

Abstract
Ligand shedding has gained increased attention as a major posttranslational modification mechanism used by cells to respond to diverse environmental conditions. The TACEadam17 protease is a critical mediator of such ligand shedding, regulating the maturation and release of an impressive range of extracellular substrates that drive diverse cellular responses. Exactly how this protease is itself activated remains unclear, in part due to the lack of available tools to measure TACE activity with temporal and spatial resolution in live cells. We have developed a FRET based biosensor for TACE activity (TSen), which is capable of reporting TACE activation kinetics in live cells with a high degree of specificity. TSen was used in combination with chemical biology to probe the dependence of various means of TACE activation on p38 and Erk kinase activities, as well as to identify a novel connection between actin cytoskeletal disruption and TACE activation. Such cytoskeletal disruption leads to rapid and robust TACE activation in some cell types and accumulation of TACE at the plasma membrane, allowing for increased cleavage of endogenous substrates. Our study highlights both the versatility of TSen as a tool to understand the mechanisms of TACE activation in live cells and the importance of actin cytoskeletal integrity as a modulator of TACE activity.
Introduction
The Tumor Necrosis Factor-α Converting Enzyme (TACE), encoded by the adam17 gene, is a trans-membrane protease that has been implicated in numerous physiological processes, including inflammation (1, 2), wound healing (3), development (4), and cancer progression (5, 6). In these diverse processes, TACE plays a common role as an extracellular sheddase that cleaves the pro-/transmembrane form of a wide variety of ligands and receptors (6). For example, TACE activity mediates auto/paracrine release of TNFα during immune response through the cleavage of pro-TNFα and release of soluble TNFα, which binds to and activates the TNF receptor (2). Similarly, TACE activity mediates auto/paracrine release of TGFα and amphiregulin (7), both of which are Epidermal Growth Factor Receptor (EGFR1) ligands that regulate cellular motility during development, wound healing, and metastasis (8–10). Interestingly, EGFR1 is also cleaved by TACE, allowing for complex feedback mechanisms in the regulation of cellular motility (11). Although ligand shedding has gained increased attention as a major posttranslational modification mechanism and significant research has been conducted in an effort to understand the consequences of auto/paracrine release of TACE substrates, relatively less is known about how TACE activity is itself regulated. Several lines of evidence suggest that TACE activity is spatially and temporally regulated within a cell or a tissue (12–14). TACE activation has been proposed to require the proteolytic cleavage of the auto inhibited pro-TACE form (15), via the protease activity of Furin (6), however, Furin deficient cells have displayed a clear ability to mature and activate TACE (16). Thus, the mechanism of proteolytic activation of TACE still remains unclear. It has been suggested that the trafficking of TACE to the plasma membrane is the primary means of TACE regulation in cells. Direct phosphorylation of TACE at Thr735 by either p38 or Erk has been proposed to facilitate trafficking of TACE to the plasma membrane, and subsequent extracellular activity (17–21). Other studies point to the importance of ER exit due to the action of iRhom2 in the activation of TACE (13). Additionally, pathophysiological mutants of Src have been shown to regulate plasma membrane display of TACE, and there is some evidence that wild type Src may also mediate this process (22, 23). Clearly, there are many outstanding questions in the field that aim to answer how the TACE protease is able to cleave 76 substrates in a cell-type dependent manner (6) and mediate various diverse physiological and pathophysiological processes. The current mechanistic understanding of TACE regulation is hampered by a lack of effective tools to measure the spatiotemporal regulation of TACE activity in live cells. In this study, we demonstrate the use of a novel FRET biosensor for TACE activity (TSen) and how its use in live cells reveals that rapid TACE activation can be achieved not only through p38 and Erk kinase activation, but also through changes in the subcellular trafficking of TACE. TSen allowed us to identify novel chemical means to both activate and repress TACE activity, which led to the discovery that chemical damage to the actin cytoskeleton causes accumulation of TACE at the plasma membrane and subsequent enhanced cellular TACE activity. Our data clearly show that TACE subcellular trafficking and p38 and/or Erk kinase activity can independently contribute to TACE activation. Our study provides insight into how individual cells respond to cytoskeletal damage by executing elevated EGFR1 ligand shedding, as well as into how groups of cells execute sustained motility during wound healing.
Results
TSen Design
Using a chimeric protein design approach, we have developed a novel genetically encoded FRET biosensor (TSen) for measuring TACE activity in live cells by fluorescence microscopy. As displayed schematically in Fig. 1A, our sensor employs an N-terminal leader sequence derived from TGFα, a TNFα cleavage site flanked by linker regions, a PDGF transmembrane domain, an eCFP FRET donor, and a YPET FRET acceptor in a similar manner as to what has been employed for the MMP14 FRET biosensor (24). A key difference between TSen and other protease cleavage biosensor designs is that two tandem valines comprise the immediate C-terminal end of the sensor, which mimic the C-terminal valines that have been shown to be required for TGFα maturation in live cells (25). The use of a TNFα cleavage site in TSen is the result of extensive research that has identified a small peptide region within TNFα as an efficient specific substrate of TACE over other metalloproteases (26, 27). Although TSen employs a TACE specific cleavage site derived from TNFα, it differs from the TNFα and TGFα proteins in the distance between the cleavage site and the transmembrane domain along the polypeptide chain (approximately 245 aa, 18 aa, and 7 aa, respectively). However, when in TSen, the TNFα cleavage site is separated from the PDGF transmembrane domain with an eCFP domain, whose N-terminus and C-terminus are positioned within 22 ± 1.8 angstroms (corresponding to a distance of approximately 6 aa) of each other (according to the eCFP PDB structure 2WSN (28)). Thus, although the TSen TCS is fairly distant from the transmembrane domain in the primary structure, these two domains are likely positioned near each other in the tertiary structure. Additionally, TSen differs from TNFα in the type of transmembrane protein it is (Type I versus Type II, respectively). Since TGFα is a Type I transmembrane protein, it does not appear that TACE takes into account the type of transmembrane protein as a parameter for selectivity. Despite the highly chimeric structure of TSen relative to the structures of endogenous TACE substrates, our study confirms that this chimeric nature does not interfere with TSen being a highly suitable substrate for TACE (discussed below).
Fig. 1. TSen Efficiently Measures TACE Catalytic Activity in Live Cells.

(a) A schematic diagram of the TSen FRET biosensor for TACE activity and the non-cleavable control sensor (NCS). (b) Activation of TACE in HelaS3 cells expressing either TSen or NCS in response to 1 hour PMA treatment. (c) Secretion of YFP by HelaS3 cells expressing either TSen or NCS in response to 3 hour PMA treatment. (d) Confocal microscopy and TACE activation in HeLaS3-TSen cell over 1 hr of PMA stimulation. (e) Activation of TACE in WT MEF-TSen cells after 1 hour PMA treatment. (f) Subcellular fractionation and western blot analysis of Pro-TGFα levels at the plasma membrane and TACE activation measured via TSen in microscopy throughout 1 hr of PMA treatment in MEF-TSen cells (g) Profiles of MEF-TSen cells to PMA titration after 1 hour for both Pro-TGFα levels at the plasma membrane and TACE activity measurements via TSen. Note that y-axes minimums are not always set to a value of 0. For microscopy experiments, N=3 trials, where each trial represents>500 cells. Brackets indicate relevant comparisons where p<0.01, t-test. All experiments are representative of at least 2 independent experiments.
TSen Measures TACE activity in Live Cells
When stably expressed in HeLa cells, TSen displays rapid enhanced activity (elevated inverse FRET Ratio, or CFP/FRET) in response to phorbol 12-myristate 13-acetate (PMA) stimulation, which is known to activate TACE (18) (Figs. 1B and S1A). This PMA dependent activity is blocked by BMS-561392 or GM6001 treatment, which are a TACE, MMP3, MMP12 and ADAMTS4 specific inhibitor (TACE IC50=122 nM) (29, 30) and a non-specific MMP inhibitor, respectively. Identical results were found in 293T-TSen cells (Fig. S1B). To determine whether PMA dependent TACE activity is contingent on the specific sequence of the TNFα cleavage site (TCS), we constructed a mutant sensor, named the non-cleavable sensor (NCS), where the TCS no longer resembles an ideal TACE substrate (Fig. 1 A). The NCS sequence is predicted to be a poor TACE substrate according to previous studies (31). When HeLa cells expressing NCS are compared to Hela-TSen cells, PMA dependent activation is not observed (Fig. 1B), validating that the TNFα cleavage site is the primary feature on the TACE sensor that is required for PMA dependent activation. Since TSen was designed to display a reduction in FRET efficiency through proteolytic cleavage, we additionally measured the YFP and CFP release from HeLaS3-TSen cells after a 3 hour period of PMA stimulation, in the presence and absence of either BMS-561392 or GM6001, in order to validate that cleavage is indeed occurring. PMA enhances YFP, but not CFP, release into the media, which can be inhibited by either type of pharmacological TACE inhibitor (Fig. 1C). When the same experiment is conducted in HelaS3-NCS cells, PMA stimulated YFP release is reduced greatly, but not completely absent (Fig. 1C). This residual PMA dependent activity in the NCS sensor is likely still dependent on TACE, as both GM6001 and BMS-561392 can inhibit it. As a result, although the majority of TACE activity measured by TSen is dependent on TACE, a small portion of activity that is detected through YFP release, but not in microscopy experiments, can be attributed to cleavage between CFP and YFP at a cryptic site independent of the TCS. These YFP secretion experiments, in conjunction with the previous microscopy experiments, demonstrate the versatile use of TSen. TSen can be used in either a secretion based fluorescent spectroscopy assay, in a similar manner to the available TACE bioassays (32), or in a FRET based live cell microscopy assay.
The use of the TSen sensor in live cells allows for the determination of the intracellular location at which TACE activation is occurring. Using confocal microscopy, we repeated our experiments with HeLaS3-TSen cells in the presence of PMA, in order to determine whether cleavage of the TSen sensor was occurring in intracellular vesicles or at the plasma membrane. We find that TSen cleavage occurs to a large degree at the plasma membrane, and to a far lesser degree in intracellular vesicles under these conditions (Figs. 1D). Although this data does not elucidate where TACE itself is activated within a cell, or where endogenous substrates may be cleaved by TACE in a cell, it does highlight how the TSen sensor works primarily as a plasma membrane TACE activity sensor in cells.
TSen Reports TACE Activity with Similar Dose and Kinetic Profiles to Plasma Membrane Pro-TGFα Cleavage
Mouse embryonic fibroblasts (MEFs) have been used in previous research to study TACE dependent cellular responses, where TACE mediates cleavage of EGFR1 ligands (4). MEF cells stably expressing TSen respond to PMA stimulation with TACE activation in a similar manner to both HeLa-TSen and 293T-TSen cells (Figs 1E, 1B and S1B, respectively). Using MEF-TSen cells, we aimed to determine whether the time and dose profiles of TACE activity in response to PMA were similar between TSen measured activation of TACE and endogenous TGFα cleavage reported by western blot analysis. Although we could not detect cleavage of endogenous Pro-TGFα in whole cell lysate (Fig. S1C), we could detect the disappearance of Pro-TGFα at the plasma membrane, using subcelluar fractionation, in a time dependent manner (Fig. 1F). The time dependent and dose dependent amount of Pro-TGFα at the plasma membrane closely resembles TACE activity as reported by TSen in fluorescent microscopy experiments (Figs. 1F and 1G, respectively). As a result, not only have we confirmed that TSen is a reporter for TACE activity in our various experimental model systems, but also that TSen is a faithful reporter of Pro-TGFα cleavage at the plasma membrane.
TSen Reports TACE Activity with High Specificity
In order to determine the specificity of TSen for measuring TACE activity, shRNA was stably expressed in HeLaS3-TSen cells to show that depletion of endogenous TACE (Fig. 2A) was capable of inhibiting PMA dependent TACE activity (Fig. 2B). Similarly, PMA stimulation of TACE −/− MEF cells expressing TSen leads to no measurable reported TACE activity, in contrast to WT MEF-TSen cells, which display a significant increase in inverse FRET ratio in response to PMA (Fig. 2C). Previous studies show that TNFα can be cleaved not only by TACE, but also by the PR3 protease (33). Additionally, the peptidomimetic gelatinase B inhibitor Regasepin1 is capable of inhibiting not only gelatinase B, but also MMP-8 and TACE to the same degree, suggesting that these proteases share overlapping substrate specificity (34). As a result, we further investigated whether depletion of either PR3 or MMP-8 by RNAi in HeLa-TSen cells (Fig. 2D) was capable of influencing basal or PMA stimulated inverse FRET ratio measurements. Depletion of either protease in these cells is insufficient to alter either basal or PMA stimulated inverse FRET ratio of TSen (Fig. 2E). Taken together, these series of experiments provide compelling evidence that TSen is a biosensor that faithfully reports TACE activity with specificity for TACE over other similar proteases.
Fig. 2. TSen Reports TACE activity with High Specificity.

(a) mRNA levels of TACE in HeLaS3-TSen upon depletion of TACE by RNAi. (b) Activation of TACE in TACE knockdown HelaS3-TSen cells after 1 hour PMA treatment. (c) Activation of TACE in WT and TACE −/− MEF-TSen cells after 1 hour PMA treatment. Inset displays a western blot analysis of TACE protein levels in TACE −/− cells. (d) mRNA levels of PR3 or MMP8 in HeLaS3-TSen upon depletion of PR3 or MMP8 by RNAi. (e) Activation of TACE in PR3 or MMP8 knockdown HelaS3-TSen cells after 1 hour PMA treatment. Note that y-axes minimums are not always set to a value of 0. For microscopy experiments, N=3 trials, where each trial represents>500 cells. Brackets indicate relevant comparisons where p<0.01, t-test, if not labeled with NS (p>0.01). All experiments are representative of at least 2 independent experiments.
TACE Activation by EGFR1 Stimulation is p38 and Erk Dependent
The ability of TACE to mediate proteolytic activation of several EGFR1 ligands is well represented in previous reports (2, 7). We determined that PMA dependent TACE activation measured through TSen depends partially upon both p38 and Erk kinases (Fig. 3A), which is consistent with findings in other studies (17, 18). Since p38 and Erk kinases are both activated upon EGFR1 stimulation and infrequent reports display clear evidence that EGFR1 stimulation can enhance TACE activation (35), we aimed to determine how widespread the phenomenon of EGFR1 activation of TACE was in several established cell lines. To this end, we created three additional stable cell lines expressing TSen (HaCaT keratinocytes, SCC13 squamous carcinoma, and VMCUB1 bladder cancer cells) and tested their responses to EGF. Although TACE does not become activated by exogenous EGF in HeLaS3-TSen cells (Fig. S2A), EGF efficiently activates TACE in three other epithelial cell lines (HaCaT, SCC13 and VMCUB1) (Figs. 3B,C and D, respectively). Interestingly, the kinetics of activation of TACE is faster in PMA stimulated cells (1 hour to saturation, Fig. S1A), compared to EGF stimulated cells (3 hours to saturation, Fig. S2B). EGF dependent TACE activation can be fully blocked in all cell lines by pharmacological inhibition of EGFR1 using Gefitinib, which is consistent with the theory that the activation of TACE by EGF occurs through EGFR1. As expected, the TACE inhibitor BMS-561392 diminishes both the basal and EGF dependent TACE activation, revealing that only a portion of the total TACE activity in all three of these cell types is dependent on EGFR1. Additionally, the effects of MEK1 or p38 inhibitors consistently show the importance of both of these kinases in EGF dependent TACE activation. Thus, EGF dependent activation of TACE in these three cell lines is similar to that of PMA dependent activation of TACE in both HeLaS3 and 293T cells (Figs. 1E and S1B), where both chemical and biochemical means of TACE activation are p38 and Erk dependent.
Fig. 3. Erk and P38 Mediate PMA and EGF Dependent Activation of TACE.
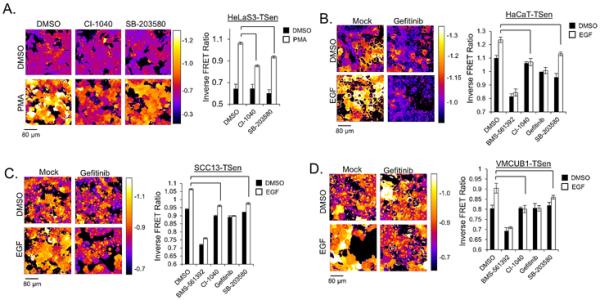
(a) Activation of TACE in HelaS3-TSen cells after 1 hour PMA treatment in the presence and absence of MEK and p38 inhibitors. (b) Activation of TACE in HaCaT-TSen cells after 3 hour EGF treatment in the presence and absence of MEK and p38 inhibitors. (c) Activation of TACE in SCC13-TSen cells after 3 hour EGF treatment in the presence and absence of MEK and p38 inhibitors. (d). Activation of TACE in VMCUB1-TSen cells after 3 hour EGF treatment in the presence and absence of MEK and p38 inhibitors. Note that y-axes minimums are not always set to a value of 0. For microscopy experiments, N=3 trials, where each trial represents>500 cells. Brackets indicate relevant comparisons where p<0.01, t-test. All experiments are representative of at least 2 independent experiments.
Chemical Screening Reveals Novel Activators and Inhibitors of TACE
In order to contribute to the understanding of cellular regulation of TACE, we conducted an unbiased screen of small molecules capable of regulating TACE using HaCaT-TSen cells. We screened 81 compounds targeting diverse cellular functions (Table S1), which represent a library of small molecules that have been either widely studied and used in research investigations, or have been clinically approved by the FDA. Thus, each compound has a known mechanism of action. Our screen revealed two candidate compounds that activate TACE (Cytochalasin D and Quinacrine) and two candidate compounds that repress TACE (Doxorubicin and Sunitinib) (Fig. 4A). All four candidates were validated in independent titration experiments (Figs. 4B, S3A, S3B, and S3C). Doxorubicin (DoxR) is a known DNA damage agent that can activate an apoptosis response in cells via activation of caspase 8, a known protease capable of activating TACE (36). Although we observe DoxR to be an inhibitor of TACE activity, we investigated whether this chemical effect on cells was independent of DoxR's known role in apoptosis. We found that DoxR cannot activate apoptosis as reported by caspase-3 cleavage under the experimental conditions used, but can cause accumulation of pro-TGFα in whole cells lysates, supporting the claim that DoxR inhibits TACE (Fig. S3D). We believe that this inability of DoxR to activate apoptosis in these cells is largely due to the high density (1200 cells/ mm2) at which cells were screened, which leads to density dependent inhibition of cellular proliferation (Fig. S3E) and presumably intercalating agent dependent DNA damage.
Fig. 4. Actin Depolymerization Activates TACE.

(a) An unbiased chemical library screen in HaCaT-TSen cells for activators and repressors of TACE. (b) Dose response to CytoD after 1 hour treatment in HaCaT-TSen cells. (c) Activation of TACE in HaCaT-TSen cells after 1 hour CytoD and LatB treatment. (d) Activation of TACE in SCC13-TSen cells after 1 hour CytoD and LatB treatment. (e) Activation of TACE in VMCUB1-TSen cells after 1 hour CytoD and LatB treatment. (f) Activation of TACE in MEF-TSen cells after 1 hour CytoD treatment. (g) Activation of TACE in MEF-TSen cells and TACE −/− MEF-TSen cells after 1 hour CytoD treatment. (h) Secretion of YFP by HaCaT-TSen cells in response to 1 hour CytoD treatment. (i) Activation of Erk in HaCaT-EKAR cells after 1 hour CytoD and LatB treatment. (j) Subcellular fractionation and western blot analysis of plasma membrane levels of TACE, Pro-TGFα, and TSen after 1 hour treatment of either PMA or CytoD in MEF-TSen cells. Note that y-axes minimums are not always set to a value of 0. For microscopy experiments, N=3 trials, where each trial represents>500 cells. Brackets indicate relevant comparisons where p<0.01 t-test, if not labeled with NS (p>0.01). All experiments are representative of at least 2 independent experiments.
Actin-Cytoskeleton Dependent Activation of TACE Relies on Accumulation of TACE at the Plasma Membrane
Since Cytochalasin D (CytoD) emerged as the strongest potential activator of TACE, we further validated the possible connection between actin cytoskeletal perturbation and TACE activity. We conducted a dose response experiment in HaCaT-TSen cells using Latrunculin B (LatB), which is an actin depolymerizing agent with a different mechanism of action as compared to CytoD, and found that LatB has a similar ability to elevate inverse FRET ratio levels (Figs. S3F, 4C, D, E, and F, respectively), with kinetics that are far faster than that of EGF (~30 min versus ~120 min, respectively) (Figs. S3G and S2B, respectively). Next, we confirmed that the drugs were capable of affecting the actin cytoskeletal structure at their respective EC50 concentrations by fixing HaCaT cells in the presence and absence of either CytoD or LatB and staining for F-actin using phalloidin-Rhodamine B (Fig. S3H). Furthermore, actin depolymerization induced elevation of inverse FRET ratio in TSen cells does not require Erk or p38 activities in HaCaT, and SCC-13 cells (Figs. 4C and D), in contrast to such dependence in VMCUB1 and MEF cells (Fig. 4E and F). Although this Erk and p38 dependence is cell type specific, the fact that actin disruption can occur completely independent of these kinase activities suggests that actin disruption is capable of regulating TACE activity through some mechanism that is distinct from TACE phosphorylation.
Three lines of evidence suggest that actin cytoskeletal disruption does indeed cause endogenous activation of TACE. First, CytoD dependent activation of inverse FRET ratio in MEF-TSen cells is completely blocked upon deletion of the adam17 gene (Fig. 4G). Second, upon measuring YFP release into the media, we found that actin depolymerizing drugs efficiently lead to YFP secretion, which is blocked by pharmacological inhibition of TACE with BMS-561392 (Fig. 4H). Third, by stably expressing a well-known Erk FRET biosensor, the EKAR sensor (37), in HaCaT, SCC-13 and VMCUB1 cells, we aimed to measure whether actin depolymerizing agents were capable of stimulating EGFR1 receptor activity, as TACE has been shown to regulate the maturation of several key EGFR1 ligands (3). Indeed, actin depolymerizing agents can efficiently activate EGFR1 dependent Erk activity in HaCaT-EKAR, SCC-13-EKAR and VMCUB1-EKAR cells, which all display elevation of FRET ratio in response to CytoD and LatB (Figs. 4I, S3I, and S3J). This enhanced Erk activity is almost completely blocked by either pre-treatment of BMS-561392 or the EGFR1 inhibitor, Gefitinib, in all three cell lines. These experiments provide compelling evidence that TSen faithfully reports that actin cytoskeletal disruption leads to TACE activation.
Since cytoskeletal disruption is variably driven by p38 and Erk kinase activities across the many cell lines investigated, we sought to explain how actin poisoning drugs were capable of activating TACE independent of p38 and Erk. The actin cytoskeleton is important for a large variety of cellular processes, including endocytosis via clatherin coated pits (38). As a result, we used subcellular fractionation by differential centrifugation to separate the plasma membrane (PM) in cell lysates from the vesicle and soluble components of the cytosol, using MEF cells. By comparing the plasma membrane fraction to the cytosol or vesicle fraction, we found that although PMA stimulation does not affect PM levels of TACE, CytoD treatment leads to a large degree of PM accumulation of TACE (Fig. 4J). However, when the same comparison is done for pro-TGFα, we clearly see that both PMA and CytoD promote decreased pro-TGFα at the PM, where CytoD elicits a greater magnitude change than PMA, which is consistent with fluorescence microscopy experiments (Figs. 4F and 2C, respectively). Upon both PMA and CytoD treatments, TSen levels at the PM remained unchanged. The accumulation of TACE at the plasma membrane is likely to be independent of p38 and Erk kinase activities, as PMA dependent activation of TACE consistently relies upon these kinase activities, but does not lead to an accumulation of TACE at the plasma membrane. Interestingly, although cleavage of the full length sensor is expected to mirror cleavage of pro-TGFα, we are unable to detect this cleavage by western blot (Fig. 4J). To explain this observation, we performed fluorescence recovery after photobleaching (FRAP) experiments on HaCaT-TSen cells, which revealed that TSen turnover at the PM occurs with a half-life of 1 minute, independent of TACE activity (Fig. S3K), in contrast to the activation of TSen measured by fluorescence microscopy occurs approximately 30 times slower (Fig. S3G). This data suggests that a combination of a relatively small portion of plasma membrane TSen being cleaved upon TACE activation and high turnover of TSen at the plasma membrane may preclude the ability to detect TSen cleavage by western blot. Nevertheless, our subcellular fractionation data suggests that only actin cytoskeletal disruption, versus PMA stimulation, leads to alteration in subcellular trafficking of TACE to and from the PM.
Discussion
Our study illustrates not only a novel technique for measuring plasma membrane TACE activity with high specificity in live cells, but also how the TSen sensor was successfully used to shed light on the complexity of TACE activation in cells. The development and use of TSen has enabled us to discover a novel actin cytoskeleton dependent mechanism for TACE activation that relies, at least in part, on increased TACE accumulation at the plasma membrane. We propose that this means for TACE activation highlights a role for the actin cytoskeleton as a sensory structure, whose integrity is functionally linked to TACE activation, EGFR1 ligand shedding, and subsequent Erk activation. Erk activation is well known to regulate cytoskeletal remodeling via cortactin activation (39–42), allowing for the possibility that cells utilize TACE mediated EGFR1 ligand shedding to respond to actin damage by executing actin remodeling and repair via Erk.
In addition to enabling us to discover a novel connection between the actin cytoskeleton and TACE activity, TSen allowed us to determine that EGF dependent activation of TACE is driven by a combination of p38 and Erk activities, unanimously, but to varying degrees, across the many cell lines investigated. This observation allows for the possibility that cells employ a positive feedback system for TACE dependent EGFR1 ligand shedding, where TACE activity can promote subsequent increases in TACE activity through EGFR1 and p38 and/or Erk activation. Such positive feedback has been proposed in previous studies (11). One physiologically relevant use of such a feedback loop may be in the wound healing response of the skin. In wound healing model systems for the skin, epithelial sheets of keratinocytes undergo sustained motility upon wounding, which depends upon sustained EGFR1 activation (43). Initiation of epithelial sheet migration may rely on a wide range of possible initial stimuli that activate p38 and/or Erk in response to direct wounding, such as has been shown for reactive oxygen species (ROS) (44). Once initiation of p38 or Erk activity has been completed, our study suggests that TACE mediated EGFR1 ligand shedding will likely continue, even when the initial stimulus has been removed. Taken together, the use of our robust and specific FRET based biosensor for TACE activity has allowed us to make observations that provide insight into not only how individual cells respond to environmental stimuli, but also how groups of cells do the same during wound healing.
Methods
Cell Culture, Stable Expression, and RT-PCR
All cells were cultured using DMEM as previously described (43). Unless explicitly stated in figures, the following doses were used for cell treatments: EGF 100 nM, BMS-561392 2.5 μM, GM6001 10 μM, CytoD 1 μM, LatB 2.5 μM, PMA 200 nM, Gefitinib 1 μM, CI-1040 500 nM, SB-203580 10 μM.
Construction of TACE Sensor
The parental vector for TSen is EKAREV described in Komatsu et al and kindly provided by Drs. Aoki and Matsuda. A cassette encoding TGF-α signal peptide (MVPSAGQLALFALGIVLAACQALENSTSPLSDPPVAAAVVSH), HA tag (YPYDVPDYA), PDGF transmembrane domain, Flag tag VV (DYKDDDDKVV) was synthesized by Genscript and subcloned in EcoRI-SalI site of EKAREV to produce pBBSR-TGFA-HA-PDGF-Flag. Oligos encoding TACE cleavage site and noncleavable site were inserted in XhoI-NotI site EKAREV to generate TACE-REV and NOC-REV respectively. The fragments encode YPet-TACE-REV-ECFP or YPet-NOC-REV-ECFP were excised by digestion with EcoRI-XbaI and inserted in between HA and PDGF. The resultant vectors were named as TSen and NCS (Fig. 1).
FRET Data Analysis and Fluorescence Spectroscopy
Live cell microscopy was conducted as previously decribed(43). Filters used for FRET measurements were the following: FRET Ex 438/24-25, Dic 520LP, Em 542/27-25 (Semrock MOLE-0189); CFP Ex 438/24-25, Dic 458LP, Em 483/32-25 (Semrock CFP-2432B-NTE-Zero). Time lapse microscopy images were analyzed and FRET calculations were performed using MATLAB as previously stated (Supplemental Data S4). Briefly, images were background corrected through subtraction using images acquired from samples with media but no cells. Pixels representing cells were identified as having an intensity 1000 units above background. Fret Ratio or Inverse Fret Ratio was calculated as either FRET intensity/CFP intensity or CFP intensity/FRET intensity, respectively, and all relevant pixels were averaged. Each image was acquired at 4× magnification for calculations, which encompasses data from at least 1000 cells per measurement. Measurements were done in triplicate. For images displayed, 40× magnification was used. A similar method of inverse FRET ratio calculations was used in confocal microscopy experiments, where a z plane of 1 μm height at 100× magnification was measured and segmentation of membrane and vesicle fractions was performed in MATLAB.
Statistical Analysis
All comparisons indicated with brackets were the result of a two-tailed t-test, from which p values were obtained, using excel. Two-tailed t-tests were conducted between data collected from triplicate trial measurements in individual experiments.
Fluorescent Protein Secretion Experiments
YFP media secretion was quantified by transfer of media supernatant to 96 well plates, followed by measurement of of YFP fluorescence using a Tecan Microplate reader (Ex. 500/20, Em. 550/20) CFP media secretion was done the same with different parameters (Ex. 438/20, Em. 485/20).
Supplementary Material
Acknowledgements
We would like to thank Drs. Carl Blobel, Kazuhiro Aoki and Yingxiao Wang for sharing reagents, and Drs Will Old, Amy Palmer, Tristan McClure-Begley, Christopher Ebmeier, and Gilson Sanchez for valuable discussions. We thank Randy Gardner-McQuade at BMS for making BMS-561392 available for this research. We thank the BioFrontiers Advance Light Microscopy Core at University of Colorado-Boulder for technical advice in microscopy experiments.
Funding This work was supported by grants from the National Institutes of Health R01CA107098, Butcher award and Cancer League of Colorado to X.L. The ImageXpress MicroXL was supported by a NCRR grant S10 RR026680 from NIH. This work was also supported in part by DARPA (Cooperative Agreement W911NF-14-2-0019 / ARO Proposal No. 64973-LS-DRP).
Footnotes
Author Contributions Experiments and analysis were performed by D.A.C and E.G. Manuscript was written by D.A.C and X.L.
Competing Interests The authors declare no competing interests.
References
- 1.Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. 2006;108:2275–2279. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 3.Maretzky T, Evers A, Zhou W, Swendeman SL, Wong PM, Rafii S, Reiss K, Blobel CP. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat Commun. 2010;2:229. doi: 10.1038/ncomms1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hassemer EL, Endres B, Toonen JA, Ronchetti A, Dubielzig R, Sidjanin DJ. ADAM17 transactivates EGFR signaling during embryonic eyelid closure. Investigative ophthalmology & visual science. 2013;54:132–140. doi: 10.1167/iovs.12-11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giricz O, Calvo V, Peterson EA, Abouzeid CM, Kenny PA. TACE-dependent TGFalpha shedding drives triple-negative breast cancer cell invasion. Int J Cancer. 2013;133:2587–2595. doi: 10.1002/ijc.28295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011;32:380–387. doi: 10.1016/j.it.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Lee DC, Sunnarborg SW, Hinkle CL, Myers TJ, Stevenson MY, Russell WE, Castner BJ, Gerhart MJ, Paxton RJ, Black RA, Chang A, Jackson LF. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann N Y Acad Sci. 2003;995:22–38. doi: 10.1111/j.1749-6632.2003.tb03207.x. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura Y, Sotozono C, Kinoshita S. The epidermal growth factor receptor (EGFR): role in corneal wound healing and homeostasis. Exp Eye Res. 2001;72:511–517. doi: 10.1006/exer.2000.0979. [DOI] [PubMed] [Google Scholar]
- 9.Wells A. Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res. 2000;78:31–101. doi: 10.1016/s0065-230x(08)61023-4. [DOI] [PubMed] [Google Scholar]
- 10.Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 11.Miller MA, Meyer AS, Beste MT, Lasisi Z, Reddy S, Jeng KW, Chen CH, Han J, Isaacson K, Griffith LG, Lauffenburger DA. ADAM-10 and -17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2074–2083. doi: 10.1073/pnas.1222387110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor alpha, L-selectin, and tumor necrosis factor alpha. Molecular biology of the cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science (New York, N.Y. 2012;335:225–228. doi: 10.1126/science.1214400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy G. Regulation of the proteolytic disintegrin metalloproteinases, the `Sheddases'. Seminars in cell & developmental biology. 2009;20:138–145. doi: 10.1016/j.semcdb.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Gonzales PE, Solomon A, Miller AB, Leesnitzer MA, Sagi I, Milla ME. Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain. The Journal of biological chemistry. 2004;279:31638–31645. doi: 10.1074/jbc.M401311200. [DOI] [PubMed] [Google Scholar]
- 16.Srour N, Lebel A, McMahon S, Fournier I, Fugere M, Day R, Dubois CM. TACE/ADAM-17 maturation and activation of sheddase activity require proprotein convertase activity. FEBS Lett. 2003;554:275–283. doi: 10.1016/s0014-5793(03)01159-1. [DOI] [PubMed] [Google Scholar]
- 17.Soond SM, Everson B, Riches DW, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- 18.Xu P, Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol Cell. 2010;37:551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diaz-Rodriguez E, Montero JC, Esparis-Ogando A. L. Yuste, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell. 2002;13:2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan H, Derynck R. Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 1999;18:6962–6972. doi: 10.1093/emboj/18.24.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gechtman Z, Alonso JL, Raab G, Ingber DE, Klagsbrun M. The shedding of membrane-anchored heparin-binding epidermal-like growth factor is regulated by the Raf/mitogen-activated protein kinase cascade and by cell adhesion and spreading. The Journal of biological chemistry. 1999;274:28828–28835. doi: 10.1074/jbc.274.40.28828. [DOI] [PubMed] [Google Scholar]
- 22.Maretzky T, Zhou W, Huang XY, Blobel CP. A transforming Src mutant increases the bioavailability of EGFR ligands via stimulation of the cell-surface metalloproteinase ADAM17. Oncogene. 2011;30:611–618. doi: 10.1038/onc.2010.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, Smithgall TE, Mills GB, Grandis JR. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouyang M, Huang H, Shaner NC, Remacle AG, Shiryaev SA, Strongin AY, Tsien RY, Wang Y. Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 2010;70:2204–2212. doi: 10.1158/0008-5472.CAN-09-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briley GP, Hissong MA, Chiu ML, Lee DC. The carboxyl-terminal valine residues of proTGF alpha are required for its efficient maturation and intracellular routing. Mol Biol Cell. 1997;8:1619–1631. doi: 10.1091/mbc.8.8.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohan MJ, Seaton T, Mitchell J, Howe A, Blackburn K, Burkhart W, Moyer M, Patel I, Waitt GM, Becherer JD, Moss ML, Milla ME. The tumor necrosis factor-alpha converting enzyme (TACE): a unique metalloproteinase with highly defined substrate selectivity. Biochemistry. 2002;41:9462–9469. doi: 10.1021/bi0260132. [DOI] [PubMed] [Google Scholar]
- 27.Jin G, Huang X, Black R, Wolfson M, Rauch C, McGregor H, Ellestad G, Cowling R. A continuous fluorimetric assay for tumor necrosis factor-alpha converting enzyme. Analytical biochemistry. 2002;302:269–275. doi: 10.1006/abio.2001.5549. [DOI] [PubMed] [Google Scholar]
- 28.Lelimousin M, Noirclerc-Savoye M, Lazareno-Saez C, Paetzold B, Le Vot S, Chazal R, Macheboeuf P, Field MJ, Bourgeois D, Royant A. Intrinsic dynamics in ECFP and Cerulean control fluorescence quantum yield. Biochemistry. 2009;48:10038–10046. doi: 10.1021/bi901093w. [DOI] [PubMed] [Google Scholar]
- 29.Qian M, Bai SA, Brogdon B, Wu JT, Liu RQ, Covington MB, Vaddi K, Newton RC, Fossler MJ, Garner CE, Deng Y, Maduskuie T, Trzaskos J, Duan JJ, Decicco CP, Christ DD. Pharmacokinetics and pharmacodynamics of DPC 333 ((2R)-2-((3R)-3-amino-3{4-[2-methyl-4-quinolinyl) methoxy] phenyl}-2-oxopyrrolidinyl)-N-hydroxy-4-methylpentanamide)), a potent and selective inhibitor of tumor necrosis factor alpha-converting enzyme in rodents, dogs, chimpanzees, and humans. Drug metabolism and disposition: the biological fate of chemicals. 2007;35:1916–1925. doi: 10.1124/dmd.107.015933. [DOI] [PubMed] [Google Scholar]
- 30.Moss ML, Sklair-Tavron L, Nudelman R. Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nature clinical practice. 2008;4:300–309. doi: 10.1038/ncprheum0797. [DOI] [PubMed] [Google Scholar]
- 31.Tucher J, Linke D, Koudelka T, Cassidy L, Tredup C, Wichert R, Pietrzik C, Becker-Pauly C, Tholey A. LC-MS based cleavage site profiling of the proteases ADAM10 and ADAM17 using proteome-derived peptide libraries. Journal of proteome research. 2014;13:2205–2214. doi: 10.1021/pr401135u. [DOI] [PubMed] [Google Scholar]
- 32.Alvarez-Iglesias M, Wayne G, O'Dea KP, Amour A, Takata M. Continuous real-time measurement of tumor necrosis factor-alpha converting enzyme activity on live cells. Lab Invest. 2005;85:1440–1448. doi: 10.1038/labinvest.3700340. [DOI] [PubMed] [Google Scholar]
- 33.Armstrong L, Godinho SI, Uppington KM, Whittington HA, Millar AB. Tumour necrosis factor-alpha processing in interstitial lung disease: a potential role for exogenous proteinase-3. Clinical and experimental immunology. 2009;156:336–343. doi: 10.1111/j.1365-2249.2009.03906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu J, Van den Steen PE, Dillen C, Opdenakker G. Targeting neutrophil collagenase/matrix metalloproteinase-8 and gelatinase B/matrix metalloproteinase-9 with a peptidomimetic inhibitor protects against endotoxin shock. Biochemical pharmacology. 2005;70:535–544. doi: 10.1016/j.bcp.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 35.Le Gall SM, Maretzky T, Issuree PD, Niu XD, Reiss K, Saftig P, Khokha R, Lundell D, Blobel CP. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J Cell Sci. 2010;123:3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Robertson JD, Walcheck B. Different signaling pathways stimulate a disintegrin and metalloprotease-17 (ADAM17) in neutrophils during apoptosis and activation. The Journal of biological chemistry. 2011;286:38980–38988. doi: 10.1074/jbc.M111.277087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K. A genetically encoded fluorescent sensor of ERK activity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nature cell biology. 2011;13:1124–1131. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelley LC, Hayes KE, Ammer AG, Martin KH, Weed SA. Cortactin phosphorylated by ERK1/2 localizes to sites of dynamic actin regulation and is required for carcinoma lamellipodia persistence. PLoS One. 2010;5:e13847. doi: 10.1371/journal.pone.0013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han MY, Kosako H, Watanabe T, Hattori S. Extracellular signal-regulated kinase/mitogen-activated protein kinase regulates actin organization and cell motility by phosphorylating the actin cross-linking protein EPLIN. Mol Cell Biol. 2007;27:8190–8204. doi: 10.1128/MCB.00661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Quiles N, Ho HY, Kirschner MW, Ramesh N, Geha RS. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP. Mol Cell Biol. 2004;24:5269–5280. doi: 10.1128/MCB.24.12.5269-5280.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelley LC, Hayes KE, Ammer AG, Martin KH, Weed SA. Revisiting the ERK/Src cortactin switch. Commun Integr Biol. 2011;4:205–207. doi: 10.4161/cib.4.2.14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapnick DA, Liu X. Leader Cell Positioning Drives Wound Directed Collective Migration in TGFbeta Stimulated Epithelial Sheets. Mol Biol Cell. 2014 doi: 10.1091/mbc.E14-01-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikolic DL, Boettiger AN, Bar-Sagi D, Carbeck JD, Shvartsman SY. Role of boundary conditions in an experimental model of epithelial wound healing. American journal of physiology. 2006;291:C68–75. doi: 10.1152/ajpcell.00411.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.