

Abstract
Cortical spreading depolarization is a metabolically costly phenomenon that affects the brain in both health and disease. Following severe stroke, subarachnoid hemorrhage, or traumatic brain injury, cortical spreading depolarization exacerbates tissue damage and enlarges infarct volumes. It is not known, however, whether spreading depolarization also occurs in the retina in vivo. We report now that spreading depolarization episodes are generated in the in vivo rat retina following retinal vessel occlusion produced by photothrombosis. The properties of retinal spreading depolarization are similar to those of cortical spreading depolarization. Retinal spreading depolarization waves propagate at a velocity of 3.0 ± 0.1 mm/min and are associated with a negative shift in direct current potential, a transient cessation of neuronal spiking, arteriole constriction, and a decrease in tissue O2 tension. The frequency of retinal spreading depolarization generation in vivo is reduced by administration of the NMDA antagonist MK-801 and the 5-HT(1D) agonist sumatriptan. Branch retinal vein occlusion is a leading cause of vision loss from vascular disease. Our results suggest that retinal spreading depolarization could contribute to retinal damage in acute retinal ischemia and demonstrate that pharmacological agents can reduce retinal spreading depolarization frequency after retinal vessel occlusion. Blocking retinal spreading depolarization generation may represent a therapeutic strategy for preserving vision in branch retinal vein occlusion patients.
Keywords: Branch retinal vein occlusion, hypoxia, NMDA antagonist, retinal ischemia, spreading depolarization
Introduction
Cortical spreading depression (CSD) was first described by Leão1 in the rabbit cortex and has since been characterized extensively.2,3 Its hallmark feature is a wave of extreme cellular depolarization that spreads over the cortical surface at a velocity of ∼3 mm/min. Since these initial observations, it has become evident that CSD is a component of a continuum of spreading depolarization (SD) phenomena. SD waves are accompanied by large shifts in transcellular ion gradients, cell swelling, and increased extracellular concentrations of neurotransmitters, particularly glutamate.1,2,4 SD is characterized electrophysiologically by a negative shift in DC (direct current) potential and a complete cessation of spiking.2
SD can affect both healthy and pathological tissue. In the healthy brain, CSD lies on the benign end of the SD continuum and is generally believed to be the mechanism that underlies migraine auras and their subsequent scotomas2,5 (see Charles and Baca6 for a more detailed discussion). The brain fully recovers from such CSD episodes without lasting consequences.7 In acute injury to the nervous system, such as severe stroke, subarachnoid hemorrhage, or traumatic brain injury, SD events are initiated at the borders of the damaged tissue and are more malignant.8–10 The combination of high extracellular levels of excitatory amino acids, increased metabolic demand, and oligemia (decreased blood flow) induced by SD waves contributes to excitotoxicity and metabolic supply-demand mismatch, ultimately leading to additional cell death and injury expansion.9,11–13 In patients, tens of SD episodes are sometimes generated in the days following an initial ischemic insult, resulting in delayed neurological deficits and secondary ischemic infarcts.11
SD also results in a complex series of blood flow changes that depend on tissue health.3 In healthy tissue affected by CSD, blood vessels initially constrict, mediated by the release of vasoconstrictors from neurons and glial cells during wave propagation.3,14,15 This preliminary drop in blood flow is followed by a period of hyperemia that lasts for several minutes, and then by a persistent oligemia lasting for over an hour after the CSD episode.16 In contrast, SD occurring in response to injury is characterized by a brief oligemia, followed by prolonged hyperemia.3
SD can occur in the retina as well as the cortex.17–19 However, retinal spreading depolarization (rSD) has previously been observed only in ex vivo preparations from species that have avascular retinas.18–22 rSD has never been described in the in vivo retina and it has been questioned whether it may be an artifact of ex vivo preparations.3
We now demonstrate for the first time that rSD occurs in the vascularized mammalian retina in vivo. rSD episodes are generated following the experimental occlusion of retinal vessels, a model of branch retinal vein occlusion (BRVO). rSD has similar properties as cortical SD and can be blocked by several drugs, including the NMDA receptor antagonist MK-801 and the 5-HT(1D) receptor agonist sumatriptan, suggesting a potential therapeutic strategy for blocking rSD and reducing vision loss in BRVO patients.
Materials and methods
Animals
All experimental procedures were approved by the University of Minnesota's Institutional Animal Care and Use Committee and conducted in accordance with the University of Minnesota guidelines, which are based on the Office of Laboratory Animal Welfare guidelines of the National Institutes of Health and the Association for Assessment and Accreditation of Laboratory Animal Care International guidelines and conducted according to the ARRIVE guidelines. Two to three month old male Long Evans rats, 275–325 g, were purchased from Harlan Sprague-Dawley Inc. Rats were housed in pairs in standard cages with corn cob bedding under a 12-hour light:dark cycle in a conventional non-SPF housing facility at the University of Minnesota. Food and water were available ad libitum. Routine care and animal husbandry were performed in the animal housing facility by authorized University of Minnesota Research Animal Resources staff, while all experiments were performed in the laboratory during daylight hours by trained personnel.
In vivo rat preparation
The in vivo rat retinal preparation has been described previously.23 Briefly, femoral artery and vein cannulation and a tracheotomy were performed under 2% isoflurane anesthesia on young adult rats. After surgery, anesthesia was switched from isoflurane to α-chloralose-HBC complex (Sigma C8849; 800 mg/kg bolus and 550 mg/kgċh infusion). α-chloralose anesthesia was chosen over isoflurane because it does not interfere with generation and propagation of rSD as isoflurane does.24,25 Animals were placed in a custom stereotaxic holder, and a contact lens was placed on the eye. Rose Bengal was administered i.v. (70 mg/kg bolus; Sigma 330000) for photothrombosis, followed by the paralytic gallamine triethiodide (Sigma G8124; 20 mg/kg bolus and 20 mg/kgċh sustained infusion) to facilitate mechanical ventilation and reduce eye movements. A heating blanket maintained the animal’s body temperature at 37° C. Animals were ventilated (SAR-1000; CWE) with a mixture of 30% oxygen and 70% nitrogen and a 2 mmHg positive end-expiratory pressure. sO2 (MouseOx; Starr Life Sciences), end-tidal CO2 (microCapStar; CWE), and blood pressure (BP-1; WPI) were monitored continuously. Ventilation parameters were adjusted to maintain physiological parameters within normal limits (mean arterial pressure: 112 ± 2 mmHg, n = 27 rats; sO2: 97.1 ± 0.2 %, n = 51 rats; end-expiratory CO2: 41.0 ± 0.7 mmHg, n = 49 rats). Animals were sacrificed at the end of experimentation with i.v. injection of potassium chloride (2 mEq/kg).
Retinal imaging
The retina was imaged with confocal microscopy (Olympus FV1000) through a 4 × dry 0.16 NA objective. Reflectance microscopy with 405 nm illumination was used to image the intrinsic optical signal (IOS) of the retina. Confocal microscopy with 488 nm illumination was used to perform angiograms following i.v. injection of fluorescein (1.5%, 0.3 mL; Sigma F2456) and to image vessels labeled with FITC-dextran (1.5%, 0.3 mL; Sigma FD2000S). Rose Bengal-labeled vessels were imaged with 559 nm illumination.
Photothrombosis and global ischemia
Primary retinal arterioles and venules were occluded by photothrombosis. Rose Bengal-filled vessels were illuminated with a 10 µm diameter, 559 nm spot for 45 min; 2–5 min of baseline retinal imaging was obtained, prior to initiating photothrombosis. Photothrombotic ischemia was maintained for 60 min, and the retina was imagined throughout this time. The photoactivation light was moved every ∼ 3 min between three and four locations along the vessel. If branch points were present, those sites were targeted to prevent vessel backfilling. Retinal imaging was continued 30–60 min after photothrombosis was terminated. Global ischemia was achieved by raising intraocular pressure. A 25-gauge needle was introduced into the vitreous humor and saline was injected using a pneumatic transducer tester (Fluke Biomedical, DPM1B) to achieve an intraocular pressure of ∼ 120 mmHg. The intraocular pressure was maintained at 120 mmHg ± 10 mmHg for 60 min.
Eyecup preparation and electrophysiology
Extracellular recordings of retinal ganglion cell (RGC) activity and the DC potential were conducted in ex vivo eyecup and in vivo preparations. Eyecups were prepared as previously described.26 Briefly, eyes from two to three month old male Long Evans rats were bisected at the equator and everted over a dome in a custom superfusion chamber.26 The eyecup was superfused at 2–3 mL/min with bicarbonate Ringers solution bubbled with 95% O2 and 5% CO2 at 33° C. Retinas were imaged by reflectance microscopy with a confocal microscope (Olympus FV1000) and a 10 × water immersion objective. Recording pipettes (∼10 µm tip diameter) were filled with bicarbonate Ringers and advanced into the RGC layer. Signals were band-pass filtered (156–1800 Hz) to record RGC spike activity and low-pass filtered (0.2 Hz) to record extracellular DC potentials. K+ gluconate ejection pipettes were filled with 150 mM K+ gluconate and positioned just above the retinal surface at the periphery of the eyecup. For oxygen glucose deprivation (OGD) experiments, eyecups were superfused with OGD Ringers solution.
For in vivo electrophysiology experiments, recording pipettes (∼0.5 µm tip diameter) were filled with 3 M NaCl. Pipettes were advanced though the sclera and vitreous within a 25-gauge guard needle attached to a custom microadvancer.27 The pipette was advanced ∼10 µm into the retina. Signals were low-pass filtered (0.5 Hz) to record extracellular DC potentials.
Oxygen partial pressure
Oxygen tension at the surface of the retina was measured with a custom optical oxygen probe (OxyLite NX pO2 Bare Fiber Beveled Sensor; Oxford Optronix Ltd.) that was advanced through the sclera and vitreous humor inside a 25-gauge guard needle. The O2-sensitive tip of the probe was beveled so that it lay flat against the surface of the retina. Control experiments demonstrated that the photothrombosis photoactivation light did not interfere with O2 measurements.
Red blood cell flux
Red blood cell (RBC) flux through arterioles was measured as previously described.28 Briefly, blood was withdrawn from the animal at the beginning of an experiment and RBCs were isolated and incubated for 5 min in the fluorescent lipophilic dye DiD (carbocyanide 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanide,4-chlorobenzenesulfonate salt solid; Invitrogen; #D-7757). Cells were washed in buffer to remove unbound dye and reinjected into the animal. Fluorescent RBCs were imaged with 635 nm confocal line scans oriented perpendicular to the vessel lumen. Line scans were acquired at 4200 Hz and labeled RBCs detected by a custom MatLab routine.
Survival surgery and immunohistochemistry
A modified in vivo preparation was adopted to permit animal survival following thrombus formation. Animals were anesthetized with 2% isoflurane and the tail vein cannulated for delivery of α-chloralose anesthetic and Rose Bengal dye. Animals breathed a mixture of 30% oxygen and 70% nitrogen. Oxygen saturation was monitored by pulse oximetry to ensure adequate ventilation. Following 60 min of photothrombosis, the animal recovered from anesthesia. Animals were sacrificed after 5 to 14 days and the retina was removed from the experimental eye and fixed in 4% paraformaldehyde for 2 h. Retinas were immunolabeled for the RGC marker Brn3a with primary goat antibodies (1:500; 24 h; sc-31984, Lot # L2414; Santa Cruz Biotechnology) and Alexa Fluor 488 donkey anti-goat secondary antibodies (1:500; 2 h; A11055; Lot # 1369678; Life Technologies) as previously described.29 The retinal vasculature was labeled with Isolectin GS-IB4-647 (1:75; 2 hours; Invitrogen I32450). Retinas were imaged with confocal microscopy. To confirm loss of RGCs, retinas were also imaged with infrared differential interference contrast microscopy.
Solutions
All pharmacological agents that were injected i.v. during in vivo experiments were dissolved in medical grade saline (Hospira, Inc., NDC 0409-4888). Bicarbonate Ringers solution used in eyecup experiments contained, in mM: 111 NaCl, 3.0 KCl, 2.0 CaCl2, 1.0 MgSO4, 0.5 NaH2PO4, 15.0 dextrose, 32 NaHCO3, bubbled with 95% O2 and 5% CO2, pH 7.4. OGD Ringers solution contained, in mM: 111 NaCl, 3.0 KCl, 2.0 CaCl2, 1.0 MgSO4, 0.5 NaH2PO4, 15.0 sucrose, 32 NaHCO3, bubbled with 95% N2 and 5% CO2, pH 7.4. All Ringers reagents and drugs were purchased from Sigma.
Image processing
rSD waves were analyzed from reflectance IOS images using a custom MatLab routine. IOS images were acquired at 0.61 Hz. (Δ intensity/Δtime)/intensity images were calculated by low pass filtering each IOS frame to reduce noise, then subtracting from each frame the mean of the preceding 20 frames and then dividing that difference by the mean. These images highlighted the leading edge of the rSD waves. Additional MatLab routines calculated the radius and velocity of rSD waves based on a user specified identification of wave origin and the distal extent of the wave. Laser speckle flowmetry images were analyzed using a custom MatLab routine as previously described.23 Ratio speckle images were calculated by first averaging all speckle images acquired before vessel occlusion and the images acquired during occlusion and dividing the latter by the former. RBC flux confocal line scan images were analyzed using a custom MatLab routine as previously described.28
Statistics
Animals were excluded from analysis if the experimental preparation was unhealthy, as determined by physiological parameters, if imaging quality was inadequate, or if data could not be collected for technical reasons (i.e.: broken recording electrodes). Data are presented as mean ± S.E.M. For each experiment, the number of observed rSD waves and the total number of animals are reported. Comparisons between two groups were performed using two-tailed Student’s t-test. The parametric version of the test was chosen when variances were equal between groups, while the non-parametric version of the test was chosen for groups with unequal variances. When multiple groups were compared to the same control, statistical differences were calculated using an ANOVA with a post hoc Dunnett’s correction. Calculations were carried out in Excel (2013) or GraphPad Prism (version 5.04). Significance was defined as P < 0.05.
Research materials related to this work can be accessed by contacting the corresponding author.
Results
We investigated whether an ischemic insult in the retina evokes SD, as it does in the cortex.2,3 A photothrombosis model of focal retinal ischemia was used to generate blood clots in primary retinal arterioles and venules in α-chloralose-anesthetized rats. Rose Bengal was injected i.v. and photoactivated with 559 nm light focused onto single vessels to form a thrombus. The Rose Bengal-filled vasculature was imaged with confocal microscopy. Concurrent laser speckle flowmetry permitted real-time visualization of blood flow changes to confirm blockage of blood vessels and to map regions of retinal ischemia. The IOS of the retina, which is produced during SD due to an increase in tissue light scattering generated by cell swelling and a concomitant decrease in interstitial volume,30,30,31 was monitored at 405 nm with reflectance microscopy to detect rSD episodes.
Photothrombosis completely blocked blood flow in the treated vessels and reduced blood flow in the adjacent tissue. Fluorescein angiography confirmed that the occluded vessels remained blocked for greater than 60 min (n = 9 rats; Figure 1(a) and (b)). Angiograms revealed complete block of the occluded vessel and reduced perfusion of capillaries near the vessel (Figure 1(b)). Laser speckle flowmetry confirmed that photothrombosis occluded the targeted vessels and that blood flow was reduced in the surrounding tissue (n = 10 rats; Figure 1(c) to (e)).
Figure 1.

Photothrombosis model of branch retinal vessel occlusion. (a) Confocal image of Rose Bengal-filled blood vessels (red) superimposed on a reflectance image of the retina shows that all vessels are patent before photothrombosis. In this and other panels, photothrombosis sites are indicated by yellow dots and the optic disc by a red circle. (b) Fluorescein angiogram of the retina in A, performed 15 min after termination of photothrombosis demonstrates that the treated venule is completely occluded. Note that capillaries near the occluded venule are not perfused. (c) Two venules in a second retina were targeted for photothrombosis. (d) Ratio laser speckle flowmetry image of the retina in C obtained 15 min after photothrombosis. Photothrombosis produced a complete block of blood flow in the two occluded vessels (dark blue) and a partial block of blood flow in the adjacent tissue (light blue). (e) Plot of blood flow in one of the occluded venules (measured at the red dot in d). Blood flow drops to zero by the end of the photothrombosis period.
A group of animals were allowed to recover following photothrombosis and the retinas were harvested after a 5 to 14 day survival period (n = 5 rats). RGCs were labeled with an anti-Brn3a antibody. In all five retinas examined, confocal images showed a clearly defined region devoid of RGCs which was centered on the occluded vessel. Across all retinas, the area of damage consistently encompassed approximately a quarter of the retina and reflected a near-total loss of RGCs. The area of damage was not correlated with survival time. One example retina is shown in Figure 2(a). When quantified, there was a total loss of Brn3a-postitive cells near the occluded vessel (Figure 2(b) and (c)). The absence of RGC labeling was due to cell loss rather than a loss of Brn3a immunoreactivity, as demonstrated by differential interference contrast microscopy images showing an absence of cells in the affected retinal region (Figure 2(d) and (e)). No loss of RGCs was observed when vessels were bleached in the absence of Rose Bengal.
Figure 2.

Retinal ganglion cell loss following vessel occlusion. (a–c) Photothrombosis of a single venule produced large-scale loss of retinal ganglion cells (RGCs) in the region supplied by the occluded vessel following a five-day survival time. Immunohistochemical labeling of the RGC marker Brn3a (a) reveals a complete absence of RGCs in the region supplied by the occluded vessel (yellow dot). Image retouched to remove dust artifacts. (b) A pseudocolor map of the density of Brn3a-positive RGCs in the retina in A, showing a decrease in RGC density in the region supplied by the occluded vessel. (c) RGC density measured along a circle 2.0 mm from the optic disc (dotted line in B). Brn3a-positive RGCs are completely absent close to the occluded vessel, but are present in high numbers in non-ischemic regions of the retina. (d–e) Differential interference contrast microscopy of a different preparation following a 14-day survival time demonstrates an absence of cells in an ischemic region (d), compared to a healthy region far from the thrombus (e).V, vessel.
Occlusion of blood vessels evokes retinal SD
rSD waves were generated in every instance following occlusion of a primary branch arteriole (n = 15 rats) or venule (n = 9 rats) (Figure 3(a) and (b), Movies 1 and 2). The properties of the rSD waves are detailed in Supplemental Table 1. The type of vessel occluded (arteriole vs venule) did not affect rSD wave properties. On average, 3.7 ± 0.6 (n = 55 waves, 15 rats) and 5.0 ± 0.9 (n = 45 waves, 9 rats) waves per animal were generated within the first hour following arteriole and venule occlusion, respectively. Latency to generation of the first wave was 7.9 ± 1.4 min following arteriole occlusion and 14.7 ± 3.0 min following venous occlusion (p = 0.07) (Figure 4(a)). The mean time interval between waves was 7.6 ± 0.6 min (data pooled between arterial and venous occlusion), while the frequency of wave generation decreased over the first hour after photothrombosis (Figure 4). Few waves (0.2 ± 0.2) were generated after 60 min following arteriole occlusion, and no waves were generated after this time following venule occlusion.
Figure 3.

Retinal spreading depolarization (rSD) occurs following vessel occlusion. (a) Intrinsic optical signal (IOS) images of a representative rSD event in response to focal thrombosis of an arteriole (yellow dot). Numbers in the upper right of each panel indicate time relative to rSD initiation. (b) Images from the series shown in A, processed with each frame representing Δ intensity/intensity (ΔI/I) to better visualize the rSD wave front. Most rSD waves are initiated near the optic disc (red circle). (c) An rSD wave initiated following onset of global ischemia, produced by increasing intraocular pressure to ∼120 mmHg. The wave was initiated near the optic disc.
Figure 4.
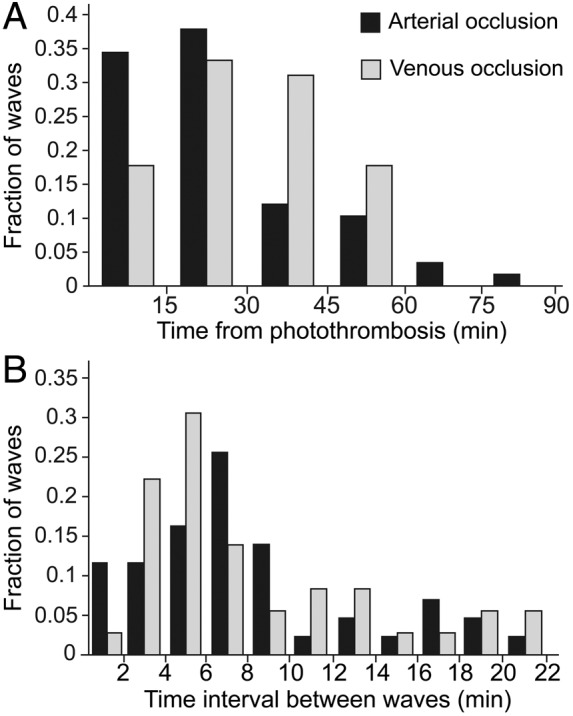
Timing of rSD wave generation following photothrombosis. (a) The number of rSD waves generated following the onset of photothrombosis, binned in 15 min intervals. The probability of rSD wave generation is highest during the first 45 min of ischemia. Few waves are generated after 60 min. Arterial occlusion: n = 58 waves, 15 rats; venous occlusion: n = 45 waves, 9 rats. (b) The time interval between rSD waves following photothrombosis. The mean interval between waves is not significantly different between arterial occlusion (7.8 ± 0.8 min; n = 43 pairs of waves; 12 rats) and venous occlusion (7.2 ± 0.8 min; n = 36 pairs of waves; 9 rats) (p = 0.58). In A and B, data are normalized to the total number of waves.
rSD waves initiated by arteriole occlusion had a propagation velocity of 3.0 ± 0.1 mm/min (n = 58 waves, 15 rats; Supplemental Table 1), similar to that observed in cortical SD. Propagation velocity of waves evoked by venule occlusion was not significantly different (2.9 ± 0.2 mm/min; n = 45 waves, 9 rats; p = 0.75). The majority of rSD waves (81.0% for arteriole occlusion and 62.2% for venule occlusion) were initiated near the optic disc (55.9 ± 12.1 µm from the edge of the disc) and propagated outward towards the retinal periphery (Movie 1). Occasionally, waves were initiated at the periphery and propagated inwards towards the central retina (12.1% for arteriole occlusion and 24.4% for venule occlusion). In a subset of these peripherally initiated episodes, the waves spiraled inwards towards the optic disc (Movie 2), reversed direction and/or generated a secondary wave after reaching the optic disc and propagated outward again. This secondary wave always propagated into areas of the retina that had not sustained the initial wave. Rarely, waves were initiated near the site of photothrombosis (6.9% for arteriole occlusion and 13.3% for venous occlusion). No waves were observed before photothrombosis was initiated.
Accumulation of free radicals has been reported to initiate SD in both the avascular ex vivo retina17,18 and the cortex.32 It could be argued that the rSD waves we observed in vivo were initiated directly by free radicals rather than by ischemia, as photothrombosis produces thrombi through free radical generation.17,18 If this were the case, then activating Rose Bengal in a retinal region containing capillaries rather than large vessels would also generate rSD. However, photothrombosis of capillaries produced no rSD events, even when photoactivation occurred over a large area (n = 3 rats), nor were rSD waves generated by photoactivation illumination in the absence of Rose Bengal in the vasculature (n = 6 rats), demonstrating that rSD is not generated by the photoactivation laser itself.
In an additional control experiment, we tested whether rSD episodes were generated following global retinal ischemia, where free radical production by photothrombosis was absent. Blood flow in the entire retinal circulation was blocked by raising intraocular pressure to ∼ 120 mmHg. An rSD wave was generated within 1.5–2.3 min (n = 3 waves in three animals) of blocking blood flow (Figure 3(c); Movie 3). Together with the observation that rSD episodes are rarely initiated at the site of photothrombosis, these data strongly suggest that rSD is a response to the acute loss of vessel perfusion.
rSD in the ex vivo eyecup
We also investigated rSD waves in an ex vivo rat eyecup preparation. rSD waves were elicited either by focal ejection of 150 mM K+ gluconate or by OGD, an ex vivo model of ischemia. rSD waves were monitored by imaging the IOS of the eyecup with reflectance microscopy. The propagation velocity of rSD waves in the eyecup evoked by either K+ gluconate ejection or OGD was 2.7 ± 0.1 mm/min (n = 19 waves; 13 waves, K+ gluconate; 6 waves, OGD; 6 rats, 3 rats per condition), which was not significantly different from the propagation velocity in vivo (p = 0.23). All 13 waves induced by K+ gluconate ejection began near the tip of the ejection pipette. In contrast, five out of six waves observed in the OGD preparation were unequivocally initiated at the optic disc, while a peripheral origin of one wave could not be excluded due to the positioning of the preparation within the imaging field. These data recapitulate the observations made in vivo, demonstrating that retinal ischemia triggers rSD and that rSD is likely to be initiated at the optic disc.
Electrophysiological properties of rSD
We characterized the electrophysiological properties of rSD in both the ex vivo rat eyecup and the in vivo preparation to determine whether it has similar electrophysiological characteristics as cortical SD. rSD waves were evoked in the eyecup by both K+ ejection and by OGD. When rSD waves were elicited by K+ gluconate ejection, a negative shift in DC potential and a brief burst of RGC spiking followed by the silencing of activity were observed when the rSD wave front reached the recording pipette (Figure 5(a) and (b); n = 3 retinas, 3 rats). Similar electrophysiological properties were observed for rSD waves elicited by OGD (Figure 5(c) and (d); n = 3 retinas, 3 rats).
Figure 5.
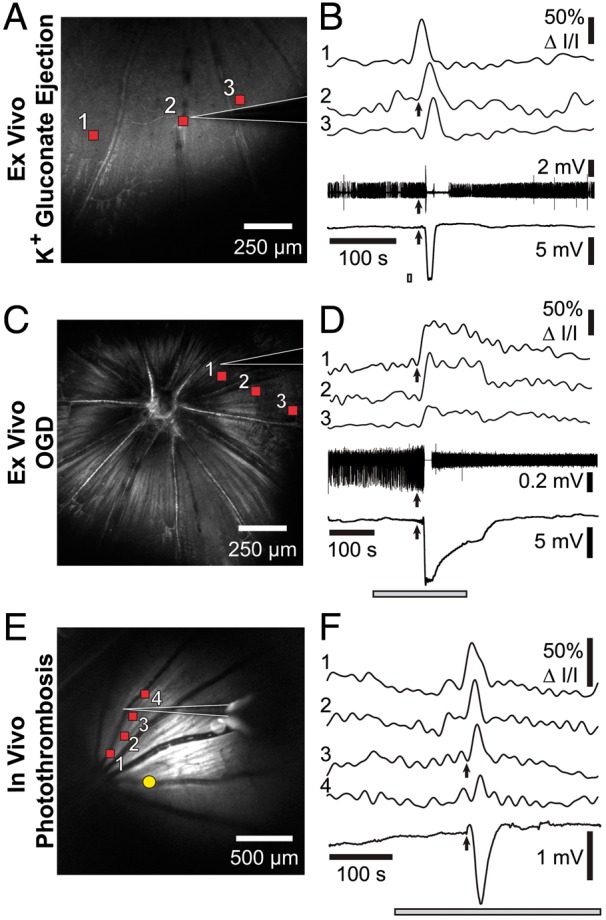
rSD waves have similar electrophysiological characteristics as cortical SD waves. Left panels show reflectance images of the retina and the recording pipette (white outlines). Right panels show IOSs of rSD waves (top traces, numbered 1–3 or 1–4) and electrophysiological responses (bottom traces). The IOS signals were recorded at the retinal locations indicated by the red squares in the left panels. Time course of the stimulus is indicated by the gray bars in the right panels. (a and b) An rSD wave in an eyecup, initiated by focal ejection of 150 mM K+ gluconate (ejection site outside of the image in A). Spike activity of a RGC (2nd from bottom trace in B) and DC potential (bottom trace in B) are shown. Upon arrival of the rSD wave at the recording pipette (arrows in B) RGC spiking is briefly stopped and a negative DC potential is generated. (c and d) An rSD wave in the eyecup, initiated by oxygen/glucose deprivation (OGD). As in B, RGC activity is halted and a negative shift in DC potential is recorded upon arrival of the rSD wave at the recording pipette. The decrease in spike amplitude after the rSD wave is due to movement of the recording pipette. (e and f) An rSD wave initiated in vivo by photothrombosis of an arteriole (yellow dot in E). Upon arrival of the rSD wave at the recording pipette a negative, DC potential is generated (bottom trace in F).
Field potential recordings were also performed in vivo, with rSD waves evoked by photothrombosis of a primary branch vessel. As in the ex vivo experiments, a negative shift in DC potential was recorded as the rSD wave passed the recording pipette (Figure 5(e) and (f); n = 3 rats). Together, these experiments demonstrate that the rSD waves observed in the retina have similar electrophysiological properties as SD waves in the cortex.
Changes in blood flow and pO2 associated with rSD
A series of blood flow changes is associated with cortical SD.3,15,33 We used a variety of techniques to determine whether similar blood flow changes are associated with rSD. Changes in the diameter of primary arterioles were monitored in vivo with confocal microscopy. A dramatic constriction of arterioles was observed as rSD waves passed the vessels (Figure 6(a) and (b)). Arteriole constriction averaged 10.6 ± 1.8% (4.7 ± 1.1 µm; n = 5 waves, 5 rats). Arteriole diameter recovered by 36 s. As described by the Hagen–Poiseuille fourth power relation,34 a constriction of 10.6% will result in a 36.1% decrease in blood flow.
Figure 6.
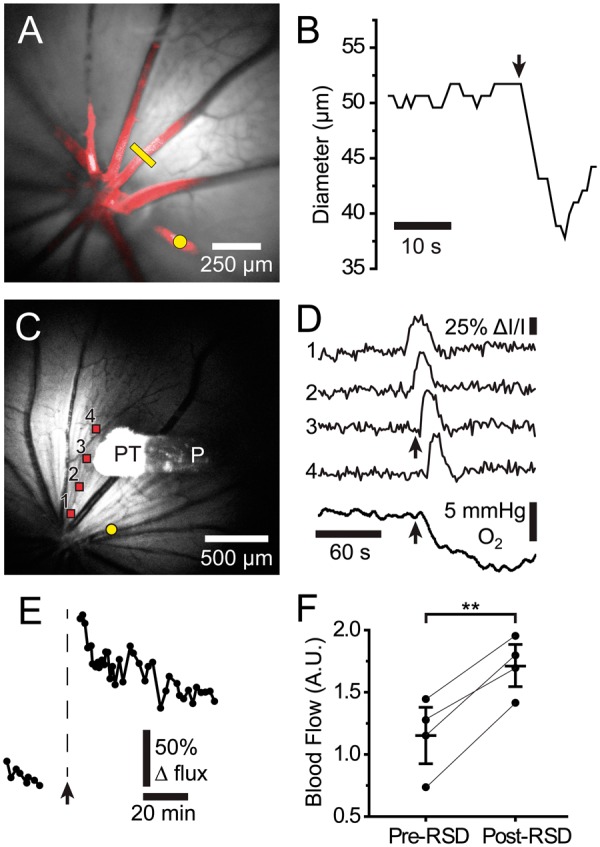
rSD-induced alterations in retinal blood flow and pO2. (a) Reflectance image of the retina with a superimposed confocal image of Rose Bengal-filled blood vessels (red). Photothrombosis of a primary arteriole (yellow dot) evoked a rSD wave. The diameter of a second arteriole was monitored with confocal line scans (yellow line). (b) Diameter of the arteriole indicated by the yellow line in A. A rapid 12% constriction of the vessel coincided with the arrival of the rSD wave (arrow). (c) Reflectance image of the retina, showing the O2 probe (P) and the O2-sensitive probe tip (PT). Photothrombosis of an arteriole (yellow dot) evoked a rSD wave. (d) IOSs (traces 1–4) show propagation of a rSD wave from the retina in C. A large, rapid decrease in retinal pO2 (bottom trace) occurs when the rSD wave reaches the O2 probe tip (arrows). (e) Blood flow in a patent primary arteriole distant from the vessel occluded by photothrombosis, measured by monitoring red blood cell (RBC) flux. When the occlusion-evoked rSD wave reached the patent vessel (arrow), blood flow increased. The blood flow increase decayed slowly over an hour. (f) Summary of blood flow changes induced by rSD, measured by monitoring RBC flux. Paired data points show mean blood flow before and after rSD in individual animals. Post-rSD flow represents the mean flow during the period 24–65 min following rSD wave generation. n = 4 rats. Mean ± S.E.M. are shown. ** p < 0.01.
Such a large drop in blood flow, coupled with the increased metabolic demand imposed by rSD, should lead to a decrease in O2 partial pressure (pO2). We tested this prediction by measuring pO2 with an optical O2 sensor at the surface of the retina. The sensor was positioned in a region far from the site of photothrombosis and outside the region directly affected by the thrombus, identified by laser speckle flowmetry. A rapid drop in pO2 of 11.6 ± 1.4 mmHg (n = 14 waves, 6 rats) was observed as the rSD wave passed the sensor (Figure 6(c) and (d)). pO2 decreases recovered slowly over 103 ± 10 s (n = 14 waves, 6 rats) and generally returned to baseline or even exhibited an overshoot above baseline for rSD episodes that occurred shortly after thrombus formation. However, with subsequent rSD waves, baseline pO2 dropped further and remained below pre-thrombus levels for the remainder of the monitoring session, in one case for over 60 min. These experiments demonstrate that rSD waves are accompanied by a transient drop in blood flow and a prolonged decrease in retinal pO2.
Longer term changes in blood flow following a rSD episode were investigated by measuring the flux of labeled RBCs as they passed through a primary arteriole that was not subjected to photothrombosis. Following the brief decrease in blood flow described above, there was a prolonged increase in flow which decayed slowly over several hours (Figure 6(e) and (f)). We did not observe an oligemic phase, which is often seen in CSD.3,15
Pharmacological block of rSD
We identified several pharmacological agents that block rSD. The experimental NMDA receptor antagonist MK-801, which blocks SD,4,35 decreased the number of rSD events by 70% and decreased the size and the velocity of the rSD waves evoked by photothrombosis by 48 and 51%, respectively (Figure 7). A second clinically approved NMDA receptor antagonist, memantine, did not have a statistically significant effect on wave number, size, or velocity, but the data trended towards a reduction in these measures. Sumatriptan, a 5-HT(1D) receptor agonist that has been shown to block rSD in the ex vivo retina,36 reduced the number rSD waves by 88%, but did not affect their size or velocity (Figure 7).
Figure 7.

rSD is blocked by the 5-HT(1D) receptor agonist sumatriptan and by the NMDA receptor antagonist MK-801. Drugs were injected i.v. 10 min before initiation of photothrombosis at concentrations of 3 mg/kg sumatriptan, 3 mg/kg MK-801, and 10 mg/kg memantine. (a) Sumatriptan and MK-801 decreased the incidence of wave generation during the first hour after photothrombosis. Memantine, another NMDA receptor antagonist, trended towards reducing the incidence of wave generation. (b) MK-801 decreased the size rSD waves. (c) MK-801 reduced rSD wave propagation velocity. (d) None of the drugs affected the site of rSD wave initiation. Regardless of treatment group, waves were predominantly initiated at the optic disc. For all panels, n = 5 animals per treatment group. Data represent mean ± S.E.M., * p < 0.05 and ** p < 0.01.
Discussion
The results of this study demonstrate that multiple SD waves are generated in the retina following occlusion of a primary retinal arteriole or venule. The rSD waves were not an artifact of the photothrombosis model of retinal ischemia, as (i) rSD waves were also generated in vivo following global ischemia produced by an increase in intraocular pressure, (ii) rSD waves occurred in the eyecup following oxygen/glucose deprivation, an ex vivo model of ischemia, and (iii) rSD waves were not evoked by photobleaching of capillaries when larger vessels were spared. rSD waves had similar properties to cortical SD waves, including their propagation velocities, changes in tissue IOSs, a negative shift in DC potential, a transient increase followed by a longer silencing of neuronal spiking, a sequence of blood flow changes, and a decrease in tissue pO2. Drugs that are effective in blocking SD in other systems, including the NMDA receptor antagonist MK-801 and the 5-HT(1D) receptor agonist sumatriptan, were also effective in reducing the generation of rSD.
In comparing the characteristics of rSD and cortical SD, it is necessary to clearly define nomenclature.3,37 Different types of SD events exist on a continuum from benign to harmful and are defined in the context of a tissue’s physiological state.37 CSD, which is thought to account for migraine auras, lies on the benign end of this continuum because it affects healthy tissue, is recurring, has no lasting effects,3,7 is blocked by NMDA receptor antagonists,4 and is characterized by a long-lasting oligemia.3 Conversely, the most harmful type of SD is anoxic depolarization (AD) which occurs in severely ischemic tissue, is a single event that is resistant to NMDA receptor antagonism38–40 and leads to irreversible tissue damage.37
In our retinal photothrombosis model, the initial rSD events we observed could be classified as AD episodes as they are triggered by tissue ischemia. Subsequent waves likely represent peri-infarct depolarizations (PIDs), a type of SD that falls between AD and CSD and is susceptible to NMDA receptor antagonism.37 Such an interpretation is supported by the observations that (i) blood flow responses followed a PID-like pattern of initial oligemia followed by prolonged hyperemia,41 and (ii) rSDs were reduced by NMDA receptor antagonists, but not eliminated entirely. The decreased frequency, size, and velocity of rSD with MK-801 blockade of NMDA receptors are likely due to an effect on PID-like rSDs, and not AD, which cannot be blocked by NMDA receptor antagonists. Indeed, MK-801 did not affect the latency to the first rSD wave following vessel occlusion. While the effects of memantine, another NMDA receptor blocker, did not reach statistical significance, they did trend towards those of MK-801. The weakness of memantine’s effects is curious and could be due to the drug having lesser bioavailability across the blood-retinal barrier. The 5-HT(1D) receptor agonist sumatriptan also reduced wave frequency but did not affect other rSD properties. Sumatriptan inhibits rSD in the ex vivo chick retina as well.36 The mechanisms underlying this effect are unclear, but are likely due to an extravascular effect since the chick retina is avascular.
In our high-intraocular pressure, global model of ischemia, only one rSD wave per animal was generated, rather than the multiple waves evoked by occlusion of a single branch vessel. This single SD wave is a property of cortical AD waves.38 However, if perfusion was reestablished by lowering intraocular pressure within a minute after rSD generation, additional waves could be evoked by subsequent increases in intraocular pressure. This finding suggests that reperfusion prior to a critical point of irreversible damage allows sufficient tissue recovery to sustain additional SD waves, consistent with observations made in the cortex.37,42
Most in vivo rSD waves were initiated near the optic disc, regardless of the site of thrombus formation. In addition, most ex vivo eyecup rSD waves evoked by OGD were initiated near the optic disc. An optic disc origin of SD waves could be due to a relative paucity of capillaries, and therefore blood flow, at the optic disc. However, this is unlikely. First, the density of blood vessels remains relatively constant across the rodent retina.43 Second, the observation that rSD waves are preferentially initiated at the optic disc in the ex vivo preparation, where blood flow does not occur, speaks against a vascular etiology. Instead, an optic disc origin of rSD waves may be related to the nature of Müller cells, the primary glial cells in the retina. Müller cells are longer and thinner near the disc, where the retina is thickest.44 Müller cells are primarily responsible for buffering extracellular levels of both glutamate45 and K+46 and thinner cells may be less effective in rapidly removing glutamate and K+ from extracellular space.44 This reduced capacity for glutamate and K+ buffering may render the region near the optic disc more sensitive to rSD initiation.47 Such a mechanism is thought to underlie the observation that K+ elevations during SD are greater in primary sensory cortices than in other brain regions, making these areas more likely to generate SD waves.48 Similarly, this mechanism may also explain why migraine auras are often initiated in the primary visual cortex,49 as the astrocyte-neuron ratio in area 17 is lower than in adjacent cortical areas.50
rSD was accompanied by a brief, yet pronounced arteriole vasoconstriction followed by a prolonged increase in blood flow lasting for an hour. The fact that blood flow increased following rSD at the same time that pO2 remained below normal is not necessarily contradictory. In the minutes following a SD episode, a large metabolic demand is imposed on retinal tissue as glutamate and K+ must be pumped back into neurons and glial cells.3,9 The increase in blood flow may result from this increased metabolic demand, but may not be sufficient to maintain O2 at normal levels. Such a pattern of an initial oligemia followed by prolonged hyperemia is also observed in cortical PID.41
It is not known whether rSD occurs in humans. To our knowledge, there have been no reports of perceptual experiences related to rSD in patients with acute retinal occlusive disorders. It is possible that such rSD perceptions would be filtered out by higher order visual processing. On the other hand, the human retina may be more resilient to ischemia-induced rSD than the rat retina. However, given our observations that multiple rSD episodes are generated in an animal model of branch retinal vein and artery occlusion, and that cortical SD occurs in analogous vessel occlusions in the human cortex, we believe it likely that rSD also occurs in patients with BRVO and related vascular disorders.
A wealth of evidence demonstrates that SD associated with pathology can expand the size of tissue damage. SD events that occur following subarachnoid hemorrhage, malignant stroke, or traumatic brain injury can produce delayed secondary ischemic infarcts in patients.11 In an animal model of ischemic stroke, SD increases the metabolic demand in the penumbra and enlarges the ischemic core.9 Blocking SD waves can limit injury expansion35,51 and cell death.35,52 In the ex vivo chick retina, SD leads to neuronal death as well.19 These observations suggest that rSD in patients may also have a deleterious effect on retinal health. Particularly following injury, such as ischemia, rSD could contribute to the evolution of retinal damage. Future experiments that test this hypothesis are critical because it may be possible to reduce cell death in BRVO patients by blocking rSD generation. Importantly, we show that an NMDA receptor blocker and a 5-HT(1D) receptor agonist reduce the frequency of rSD wave generation. Drugs with these mechanisms of action are already clinically approved and may prove to be effective therapeutic agents for reducing vision loss in BRVO patients.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH RO1 EY004077 (EAN), NIH P30-EY011374 (EAN), the Winston and Maxine Wallin Neuroscience Discovery Fund (EAN), American Heart Association Predoctoral Fellowship 13PRE16960081 (AIS), and CTSI TRDP Award NIHUL1TR000114 (AIS).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AIS, KRB, and EAN designed the experiments, AIS and JK conducted the in vivo experiments, KRB conducted the eyecup experiments, and AMS conducted the immunohistochemistry experiments. AIS and KRB analyzed the data, AIS and EAN wrote the manuscript, and EAN supervised the study.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Leão AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol 1944; 7: 359–390. [DOI] [PubMed] [Google Scholar]
- 2.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 2001; 81: 1065–1096. [DOI] [PubMed] [Google Scholar]
- 3.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015; 95: 953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou N, Rungta RL, Malik A, et al. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J Cereb Blood Flow Metab 2013; 33: 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain 1994; 117: 199–210. [DOI] [PubMed] [Google Scholar]
- 6.Charles AC, Baca SM. Cortical spreading depression and migraine. Nat Rev Neurol 2013; 9: 637–644. [DOI] [PubMed] [Google Scholar]
- 7.Anderson TR, Andrew RD. Spreading depression: imaging and blockade in the rat neocortical brain slice. J Neurophysiol 2002; 88: 2713–2725. [DOI] [PubMed] [Google Scholar]
- 8.Lauritzen M, Dreier JP, Fabricius M, et al. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 2011; 31: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Bornstädt D, Houben T, Seidel JL, et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron 2015; 85: 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nedergaard M, Hansen AJ. Characterization of cortical depolarizations evoked in focal cerebral ischemia. J Cereb Blood Flow Metab 1993; 13: 568–574. [DOI] [PubMed] [Google Scholar]
- 11.Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006; 129: 3224–3237. [DOI] [PubMed] [Google Scholar]
- 12.Hinzman JM, DiNapoli VA, Mahoney EJ, et al. Spreading depolarizations mediate excitotoxicity in the development of acute cortical lesions. Exp Neurol 2015; 267: 243–253. [DOI] [PubMed] [Google Scholar]
- 13.Takano T, Tian GF, Peng W, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 2007; 10: 754–762. [DOI] [PubMed] [Google Scholar]
- 14.Girouard H, Bonev AD, Hannah RM, et al. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A 2010; 107: 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fordsmann JC, Ko RWY, Choi HB, et al. Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci 2013; 33: 2562–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lauritzen M, Jørgensen MB, Diemer NH, et al. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol 1982; 12: 469–474. [DOI] [PubMed] [Google Scholar]
- 17.Netto M, Martins-Ferreira H. Elicitation of spreading depression by rose bengal photodynamic action. Photochem Photobiol 1989; 50: 229–234. [DOI] [PubMed] [Google Scholar]
- 18.Netto M, do Carmo RJ, Martins-Ferreira H. Retinal spreading depression induced by photoactivation: involvement of free radicals and potassium. Brain Res 1999; 827: 221–224. [DOI] [PubMed] [Google Scholar]
- 19.Yu Y, Santos LM, Mattiace LA, et al. Reentrant spiral waves of spreading depression cause macular degeneration in hypoglycemic chicken retina. Proc Natl Acad Sci U S A 2012; 109: 2585–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori S, Miller WH, Tomita T. Muller cell function during spreading depression in frog retina. Proc Natl Acad Sci U S A 1976; 73: 1351–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higashida H, Sakakibara M, Mitarai G. Spreading depression in isolated carp retina. Brain Res 1977; 120: 67–83. [DOI] [PubMed] [Google Scholar]
- 22.Olsen JS, Miller RF. Spontaneous slow potentials and spreading depression in amphibian retina. J Neurophysiol 1977; 40: 752–767. [DOI] [PubMed] [Google Scholar]
- 23.Srienc AI, Kurth-Nelson ZL, Newman EA. Imaging retinal blood flow with laser speckle flowmetry. Front Neuroenerg 2010; 2: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takagaki M, Feuerstein D, Kumagai T, et al. Isoflurane suppresses cortical spreading depolarizations compared to propofol–implications for sedation of neurocritical care patients. Exp Neurol 2014; 252: 12–17. [DOI] [PubMed] [Google Scholar]
- 25.Kudo C, Nozari A, Moskowitz MA, et al. The impact of anesthetics and hyperoxia on cortical spreading depression. Exp Neurol 2008; 212: 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman EA, Bartosch R. An eyecup preparation for the rat and mouse. J Neurosci Methods 1999; 93: 169–175. [DOI] [PubMed] [Google Scholar]
- 27.Hultman D, Newman EA. A micro-advancer device for vitreal injection and retinal recording and stimulation. Exp Eye Res 2011; 93: 767–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornfield TE, Newman EA. Measurement of retinal blood flow using fluorescently labeled red blood cells. Eneuro 2015; 2: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mead B, Thompson A, Scheven BA, et al. Comparative evaluation of methods for estimating retinal ganglion cell loss in retinal sections and wholemounts. PLoS One 2014; 9: e110612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dahlem YA, Hanke W. Intrinsic optical signal of retinal spreading depression: second phase depends on energy metabolism and nitric oxide. Brain Res 2005; 1049: 15–24. [DOI] [PubMed] [Google Scholar]
- 31.Hanke W, de Lima VM. Central nervous tissue: an excitable medium. a study using the retinal spreading depression as a tool. Philos Trans A Math Phys Eng Sci 2008; 366: 359–368. [DOI] [PubMed] [Google Scholar]
- 32.Malkov A, Ivanov AI, Popova I, et al. Reactive oxygen species initiate a metabolic collapse in hippocampal slices: potential trigger of cortical spreading depression. J Cereb Blood Flow Metab 2014; 34: 1540–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 2009; 29: 1517–1527. [DOI] [PubMed] [Google Scholar]
- 34.Pries AR, Secomb TW, Gaehtgens P, et al. Blood flow in microvascular networks. Experiments and simulation. Circ Res 1990; 67: 826–834. [DOI] [PubMed] [Google Scholar]
- 35.Gill R, Andine P, Hillered L, et al. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab 1992; 12: 371–379. [DOI] [PubMed] [Google Scholar]
- 36.Maranhao-Filho PA, Martins-Ferreira H, Vincent MB, et al. Sumatriptan blocks spreading depression in isolated chick retina. Cephalalgia 1997; 17: 822–825. [DOI] [PubMed] [Google Scholar]
- 37.Dreier JP, Reiffurth C. The stroke-migraine depolarization continuum. Neuron 2015; 86: 902–922. [DOI] [PubMed] [Google Scholar]
- 38.Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex 2001; 11: 249–259. [DOI] [PubMed] [Google Scholar]
- 39.Joshi I, Andrew RD. Imaging anoxic depolarization during ischemia-like conditions in the mouse hemi-brain slice. J Neurophysiol 2001; 85: 414–424. [DOI] [PubMed] [Google Scholar]
- 40.Murphy TH, Li P, Betts K, et al. Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci 2008; 28: 1756–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hashemi P, Bhatia R, Nakamura H, et al. Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leão's spreading depression. J Cereb Blood Flow Metab 2009; 29: 166–175. [DOI] [PubMed] [Google Scholar]
- 42.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011; 17: 439–447. [DOI] [PubMed] [Google Scholar]
- 43.Ivanova E, Toychiev AH, Yee CW, et al. Intersublaminar vascular plexus: the correlation of retinal blood vessels with functional sublaminae of the inner plexiform layer. Invest Ophthalmol Vis Sci 2014; 55: 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reichenbach A and Wohlrab F Morphometric parameters of Muller (glial) cells dependent on their topographic localization in the nonmyelinated part of the rabbit retina. A consideration of functional aspects of radial glia. J Neurocytol 1986; 15: 451–459. [DOI] [PubMed]
- 45.Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature 1987; 327: 707–709. [DOI] [PubMed] [Google Scholar]
- 46.Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 1984; 225: 1174–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seidel JL, Escartin C, Ayata C, et al. Multifaceted roles for astrocytes in spreading depolarization: a target for limiting spreading depolarization in acute brain injury? Glia 2015; 64: 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogdanov VB, Middleton NA, Theriot JJ, et al. Susceptibility of primary sensory cortex to spreading depolarizations. J Neurosci 2016; 36: 4733–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen JM, Baca SM, Vanvalkenburgh P, et al. Distinctive anatomical and physiological features of migraine aura revealed by 18 years of recording. Brain 2013; 136: 3589–3595. [DOI] [PubMed] [Google Scholar]
- 50.Leuba G, Garey LJ. Comparison of neuronal and glial numerical density in primary and secondary visual cortex of man. Exp Brain Res 1989; 77: 31–38. [DOI] [PubMed] [Google Scholar]
- 51.Dreier JP, Drenckhahn C, Woitzik J, et al. Spreading ischemia after aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl 2013; 115: 125–129. [DOI] [PubMed] [Google Scholar]
- 52.Sadeghian H, Jafarian M, Karimzadeh F, et al. Neuronal death by repetitive cortical spreading depression in juvenile rat brain. Exp Neurol 2012; 233: 438–446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.