

Abstract
The CRISPR/Cas9 system is a great revolution in biology. This technology allows the modification of genes in vitro and in vivo in a wide variety of living organisms. In most Duchenne muscular dystrophy (DMD) patients, expression of dystrophin (DYS) protein is disrupted because exon deletions result in a frame shift. We present here the CRISPR-induced deletion (CinDel), a new promising genome-editing technology to correct the DMD gene. This strategy is based on the use of two gRNAs targeting specifically exons that precede and follow the patient deletion in the DMD gene. This pair of gRNAs induced a precise large additional deletion leading to fusion of the targeted exons. Using an adequate pair of gRNAs, the deletion of parts of these exons and the intron separating them restored the DMD reading frame in 62% of the hybrid exons in vitro in DMD myoblasts and in vivo in electroporated hDMD/mdx mice. Moreover, adequate pairs of gRNAs also restored the normal spectrin-like repeat of the dystrophin rod domain; such restoration is not obtained by exon skipping or deletion of complete exons. The expression of an internally deleted DYS protein was detected following the formation of myotubes by the unselected, treated DMD myoblasts. Given that CinDel induces permanent reparation of the DMD gene, this treatment would not have to be repeated as it is the case for exon skipping induced by oligonucleotides.
Keywords: Cas9, CRISPR, DMD gene, Duchenne muscular dystrophy, dystrophin, gene therapy, gRNA
Introduction
Duchenne muscular dystrophy (DMD) is a monogenic hereditary disease linked to the X chromosome, which affects a boy in about 3,500 births.1 The cause of the disease is the inability of the body to synthesize the dystrophin (DYS) protein, an elongated cytoskeletal protein of 427 kDa mostly containing a central rod domain of 24 spectrin-like repeats. Dystrophin plays a fundamental role in maintaining the integrity of the sarcolemma.2,3 The absence of this protein is secondary to a mutation of the DMD gene.4 The most frequently encountered mutations, found in about 45% of DMD patients, are deletions of one or more exons in the region between exons 45 and 55, called the hot region of DMD gene.5 Most of these deletions induce a codon frameshift of the mRNA transcript leading to the production of a truncated DYS protein. Since the latter is rapidly degraded, the absence of DYS at the sarcolemma increases its fragility and leads to muscle weakness characteristic of DMD. Naturally occurring in-frame deletions generate an internally deleted DYS protein and result in the milder Becker muscular dystrophy (BMD) phenotype.6 For DMD patients, skeletal muscular weaknesses will unfortunately lead to death, between 18 and 30 years of age,7,8 while some BMD patients can have a normal life expectancy.6 To date, there is no cure for DMD and BMD.
The identification of the molecular basis for the DMD and BMD phenotypes established the foundation for DMD gene therapy.9,10,11,12,13 Different strategies for DMD gene therapy are currently under development, i.e., restoration of the full-length transcript or expression of a truncated in-frame DMD gene. Since the 2.4-Mb DMD gene contains 79 exons and encodes a 14 kb mRNA,14,15 it is difficult to develop a gene therapy to deliver efficiently the full-length gene or even its cDNA in muscle precursor cells in vitro or in the muscle fibers in vivo. Shorter versions of the DMD cDNA, called micro-dystrophin (of about 4 kb), have been delivered in vivo using adeno-associated virus (AAV) vector but failed to translate therapeutic efficacy from mice and dogs to human patients.12,16,17,18,19
An alternative to gene replacement is to modify directly within the cells the DMD mRNA or the DMD gene itself. Correction of the reading frame of the mRNA can be obtained by exon skipping using a synthetic antisense oligonucleotide interacting with the primary transcript with the splice donor or splice acceptor of the exon, which precedes or follows the patient deletion20,21,22 or by targeting internal sites in the exons.23 Such skipping of one, or if necessary several complete exons, restores a reading frameshift and generates an internally deleted DYS protein similar to the one found in BMD patients.6 Exon skipping has yielded very encouraging results in vitro,24 in vivo in animal models25,26 and in DMD patients.27,28,29 However, a phase 3 clinical trial was recently abandoned by Glaxo Smith Kline because it did not show statistically significant functional improvements.30 The recent development of sequence-specific nucleases (meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the CRISPR/Cas9 system) now permits to correct the DMD gene itself.31,32,33,34,35,36,37
The CRISPR system is a defense mechanism identified in many bacterial species.38,39,40,41,42,43 It has been modified to allow gene editing in mammalian cells. That modified system still uses a Cas9 nuclease to generate double-strand breaks (DSB) at a specific DNA target sequence.44,45 The recognition of the cleavage site is determined by base pairing of the gRNA with the target DNA and the presence of a trinucleotide called PAM (protospacer adjacent motif) juxtaposed to the targeted DNA sequence.46 This PAM is NGG for the Cas9 of Streptococcus pyogenes, the most commonly used enzyme.47,48
In this study, we used two gRNAs targeting exons 50 and 54 of the DMD gene. The in vitro experiments were done in 293T cells or in myoblasts of a DMD patient having a deletion of exons 51–53 inducing a frame shift. Preliminary in vivo experiments were also performed in the hDMD/mdx mouse that contains a full-length human DMD gene. Our results show that both in vitro and in vivo, two gRNAs allowed precise DSB and induced a large deletion (i.e., more than 160 kb in 293T cells). The junction between the remaining DNA sequences was achieved exactly as predicted. Depending on the pairs of gRNAs, it was possible to restore the reading frame resulting in the synthesis of an internally deleted DYS protein by the myotubes formed by the corrected myoblasts of a DMD patient with an out-of-frame deletion. We also identified pairs of gRNAs that not only restored the frame shift but also restored the spectrin-like repeats structural characteristics. We expect that this approach could confer more functionality to the DYS protein and will be more beneficial to the patient. The CRISPR-induced deletion (CinDel) therapeutic approach could eventually be used to restore directly in vivo the reading frame for most deletions observed in DMD patients. This is therefore a practical feasible therapeutic approach.
Results
Tests of individual gRNAs in 293T cells and in DMD myoblasts
Twenty-four different pSpCas(BB)-2A-GFP-gRNA plasmids were made: 10 containing gRNAs targeting different sequences of the exon 50 of the DMD gene and 14 containing gRNAs targeting the exon 54 (Table 1 and Figure 1a,b). For screening purposes in order to test the activity of these gRNAs, these plasmids were first transfected in 293T cells. Under standard transfection conditions, 80% of 293T cells showed expression of the GFP confirming the effectiveness of the transfection (Figure 1c, 293T LF). The DNA from those cells was extracted 48 hours after transfection. The exon 50 of the DMD was amplified by PCR using primers Sense 49 and Antisense 50 and exon 54 was amplified with primers Sense 53 and Antisense 54. The presence of INDELs, produced by nonhomologous end-joining following the DSBs generated by the gRNAs and the Cas9, was detected using the Surveyor/Cel1 enzymatic assay (Figure 2a,b). An expected pattern of three bands was detected with most gRNAs; the upper band representing the uncut PCR product and the two lowest bands the Cel1 products whose lengths are related to the guide used to induce the DSB.
Table 1. gRNAs targeting the DMD gene used in this study.
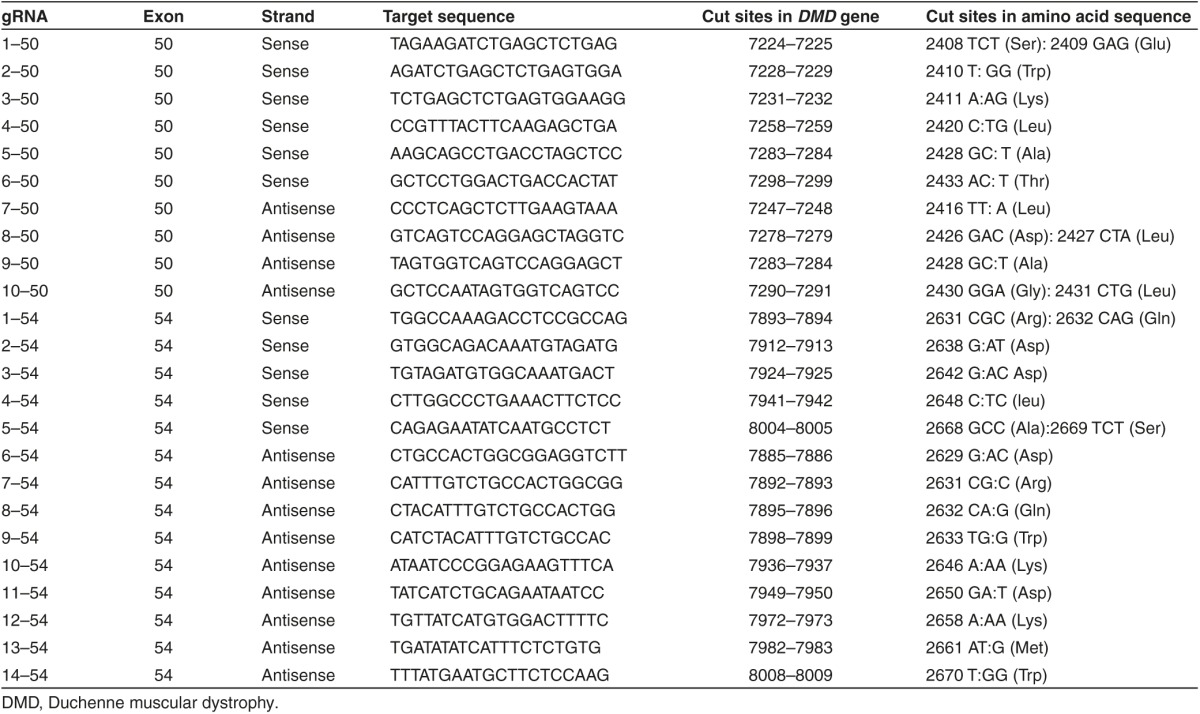
Figure 1.

gRNA screening. (a and b) The figure illustrates the sequence of exons 50 and 54 of the human DMD gene. For exon 50, 10 different PAMs (numbered 1 to 10) are identified: 6 are in the sense strand and 4 in the antisense strand. For exon 54, 14 PAMs are identified, 5 in the sense strand and 9 in the antisense strand. In the sense strand the GG of the PAMs are over-lighted in yellow, while they are over-lighted in green in the antisense strand. The third nucleotide of the PAMs is over-lighted in blue in both strands. (c) The eGFP expression was monitored in 293T and in DMD myoblasts after transfection of the pSpCas(BB)-2A-GFP-gRNA with lipofectamine 2000 using two different protocols (standard LF and improved LF+).
Figure 2.

Surveyor assay for gRNA screening in 293T cells and in myoblasts. The assay was performed on genomic DNA extracted from (a and b) 293T cells or (c and d) myoblasts transfected individually with different gRNAs. Screening was performed separately for (a and c) exon 50 and (b and d) exon 54. Genomic DNA of nontransfected cells was used for negative control (NC) for the Surveyor assay. The gRNA numbers correspond with the PAM identified in Figure 1a and the targeted sequences in Table 1. MW, molecular weight marker.
The gRNAs were also subsequently tested individually in immortalized myoblasts from a DMD patient having a deletion of exons 51 through 53. Unfortunately, transfection efficiency was very low in myoblasts under the standard lipofectamine 2000 transfection14 (Figure 1c, myoblast LF). However, we improved the protocol (see Material and Methods section), and we were able to see ~20–25% of myoblasts expressing GFP (Figure 1c, myoblast LF+). The Surveyor assay revealed the presence of INDELs in amplicons of exons 50 (Figure 2c) and 54 (Figure 2d) obtained from these myoblasts.
Tests of gRNA pairs
Given that the CRISPR/Cas9 induces a DSB at exactly 3 bp from the PAM in the 5′ direction, it was possible to predict the consequence of cutting exons 50 and 54 with various pairs of gRNAs. This analysis predicted four possibilities, as illustrated in Figure 3a and detailed in Supplementary Table S1). When the total number of coding nucleotides, which are deleted (i.e., the sum of the nucleotides of exons 51, 52, and 53 and the portions of exons 50 and 54, which are deleted), is a multiple of three, there are three different results possible. (i) The junction of the remains of 50 exons and 54 does not generate a codon for a new amino acid. (ii) A new codon, derived from the junction of the remains of 50 exons and 54, encodes a codon for a new amino acid. (iii) The new codon formed by the junction of the remaining parts of exons 50 and 54 is a stop codon. Finally, the most frequent result is (iv) the total number of coding nucleotides, which are deleted, is not a multiple of three resulting in an incorrect reading frame of the DMD gene.
Figure 3.

The CinDel approach can generate four possible DMD gene modifications. (a) Double-strand breaks created by the Cas9 and different gRNA pairs can theoretically modify the DMD gene four different ways: (i) in blue, correct junction of the normal codons of exons 50 and 54; (ii) in green, the junction of the nucleotides of exons 50 and 54 generates the codon for a new amino acid at the junction site but the remaining codons of exon 54 are normal; (iii) in white, junction of the nucleotides of exons 50 and 54 results in an incorrect reading frame that changes the remaining codons of exon 54; and (iv) in red, the junction of the nucleotides of exons 50 and 54 generates a new stop codon at the junction site. (b and c) Different gRNA pairs were experimentally tested in 293T cells and in myoblasts and PCR amplification generated amplicons of the expected sizes. The pairs tested (those with a X in a) produced the expected mutations, which were confirmed by sequencing. MW, molecular weight markers.
The deletion of part of the DMD gene was investigated by transfecting 293T cells and human DMD myoblasts with different pairs of plasmids encoding gRNAs: one targeting exon 50 and the other the exon 54 (Figure 3). To detect successful deletions, genomic DNA was extracted from these transfected and nontransfected cells 48 hours later and amplified by PCR using primers sense 49 and antisense 54. No amplification was obtained from DNA extracted from untransfected cells (Figure 3c, NC lanes 1 and 6) because of the expected amplicon size (about 160 Kbp) of the wild-type DMD gene (i.e., exon 50 to exon 54) is too big. However, amplicons, containing a hybrid exon, of the expected sizes were obtained when a pair of gRNAs was used (Figure 3c, lanes 2 to 5 and lanes 7 to 10), confirming the excision of a sequence of about 160,100 bp in 293T cells and of a smaller number of nucleotides in the DMD myoblasts.
As shown in Figure 3, we have tested several different gRNA pairs (targeting exons 50 and 54), and all produced exactly the expected modification of the DMD gene according to the four possibilities explained above.
Characterization of the hybrid exon 50–54 in 293T cells
The amplicons obtained following transfection of the gRNA pairs were gel purified and cloned into the pMiniT plasmid, transformed in bacteria, and clones were screened for successful insertions. Positive clones, according to the digestion pattern (data not shown), were sent for sequencing to demonstrate the presence of a hybrid exon formed by the fusion of a part of exon 50 with a portion of exon 54. For example, in 100% (7/7) of sequences obtained for the gRNA5–50 and gRNA1–54 pair, the DMD gene was cut in both exons at exactly three nucleotides in the 5′ direction from the PAM (data not shown). This exercise was repeated with different pairs of gRNAs and for each functional gRNA pair, the CinDel technique removed successfully a portion of about 160,100 bp in the DMD gene of 293T cells.
Characterization of the hybrid exon 50–54 in myoblasts
We also wanted to confirm the accuracy of cuts produced by the Cas9 from our expression plasmids in the myoblasts of a DMD patient already having a deletion of exons 51 to 53. We thus transfected in these cells either the pair of plasmids coding for gRNA2–50 and for gRNA2–54, or the pair of plasmids coding for gRNA gRNA1–50 and gRNA5–54. Both of these pairs were previously characterized to produce a deletion in the DMD gene restoring the reading frame. As control, we also used a pair of gRNAs (i.e., gRNA5–50 and gRNA1–54) that should not restore the reading frame. As in 293T, genomic DNA of these myoblasts was extracted 48 hours after transfection and amplified with primers sense 49 and antisense 54, and amplicons were cloned into the plasmid pMiniT. The plasmids were extracted from bacterial clones, screened for the presence of an insert according to the digestion pattern (data not shown), and positive clones were sequenced. The sequences of 45 clones were analyzed for the gRNA2–50/2–54 pair, and the most abundant product (25/45, i.e., 56%) contained exactly the expected junction between the remaining parts exons 50 and 54 to produce a 141 bp hybrid exon (Figure 4). For 60% (27/45), a new codon (Y) was created (Figure 4a). A total of 62% (28/45) was detected as in-frame hybrid exons (Figure 4b) and 38% (17/45) as out-of-frame hybrid exons (Figure 4b). For the second pair (gRNA1-50/5-54), similar results were obtained: 70% (7/10) contained the expected junction with no additional amino acid (data not shown).
Figure 4.

gRNA pairs can induce deletions that restore the reading frame in the DMD gene in DMD myoblasts. (a) Sequence obtained from the amplification of the hybrid exon 50–54 following transfection of the gRNA2–50 and gRNA2–54 pair shows a newly formed codon TAT (coding for tyrosine) at the junction site. This new codon is formed by the nucleotide T from the remaining exon 50 and nucleotides AT from the remaining exon 54. (b) Other in-frame and out-of-frame sequences were also found.
For the third gRNA pair (gRNA5–50/1–54), the plasmids were extracted from eight bacterial clones and sequenced. The sequence of these clones also demonstrated that 75% (6/8) of these hybrid exons 50–54 (amplicon 655 bp) contained the expected reading frame shift (data not shown). One of the two remaining clones showed a 1 bp insertion in addition of the expected deletion, this restored the DMD reading frame. Another clone showed an additional deletion of 11 bp that did not restore the reading frame.
Since the CinDel method was producing the expected results in 293T cells and in DMD myoblasts in culture, plasmids coding for a pair of gRNAs (gRNA2–50/2–54) were electroporated in the tibialis anterior of a hDMD/mdx mouse to confirm CinDel effects in vivo. Genomic DNA was extracted 7 days later from the electroporated tibialis anterior and from a nonelectroporated tibialis anterior. The exons 50 and 54 of the human dystrophin gene were PCR amplified. Expected bands were observed following digestion of the amplicon of these exons by the Cel1 enzyme of the Surveyor assay (Supplementary Figure S1a, CinDel lanes). These results confirmed that both gRNAs were able to induce mutations of their targeted exon in vivo. Moreover, the hybrid exon 50–54 was also PCR amplified (Supplementary Figure S1b), demonstrating that both gRNAs were able to cut simultaneously in vivo leading to a large deletion of more than 160 kb. The amplicons of the hybrid exon 50–54 were cloned in bacteria and 11 clones were sequenced. The sequences of 7 of these clones were the same as those obtained for in vitro experiments with the same gRNA pair (Figure 4b, line 1), thus 64% (7 out of 11) of the sequences showed a correct restoration of the reading frame in vivo.
DYS expression in myotubes formed by genetically corrected myoblasts
In order to verify whether the CinDel gene therapy method was efficient in restoring the expression of the DYS protein, normal wild-type (WT) myoblasts and DMD myoblasts transfected or not with the gRNA pair 2–50/2–54 were differentiated into myotubes in vitro. Myoblast fusion was obtained from all conditions tested (Figure 5a), and the proteins from the resulting myotubes were extracted after 7 days in the fusion medium. Western blots confirmed the presence of a truncated DYS protein in CinDel-corrected cells with an expected molecular weight of about 400 kDa (Figure 5b CinDel induced with ARNg2-50/2-54 and 6c CinDel induced with ARNg1-50/5-54). The size of this protein corresponds to the weight expected in the absence of exons 51–53 and of portions of exons 50 and 54, while the molecular weight of the full-length DYS protein is 427 kDa in WT myotubes (Figure 5b,c, CTL+). No DYS protein was detected in proteins extracted from the DMD myotubes that had not been genetically corrected (Figure 5b,c, CTL-). This result indicates that myotubes formed in vitro by myoblasts of a DMD patient in which the reading frame has been restored by the CinDel are able to express an internally truncated DYS protein of about 400 kDa.
Figure 5.

CinDel correction in myoblasts restored the DYS protein expression in myotubes. (a) Normal wild-type myoblasts (CTL+), uncorrected DMD myoblasts with a deletion of exons 51–53 (CTL-) as well as CinDel-corrected DMD myoblasts (CinDel) were allowed to fuse to form abundant myotubes containing multiple nuclei. Proteins were extracted from these three types of myotubes. The DMD myoblasts (Δ51–53) were genetically corrected with (b) gRNA2–50 and gRNA2–54 and (c) with gRNA1–50 and gRNA5–54. In b and c, western blot detected no DYS protein in uncorrected DMD myotubes (CTL-), a 427 kDa DYS protein was detected in the wild-type myotubes (CTL+), and a truncated DYS protein (about 400 kDa) was detected in the CinDel-corrected DMD myotubes (CinDel).
3-D spectrin-like models resulting from skipping of exon 50 and formation of hybrid exons 2–50/2–54 and 1–50/4–54
The DYS protein is important to maintain the integrity of the sarcolemma. The central part of this protein called the rod domain in made of 24 spectrin-like repeats. According to homology models available from the eDystrophin Website (http://edystrophin.genouest.org/), the dystrophin spectrin-like repeat predicted structures are composed of the typical triple coiled-coil structure, i.e., three α-helices (A, B, and C) linked by two loops (AB and BC) (Figure 6 and Supplementary Figure S2).49 The integral structure of the spectrin-like repeat 19 (R19) is illustrated in Figure 6b. The deletion of exons 51–53 results in the complete removal of parts of helices A, B, and C of R20 and of helix A of R21. This deletion results in a frameshift and thus in the absence of the dystrophin C terminal. It is possible to restore the reading frame by skipping exon 50. The structure of the resulting hybrid spectrin-like repeat (R19-R21) is abnormal the beginning of helix C of R19 is linked with helix B of R21 (Figure 6c). However, for the same deletion of exons 51 to 53, it is also possible to restore the reading frame by using pairs of gRNAs (either pair 2–50/2–54 or pair 1–50/4–54) resulting in the formation of an hybrid exon 50–54. These hybrid exons involve the partial deletion of residues belonging to spectrin-like repeats R19 and R21. However, in order to generate a correct integral spectrin-like repeat, helix from R19 should be linked to an adequate helix of R21. Structural analysis of hybrid exons 2–50/2–54 and 1–50/4–54 was achieved through primary sequence alignment and homology modeling. In the hybrid exon 2–50/2–54, the helix C of R19 is linked directly to the helix B of R21 (i.e., absence of an helix A between helices C and B). However, hybrid exon 1–50/4–54 involves a link between the helix C of repeat R19 and helix C of R21.
Figure 6.

Structural representations of integral spectrin-like repeat R19 and of various hybrid spectrin-like repeats. (a) Primary structure alignments for spectrin-like repeats R19, R20 and R21. Exons associated with these spectrin repeats are identified in gray (below the sequences). The secondary structure for spectrin repeats is represented above the sequences, H for alpha helices and C for the loop segments. Residues between pairs of arrows of the same color are deleted in the resulting hybrid spectrin-like repeats R19–R21. For a patient with a deletion of exons 51–53, the reading frame may be restored by skipping exon 50, thus linking directly exon 49–54. Linking points of deletion of exons 49–54 are highlighted in red. The hybrid exons 2–50/2–54 linking points are highlighted blue and those of hybrid exons 1–50/4–54 in green. (b) Homology models for integral spectrin repeat R19 was obtained from eDystrophin Website (http://edystrophin.genouest.org/). (c) The homology model for the deletion of exons 50–53 (obtained by skipping of exon 50 in a patient with a deletion of exons 51–53). The homology models for (d) hybrid exon 2–50/2–54 and (e) hybrid exon 1–50/4–54 are also illustrated. Structural motifs, as identified in the primary sequence alignment, are colored as follows: helix A is in green, helix B is in orange, and helix C is in blue. Loops AB and BC are in light gray. Colors are darker for spectrin repeat R19 and lighter for spectrin repeat R21.
The homology model resulting from the skipping of exon 50 (deletion of exons 50–53) illustrated in Figure 6c is diverging from the original spectrin-like structure (Figure 6b). The homology model for hybrid exon 2–50/2–54 (Figure 6d) is also diverging from the original spectrin-like structure (Figure 6b). On the other hand, the homology model for hybrid exon 1–50/4–54 corresponds to the sequence length and structure of the original spectrin-like repeat (Figure 6b,e, respectively). Thus, the dystrophin produced following the formation of a hybrid exon with gRNA1–50 and gRNA4–54 would have a similar structure as the integral dystrophin and would probably function better than the dystrophin protein formed by skipping of exon 50 or formed by gRNA2–50 and gRNA2–54.
Discussion
The aim of this study was to develop a new therapeutic approach for DMD, which could eventually because of its simplicity and efficacy be done directly in vivo. This potential therapy is based on the permanent restoration of the DMD reading frame and the dystrophin protein structure by generating additional deletions in exons flanking the deletion already present in the patient DMD gene, as summarized in the Figure 7. As mentioned above, these additional deletions can produce predictably four types of results. Indeed, our results confirmed that gRNAs and Cas9 cut DNA most of the times at exactly three nucleotides from the PAM in the 5′ direction45 and produced the predicted results.
Figure 7.

Summary of the CinDel therapeutic approach. DMD gene of a DMD patient has a deletion of exons 51, 52, and 53 compared to the wild-type dystrophin. This produces a reading frame shift when the DNA is translated into a mRNA that results into a stop codon in exon 54 and aborts transcription. When the exons 50 and 54 are cut by the CinDel treatment, a hybrid exon 50/54 is formed and the reading frame is restored, allowing the normal transcription of the mRNA.
Deletions of genomic DNA with the CRISPR system have already been shown to be very effective in other genes.45,50 We suggest to call Cindel, the CRISPR-Induced Deletion therapeutic approach. This therapeutic approach has some similarity with exon skipping. However, the exon skipping therapeutic approach has three disadvantages: (i) since the mRNA is targeted by the treatment, the oligonucleotides must be administered repeatedly during the lifetime of the patient; (ii) it removes complete exons, or (iii) it sometimes requires (depending of the patient deletion) skipping more than one exon to restore the reading frame. This last situation requires the simultaneous use of several different oligonucleotides, which increases the cost of treatment and also reduces its effectiveness.51,52 Based on our analysis of the positions of the cuts induced by the potential gRNAs targeting the different DMD exons, the CinDel approach should always require only two gRNAs for the most frequent deletions observed in DMD patients.
Our research group has initially made a proof-of-principle that it was possible to restore the reading frame of the DMD gene by inducing micro-insertion or micro-deletions (INDELs) with meganucleases.32 However, with that technology it was impossible to design a meganuclease targeting exactly the desired sequence. In collaboration with the Gersbach's group, we then showed that it was possible to induce INDELs with TALENs.34 Such a restoration of the reading frame with INDELs induced by TALENs was recently also done by the Li et al.33 However, the INDELs restored the reading frame by spontaneous nonhomologous end-joining in only one out-of-three DSB and formed a codon for an abnormal amino acid that may affect the dystrophin protein structure. The Gersbach's group also restored the DMD reading frame in the cells of DMD patients by deleting complete exons (rather than part of exons) using either zinc finger nucleases or the CRISPR system.31,37 With this approach, rather than with the CinDel approach, depending on the patient deletion, it is sometime necessary to delete two or more complete exons to restore the reading frame, but the normal spectrin-like repeat structure is not restored. Another group recently used the CRISPR system to delete exons 21–23 in mdx mouse and showed that the rescued dystrophin expression restored the sarcolemma integrity in muscle cells infected with an adenovirus coding for Cas9 and 2 gRNAs.53 However, adenovirus delivery works only in newborn mouse muscles. Finally, Li et al.33 restored the DMD gene by knock-in the missing exon following a DSB induced with TALENs or the CRISPR/Cas9. Although this approach seems ideal, the knock-in, which occurred only following homology-directed repair, was obtained only at a very low frequency. Thus, the muscle precursor cells would have to be genetically corrected in vitro, selected, expanded, and transplanted in every skeletal muscle of the patients. Our group has been working on myoblast transplantation since several years,54,55 and we know that this is a very labor-intensive therapy. On the contrary, the correction of the DMD reading frame with the CinDel therapy could be done directly in vivo. It would thus be a much easier therapeutic approach since it would not require culture and transplantation of hundred of millions of cells.
The CinDel approach, as well as any other gene correction approaches, will not restore the reading frame in all DMD gene copies of a muscle fiber. Indeed, several DMD genes will probably not be cut by Cas9 or be cut in only one of the two exons. However, our results indicated that when there is a deletion induced by cutting the gene in both exons, the reading frame is restored correctly in 64% of the cases in vivo. This should be sufficient to permit dystrophin expression on whole length of the muscle fibers. Indeed, the DMD gene has a nuclear domain of 549 microns (i.e., a single competent nucleus produces DYS protein that diffuses on about 549 µm of sarcolemma). This 549 µm segment of a human muscle fiber contains 25–35 nuclei.56,57,58 The restoration of the reading frame in the DMD gene of only 1 out of 30 nuclei (around 3.3% of the nuclei) would thus be enough to insure the presence of DYS protein on the complete sarcolemma and would thus be therapeutic.
The CinDel therapy could eventually be applied to all the deletions observed in the rod domain of various DMD patients by selecting the appropriate gRNAs targeting the exons that precede and follow the patient deletion. To apply this therapeutic approach to DMD patients there are several aspects that need further experimentation. First, it will be important to verify that the gRNAs used to restore the DMD reading frame are not also mutating other genes resulting in adverse effects. All the gRNAs listed in Table 1 have potential off-targets identified by various software programs and could thus in principal mutate other sites. However, the specificity of the SpCas9 nuclease has recently been improved by engineering its gene,59 and this does reduce the possibility of off-target mutations. Thus rather than trying to identify with a software the potential off-targets, the real off-target double-strand breaks will have to be identified by the new third-generation strategies based on high-throughput sequencing such as a integrase-deficient lentivirus vectors, BLESS or Guide-Seq.38,60,61 Second, for each type of exon deletion, there are several pairs of gRNAs (especially when using the S. aureus Cas9), which will be able to restore the expression of an internally deleted DYS protein. It will be necessary to determine which pair of gRNAs restores a correct spectrin-like repeat. This possibility does not exist when complete exons are removed by exon skipping or by deletion of complete exons with the CRISPR system as done by Gersbach's team.37 Third, the cutting of the gene with the CinDel approach requires the expression of Cas9. This protein should be expressed only transitionally to avoid increasing the mutation of off-target genes and an immune response against this foreign protein. Fourth, the best delivery method for in vivo modification of the DMD gene should be identified. The best vector seems to be an AAV vector, which permits the delivery of the Cas9 of S. aureus and of two gRNAs.62
For a patient with a deletion of exons 51–53, our results demonstrated that the pair of gRNA pairs 1–50 and 4–54. The exact dystrophin sequences targeted by these gRNAs are not present elsewhere in the human genome and therefore using the new engineered eSpCas9 off-target mutations may be rare.59 This pair of gRNAs induce a deletion that not only restored restore the reading frame of the dystrophin coding gene but also permit the production of a dystrophin protein that would have a correct structure since the spectrin-like repeat of helix C of repeat 19 will be fused with helix C of repeat 21. Our team is currently working on identifying the best pairs of gRNAs to correct the 14 most frequent deletion observed in DMD patients.
Materials and methods
Identification of targets and gRNA cloning. The plasmid pSpCas(BB)-2A-GFP (pX458), a gift from Feng Zhang (Addgene plasmid # 48138)63 contains two BbsI restriction sites necessary for insertion of a protospacer under the control of the U6 promoter was used in our study. The pSpCas(BB)-2A-GFP plasmid also contains the Cas9 of S. pyogenes, and eGFP genes under the control of the CBh promoter; both genes are separated by a sequence encoding the peptide T2A.
The nucleotide sequences targeted by the gRNAs in exons 50 and 54 were identified using the Leiden muscular dystrophy website http://www.dmd.nl/seqs/murefDMD.html by screening for PAM (i.e., NGG sequence for S. pyogenes Cas9) in the sense and antisense strands of each exon sequence (Figure 1). The gRNAs were assembled following the instruction on Zhang's website http://www.broadinstitute.org/history-leadership/scientific-leadership/coremembers/feng-zhang. Oligonucleotides coding for the target sequences and their complementary sequence were synthesized by Integrated DNA Technologies (Coralville, IA) and cloned into BbsI sites as protospacers leading to the production of 10 gRNAs targeting exon 50 and 14 gRNAs targeting exon 54. Briefly, the oligonucleotides were phosphorylated using T4 PNK (NEB, Ipwisch, MA) then annealed and cloned into the BbsI sites of the plasmid pSpCas(BB)-2A-GFP using the Quickligase (NEB, Ipwisch, MA). Following clone isolation and DNA amplification, samples were sent for sequencing using the primer U6F (5′-GTCGGAACAGGAGAGCGCACGAGGGAG) to the Genotyping Platform CHUL/CHUQ, and sequencing results were analyzed using the NCBI BLAST platform (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Cell culture
Transfection of the expression plasmid in 293T cells and in DMD patient myoblasts. The gRNA activities were tested individually or in pairs by transfection of the pSpCas(BB)-2A-GFP-gRNA plasmid encoding each gRNA in 293T cells and in DMD myoblasts having a deletion of exons 51 to 53. The 293T cells were grown in Dulbecco's modified Eagle medium (Invitrogen, Grand Island, NY) containing 10% fetal bovine serum (FBS) and antibiotics (penicillin 100 U/ml/streptomycin 100 µg/ml). DMD patient myoblasts and normal myoblasts from healthy donor (WT) were grown in MB1 medium (Hyclone; Thermo Scientific, Logan, UT) containing 15% FBS, without antibiotics. Cells in either 24-well or 6-well plates were transfected at 70–80% confluence using, respectively, 1 or 5 µg of plasmid DNA and 2 or 10 µl of lipofectamine 2000 (Invitrogen, Carlsbad, CA) previously diluted in Opti-Mem (Invitrogen, Grand Island, NY). For gRNA pair transfection, half of the DNA mixture was coming from the plasmid encoding the gRNA-50 and half from the gRNA-54. The cells were incubated at 37 °C in the presence of 5% CO2 for 48 hours. The transfection success was evaluated by the GFP expression in the transfected cells under microscopy with a Nikon TS 100 (Eclipse, Japan).
Myoblast transfection with lipofectamine 2000 following the previous standard protocol was not sufficiently effective and was improved as follows. The MB1 medium was aspirated before transfection and myoblasts were washed once with 500 µl of 1× Hanks Balanced Salt Solution (Invitrogen, Grand Island, NY). The complex lipofectamine 2000 plasmid DNA (diluted in Opti-Mem as above) was then poured directly on cells, instead of being in media, and the cells/DNA complex was incubated at 37 °C during 15 minutes. After this time, the antibiotic-free medium was added to the cells, and the plate was returned to the incubator for 18–24 hours. After that time, the medium was aspired and replaced with the fresh medium. The plate was incubated for another 24 hours.
Myoblast differentiation in myotubes and dystrophin expression. The DMD myoblasts were allowed to fuse in myotubes to induce the expression of dystrophin. To permit this myoblast fusion, the MB1 medium was replaced by the minimal Dulbecco's modified Eagle medium containing 2% FBS. Myoblasts were incubated at 37 °C in 5% CO2 for 7 days. Untransfected myoblasts (negative control) of the DMD patient and WT myoblasts from a healthy donor (positive control) were also grown under the same conditions to induce their differentiation in myotubes.
Genomic DNA extraction and analysis The genomic DNA was extracted from the 293T or myoblasts using a standard phenol–chloroform method 48 hours after transfection with the pSpCas(BB)-2A-GFP-gRNA plasmid(s). Briefly, the cell pellet was resuspended in 100 µl of lysis buffer containing 10% sarcosyl and 0.5 mol/l, pH 8, ethylene diamine tetra acetic acid. Twenty microliters of proteinase K (10 mg/ml) were added. The suspension was mixed by up down, incubated 10 minutes at 55 °C and centrifuged at 13,200 rpm for 2 minutes. The supernatant was collected in a new Eppendorf. One volume of phenol–chloroform was added and following centrifugation, the aqueous phase was recovered in a new Eppendorf and ethanol-precipitated with 1/10 volume of NaCl 5 mol/l and two volumes of 100% ethanol. The pellet was washed with 70% ethanol, centrifuged, and the DNA was resuspended in 50 µl of double-distilled water. The genomic DNA concentration was assayed with a Nanodrop (Thermo Scientific, Logan, UT).
Exons 50 and 54 and the hybrid exon 50–54 were amplified by PCR to confirm the successful individual cuts or deletions. For exon 50, the sense primer targeted the end of intron 49 (called sense 49 5′-TTCACCAAATGGATTAAGATGTTC) and the antisense primer targeted the start of intron 50 (called antisense 50 5′- ACTCCCCATATCCCGTTGTC). For exon 54, the forward and reverse primers targeted respectively the end of the intron 53 (called sense 53 5′- GTTTCAAGTGATGAGATAGCAAGT) and the start of intron 54 (called antisense 54 5′-TATCAGATAACAGGTAAGGCAGTG). For the hybrid exon 50–54, the forward sense 49 and reverse antisense 54 were used. All PCR amplifications were performed in a thermal cycler C1000 Touch of BIO RAD (Hercules, CA) with the Phusion high-fidelity polymerase (Thermo Scientific, Vilnius, Lithuania) using the following program for exon 50, exon 54 and the hybrid exon 50–54: 98 °C/10 seconds 58 °C/20 seconds, 72 °C/1 minute for 35 cycles.
The amplicons of individual exons 50 and 54 were used to perform the Surveyor assay. The first part of the test was the hybridization of amplicons using the slow program (denaturation at 95 °C followed by gradual cooling of the amplicons) with BIO RAD thermal cycler C1000Touch. Subsequently, the amplicons were digested with nuclease Cel (Integrated DNA Technologies, Coralville, IA) in the thermal cycler at 42 °C for 25 minutes. The digestion products were visualized on agarose gel 1.5%.
Cloning and sequencing of the hybrid exons. The amplicon of hybrid exons obtained by the amplification of genomic DNA extracted from 293T cells or myoblasts transfected with two different pSpCas(BB)-2A-GFP-gRNAs was purified by gel extraction (Thermo Scientific, Lithuania). The bands of about 480 to 655 bp were cloned into the linearized cloning vector pMiniT (NEB, Ipwisch, MA). On day 3, the plasmid DNA was extracted with the Miniprep Kit (Thermo Scientific, Lithuania), and the cloning vector was digested simultaneously with EcoRI and PstI to confirm the insertion of the amplicon. The insert was flanked by two EcoRI restriction sites in the cloning vector pMiniT. Digestion with EcoRI generated two fragments of 2,500 bp (plasmid without insert) and of 480 to 655 bp (amplicon inserted). It should be noted that there was a PstI restriction site in the remaining part of exon 54. Thus, a PstI digestion generated two fragments. The clones, which gave after double digestion with EcoRI and Pstl these two fragments, were sent for sequencing using primers provided by the manufacturer (NEB). Sequencing results were analyzed with the NCBI BLAST platform (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and the Expert Protein Analysis System platform (htt://www.expasy.org). This software allowed the visualization both the nucleotide sequences of the hybrid exon 50–54 and of the corresponding amino acid sequences.
Molecular modeling. Homology models of WT DMD gene corresponding to spectrin repeats R19, R21, R18–19 and R21–22 were obtained from the eDystrophin web site (http://edystrophin.genouest.org/).49 Primary sequence alignment was realized using the TM-Align64 (http://zhanglab.ccmb.med.umich.edu/TM-align/) web server based on the structural alignment of R19 and R21 homology models. Homology models were realized using the iTasser web server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/)65,66,67,68 using primary sequences of hybrid exon for deletion of exons 51–53 and hybrid exons 2–50/2–54 and 1–50/4–54.
Proteins analysis. Myotubes were harvested and proteins were extracted with the methanol–chloroform method. Briefly, cell pellets were resuspended in lysis buffer containing 75 mmol/l Tris–HCl, pH 7.4, 1 mmol/l dithiotreitol, 1 m mmol/l phenylmethylsulfonyl fluoride, and 1% sodium dodecyl sulfate. Protein extracts were dried with the speed vacuum Univapo 100 ECH (Uniequip, Martinsried, Germany) to remove all traces of methanol. Samples were then diluted in a buffer containing 0.5% mercaptoethanol and heated at 95 °C for 5 minutes. The protein concentrations were assayed by Amido Black using Imager2200 AlphaDigiDoc (Alpha Innotech, Fisher Scientific, Suwanee, GA).
Seventy-five micrograms of protein of each sample were separated on a 7% polyacrylamide gel and transferred onto nitrocellulose membrane at 4 °C for 16 hours. In order to detect dystrophin on the membrane, a primary mouse monoclonal antibody (cat# NCL-DYS2; Leica Biosystems, Newcastle, UK) recognizing the C-terminus of the human dystrophin was used. The antibody was diluted 1:25 in 0.1× PBS containing 5% milk and 0.05% Tween-20 and incubated at 4 °C for 16 hours.
SUPPLEMENTARY MATERIAL Figure S1. CinDel correction is effective in vivo in the hDMD/mdx mouse model. Figure S2. Primary sequence DMD exons 49 to 54 and spectrin-like repeat positions. Table S1. Expected results of cutting exons 50 and 54 with various gRNA pairs.
Acknowledgments
This work was supported by grants from the Canadian Institute of Health Research (CIHR). J.P.I.E. has a studentship from the Canadian Francophonie Scholarship Program (CFSP), financed by Foreign Affairs, Trade and Development Canada (FATDC).
The authors declare no conflict of interest related to this work.
Supplementary Material
References
- Engel, A, and Banker, BQ (1986). Myology: Basic and Clinical. McGraw-Hill: New York. [Google Scholar]
- Rybakova, IN, Patel, JR and Ervasti, JM (2000). The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol 150: 1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, EP, Brown, RH Jr and Kunkel, LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928. [DOI] [PubMed] [Google Scholar]
- Hoffman, EP, Brown, RH and Kunkel, LM (1992). Dystrophin: the protein product of the Duchene muscular dystrophy locus. 1987. Biotechnology 24: 457–466. [PubMed] [Google Scholar]
- Bladen, CL, Salgado, D, Monges, S, Foncuberta, ME, Kekou, K, Kosma, K et al. (2015). The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36: 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, M, Beggs, AH, Moyer, M, Scherpf, S, Heindrich, K, Bettecken, T et al. (1989). The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 45: 498–506. [PMC free article] [PubMed] [Google Scholar]
- Hoffman, EP (1993). Genotype/phenotype correlations in Duchenne/Becker dystrophy. Mol Cell Biol Hum Dis Ser 3: 12–36. [DOI] [PubMed] [Google Scholar]
- Emery, AE (2002). The muscular dystrophies. Lancet 359: 687–695. [DOI] [PubMed] [Google Scholar]
- Duan, D (2011). Duchenne muscular dystrophy gene therapy: lost in translation? Res Rep Biol 2011: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyenvalle, A, Seto, JT, Davies, KE and Chamberlain, J (2011). Therapeutic approaches to muscular dystrophy. Hum Mol Genet 20: R69–R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny, P, Swiderski, K and Chamberlain, JS (2013). Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve 47: 649–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell, JR, Rodino-Klapac, L, Sahenk, Z, Malik, V, Kaspar, BK, Walker, CM et al. (2012). Gene therapy for muscular dystrophy: lessons learned and path forward. Neurosci Lett 527: 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaart, IE and Aartsma-Rus, A (2012). Gene therapy for Duchenne muscular dystrophy. Curr Opin Neurol 25: 588–596. [DOI] [PubMed] [Google Scholar]
- Monaco, AP, Neve, RL, Colletti-Feener, C, Bertelson, CJ, Kurnit, DM and Kunkel, LM (1986). Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 323: 646–650. [DOI] [PubMed] [Google Scholar]
- Kunkel, LM, Hejtmancik, JF, Caskey, CT, Speer, A, Monaco, AP, Middlesworth, W et al. (1986). Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature 322: 73–77. [DOI] [PubMed] [Google Scholar]
- Gregorevic, P, Blankinship, MJ, Allen, JM, Crawford, RW, Meuse, L, Miller, DG et al. (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med 10: 828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic, P, Allen, JM, Minami, E, Blankinship, MJ, Haraguchi, M, Meuse, L et al. (2006). rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 12: 787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z, Kuhr, CS, Allen, JM, Blankinship, M, Gregorevic, P, Chamberlain, JS et al. (2007). Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol Ther 15: 1160–1166. [DOI] [PubMed] [Google Scholar]
- Qiao, C, Koo, T, Li, J, Xiao, X and Dickson, JG (2011). Gene therapy in skeletal muscle mediated by adeno-associated virus vectors. Methods Mol Biol 807: 119–140. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus, A (2012). Overview on DMD exon skipping. Methods Mol Biol 867: 97–116. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus, A and van Ommen, GJ (2007). Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA 13: 1609–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus, A and van Ommen, GJ (2009). Less is more: therapeutic exon skipping for Duchenne muscular dystrophy. Lancet Neurol 8: 873–875. [DOI] [PubMed] [Google Scholar]
- Bremmer-Bout, M, Aartsma-Rus, A, de Meijer, EJ, Kaman, WE, Janson, AA, Vossen, RH et al. (2004). Targeted exon skipping in transgenic hDMD mice: a model for direct preclinical screening of human-specific antisense oligonucleotides. Mol Ther 10: 232–240. [DOI] [PubMed] [Google Scholar]
- Dunckley, MG, Manoharan, M, Villiet, P, Eperon, IC and Dickson, G (1998). Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum Mol Genet 7: 1083–1090. [DOI] [PubMed] [Google Scholar]
- Lu, QL, Mann, CJ, Lou, F, Bou-Gharios, G, Morris, GE, Xue, SA et al. (2003). Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med 9: 1009–1014. [DOI] [PubMed] [Google Scholar]
- Mann, CJ, Honeyman, K, McClorey, G, Fletcher, S and Wilton, SD (2002). Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med 4: 644–654. [DOI] [PubMed] [Google Scholar]
- Takeshima, Y, Yagi, M, Wada, H, Ishibashi, K, Nishiyama, A, Kakumoto, M et al. (2006). Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr Res 59: 690–694. [DOI] [PubMed] [Google Scholar]
- van Deutekom, JC, Janson, AA, Ginjaar, IB, Frankhuizen, WS, Aartsma-Rus, A, Bremmer-Bout, M et al. (2007). Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 357: 2677–2686. [DOI] [PubMed] [Google Scholar]
- Kinali, M, Arechavala-Gomeza, V, Feng, L, Cirak, S, Hunt, D, Adkin, C et al. (2009). Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol 8: 918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, QL, Cirak, S and Partridge, T (2014). What can we learn from clinical trials of exon skipping for DMD? Mol Ther Nucleic Acids 3: e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout, DG, Kabadi, AM, Thakore, PI, Perez-Pinera, P, Brown, MT, Majoros, WH et al. (2015). Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther 23: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau, J, Chapdelaine, P, Boisvert, S, Almeida, LP, Corbeil, J, Montpetit, A et al. (2011). Endonucleases: tools to correct the dystrophin gene. J Gene Med 13: 522–537. [DOI] [PubMed] [Google Scholar]
- Li, HL, Fujimoto, N, Sasakawa, N, Shirai, S, Ohkame, T, Sakuma, T et al. (2015). Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports 4: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout, DG, Perez-Pinera, P, Thakore, PI, Kabadi, AM, Brown, MT, Qin, X et al. (2013). Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther 21: 1718–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, C, McAnally, JR, Shelton, JM, Mireault, AA, Bassel-Duby, R and Olson, EN (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345: 1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, K, Fujii, W, Tsuboi, M, Tanihata, J, Teramoto, N, Takeuchi, S et al. (2014). Generation of muscular dystrophy model rats with a CRISPR/Cas system. Sci Rep 4: 5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousterout, DG, Kabadi, AM, Thakore, PI, Majoros, WH, Reddy, TE and Gersbach, CA (2015). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 6: 6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Cong, L, Yan, WX, Scott, DA, Gootenberg, JS, Kriz, AJ et al. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature 520: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L, Ran, FA, Cox, D, Lin, S, Barretto, R, Habib, N et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M, East, A, Cheng, A, Lin, S, Ma, E and Doudna, J (2013). RNA-programmed genome editing in human cells. Elife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, JD and Joung, JK (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P, Yang, L, Esvelt, KM, Aach, J, Guell, M, DiCarlo, JE et al. (2013). RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, SW, Kim, S, Kim, JM, and Kim, JS (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232. [DOI] [PubMed] [Google Scholar]
- Doudna, JA and Charpentier, E (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. [DOI] [PubMed] [Google Scholar]
- Zheng, Q, Cai, X, Tan, MH, Schaffert, S, Arnold, CP, Gong, X et al. (2014). Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 57: 115–124. [DOI] [PubMed] [Google Scholar]
- Deltcheva, E, Chylinski, K, Sharma, CM, Gonzales, K, Chao, Y, Pirzada, ZA et al. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471: 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini, LA and Sontheimer, EJ (2010). CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11: 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M, Chylinski, K, Fonfara, I, Hauer, M, Doudna, JA and Charpentier, E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas, A, Lucchetti-Miganeh, C, Yaou, RB, Kaplan, JC, Chelly, J, Leturcq, F et al. (2012). Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J Rare Dis 7: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver, MC, Bauer, DE, Dass, A, Yien, YY, Chung, J, Masuda, T et al. (2014). Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem 289: 21312–21324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus, A, Kaman, WE, Weij, R, den Dunnen, JT, van Ommen, GJ and van Deutekom, JC (2006). Exploring the frontiers of therapeutic exon skipping for Duchenne muscular dystrophy by double targeting within one or multiple exons. Mol Ther 14: 401–407. [DOI] [PubMed] [Google Scholar]
- Béroud, C, Tuffery-Giraud, S, Matsuo, M, Hamroun, D, Humbertclaude, V, Monnier, N et al. (2007). Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum Mutat 28: 196–202. [DOI] [PubMed] [Google Scholar]
- Xu, L, Park, KH, Zhao, L, Xu, J, El Refaey, M, Gao, Y, et al. (2015). CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Skuk, D and Tremblay, JP (2014). Clarifying misconceptions about myoblast transplantation in myology. Mol Ther 22: 897–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuk, D and Tremblay, JP (2011). Intramuscular cell transplantation as a potential treatment of myopathies: clinical and preclinical relevant data. Expert Opin Biol Ther 11: 359–374. [DOI] [PubMed] [Google Scholar]
- Bruusgaard, JC, Liestøl, K, Ekmark, M, Kollstad, K and Gundersen, K (2003). Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo. J Physiol 551: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, I, Vilquin, JT, Asselin, I, Chamberlain, J and Tremblay, JP (1998). Transplantation of myoblasts from a transgenic mouse overexpressing dystrophin prduced only a relatively small increase of dystrophin-positive membrane. Muscle Nerve 21: 91–103. [DOI] [PubMed] [Google Scholar]
- Pavlath, GK, Rich, K, Webster, SG and Blau, HM (1989). Localization of muscle gene products in nuclear domains. Nature 337: 570–573. [DOI] [PubMed] [Google Scholar]
- Slaymaker, IM, Gao, L, Zetsche, B, Scott, DA, Yan, WX and Zhang, F (2015). Rationally engineered Cas9 nucleases with improved specificity. Science (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Tsai, SQ, Zheng, Z, Nguyen, NT, Liebers, M, Topkar, VV, Thapar, V et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X, Wang, Y, Wu, X, Wang, J, Wang, Y, Qiu, Z et al. (2015). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol 33: 175–178. [DOI] [PubMed] [Google Scholar]
- Friedland, AE, Baral, R, Singhal, P, Loveluck, K, Shen, S, Sanchez, M et al. (2015). Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 16: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, FA, Hsu, PD, Wright, J, Agarwala, V, Scott, DA and Zhang, F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y and Skolnick, J (2005). TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33: 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J, Yan, R, Roy, A, Xu, D, Poisson, J and Zhang, Y (2015). The I-TASSER Suite: protein structure and function prediction. Nat Methods 12: 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J and Zhang, Y (2015). I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43: W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, A, Kucukural, A and Zhang, Y (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.