP P448Lneo− muscles. We stained muscle cross-sections with hematoxylin and eosin (Figure 3A). Stained sections were then scored for the percentage of myofibers with centrally located nuclei, myofiber diameter, CV in myofiber diameter, myofiber uptake of IgG, and the percentage of nonmuscle area (Figure 3, Figure 4, and 5). The percentage of myofibers with central nuclei is used as a measure of cycles of muscle degeneration and regeneration, because dystrophic mouse muscles maintain nuclei within the center of the muscle at much higher incidence once regeneration has occurred after muscle injury or loss.58 Likewise, dystrophic processes lead to both hypertrophy and the presence of smaller regenerating muscles, which lowers the average measure of myofiber diameter and increases its variance.58 In addition, skeletal myofibers can take up serum IgG on membrane rupture, and the percentage of nonmuscle area in the muscle is increased as muscle fibers are lost and replaced with extracellular matrix or fat. Age-matched WT (C57Bl/6) muscles were used for comparison in some instances.
Figure 3.

Inhibition of muscle pathology by GALGT2 overexpression in FKRP P448Lneo− mice. The gastroc, quad, and TA muscles of FKRP P448Lneo− mice were treated with rAAVrh74.MCK.GALGT2 at 2 to 4 weeks of age, analyzed at 1, 3, and 6 months after injection, and compared with age-matched C57/BL6 (wild-type) controls. A: H&E staining of cross-sections of rAAVrh74.MCK.GALGT2- (treated) or PBS- (untreated) injected FKRP P448Lneo− mouse and wild-type mouse muscles. B–D: The percentage of centrally located myofiber nuclei (B), the average myofiber diameter (C), and the variance in myofiber diameter (D) were compared for GALGT2-expressing (positive), nonexpressing myofibers within treated muscle (ipsilateral negative), and nonexpressing myofibers in untreated muscles (contralateral negative). Line at 250 (or below) indicates a nondystrophic level of variance. E: The percentage of nonmuscle area in muscle sections was compared in treated and untreated FKRP P448Lneo− muscles and in unaffected wild-type muscles. Error bars represent SEM. n = 21 (B), 24 (C), 18 (D), or 15 (E) measurements from three muscles per group. Scale bar = 50 μm. *P < 0.05, **P < 0.01, and ***P < 0.001. GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; H&E, hematoxylin and eosin; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; quad, quadriceps; rAAV, recombinant adeno-associated virus; TA, tibialis anterior.
Figure 4.

Change in myofiber size distribution after overexpression of GALGT2 in FKRP P448Lneo− mice. The gastroc, quad, and TA muscles of rAAVrh74.MCK.GALGT2-treated and -untreated FKRP P448Lneo− mice were analyzed at 1, 3, and 6 months after injection. GALGT2 overexpressing (positive), non-GALGT2 overexpressing myofibers within rAAVrh74.MCK.GALGT2-injected muscle (ipsilateral negative) and non-GALGT2 overexpressing myofibers in PBS-injected muscle (contralateral negative) were identified via WFA staining. Mini-Feret myofiber diameters were quantified and plotted in terms of frequency of size distribution. Data points are averages of 21 (gastroc), 24 (quad), or 18 (TA) measurements from three muscles per group. GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; quad, quadriceps; rAAV, recombinant adeno-associated virus; TA, tibialis anterior; WFA, Wisteria floribunda agglutinin.
Figure 5.

Quantification of immunoglobulin G (IgG) uptake after overexpression of GALGT2 in FKRP P448Lneo− mice. The gastroc, quad, and TA muscles of FKRP P448Lneo− mutant mice were injected with rAAVrh74.MCK.GALGT2 (treated) or PBS (untreated) and compared at 1, 3, and 6 months after injection. A: Gastroc muscle cross-sections were stained with WFA (green) to label CT glycan overexpression and an anti-mouse IgG antibody to identify myofibers with IgG uptake (red). Nuclei were stained with DAPI (blue). B: Quantification of the percentage of total IgG+ area in muscle cross-sections. Error bars represent SEM. n = 15 measurements from three muscles per group. *P < 0.05, **P < 0.01, and ***P < 0.001. Scale bar = 100 μm. CT, cytotoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; quad, quadriceps; rAAV, recombinant adeno-associated virus; TA, tibialis anterior; WFA, Wisteria floribunda agglutinin.
First, muscle sections were quantified for the percentage of myofibers with centrally located nuclei (Figure 3B). Multiple published studies have shown that this level is not elevated beyond 5% and is often well below that in WT muscles.36, 58 At 1 month after treatment (between 6 and 8 weeks of age), most FKRP P448Lneo− muscles have not yet undergone a significant extent of myofiber damage, but in treated muscles, only nonexpressing myofibers had central nuclei. By 3 months after treatment, all contralateral (untreated) FKRP P448Lneo− muscles had significant elevations in percentages of myofibers with central nuclei relative to 1 month after treatment (P < 0.001 for all; not shown), and myofibers expressing the GALGT2 transgene had significantly reduced percentages relative to both contralateral untreated myofibers and ipsilateral nonexpressing myofibers in the treated muscle. The same findings were present at 6 months after treatment, although the values for expressing myofibers increased somewhat relative to 3 months after treatment. These findings show that rAAVrh74.MCK.GALGT2 treatment of FKRP P448Lneo− muscles at an early age can inhibit the cycles of skeletal myofiber degeneration and regeneration that arise from MD.
Measures of myofiber diameter likewise showed that GALGT2-overexpressing FKRP P448Lneo− myofibers had significantly increased average myofiber diameter (Figure 3C) and reduced CV in myofiber diameter (Figure 3D) relative to untreated contralateral muscles, again indicating a lack of muscle damage and normal muscle growth. The CV in myofiber diameter is considered normal if at 250 or below. Here again, at 1 month after treatment, some nonexpressing ipsilateral myofibers showed reduced diameters, which would be consistent with increased numbers of smaller regenerating myofibers. We additionally plotted the frequency distribution of myofiber diameters for each condition in treated myofibers and in ipsilateral and contralateral untreated myofibers (Figure 4). These data showed uniform distributions of increased myofiber diameters over a range of sizes at 3 and 6 months after treatment in treated muscles and that myofiber diameters were increased in all rAAVrh74.MCK.GALGT2-treated FKRP P448Lneo− muscles.
Finally, we assessed the percentage of nonmuscle area in muscle sections (Figure 3E). This is a measure of muscle loss due to muscle wasting, which is the single most profound cause of muscle weakness in the MDs. As expected, WT muscles showed a baseline level of nonmuscle area below 5%. This 5% value includes intramuscular blood vessels, ECM, and other nonmuscle structures (eg, muscle spindles and intramuscular peripheral nerves). In untreated FKRP P448Lneo− muscles, the extent of nonmuscle area was elevated twofold (to 10% of total area) at 1 month after treatment (6 to 8 weeks of age) and increased further to 15% to 20% by 3 or 6 months after treatment. Treated FKRP P448Lneo− muscles showed significant reductions in nonmuscle area in the gastroc, quad, and TA muscles, such that the percentage of nonmuscle area was reduced by one-half or more relative to untreated muscles at all ages. The trend toward elevated muscle loss with age, however, was still present in treated muscles as one went from 1 to 3 to 6 months after treatment. Thus, treated FKRP P448Lneo− muscles still showed elevated muscle loss relative to WT, and this was consistent with an only partial therapeutic effect and with the slow loss of muscle GALGT2 expression over time (Figure 1, A–C).
The reduction in all of these pathology measures in GALGT2-treated muscles strongly argues that GALGT2 overexpression protects FKRP P448Lneo− myofibers from damage. When myofiber membranes are ruptured, they release cytoplasmic muscle proteins to the exterior of the cell that can enter the serum, for example, creatine kinase, and take up serum proteins into the muscle cell, including IgG. To assess damage resulting in IgG uptake, we stained GALGT2-treated and control FKRP P448Lneo− muscles with an anti-mouse IgG antibody and scored the area of IgG+ staining relative to total muscle area (Figure 5). There was increased IgG uptake in untreated FKRP P448Lneo− gastroc, quad, and TA muscles at 3 and 6 months after treatment, with a particular spike in damage at 3 months. This heightened membrane damage at 3 months was significantly reduced, by 70% to 80%, in all GALGT2-treated muscles.
Effect of rAAVrh74.MCK.GALGT2 Treatment on α Dystroglycan Expression and Glycosylation
FKRP P448Lneo− muscles, like human LGMD2I muscles, maintain some, albeit reduced, expression of natively glycosylated α dystroglycan, which is the form of the protein most competent to bind to ECM proteins, including laminin 211.7, 16, 19 Natively glycosylated α dystroglycan is most readily observed by binding of the IIH6 monoclonal antibody, which requires native glycosylation for binding.59 IIH6 also can block laminin binding to the O-mannose-phosphate–linked glycans present on the α dystroglycan protein.7 Consistent with the findings reported by Lu and colleagues,60 we observed IIH6 expression predominantly on small regenerating myofibers in untreated FKRP P448Lneo− muscles (Figure 6A); however, increased numbers of myofibers stained with IIH6 were observed in rAAVrh74.MCK.GALGT2-treated FKRP P448Lneo− muscle sections; these myofibers also expressed embryonic myosin (Figure 6A). Expression of IIH6, however, was not elevated on most myofibers overexpressing WFA, although a few such myofibers did contain increased IIH6 staining. Costaining for IIH6 and embryonic myosin showed that positively stained myofibers expressed both markers (Figure 6B). Thus, rAAVrh74.MCK.GALGT2-treated muscles had increased expression of both IIH6 and embryonic myosin, indicating the presence of increased numbers of regenerating myofibers expressing natively glycosylated α dystroglycan.
Figure 6.

Immunostaining for natively glycosylated α-dystroglycan and for embryonic myosin in GALGT2-overexpressing FKRP P448Lneo− muscles. Data shown are from rAAVrh74.MCK.GALGT2-injected (treated) or PBS-injected (untreated) gastrocnemius muscles of FKRP P448Lneo− mice at 3 months after injection. A: rAAVrh74.MCK.GALGT2-treated and untreated muscle cross-sections were immunostained with antibodies to IIH6 (red) or E. myosin (red) and WFA (green). Merged channels are shown on the right. B: rAAVrh74.MCK.GALGT2-treated muscle cross-section was triple-stained with WFA (green), IIH6 (red), and E. myosin (white) to demonstrate IIH6-E.myosin colocalization. Nuclei were stained with Hoechst 33342 (blue). Scale bars: 100 μm (A); 50 μm (B). E. myosin, embryonic myosin; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; IIH6, natively glycosylated α dystroglycan; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; rAAV, recombinant adeno-associated virus; WFA, Wisteria floribunda agglutinin.
We next performed lectin precipitations and dystroglycan immunoprecipitations to identify the extent of CT glycan on α dystroglycan (Figure 7, respectively). We used WFA, a βGalNAc binding lectin that recognizes the CT glycan made by GALGT2,27 and WGA, a non-GalNAc binding lectin known to precipitate non-CT glycosylated α dystroglycan.6 We precipitated differing amounts of NP-40–solubilized muscle protein lysate to determine the extent of GALGT2-dependent glycosylation in treated and untreated FKRP P448Lneo− muscles. Precipitates were probed by Western blot analysis with antibodies that recognize the dystroglycan polypeptide (αDG), natively glycosylated IIH6, and the CT glycan (CT2). In WT muscles, a modest amount of GALGT2-dependent glycosylation of α dystroglycan with the CT glycan can be observed when 3 mg of muscle protein is used for precipitation, but little or no signal is present when lesser amounts are used unless GALGT2 is overexpressed in the muscle.49 Here, we observed a slight increase in WFA precipitated material even in untreated FKRP P448Lneo− muscles, but this material contained little or no CT glycan, suggesting it may be recognizing incompletely glycosylated structures of α dystroglycan such as GalNAcβ1,3GlcNAcβ1,4Man(6P)-O that would normally be modified by FKRP but that are not the CT glycan.24 Treated FKRP P448Lneo− muscles showed a slight increase in WFA precipitated material, with α dystroglycan identified when 0.5 mg of protein was used for the precipitation (Figure 7A). Some WFA precipitated protein could be immunoblotted with CT2, but the extent of increased glycosylation was modest. Because antipeptide antibodies such as αDG recognize incompletely glycosylated α dystroglycan far better than natively glycosylated protein, we identified predominantly low molecular weight underglycosylated forms of α dystroglycan with the αDG antibody in WFA precipitates, whereas CT2 and IIH6 blotted higher molecular weight proteins, consistent with natively glycosylated protein.
Figure 7.
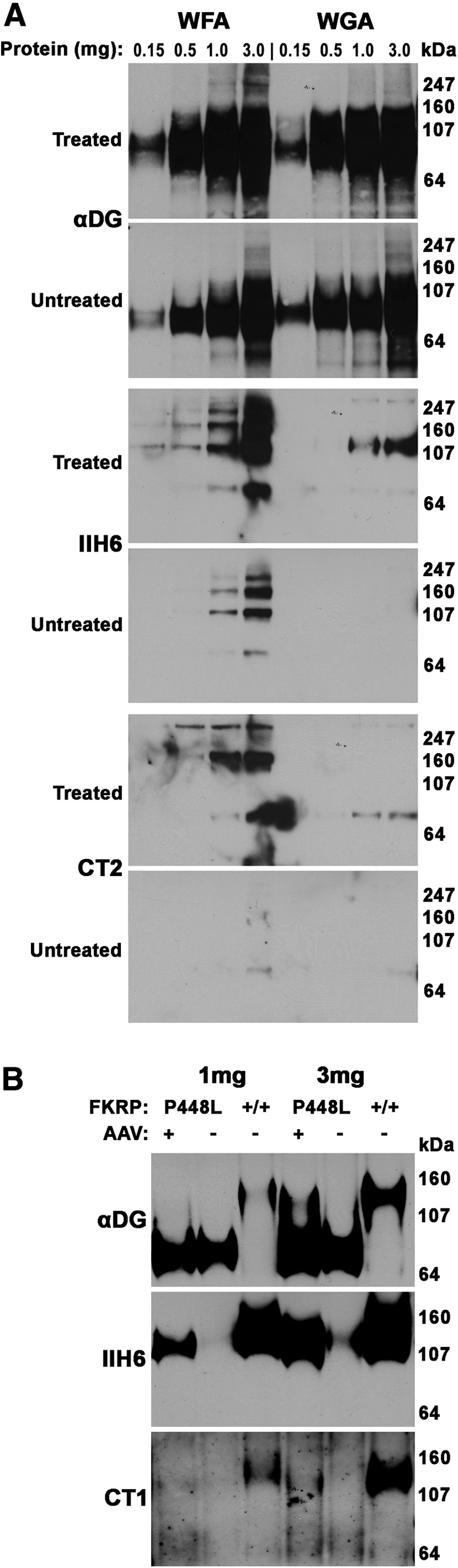
Assessment of α-dystroglycan glycosylation after GALGT2 overexpression in FKRP P448Lneo− muscle. A: Gastrocnemius muscles from FKRP P448Lneo− mice were treated with rAAVrh74.MCK.GALGT2 (treated) or PBS (untreated). At 3 months after treatment, muscles were solubilized in nonionic detergent and subjected to lectin precipitation with WFA, to identify GALGT2-dependent glycosylation of α dystroglycan, or WGA, a control, using different amounts of solubilized protein. Precipitated proteins were separated by SDS-PAGE and blotted for αDG, IIH6, which recognizes natively glycosylated αDG, or CT2, which recognizes the CT glycan. B: NP-40 protein lysates were immunoprecipitated with an antibody to β dystroglycan (that also coprecipitates α dystroglycan), followed by immunoblotting for with αDG, IIH6, or CT1 antibodies. CT1 and CT2 antibody both recognize the CT glycan. Note, although molecular weights are different for certain panels, these are the only protein bands identified by each reagent on such blots. CT, cytotoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; IIH6, natively glycosylated α dystroglycan; MCK, muscle creatine kinase; PBS, phosphate-buffered saline; rAAV, recombinant adeno-associated virus; WFA, Wisteria floribunda agglutinin; WGA, wheat germ agglutinin; αDG, α dystroglycan polypeptide.
To assess dystroglycan glycosylation further, we also immunoprecipitated dystroglycan from NP-40–extracted muscle cell lysates using MANDAG2, an antibody to β dystroglycan that coprecipitates α dystroglycan.41, 55, 61 Here, we were better able to recognize both incompletely glycosylated and more natively glycosylated forms of α dystroglycan using the αDG antibody. GALGT2-treated FKRP P448Lneo− muscle precipitates showed increased IIH6 expression relative to untreated muscle precipitates; however, CT glycan immunoblots (here using CT1, which, like CT2, recognizes CT glycans) showed little, if any, enrichment. Taken together, these data suggest that GALGT2 overexpression in FKRP P448Lneo− mutant muscles results in little to no increase in α dystroglycan glycosylation with the CT glycan.
That IIH6 was primarily concentrated on regenerating myofibers (Figure 6A) suggested that this GALGT2-dependent glycosylation of α dystroglycan predominantly occurred in these cells. Endogenous GALGT2 expression is elevated in such regenerating myofibers, which would be consistent with this notion.49 We measured endogenous mouse Galgt2 gene expression in untreated FKRP P448Lneo− muscle, relative to age-matched WT muscle, in pooled TA, gastroc, and quad samples, and found that it was elevated 2.0- ± 0.3-fold at 6 weeks, 2.1- ± 0.6-fold at 14 weeks, and 4.1- ± 1.7-fold at 26 weeks of age (errors are SEM; n = 9 per group). Thus, endogenous mouse Galgt2 gene expression is also elevated in FKRP P448Lneo− muscles.
We had previously shown that GALGT2 overexpression in muscle cells not only leads to glycosylation of α dystroglycan but also to glycosylation of a glycolipid.37 To determine whether lipid glycosylation accounted for some of the increased WFA staining in GALGT2-overexpressing myofibers, we stained rAAVrh74.MCK.GALGT2-treated muscle with WFA both before and after lipid extraction of muscle sections (Figure 8). A significant fraction of the WFA staining in FKRP P448Lneo− muscle was eliminated after lipid extraction, suggesting that most, although not all, of the WFA staining in GALGT2-overexpressing FKRP P448Lneo− muscle was due to lipid glycosylation.
Figure 8.
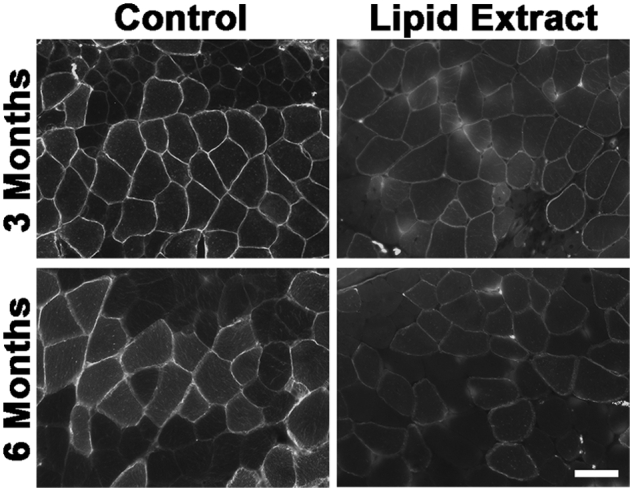
Comparison of WFA staining before and after lipid extraction in rAAVrh74.MCK.GALGT2-treated FKRP P448Lneo− muscles. FKRP P448Lneo− mice were treated with rAAVrh74.MCK.GALGT2 and muscles were stained for WFA before (control) or after lipid extraction at 3 or 6 months after injection. Time-matched images are shown. Scale bar = 50 μm. GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; MCK, muscle creatine kinase; rAAV, recombinant adeno-associated virus; WFA, Wisteria floribunda agglutinin.
Effect of rAAVrh74.MCK.GALGT2 on Expression of Dystrophin and Laminin α2 Surrogates in FKRP P448Lneo− Mutant Muscles
In WT, mdx, Sgca−/−, and dyW muscles, GALGT2 overexpression can induce the expression of dystrophin surrogates, particularly utrophin and plectin 1, and laminin α2 surrogates, including agrin and laminin α5, in treated myofibers.36, 37, 39, 43 In addition, other molecules known to be therapeutic in various forms of MD, including integrin α7, can also be induced by GALGT2 overexpression.42 Such inductions can occur at both the protein and mRNA level in most instances. We therefore next determined changed protein and mRNA expression for these surrogates using Western blot analysis and quantitative RT-PCR (Figure 9). For Western blot analysis, glyceraldehyde-3-phosphate dehydrogenase was used as control for protein loading and transfer, whereas fold-changes relative to WT for treated and untreated (contralateral) muscles were compared by quantitative RT-PCR with 18S mRNA expression as an internal control. Immunoblots using an antibody to the αDG identified dystroglycan protein in WT, FKRP P448Lneo− untreated, and FKRP P448Lneo− treated muscles. As reported previously,16, 19 the molecular weight of αDG in FKRP P448Lneo− muscles was reduced relative to WT. As before, IIH6 expression was largely absent in FKRP P448Lneo− muscles,25 although it was slightly elevated in treated compared with untreated muscles (Figure 6A). For most dystrophin and laminin α2 surrogates, protein and mRNA levels were elevated in both treated and untreated FKRP P448Lneo− muscles relative to WT, and GALGT2, embryonic myosin, utrophin, and agrin protein were further elevated in treated FKRP P448Lneo− muscles relative to untreated FKRP P448Lneo− muscles (Figure 9A). Elevated mRNA levels in each instance trended higher as well (Figure 9, B and C). The one instance in which this reached significance was for mRNA expression of embryonic myosin (Figure 9C).
Figure 9.

Protein and mRNA expression of dystrophin and laminin α2 surrogates after rAAVrh74.MCK.GALGT2 treatment of FKRP P448Lneo− muscles. A: Forty microgram of whole muscle tissue protein lysate from WT or FKRP P448Lneo− muscles, treated or untreated with rAAVrh74.MCK.GALGT2, was separated on 4% to 12% gradient SDS-PAGE gels and immunoblotted with the indicated antibodies. IIH6 is an antibody to α dystroglycan that identifies native glycans needed for laminin binding, whereas αDG is a sheep polyclonal antibody that recognizes the αDG polypeptide. B: mRNA isolated from treated and untreated FKRP P448Lneo− muscle and WT muscle were subjected to semiquantitative real-time PCR. Fold-change in mRNA expression for the indicated genes in treated and untreated FKRP P448Lneo− muscles is shown relative to WT muscle set at 1 (black line at y = 1 indicates WT mRNA expression). C: Comparison of fold-change in gene expression for E. myosin, relative to WT, in rAAVrh74.MCK.GALGT2-treated and untreated FKRP P448Lneo− muscles. Error represent are SEM. n = 6 (B) or 12 (C) measurements from three muscles per group. ***P < 0.001. E., embryonic; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IIH6, natively glycosylated α dystroglycan; MCK, muscle creatine kinase; rAAV, recombinant adeno-associated virus; WT, wild-type; αDG, α dystroglycan polypeptide.
We also compared immunostaining for WFA with staining for agrin, dystroglycan (polypeptide), dystrophin, integrin α7B, laminin α4, laminin α5, plectin 1, and utrophin (Figure 10). Here again, much as for IIH6 staining shown previously (Figure 6A), elevated agrin, integrin α7B, laminin α4, laminin α5, and staining was primarily present on small regenerating myofibers. Some elevated extrasynaptic staining of laminin α4 and laminin α5, however, was present on larger, WFA-stained myofibers as well, but unlike previous studies in other MD models, expressing myofibers were not the predominant cell type with altered expression.36, 37, 39
Figure 10.

Immunostaining for dystrophin and laminin α2 surrogates in GALGT2-overexpressing FKRP P448Lneo− muscle. FKRP P448Lneo− muscles (gastroc) were injected with rAAVrh74.MCK.GALGT2 and analyzed at 3 months after injection. Cross-sections of muscle were immunostained with antibodies to the indicated proteins (red) and counterstained with WFA (green). Nuclei are stained with Hoechst 33342 (blue). Scale bar = 50 μm. GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; MCK, muscle creatine kinase; rAAV, recombinant adeno-associated virus; WFA, Wisteria floribunda agglutinin.
Lack of Therapeutic Effect in rAAVrh74.MCK.GALGT2-Treated Dystroglycan-Deficient Muscles
To further assess the importance of dystroglycan for GALGT2 gene therapy, we compared experiments performed on FKRP P448Lneo− muscles with muscles from P3ProCreDag1lox/lox mice, which lack dystroglycan protein in adult hindlimb muscles, including the TA, gastroc, and quad muscles, but are viable due to caudally directed expression of the P3ProCre transgene, which allows for dystroglycan expression in the diaphragm and in more rostral limb muscles, including arm muscles such as the biceps and triceps.50 For these experiments, we used an AAV vector with the CMV promoter, which is generally stronger than MCK, and a mouse Galgt2 cDNA (rAAV1.CMV.Galgt2). We stained cross-sections of treated muscles with WFA, as before, to assess GALGT2 gene overexpression at either 3 weeks or 3 months (12 weeks) after treatment (Figure 11). At 3 weeks after treatment, most muscle cells in treated hindlimb muscles showed high expression of transgene and CT glycan. Thus, dystroglycan is not required for GALGT2 expression or for expression of the CT glycan. CT glycan overexpression, however, was largely lost by 12 weeks in dystroglycan-deficient muscles (Figure 11A). By contrast, mdx muscles treated in the same way maintained high expression at 12 weeks after treatment (Figure 11B), as much as treated FKRP P448Lneo− muscles (Figure 1C). Quantification of the percentage of myofibers overexpressing CT glycan showed that both the TA and gastroc muscles had significant reductions in muscle transduction at 12 weeks versus the 3 weeks after treatment (Figure 11C). Thus, CT glycan overexpression was not maintained, suggesting that GALGT2 overexpression does not protect dystroglycan-deficient muscles from damage that would then eliminate GALGT2 expression. Consistent with this notion, we saw no significant change at 12 weeks after treatment in treated versus untreated Dag1-deficient muscles for either measures of percentage of myofibers with centrally located nuclei (Figure 12A) or variance in myofiber diameter (Figure 12B). In addition, although we could easily identify GALGT2-glycosylated α dystroglycan with the CT glycan by precipitation with WFA using only 150 μg of total muscle protein in treated WT or in mdx skeletal muscles, we did not identify any such protein, or any other CT glycosylated protein, in Dag1-deficient muscles (Figure 12C).
Figure 11.

Quantification of skeletal muscle pathology in GALGT2-overexpressing Dag1+/+ and Dag1−/− muscles. P3ProCreDag1lox/lox mice or mdx mice were treated by intramuscular injection with rAAV1.CMV.Galgt2 for 3 or 12 weeks. A: Muscles in P3ProCreDag1lox/lox mice that express dystroglycan [Dag1+, biceps (top)/triceps (bottom)] and that do not express dystroglycan (Dag1−, gastroc, TA) were compared at 3 or 12 weeks after rAAV1.CMV.Galgt2 treatment for expression using WFA staining. B: WFA staining of rAAV1.CMV.Galgt2-treated mdx muscles at 12 weeks after treatment. C: The percentage of total myofibers with membrane overexpression of the CT glycan was quantified using WFA staining at 3 or 12 weeks after treatment. Error bars represent SEM. n = 6 to 12 muscles per group (C). ***P < 0.001 between 3 and 12 weeks after infection for Dag1− versus Dag1+ gastroc and TA comparisons. Scale bars = 250 μm (A and B). CMV, cytomegalovirus; CT, cytoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; rAAV, recombinant adeno-associated virus; TA, tibialis anterior; WFA, Wisteria floribunda agglutinin.
Figure 12.

Quantification of muscle morphometry and lectin precipitation in GALGT2-overexpressing WT, mdx, and dystroglycan-deficient skeletal muscles. A and B: Percentages of myofibers with centrally located nuclei (A) and variance in myofiber diameter (B) were measured for rAAV1.CMV.Galgt2-treated and mock-treated muscles from P3Pro-Cre Dag1lox/lox mice. Line at 250 (or below) indicates variance levels expected for nondystrophic muscle, whereas normal percentages of central nuclei are typically below 5%. In this mouse strain, gastroc, TA, and quad muscles fail to express dystroglycan, whereas triceps muscles express dystroglycan. C: Protein lysate (150 μg) extracted in 1% NP-40 was precipitated with WFA or WGA. Precipitated proteins were immunoblotted for α dystroglycan (using IIH6), β dystroglycan (using 43DAG), or the CT glycan (using CT2). The gastroc muscles from WT, mdx, or P3ProCreDag1lox/lox (Dag1−/−) mice that had been treated with rAAV1.CMV.Galgt2 for 0, 3, or 12 weeks are compared. Error bars represent SEM (A and B). n = 4 to 8 muscles per condition (A and B). CMV, cytomegalovirus; CT, cytotoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; IIHA, IIH6, natively glycosylated α dystroglycan; quad, quadriceps; rAAV, recombinant adeno-associated virus; TA, tibialis anterior; WFA, Wisteria floribunda agglutinin; WGA, wheat germ agglutinin; WT, wild-type.
Discussion
LGMD2I is a dystroglycanopathy caused by FKRP mutations that is associated with abnormal glycosylation of α dystroglycan.15, 16, 19 Abnormal glycosylation includes the reduced expression of O-mannose-phosphate–linked glycans needed for ECM protein binding,16, 19 secondary to reductions in ribitol 5-phosphate synthesized on α dystroglycan by FKRP and fukutin.24 Here, we have used the FKRP P448Lneo− mouse model made by Lu and colleagues25 to test a surrogate gene therapy, rAAVrh74.MCK.GALGT2, that is known to inhibit muscle pathology in the mdx mouse model for Duchenne MD, the dyW mouse model for congenital MD 1A, and the Sgca−/− mouse model for LGMD2D.36, 37, 38, 39 Because GALGT2 overexpression normally induces the glycosylation of α dystroglycan in skeletal muscle29, 36 and because the full therapeutic potential of GALGT2 requires the expression of dystroglycan,49 it was unclear whether this approach would be of any benefit in a disease in which α dystroglycan glycosylation, but not α dystroglycan expression, is affected.16, 19 Here, we show that GALGT2 overexpression can inhibit the development of muscle pathology in FKRP P448Lneo− mice. rAAVrh74.MCK.GALGT2 treatment of limb muscles inhibited the development of cycles of muscle degeneration and regeneration, as evidenced by a reduction in central nucleation of myofibers; changed the distribution of myofiber size toward muscle cells with larger, more uniform, diameters; and decreased the degree of muscle wasting by one-half relative to untreated muscles. GALGT2 gene therapy also reduced serum IgG uptake into damaged myofibers to a very significant degree, demonstrating a protection of FKRP mutant muscles from injury. GALGT2 gene expression and the number of rAAVrh74.MCK.GALGT2 vgs in each muscle, however, were reduced over the span of the 6-month treatment interval, indicating the gradual loss of expressing myofibers with time. This appeared to correlate with an increased peak of muscle damage at 3 months of age in FKRP mutant mice, because treatment at a later age did not lead to a similar reduction in expression. These data are consistent with the notion that GALGT2 overexpression has a partial impact on the development of muscle pathology in FKRP P448Lneo− mice, but that it may not be as effective as previous studies in mdx mice, where GALGT2 overexpression can arrest disease pathology for almost the entire life span of the animal.40
Aside from its impact of muscle pathology, there were several additional findings of interest in this study. Foremost among these was that rAAVrh74.MCK.GALGT2-treated FKRP P448Lneo− muscles showed an increased number of regenerating myofibers that suggested a treatment-induced stimulation of muscle regeneration. Elevated numbers of regenerating myofibers were evidenced not only by increased numbers of myofibers stained with antibodies to embryonic myosin but also by increased embryonic myosin gene and protein expression in GALGT2-treated muscles. Although this increase could be due to the delay in muscle damage that then caused a more coordinated peak of regeneration to occur as the therapeutic effects of GALGT2 wore off, it could also be due to a trans effect from GALGT2 overexpressing myofibers on nonexpressing myofibers in regions where regeneration was occurring. The increased regeneration present in GALGT2-treated muscles did not increase the overall percentage of myofibers with central nuclei, because this regeneration occurred within the context of a largely protected muscle, where regeneration therefore was occurring only in a small percentage of the overall muscle area. This is the first evidence to our knowledge to suggest that GALGT2 overexpression may increase muscle regeneration in dystrophic muscles. Such a finding would be consistent with the fact that endogenous muscle GALGT2 expression is elevated after acute injury in the period when embryonic myosin is also most elevated49, 53 and with the fact that GALGT2-deficient muscles show reduced muscle growth in response to acute muscle injury.49 A recent study by Brown and colleagues62 has shown that FKRP mutant mice have a reduction in α dystroglycan glycosylation that occurs during embryonic muscle development and leads to a reduction in laminin α2 expression and early deficits in Pax7+ stem cells in some muscles.62 A therapy that induces regeneration in trans while protecting mature myofibers through expression would perhaps be ideally suited to overcome these deficits.
Another finding of interest is that, unlike GALGT2 overexpression in mdx, dyW, and Sgca−/− muscles,36, 37, 39 GALGT2 overexpression in FKRP P448Lneo− muscles did not dramatically increase the expression of dystrophin or laminin α2 surrogates in treated myofibers or the glycosylation of α dystroglycan. Although both agrin and utrophin showed elevated gene and protein expression in treated FKRP P448Lneo− muscles, the levels induced did not approach the twofold to fourfold increase in gene and protein expression, for example, observed in GALGT2-treated macaque muscle.43 Moreover, some, perhaps most, of this increased expression was due to the increased presence of regenerating myofibers in treated muscles, where these proteins normally would be expected to have higher expression. Further, other dystrophin and laminin α2 surrogates, including plectin 1, laminin α4, laminin α5, and integrin α7, were not increased by GALGT2 overexpression. Thus, although all of these surrogate proteins were increased in FKRP P448Lneo− muscles relative to nondystrophic WT muscle, full induction of these surrogates by GALGT2 overexpression likely requires the native glycosylation of α dystroglycan. The ability of rAAVrh74.MCK.GALGT2 to induce glycosylation of α dystroglycan with the CT glycan in FKRP P448Lneo− muscle was also reduced relative to mdx or WT muscles. Thus, native glycosylation of α dystroglycan may be required for effective glycosylation by GALGT2, and this is consistent with previous studies in Chinese hamster ovary cells showing that GALGT2 glycosylation only occurred when like-acetylglucosaminyltransferase was also overexpressed.42 Thus, improper glycosylation of α dystroglycan resulting from FKRP deficiency negatively affected a number of expected GALGT2-induced molecular changes in skeletal muscle.
These studies further showed that dystroglycan is required for rAAVrh74.MCK.GALGT2 therapy to be fully effective, because therapeutic efficacy was completely lost in dystroglycan-deficient muscles by 3 months after treatment. By contrast, GALGT2 gene therapy remained highly effective at 3 months after treatment in both mdx and FKRP P448Lneo− muscles. Interestingly, in GALGT2-overexpressing Dag1−/− and FKRP P448Lneo− muscles, there still remains CT glycan expression in the muscle membrane due to GALGT2 glycosylation of a glycolipid, and this glycosylation may affect the extent of MD. This notion is also consistent with the fact that GALGT2 transgenic Dag1−/− muscles, where GALGT2 overexpression is higher than is normally found with gene therapy, show reduced muscle pathology at 2 months of age that is lost by 9 months of age.49 In other studies, this glycolipid GALGT2 substrate has been shown to be a sialylpentosylceramide.63 Although α dystroglycan is expressed as multiple glycoforms of differing molecular weights in various tissues due to its complex glycosylation pattern,3 we identified a 300- to 350-kDa CT+ protein in WFA precipitates of GALGT2-treated muscles that is unlikely to be α dystroglycan. This protein may also contribute to the therapeutic mechanism of GALGT2. Finally, we observed elevated IIH6 expression in GALGT2-treated FKRP P448Lneo− muscles, including on α dystroglycan. However, this was not evident on most treated myofibers by immunostaining, as would be expected, for example, in FKRP gene replacement therapies.64, 65 Nevertheless, changed functional glycosylation of α dystroglycan may still also be a factor in the therapeutic mechanism of GALGT2.
The data presented here demonstrate that GALGT2 overexpression can inhibit the extent of muscle pathology that occurs in the FKRP P448Lneo− mutant model for LGMD2I and that these effects occur despite a lack of functional correction of FKRP-deficient glycosylation in all treated muscles. Such experiments point to the possibility that alternatives to gene correction may be viable therapeutic approaches to the treatment of LGMD2I.
Acknowledgments
We thank Jeffery Miner (Washington University, St Louis, MO) for the gifts of laminin α4 and laminin α5 antibodies and for P3Pro-Cre Dag1lox/lox mice, Kevin Campbell (University of Iowa, Iowa City, IA) for generating and allowing us to use Dag1lox/lox mice, John Epstein (University of Pennsylvania, Philadelphia, PA) for generating and allowing us to use P3Pro-Cre mice, Qi Long Lu (Charlotte Health Sciences Center, Charlotte, NC) for FKRP P448L mice, Eva Engvall (Burnham Institute, La Jolla, CA) for rabbit polyclonal antibody to integrin α7B, and Reed Clark and Scott Loiler (Nationwide Children's Hospital, Columbus, OH) and members of the Viral Vector Core for rAAVrh74.MCK.GALGT2 production.
Footnotes
Supported by NIH grant R01 AR049722 (P.T.M.).
Disclosures: P.T.M. is the inventor of rAAVrh74.MCK.GALGT2, which is a licensed product. Several patent applications have been filed on rAAVrh74.MCK.GALGT2.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.05.021.
Supplemental Data
Supplemental Figure S1.
rAAVrh74.MCK.GALGT2 treatment of adult FKRP P448Lneo− muscle induces CT glycan overexpression. The gastroc, quad, and TA muscles of an FKRP P448Lneo− mutant mouse were injected with rAAVrh74.MCK.GALGT2 (treated) or PBS (untreated) at 3 months and analyzed 6 months after injection. A: Muscle cross-sections were stained with WFA (green) to label CT glycan overexpression and with an antibody to laminin α2 (red) to label the muscle sarcolemma. Nuclei were stained with DAPI (blue). B: Quantification of the percentage of total myofibers with membrane overexpression of the CT glycan. Error bars represent SEM. n = 5 measurements per muscle from one animal. Scale bar = 100μm. CT, cytotoxic T cell; GALGT2, β-1,4 N-acetylgalactosaminyltransferase 2; gastroc, gastrocnemius; PBS, phosphate-buffered saline; quad, quadriceps; TA, tibialis anterior; WFA, Wisteria floribunda agglutinin.
References
- 1.Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H., Straub V., Mathews K.D., Moore S.A., Campbell K.P. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida-Moriguchi T., Yu L., Stalnaker S.H., Davis S., Kunz S., Madson M., Oldstone M.B., Schachter H., Wells L., Campbell K.P. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida-Moriguchi T., Campbell K.P. Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology. 2015;25:702–713. doi: 10.1093/glycob/cwv021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobson C.M., Hempel S.J., Stalnaker S.H., Stuart R., Wells L. O-Mannosylation and human disease. Cell Mol Life Sci. 2013;70:2849–2857. doi: 10.1007/s00018-012-1193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin P.T. The dystroglycanopathies: the new disorders of O-linked glycosylation. Semin Pediatr Neurol. 2005;12:152–158. doi: 10.1016/j.spen.2005.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 7.Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohlendieck K., Ervasti J.M., Matsumura K., Kahl S.D., Leveille C.J., Campbell K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- 9.Henry M.D., Campbell K.P. Dystroglycan inside and out. Curr Opin Cell Biol. 1999;11:602–607. doi: 10.1016/s0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 10.Sanes J.R., Engvall E., Butkowski R., Hunter D.D. Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J Cell Biol. 1990;111:1685–1699. doi: 10.1083/jcb.111.4.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engvall E., Earwicker D., Haaparanta T., Ruoslahti E., Sanes J.R. Distribution and isolation of four laminin variants; tissue restricted distribution of heterotrimers assembled from five different subunits. Cell Regul. 1990;1:731–740. doi: 10.1091/mbc.1.10.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore S.A., Saito F., Chen J., Michele D.E., Henry M.D., Messing A., Cohn R.D., Ross-Barta S.E., Westra S., Williamson R.A., Hoshi T., Campbell K.P. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- 13.Godfrey C., Foley A.R., Clement E., Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev. 2011;21:278–285. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Jimenez-Mallebrera C., Brown S.C., Sewry C.A., Muntoni F. Congenital muscular dystrophy: molecular and cellular aspects. Cell Mol Life Sci. 2005;62:809–823. doi: 10.1007/s00018-004-4510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockington M., Blake D.J., Prandini P., Brown S.C., Torelli S., Benson M.A., Ponting C.P., Estournet B., Romero N.B., Mercuri E., Voit T., Sewry C.A., Guicheney P., Muntoni F. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198–1209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockington M., Yuva Y., Prandini P., Brown S.C., Torelli S., Benson M.A., Herrmann R., Anderson L.V., Bashir R., Burgunder J.M., Fallet S., Romero N., Fardeau M., Straub V., Storey G., Pollitt C., Richard I., Sewry C.A., Bushby K., Voit T., Blake D.J., Muntoni F. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 17.Topaloglu H., Brockington M., Yuva Y., Talim B., Haliloglu G., Blake D., Torelli S., Brown S.C., Muntoni F. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology. 2003;60:988–992. doi: 10.1212/01.wnl.0000052996.14099.dc. [DOI] [PubMed] [Google Scholar]
- 18.Beltran-Valero de Bernabe D., Voit T., Longman C., Steinbrecher A., Straub V., Yuva Y., Herrmann R., Sperner J., Korenke C., Diesen C., Dobyns W.B., Brunner H.G., van Bokhoven H., Brockington M., Muntoni F. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown S.C., Torelli S., Brockington M., Yuva Y., Jimenez C., Feng L., Anderson L., Ugo I., Kroger S., Bushby K., Voit T., Sewry C., Muntoni F. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol. 2004;164:727–737. doi: 10.1016/s0002-9440(10)63160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M., Hamano K., Sakakihara Y., Nonaka I., Nakagome Y., Kanazawa I., Nakamura Y., Tokunaga K., Toda T. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y.K., Ogawa M., Tagawa K., Noguchi S., Ishihara T., Nonaka I., Arahata K. Selective deficiency of alpha-dystroglycan in Fukuyama-type congenital muscular dystrophy. Neurology. 2001;57:115–121. doi: 10.1212/wnl.57.1.115. [DOI] [PubMed] [Google Scholar]
- 22.Toda T., Chiyonobu T., Xiong H., Tachikawa M., Kobayashi K., Manya H., Takeda S., Taniguchi M., Kurahashi H., Endo T. Fukutin and alpha-dystroglycanopathies. Acta Myol. 2005;24:60–63. [PubMed] [Google Scholar]
- 23.Mendell J.R., Boue D.R., Martin P.T. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol. 2006;9:427–443. doi: 10.2350/06-07-0127.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanagawa M., Kobayashi K., Tajiri M., Manya H., Kuga A., Yamaguchi Y., Akasaka-Manya K., Furukawa J., Mizuno M., Kawakami H., Shinohara Y., Wada Y., Endo T., Toda T. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. 2016;14:2209–2223. doi: 10.1016/j.celrep.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 25.Blaeser A., Keramaris E., Chan Y.M., Sparks S., Cowley D., Xiao X., Lu Q.L. Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum Genet. 2013;132:923–934. doi: 10.1007/s00439-013-1302-7. [DOI] [PubMed] [Google Scholar]
- 26.Chan Y.M., Keramaris-Vrantsis E., Lidov H.G., Norton J.H., Zinchenko N., Gruber H.E., Thresher R., Blake D.J., Ashar J., Rosenfeld J., Lu Q.L. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. 2010;19:3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- 27.Martin P.T., Scott L.J., Porter B.E., Sanes J.R. Distinct structures and functions of related pre- and postsynaptic carbohydrates at the mammalian neuromuscular junction. Mol Cell Neurosci. 1999;13:105–118. doi: 10.1006/mcne.1999.0737. [DOI] [PubMed] [Google Scholar]
- 28.Martin P.T. Glycobiology of the synapse. Glycobiology. 2002;12:1R–7R. doi: 10.1093/glycob/12.1.1r. [DOI] [PubMed] [Google Scholar]
- 29.Xia B., Hoyte K., Kammesheidt A., Deerinck T., Ellisman M., Martin P.T. Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev Biol. 2002;242:58–73. doi: 10.1006/dbio.2001.0530. [DOI] [PubMed] [Google Scholar]
- 30.Hoyte K., Kang C., Martin P.T. Definition of pre- and postsynaptic forms of the CT carbohydrate antigen at the neuromuscular junction: ubiquitous expression of the CT antigens and the CT GalNAc transferase in mouse tissues. Brain Res Mol Brain Res. 2002;109:146–160. doi: 10.1016/s0169-328x(02)00551-x. [DOI] [PubMed] [Google Scholar]
- 31.Smith P.L., Lowe J.B. Molecular cloning of a murine N-acetylgalactosamine transferase cDNA that determines expression of the T lymphocyte-specific CT oligosaccharide differentiation antigen. J Biol Chem. 1994;269:15162–15171. [PubMed] [Google Scholar]
- 32.Lefrancois L., Bevan M.J. Novel antigenic determinants of the T200 glycoprotein expressed preferentially by activated cytotoxic T lymphocytes. J Immunol. 1985;135:374–383. [PubMed] [Google Scholar]
- 33.Lefrancois L., Bevan M.J. Functional modifications of cytotoxic T-lymphocyte T200 glycoprotein recognized by monoclonal antibodies. Nature. 1985;314:449–452. doi: 10.1038/314449a0. [DOI] [PubMed] [Google Scholar]
- 34.Dohi T., Ohta S., Hanai N., Yamaguchi K., Oshima M. Sialylpentaosylceramide detected with anti-GM2 monoclonal antibody. Structural characterization and complementary expression with GM2 in gastric cancer and normal gastric mucosa. J Biol Chem. 1990;265:7880–7885. [PubMed] [Google Scholar]
- 35.Dall'Olio F., Malagolini N., Chiricolo M., Trinchera M., Harduin-Lepers A. The expanding roles of the Sd(a)/Cad carbohydrate antigen and its cognate glycosyltransferase B4GALNT2. Biochim Biophys Acta. 2014;1840:443–453. doi: 10.1016/j.bbagen.2013.09.036. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen H.H., Jayasinha V., Xia B., Hoyte K., Martin P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu R., Chandrasekharan K., Yoon J.H., Camboni M., Martin P.T. Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am J Pathol. 2007;171:181–199. doi: 10.2353/ajpath.2007.060927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin P.T., Xu R., Rodino-Klapac L.R., Oglesbay E., Camboni M., Montgomery C.L., Shontz K., Chicoine L.G., Clark K.R., Sahenk Z., Mendell J.R., Janssen P.M. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am J Physiol Cell Physiol. 2009;296:C476–C488. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu R., DeVries S., Camboni M., Martin P.T. Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am J Pathol. 2009;175:235–247. doi: 10.2353/ajpath.2009.080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu R., Camboni M., Martin P.T. Postnatal overexpression of the CT GalNAc transferase inhibits muscular dystrophy in mdx mice without altering muscle growth or neuromuscular development: evidence for a utrophin-independent mechanism. Neuromuscul Disord. 2007;17:209–220. doi: 10.1016/j.nmd.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoon J.H., Johnson E., Xu R., Martin L.T., Martin P.T., Montanaro F. Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J Proteome Res. 2012;11:4413–4424. doi: 10.1021/pr300328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon J.H., Chandrasekharan K., Xu R., Glass M., Singhal N., Martin P.T. The synaptic CT carbohydrate modulates binding and expression of extracellular matrix proteins in skeletal muscle: partial dependence on utrophin. Mol Cell Neurosci. 2009;41:448–463. doi: 10.1016/j.mcn.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chicoine L.G., Rodino-Klapac L.R., Shao G., Xu R., Bremer W.G., Camboni M., Golden B., Montgomery C.L., Shontz K., Heller K.N., Griffin D.A., Lewis S., Coley B.D., Walker C.M., Clark K.R., Sahenk Z., Mendell J.R., Martin P.T. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol Ther. 2014;22:713–724. doi: 10.1038/mt.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deconinck N., Tinsley J., De Backer F., Fisher R., Kahn D., Phelps S., Davies K., Gillis J.M. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997;3:1216–1221. doi: 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- 45.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 46.Rafael J.A., Tinsley J.M., Potter A.C., Deconinck A.E., Davies K.E. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 47.Bentzinger C.F., Barzaghi P., Lin S., Ruegg M.A. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J. 2005;19:934–942. doi: 10.1096/fj.04-3376com. [DOI] [PubMed] [Google Scholar]
- 48.Moll J., Barzaghi P., Lin S., Bezakova G., Lochmuller H., Engvall E., Muller U., Ruegg M.A. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- 49.Xu R., Singhal N., Serinagaoglu Y., Chandrasekharan K., Joshi M., Bauer J.A., Janssen P.M., Martin P.T. Deletion of Galgt2 (B4Galnt2) reduces muscle growth in response to acute injury and increases muscle inflammation and pathology in dystrophin-deficient mice. Am J Pathol. 2015;185:2668–2684. doi: 10.1016/j.ajpath.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarad G., Miner J.H. The Pax3-Cre transgene exhibits a rostrocaudal gradient of expression in the skeletal muscle lineage. Genesis. 2009;47:1–6. doi: 10.1002/dvg.20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohn R.D., Henry M.D., Michele D.E., Barresi R., Saito F., Moore S.A., Flanagan J.D., Skwarchuk M.W., Robbins M.E., Mendell J.R., Williamson R.A., Campbell K.P. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- 52.Li J., Chen F., Epstein J.A. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 53.Singhal N., Martin P.T. A role for Galgt1 in skeletal muscle regeneration. Skeletal Muscle. 2015;5:3. doi: 10.1186/s13395-014-0028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mundegar R.R., Franke E., Schäfer R., Zweyer M., Wernig A. Reduction of high background staining by heating unfixed mouse skeletal muscle tissue sections allows for detection of thermostable antigens with murine monoclonal antibodies. J Histochem Cytochem. 2008;56:969–975. doi: 10.1369/jhc.2008.950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pereboev A.V., Ahmed N., thi Man N., Morris G.E. Epitopes in the interacting regions of beta-dystroglycan (PPxY motif) and dystrophin (WW domain) Biochim Biophys Acta. 2001;1527:54–60. doi: 10.1016/s0304-4165(01)00147-7. [DOI] [PubMed] [Google Scholar]
- 56.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 57.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De la Porte S., Morin S., Koenig J. Characteristics of skeletal muscle in mdx mutant mice. Int Rev Cytol. 1999;191:99–148. doi: 10.1016/s0074-7696(08)60158-8. [DOI] [PubMed] [Google Scholar]
- 59.Michele D.E., Campbell K.P. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- 60.Awano H., Blaeser A., Keramaris E., Xu L., Tucker J., Wu B., Lu P., Lu Q.L. Restoration of Functional Glycosylation of alpha-Dystroglycan in FKRP Mutant Mice Is Associated with Muscle Regeneration. Am J Pathol. 2015;185:2025–2037. doi: 10.1016/j.ajpath.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 61.Johnson E.K., Li B., Yoon J.H., Flanigan K.M., Martin P.T., Ervasti J., Montanaro F. Identification of new dystroglycan complexes in skeletal muscle. PLoS One. 2013;8:e73224. doi: 10.1371/journal.pone.0073224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim J., Hopkinson M., Kavishwar M., Fernandez-Fuente M., Brown S.C. Prenatal muscle development in a mouse model for the secondary dystroglycanopathies. Skeletal Muscle. 2015;6:3. doi: 10.1186/s13395-016-0073-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawamura Y.I., Kawashima R., Fukunaga R., Hirai K., Toyama-Sorimachi N., Tokuhara M., Shimizu T., Dohi T. Introduction of Sd(a) carbohydrate antigen in gastrointestinal cancer cells eliminates selectin ligands and inhibits metastasis. Cancer Res. 2005;65:6220–6227. doi: 10.1158/0008-5472.CAN-05-0639. [DOI] [PubMed] [Google Scholar]
- 64.Xu L., Lu P.J., Wang C.H., Keramaris E., Qiao C., Xiao B., Blake D.J., Xiao X., Lu Q.L. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of alpha-dystroglycan and improves muscle functions. Mol Ther. 2013;21:1832–1840. doi: 10.1038/mt.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qiao C., Wang C.H., Zhao C., Lu P., Awano H., Xiao B., Li J., Yuan Z., Dai Y., Martin C.B., Li J., Lu Q., Xiao X. Muscle and heart function restoration in a limb girdle muscular dystrophy 2I (LGMD2I) mouse model by systemic FKRP gene delivery. Mol Ther. 2014;22:1890–1899. doi: 10.1038/mt.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]