

Abstract
There is a conserved ATPase domain in topoisomerase II (topo II) and heat shock protein 90 (Hsp90) which belong to the GHKL (gyrase, Hsp90, histidine kinase, and MutL) family. The inhibitors that target each of topo II and Hsp90 are intensively studied as anti-cancer drugs since they play very important roles in cell proliferation and survival. Therefore the development of dual targeting anti-cancer drugs for topo II and Hsp90 is suggested to be a promising area. The topo II and Hsp90 inhibitors, known to bind to their ATP binding site, were searched. All the inhibitors investigated were docked to both topo II and Hsp90. Four candidate compounds as possible dual inhibitors were selected by analyzing the molecular docking study. The pharmacophore model of dual inhibitors for topo II and Hsp90 were generated and the design of novel dual inhibitor was proposed.
Keywords: Topoisomerase II, Heat shock protein 90, Molecular docking study, Design of dual inhibitor
INTRODUCTION
Topoisomerase II (topo II) and heat shock protein 90 (Hsp90) both contain a conserved ATPase domain and belong to the same family, namely, GHKL (gyrase, Hsp90, histidine kinase, and MutL) domain (Dutta and Inouye, 2000; Chene, 2002). ATPase domain in both of these proteins requires ATP to exert important cellular functions such as cell cycle progression, proliferation and survival. Therefore, inhibitors targeting the ATP binding site of these two proteins through binding in an ATP-competitive manner were searched and characterized in this study. Another important biological implication in topo II and Hsp90 is that they are both overexpressed in proliferating cancer cells and have been attractive targets for the development of anti-cancer drugs (Neckers, 2002; Nitiss, 2009a).
Topo II is very important in cellular processes such as transcription and replication by introducing transient breaks in DNA double strand (Nitiss, 2009b). Topo II requires ATP binding for its conformational change to solve topological problems in DNA. Recently, there are much efforts in developing catalytic inhibitors of topo II in order to overcome the side-effects of topo II poisons such as etoposide (Pogorelcnik et al., 2013). Hsp90 is a molecular chaperone which has diverse client proteins involved in tumor growth and survival. Therefore, Hsp90 also has been an attractive target for chemotherapeutic development and phase II clinical trial was conducted for Hsp90 inhibitor, 17-allyaminogeldanamycin (17-AAG) (Sidera and Patsavoudi, 2014). In 2006, Jenkins and coworkers reported that the topo II and Hsp90 form a complex, and co-treatment of 17-AAG showed synergistic efficacy by enhancing the activity of topo II poison (Barker et al., 2006; Yao et al., 2007). From these findings, development of inhibitors that target both ATPase domains of topo II and Hsp90 can be a promising research area. There are many advantages of multi-target drugs since they can simultaneously inhibit multiple pathways and escape an undesirable drug-drug interaction which may encounter with co-treatment of single-target drugs (Petrelli and Giordano, 2008).
In this review, the topo II and Hsp90 inhibitors that bind to the ATPase domain of each of topo II and Hsp90 are analyzed and the possibility of designing dual inhibitor is explored through molecular modelling studies.
METHODS
Molecular docking studies
The 3D structures of the inhibitors of topo II and Hsp90 were sketched using Sybyl X-2.1.1 (Certara L.P., St. Louis, MO, USA). All the structures were energetically minimized using Tripos force field and Gasteiger-Hückel charges. The structures of ATPase domain of topo II and Hsp90 were retrieved from RCSB Protein Data Bank (PDB entry code: 1ZXM and 3EKR) (Wei et al., 2005; Kung et al., 2008). The ligands were extracted and water molecules were removed from the initial x-ray crystal structure. The docking was carried out for both of the topo II and Hsp90 inhibitors to topo II and Hsp90 using Surflex-Dock (Jain, 2003). The protomol was generated using ligand mode, which used the ligand, extracted from the crystal structure occupying the ATP binding site, to ensure that the inhibitor could bind to the ATP binding site. Then polar hydrogens were added to the structure. After the protomol generation, ligands were docked using Surflex-Dock Geom and GeomX modes using default parameters.
Pharmacophore hypothesis generation
Compounds PU3, 3t, AUY922 and comp. 14 were used as input molecules to generate pharmacophore model using GASP module implemented in Sybyl X-2.1.1. Four molecules selected were used as the data set for the pharmacophore model generations. All the features on each of the molecules were used and the default GA parameters were used. The parameters used for the calculations were as follows; 100 population size, 1.1 selection pressure, 100000 max operations, 6500 operation increment and 0.01 fitness.
RESULTS
Comparison of the structural similarities of ATPase domain of topo II and Hsp90
Among structures of ATPase domain of topo II and Hsp90 deposited in Protein Data Bank (PDB), 1ZXM for topo II and 3EKR and 1BYQ for Hsp90 were chosen for structure comparison and docking. The length of the two proteins is 376 and 217 amino acid residues for topo II and Hsp90, respectively. The similarity of the two proteins were compared by sequence alignment using BioEdit (Fig. 1) (Hall, 1999). Although the sequence identity between the two ATPase domains is 15.8 %, which is rather small value, the overall fold has high similarity where they are superimposable (Fig. 2).
Fig. 1.

The sequence alignment of the ATPase domain of topo II (1ZXM) and Hsp90 (3EKR). The alignment were generated using BioEdit.
Fig. 2.

The structure of ATPase domain of (A) topo II and (B) Hsp90. AMPPNP and ADP bound to topo II and Hsp90, respectively are represented in space-filling model colored by atom type (gray: carbon; red: oxygen; blue: nitrogen; orange: phosphorus). The proteins are represented in ribbon.
The ATP binding sites of the two proteins can be suggested to have similar environment. The amino acid residues involved in binding with the ligand adenylyl-imidophosphate (AMPPNP) or ADP for topo II and Hsp90, respectively, do not coincide exactly, however the properties of each amino acids are conserved. For example, in topo II, Asn120 forms hydrogen bond with the N6 amino group of adenine ring, whereas in Hsp90, Asp93 is involved in the hydrogen bond interaction. The hydrophobic residue Ile125 in topo II corresponds to the residue of Met98 in Hsp90 which is also hydrophobic. Additionally, the size of the ATP binding site of each protein was calculated with Computed Atlas of Surface Topography of proteins (CASTp, http://sts.bioe.uic.edu/castp/) (Liang et al., 1998). As listed in Table 1 and shown in Fig. 3, the calculated area and volume of topo II ATP binding site were 792.2 Å2 and 1077.6 Å3, respectively. The ATP binding site’s area and volume of Hsp90 were slightly smaller than topo II, 628.9 Å2 and 971.0 Å3, respectively. The mouth opening of the binding pocket was also identified and characterized with CASTp. Although the overall pocket size was slightly larger for topo II, the area and the circumcircle of the mouth opening of Hsp90 were larger than topo II, with the values of 167.6 Å2, 78.6 Å and 70.9 Å2, 52.6 Å, respectively.
Table 1.
The characterization of the active sites of topo II and Hsp90 by CASTp
| Protein | Mouth | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Area (Å2) | Volume (Å3) | Number* | Area (Å2) | Circumcircle (Å) | |
| topo II | 792.2 | 1077.6 | 2 | 70.9 | 52.6 |
| Hsp90 | 628.9 | 971.0 | 1 | 167.6 | 78.6 |
Number of mouth openings for the pocket. Each has to be large enough to allow the solvent probe to pass through.
Fig. 3.

The comparison of ATP binding site of (A) topo II and (B) Hsp90. The channel was created using MOLCAD implemented in Sybyl, colored by electrostatic potential. The color ramp ranges from red (most positive) to purple (most negative). AMPPNP and ADP bound to topo II and Hsp90, respectively are represented in sticks colored by atom type (gray: carbon; red: oxygen; blue: nitrogen; orange: phosphorus). The proteins are represented in ribbon (blue: β-strand; red: α-helix).
Topo II inhibitors that bind to the ATPase domain
The topo II inhibitors that bind to the topo II ATPase domain were searched. The inhibitors can be largely divided into two categories, purine analogues and non-purine analogues. Table 2 lists the topo II inhibitors and gives information about their structures and ATPase inhibition activity where applicable.
Table 2.
Topo II inhibitors that bind to the ATPase domain
| Name | Structure | IC50* | Type | Reference |
|---|---|---|---|---|
| Comp. 1 |
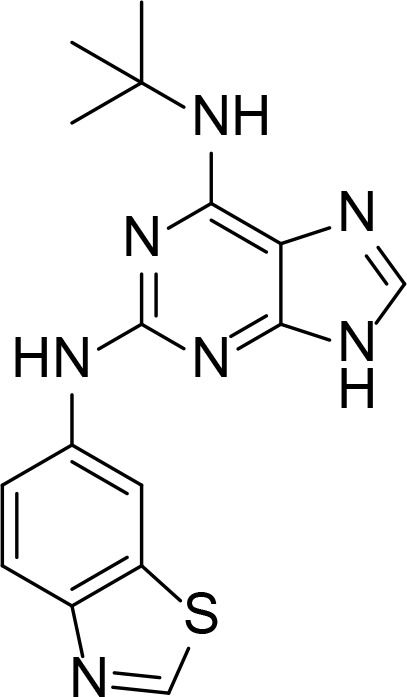
|
1.7 µM | Purine analog | Furet et al., 2009 |
| Comp. 2 |
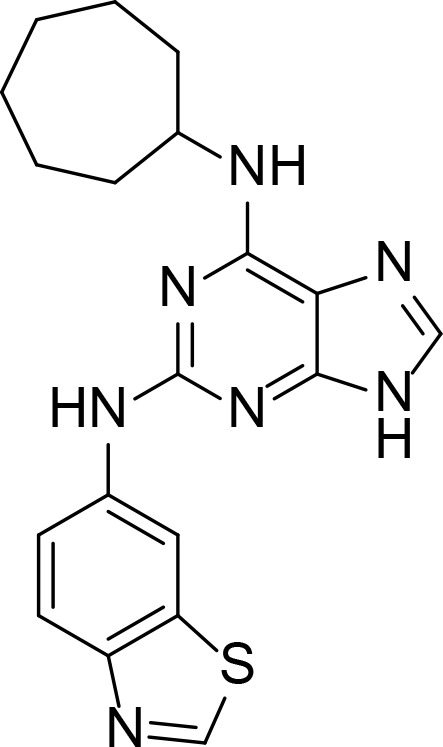
|
8.4 µM | Purine analog | Furet et al., 2009 |
| NSC35866 |
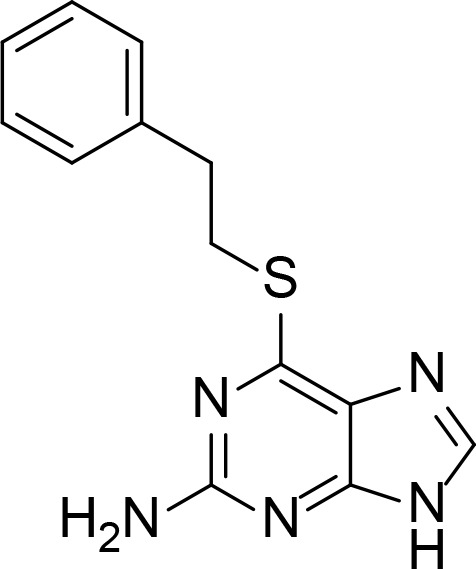
|
50 µM | Purine analog | Jensen et al., 2005 |
| NCS348400 |
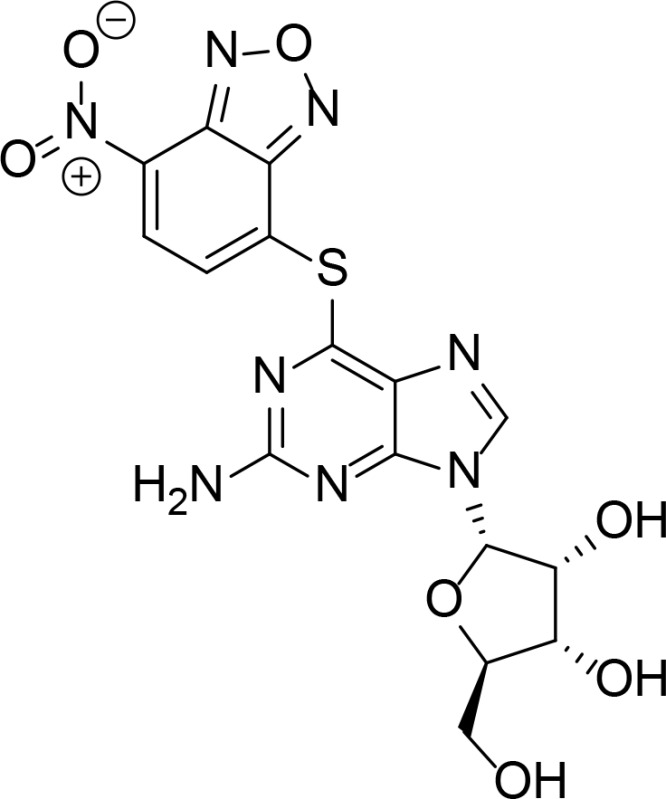
|
0.39 µM | Purine analog | Jensen et al., 2006 |
| QAP1 |
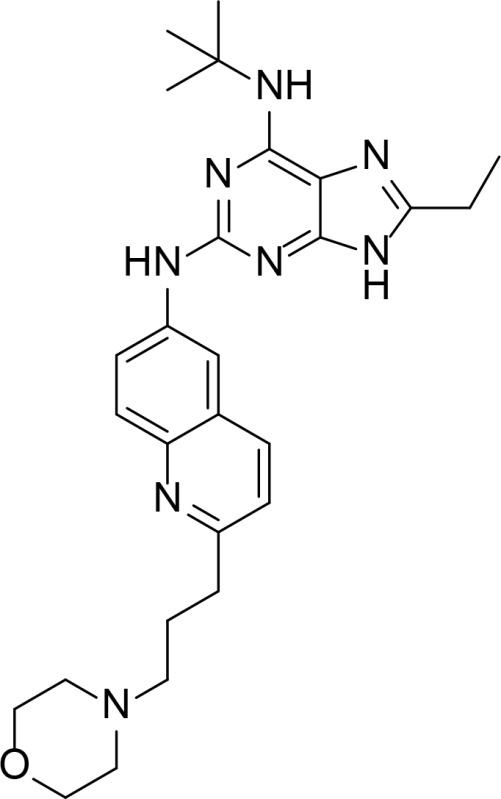
|
128 nM | Purine analog | Chene et al., 2009 |
| 2c |
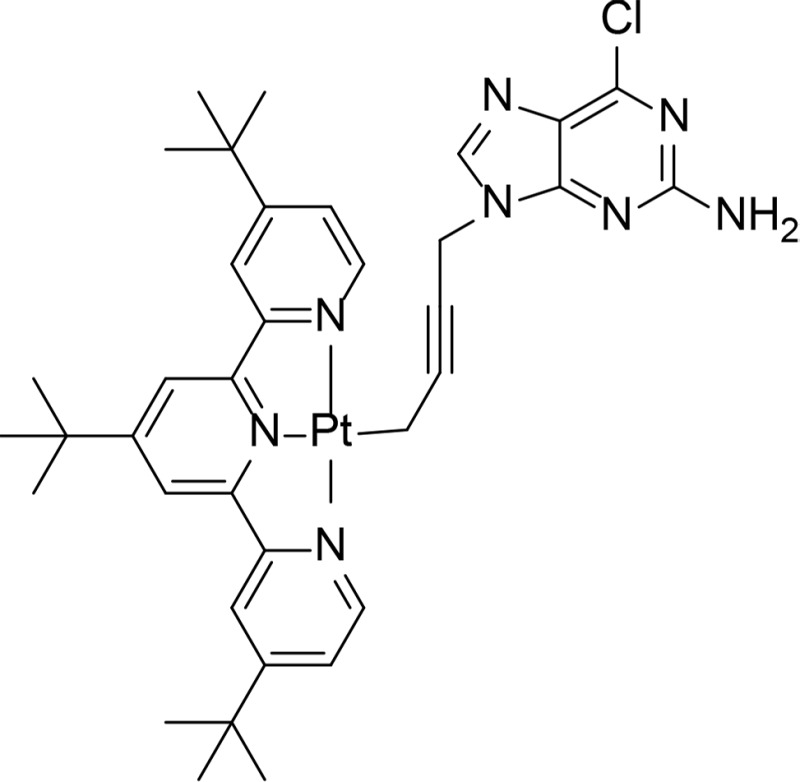
|
Ki=1.25 µM in ATP competition assay | Purine analog with platinum | Wang et al., 2010 |
| 8-Cl-ATP |
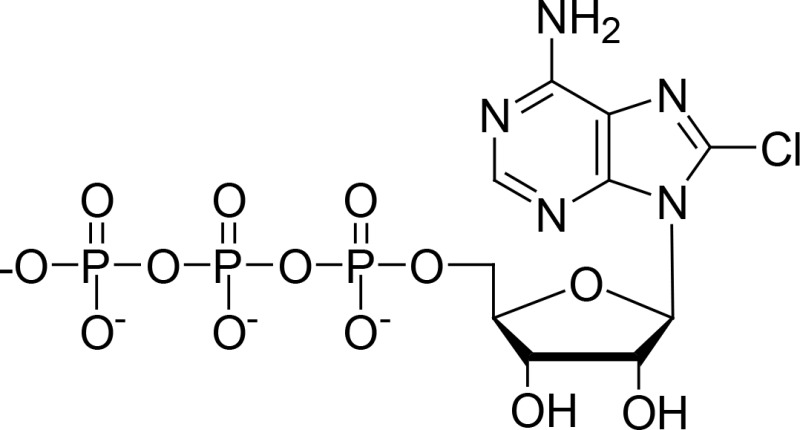
|
Yang et al., 2009 | ||
| 3t |
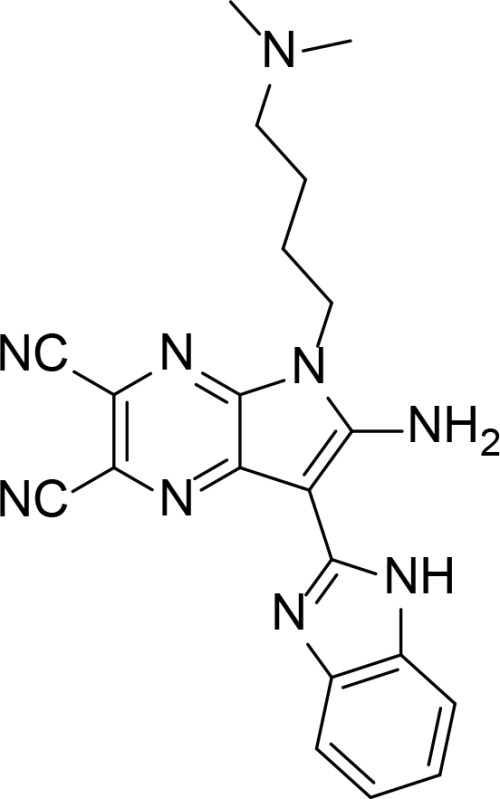
|
1,3-benzoazolyl-substituted pyrrolo[2,3-b]pyrazine derivatives | Li et al., 2016 | |
| Daurinol |
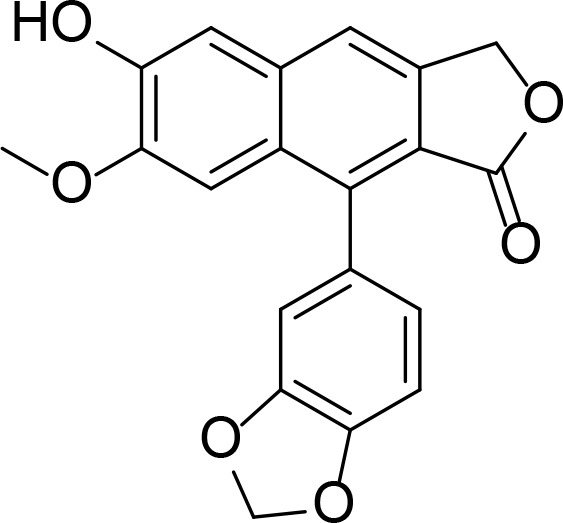
|
Natural product | Wang et al., 2010 | |
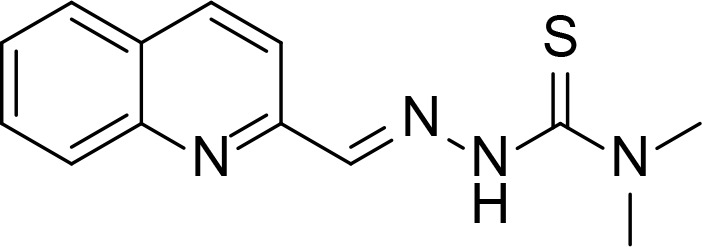
| TSC24 |
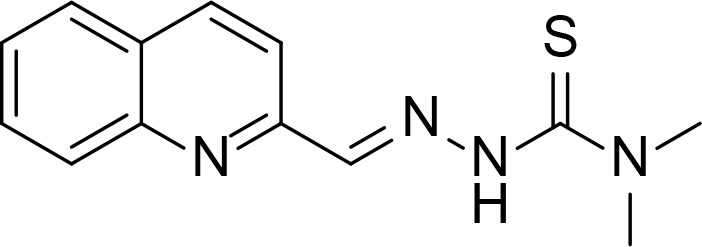
|
Kd=18.3 µM compared to ATP (615 µM) | Thiosemicarbazone | Huang et al., 2010 |
| Comp. 5 |
|
52.77 µM, Ki=75 µM | N-fused imidazole | Baviskar et al., 2011 |
| Comp. 14 |
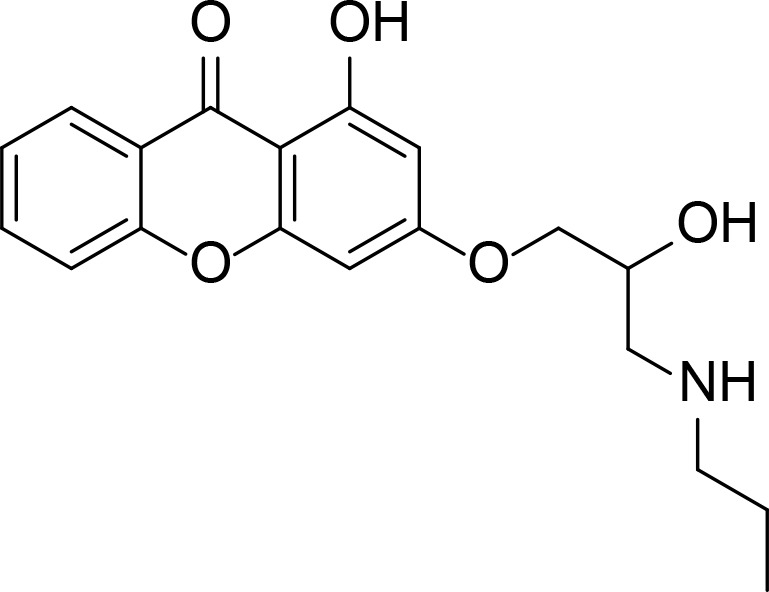
|
Xanthone | Jun et al., 2011 | |
| Comp. 14mod |
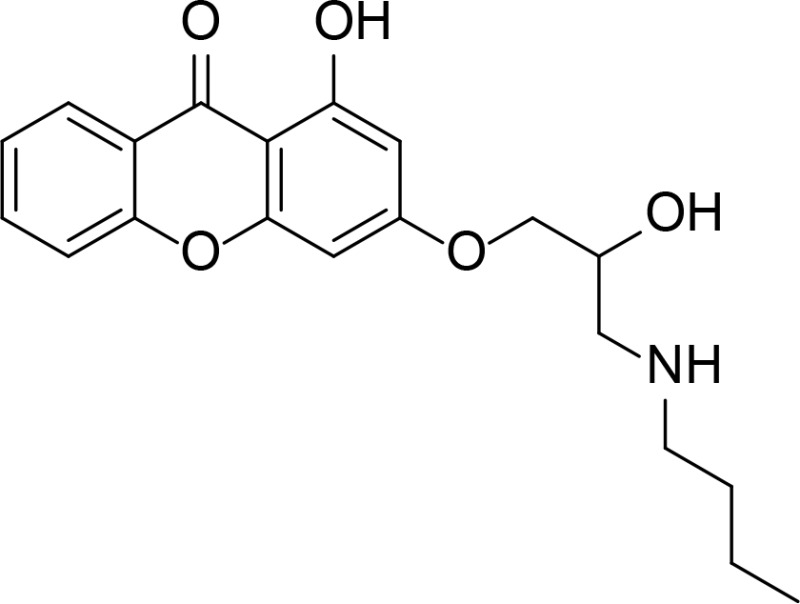
|
Xanthone | Park et al., 2013 | |
| Comp. 18 |
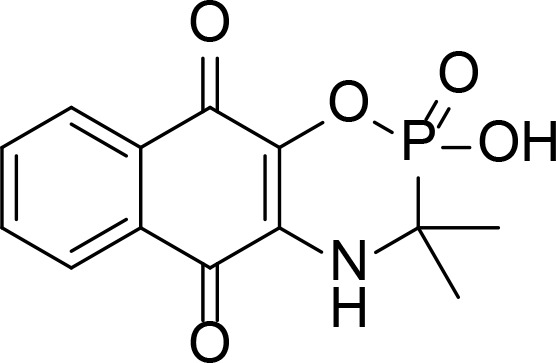
|
Naphthoquinone fused cyclic aminoalkyl-phosphonates and aminoalkyl-phosphonic monoester | Wang et al., 2008 | |
| Salvicine |
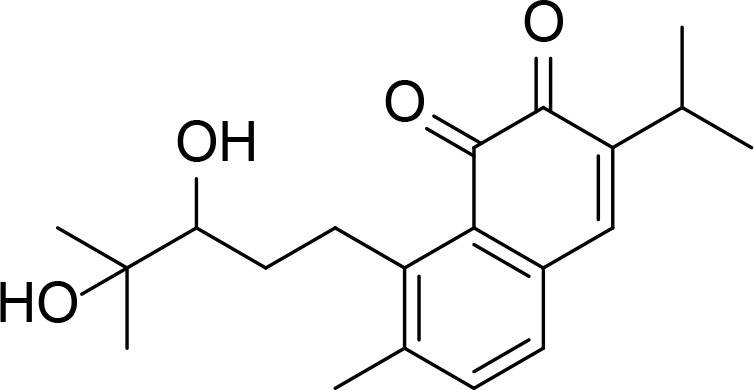
|
Kd=74.3 µM | Natural product | Hu et al., 2006 |
| D11 |
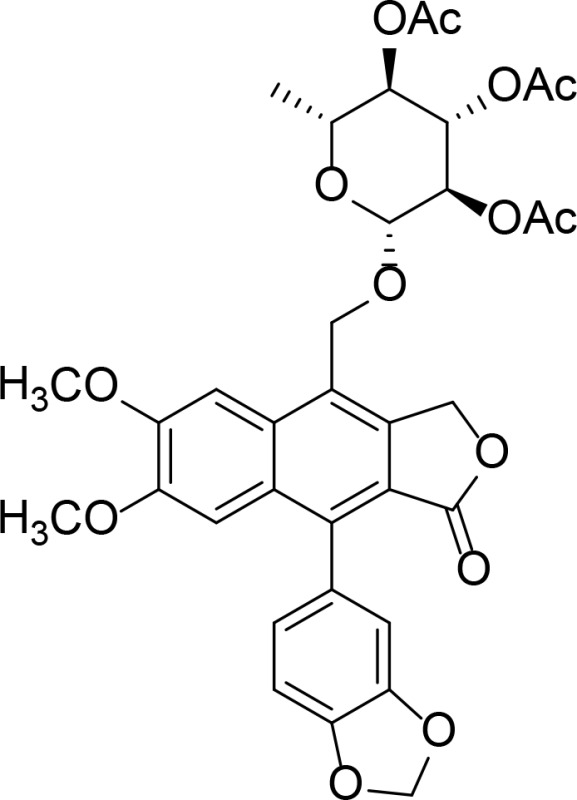
|
Kd=37.7 µM | Diphyllin glycoside | Gui et al., 2011 |
| Gambogic acid |
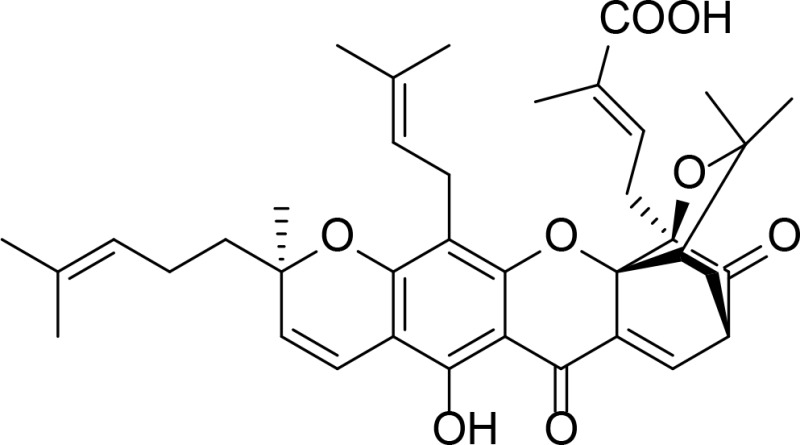
|
Kd=3.32 µM | Natural product | Qin et al., 2007 |
| Emodin |
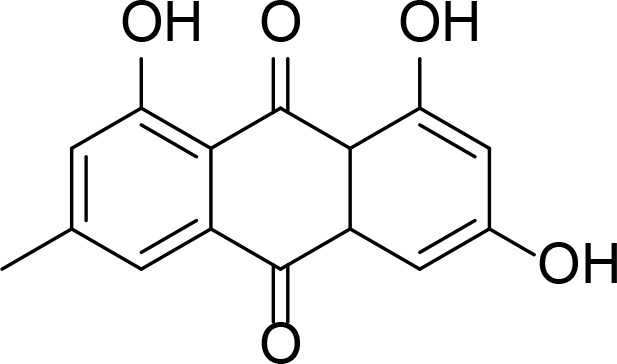
|
Anthraquinone analog | Li et al., 2010 |
IC50 values for the compound otherwise noted; inhibition constant (Ki), dissociation constant (Kd).
Purine analogue inhibitors contain the purine ring and have substitutions on the 2, 6, or 9 positions. In order to develop novel topo II catalytic inhibitors, 1,990 compounds from the National Cancer Institute (NCI) diversity set library was screened and S6-substituted thioguanine analog, NSC35866 was identified (Jensen et al., 2005). This finding was further expanded to discover more potent ATPase inhibitors by screening 40 substituted purine or purine-like compounds in the NCI database and several compounds including NSC348400 were identified from this screening (Jensen et al., 2006). Compounds 1 and 2 were searched from the Novartis compound collection to specifically target the ATP binding site of topo II (Furet et al., 2009). The hydrogen bond forming residues of topo II were Asn120 and Asn91 which were identical in three topo II complexes with compounds 1, 2 and AMPPNP, however the purine ring of compounds 1 and 2 adopted different orientation compared to that of ATP. Compounds 1 and 2 were further optimized by considering these interactions with the binding site and obtained a purine analogue with substitution of an ethyl group at position C6 and a morpholino-ethoxy group in the quinolone substituted on position N2 (called quinoline amino-purine, QAP1) (Chene et al., 2009). QAP1 showed improvement in topo II ATPase inhibitory activity with the half maximal inhibitory concentration (IC50) of 128 ± 21 nM. 3t has a new scaffold, aloisine moiety, which is similar to purine ring (Li et al., 2016). In contrast to compounds 1 and 2, the aloisine ring was aligned with the purine ring of ATP from docking study. 2c is a organoplatinum(II) complex with an attachment of 2-amino-6-chloropurine (Wang et al., 2010). 2c inhibited topo II by preventing ATP entering into the ATPase domain. Although 2c is a purine analog, its purine moiety did not occupy the ATP purine ring binding site, but the tert-butyl groups of the terpyridine scaffold occupied on it, determined by molecular docking study. 8-chloro-adenosine (8-Cl-Ado) is an anti-cancer agent currently undergoing phase I/II clinical trial. 8-Cl-Ado converted into 8-Cl-ATP in cells and it competed with ATP to inhibit topo II (Yang et al., 2009).
There are some topo II inhibitors originated from natural products. Daurinol is a lignan isolated from Haplophyllum dauricum, whose structure is similar to etoposide (Kang et al., 2014). Etoposide is a well-known cytotoxic anti-cancer drug functioning as a topo II poison. Daurinol occupied the same binding site with AMPPNP, which was shown by molecular docking study, suggesting it inhibited topo II by targeting the ATPase domain. Gambogic acid (GA) is a natural product isolated from Garcinia hanburi tree. GA was shown to be a catalytic inhibitor of topo II by binding to the ATPase domain, determined by surface plasmon resonance (SPR) analysis and by molecular docking (Qin et al., 2007). Diphyllin was extracted from Justicia procumbens, which showed tumoricidal effects. D11 is a novel acetylated D-quinovose diphyllin analogue exhibiting potent topo II inhibitory activity and binding to the ATPase domain (Gui et al., 2011). When the compound binds to the ATP binding site it is not always a topo II catalytic inhibitor, as it is the case with salvicine and emodin. Salvicine is a derivative of diterpenequinone isolated from Salvia prionitis, which bound to the ATPase domain validated by SPR and molecular docking, and acted as a topo II poison generating double strand breaks (Hu et al., 2006). Emodin is an anthraquinone isolated from Rheum emodi and also from molds, lichens and fungi. Emodin also generated DNA double strand breaks and stabilized the topo II-DNA cleavage complex (Li et al., 2010).
Last group of topo II inhibitors are synthesized compounds designed from different scaffolds known to be biologically active or potent anti-cancer agents. Thiosemicarbazone (TSC) is one of the scaffolds, and their derivatives including TSC24 showed catalytic inhibition of topo II (Huang et al., 2010). TSC24 directly bound to the ATPase domain which was confirmed by competitive inhibition assay, SPR and molecular docking studies. Baviskar and coworkers designed and synthesized bicyclic N-fused aminoimidazole which had similar structure to reported topo II inhibitors and marketed drugs such as zolpidem and zolimidine (Baviskar et al., 2011). From the synthesized compounds, comp. 5 is a non-intercalating topo II catalytic inhibitor that bound to the ATP binding site. Compounds 14 and 14mod are xanthone derivatives that bound to the topo II ATPase domain, which was confirmed by ATPase competitive inhibition assay and molecular docking (Jun et al., 2011; Park et al., 2013). There were several topo II catalytic inhibitors containing quinone moiety that bound to the ATP binding site. Pyranonaphthoquinone, comp. 3a, was shown to be a topo II catalytic inhibitor and suggested to bind to the ATPase domain through docking (Jimenez-Alonso et al., 2008). Naphthoquinone fused cyclic aminoalkyl-phosphonates and aminoalkyl-phosphonic monoester were synthesized and tested for their topo II activity (Wang et al., 2008). Some of them including comp. 18 were catalytic topo II inhibitors and these compounds were docked into the ATP binding site of topo II (Ma et al., 2011).
Hsp90 inhibitors that bind to the N-terminal ATPase domain
Geldanamycin (GDA) and radicicol (RDC), antibiotic isolated from natural product (Roe et al., 1999) were the first discovered Hsp90 inhibitors that target the N-terminal ATPase domain. Due to poor solubility and hepatotoxicity of GDA and RDC, GDA and RDC derivatives were designed and synthesized to have good physical properties and stability with improved potency. 17-AAG is a GDA derivative that improved the toxicity and stability of GDA itself (Schulte and Neckers, 1998). The co-crystal structure of 17-DMAG and Hsp90 N-terminal ATPase domain was solved (Jez et al., 2003). GDA derivatives were also genetically engineered to produce GDA analogs, such as KOSN1559, showing better binding affinity than GDA (Patel et al., 2004).
Another group of Hsp90 inhibitors are RDC analogs. KF25706, KF29163 and KF58333 were chemically synthesized and their biological activities were assessed (Soga et al., 1999; Agatsuma et al., 2002). Various RDC analogs were further synthesized such as aigialmycin D, c-RDC, pochonin A, pochonin D and O-(piperidinocarbonyl) methyloxime derivative of RDC.
The GDA and RDC analogs are rather big in size and their poor properties in solubility and toxicity led to designing and synthesizing purine analogs for inhibiting Hsp90 by binding to the ATP binding site. PU3 is one of them and it competed with GDA for Hsp90 binding and when treated in cancer cells, HER2 level decreased (Chiosis et al., 2001). Other small Hsp90 inhibitors include pyrazole analogs such as CCT018159 and G3130. CCT018159 was searched from high-throughput screening compound collection of more than 56,000 compounds utilizing the ATPase activity assay (Rowlands et al., 2004). The crystal structure of G3130 bound to the N-terminal ATP binding domain of Hsp90 was solved and the value of Kd was 280 nM determined by SPR (Kreusch et al., 2005). SNX0723 is one of the synthetic compound having a novel scaffold containing benzamide moiety which was discovered to bind to the ATPase domain of Hsp90 by screening a compound library (Putcha et al., 2010). Resorcinol moiety was also identified to be an important scaffold for ATPase binding in Hsp90. AUY922, AT-13387 and CPUY201112 are the Hsp90 inhibitors that have resorcinol moiety which plays an important role in hydrogen bonding and hydrophobic interactions with the receptor (Dutta Gupta et al., 2014). Hsp90 inhibitors targeting its N-terminal ATP binding site reviewed in the current study are listed in Table 3.
Table 3.
Hsp90 inhibitors that bind to the ATPase domain
| Name | Structure | IC50 | Type | Reference |
|---|---|---|---|---|
| GDA |
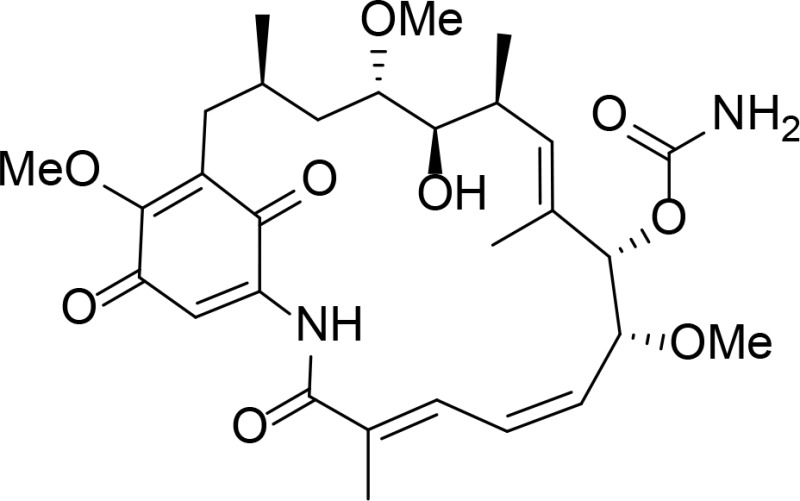
|
Kd=1.2 µM (determined from isothermal calorimetry (ITC)) | Roe et al., 1999 | |
| Radicicol |
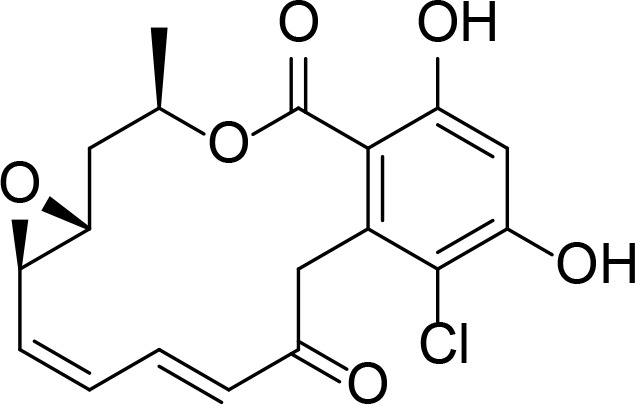
|
23 nM, Kd=19 nM (ITC) | Roe et al., 1999 | |
| 17-AAG |
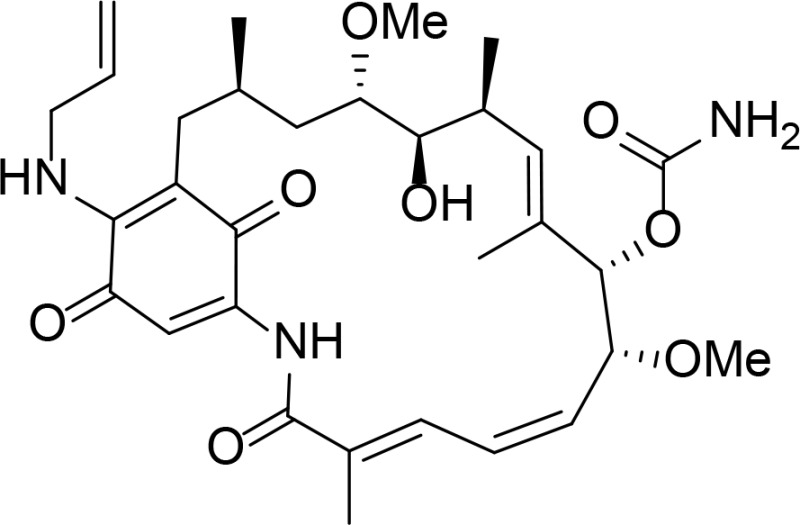
|
Geldanamycin derivative | Schulte and Neckers, 1998 | |
| 17-DMAG |
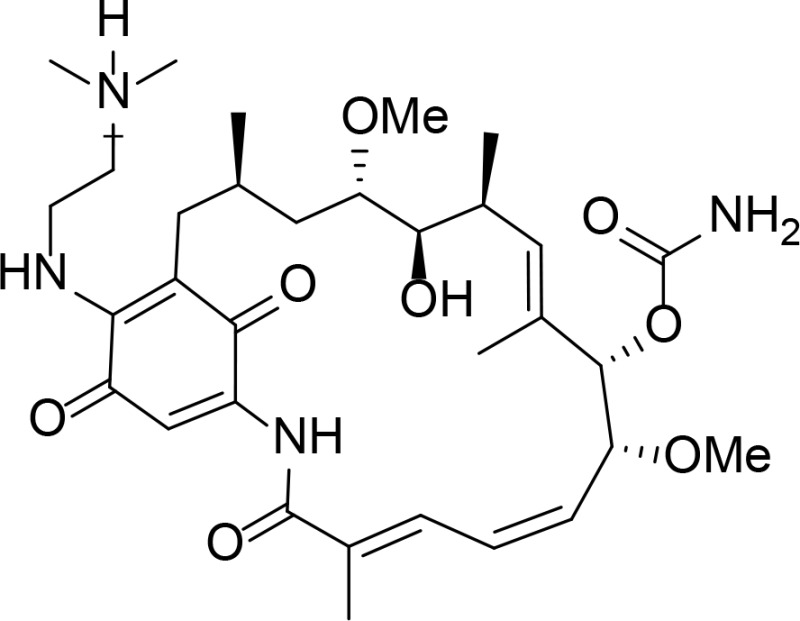
|
Geldanamycin derivative | Jez et al., 2003 | |
| KOSN1559 |
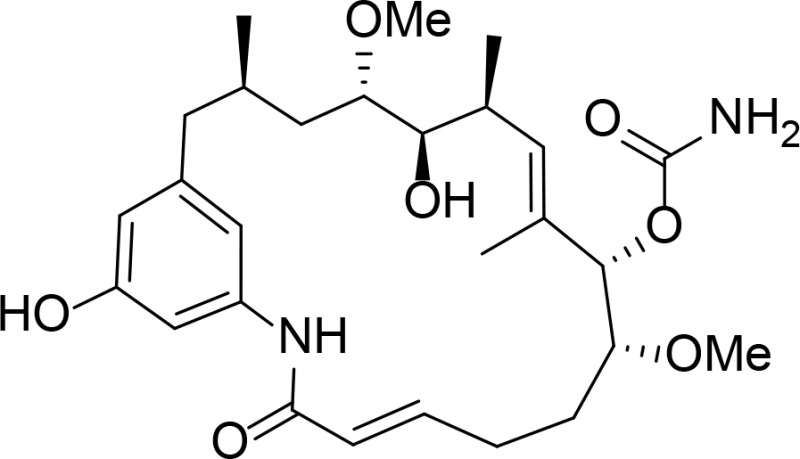
|
Kd=16 nM | Geldanamycin derivative | Patel et al., 2004 |
| KF25706 |
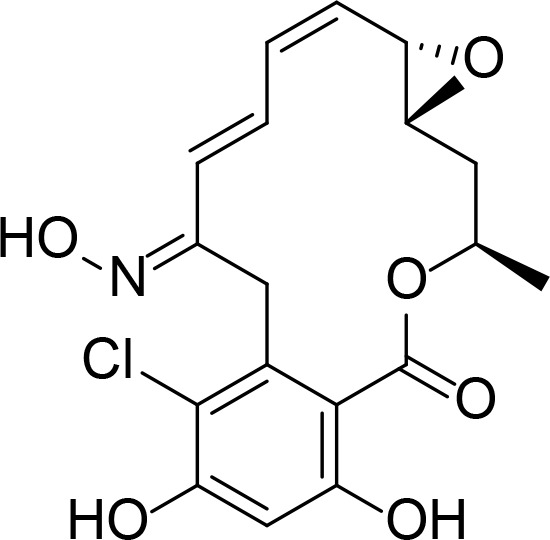
|
RDC analog | Soga et al., 1999 | |
| KF29163 |
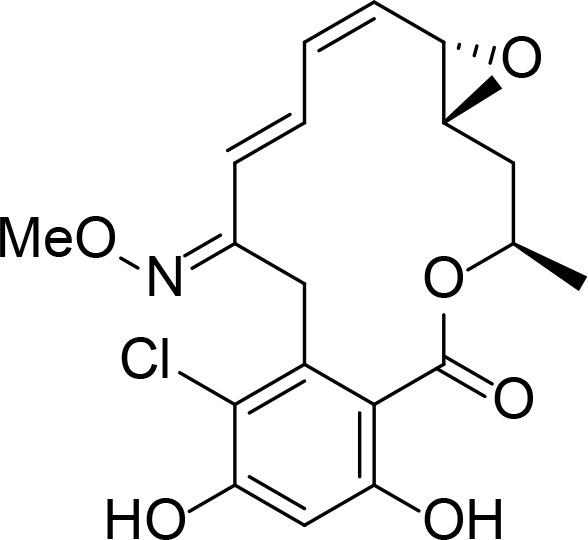
|
RDC analog | Agatsuma et al., 2002 | |
| c-RDC |
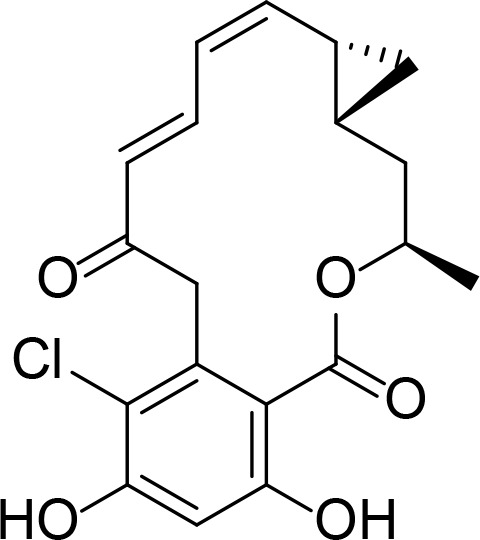
|
RDC analog | Yang et al., 2004 | |
| Aigialmycin D |
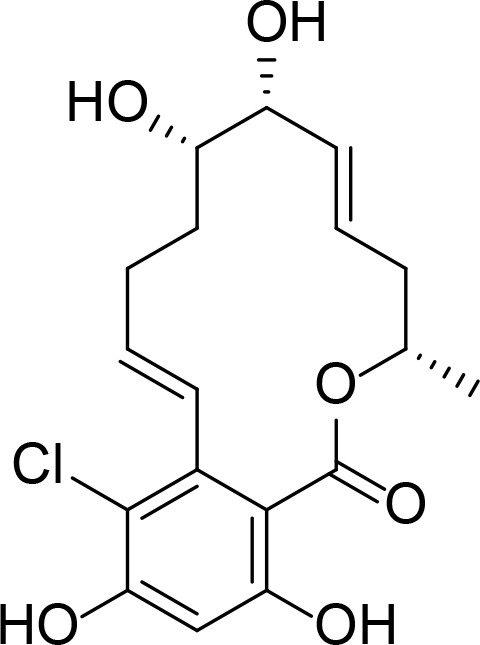
|
RDC analog | Yang et al., 2004 | |
| Pochonin A |
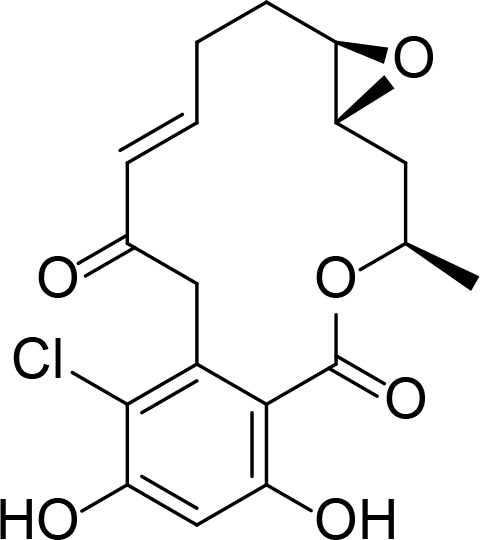
|
90 nM | RDC analog | Moulin et al., 2005a |
| Pochonin D |
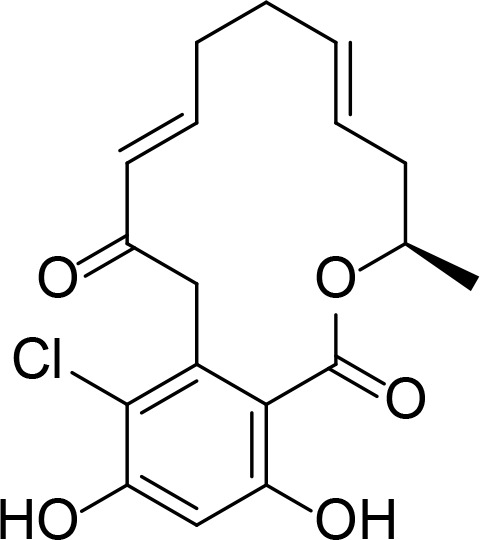
|
RDC analog | Moulin et al., 2005b | |
| KF58333 |
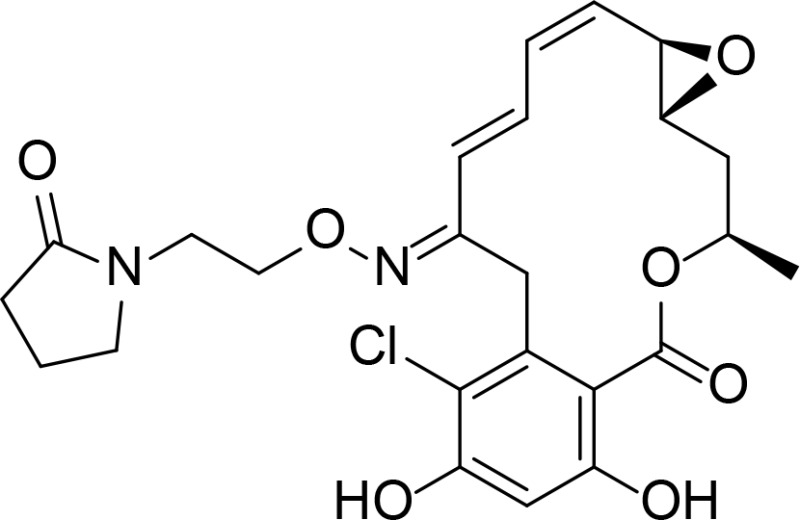
|
RDC analog | Soga et al., 2001 | |
| o-(piperidinocarbonyl) methyloxime derivative of RDC |
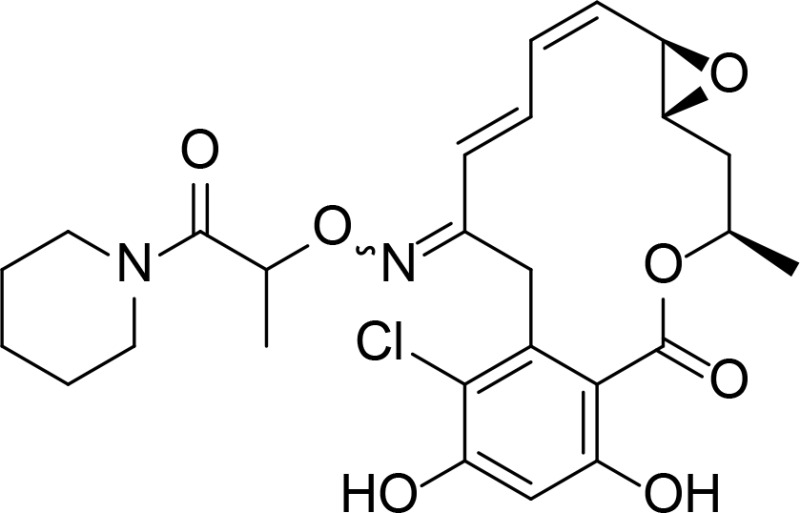
|
RDC analog | Ikuina et al., 2003 | |
| PU3 |
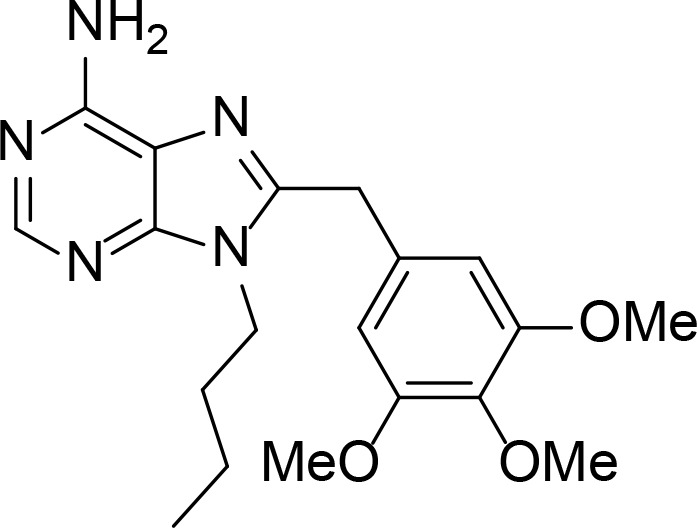
|
Kd=15∼20 µM | Purine derivative | Chiosis et al., 2001 |
| PU3 |
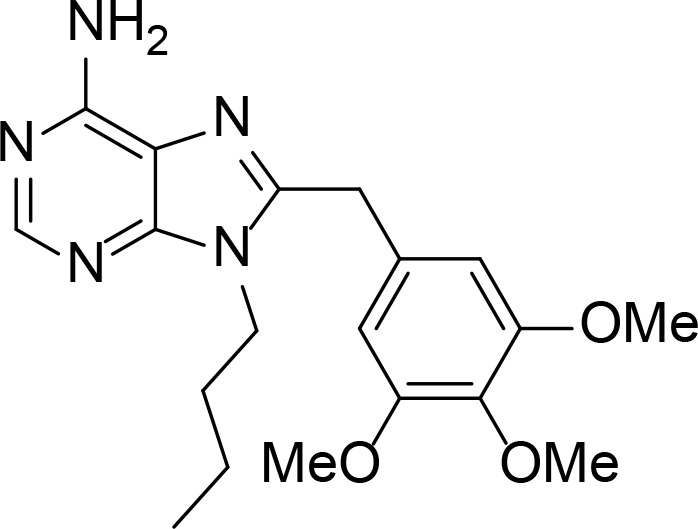
|
Kd=15∼20 µM | Purine derivative | Chiosis et al., 2001 |
| CCT018159 |
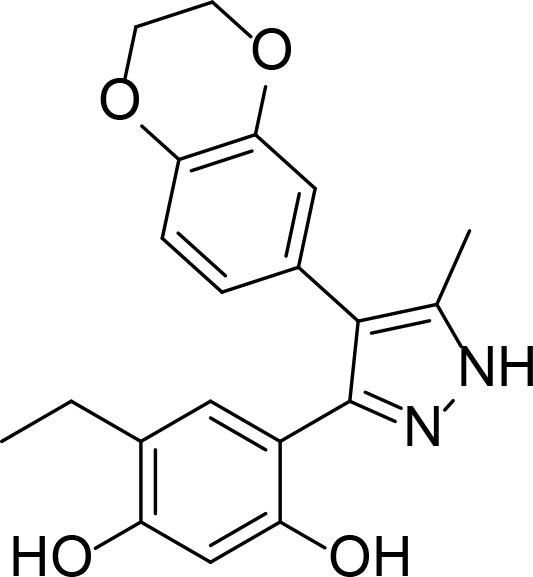
|
8.9 µM | Pyrazole | Rowlands et al., 2004 |
| G3130 |
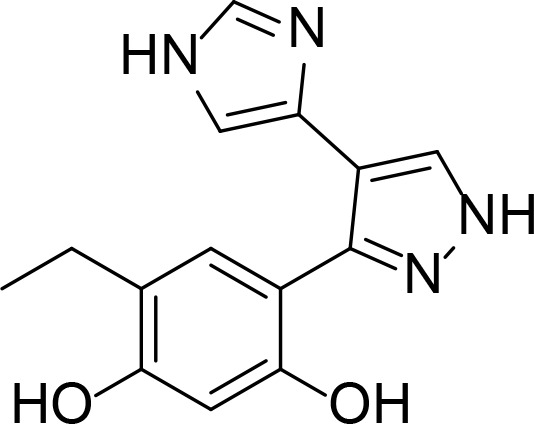
|
Kd =280 nM (SPR) | Pyrazole | Kreusch et al., 2005 |
| SNX0723 |
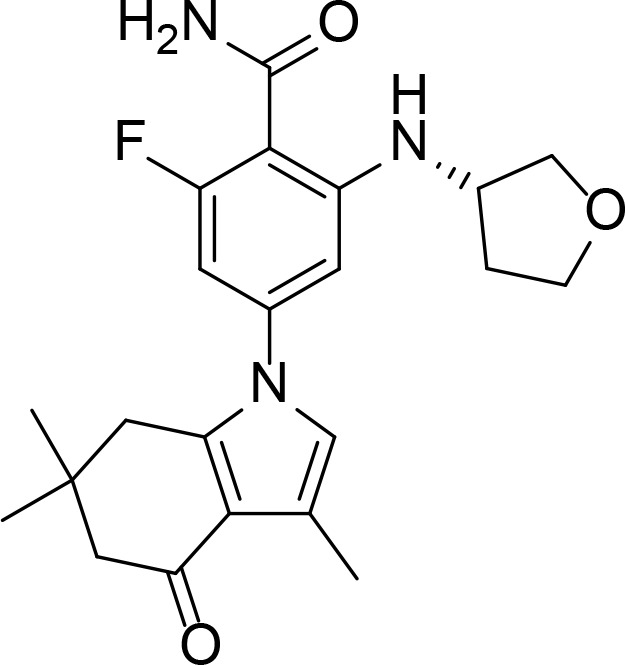
|
Benzamide | Putcha et al., 2010 | |
| AUY922 |
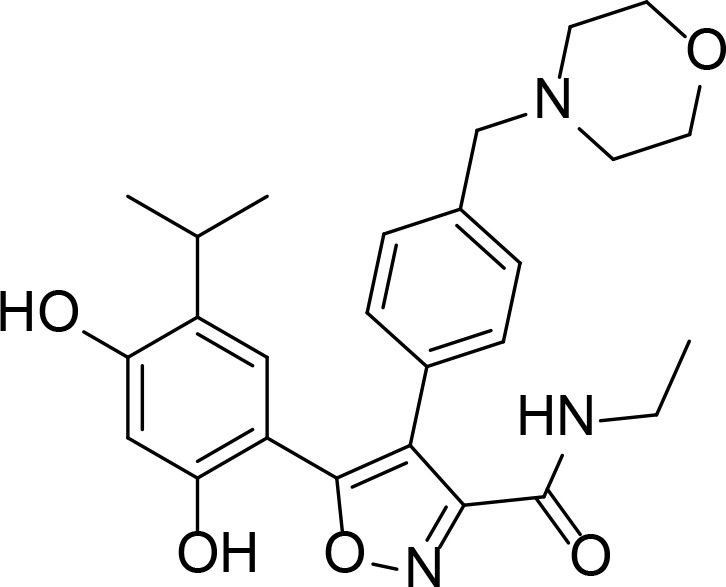
|
Resorcinol | Brough et al., 2008 | |
| AT-13387 |
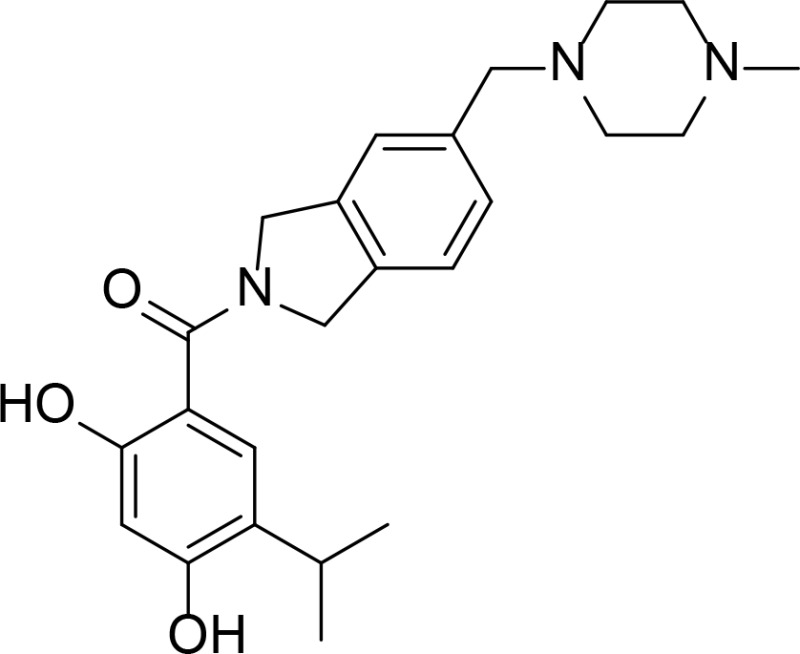
|
Resorcinol | Murray et al., 2010 | |
| CPUY201112 |
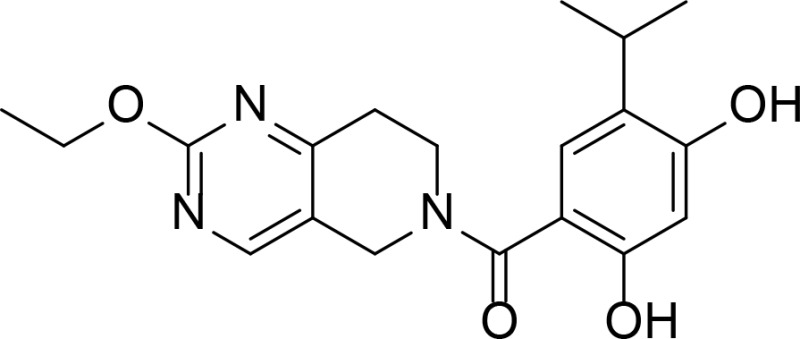
|
Resorcinol | Xu et al., 2016 |
Molecular docking studies
The similar molecular environment in the ATP binding sites of topo II and Hsp90 led us to assess whether reported inhibitors targeting either topo II or Hsp90 could function as a dual inhibitor. The listed topo II and Hsp90 inhibitors mentioned above were subjected for docking against both topo II and Hsp90. Tables 4 and 5 list the docking results of topo II and Hsp90, respectively. Surflex-Dock gives total score, crash and polar values for each of conformers. Generally, the inhibitors targeting their own binding partner scored high total score. Interestingly, the best scoring inhibitor for topo II was PU3, which was reported as an Hsp90 inhibitor with purine ring. The inhibitors showing good docking score for Hsp90 did not perform well with topo II ATP binding site. This may be due to smaller mouth opening in ATP binding pocket of topo II than Hsp90 which was calculated from CASTp. The typical Hsp90 inhibitors are bulkier compared to topo II inhibitors, therefore it would be difficult for bulky Hsp90 inhibitors to enter into the topo II ATP binding pocket.
Table 4.
Topo II docking results of combined inhibitors
| Name | Target | Total Score1 | Crash2 | Polar3 | Similarity4 |
|---|---|---|---|---|---|
| PU3 | Hsp90 | 13.1154 | −0.9961 | 4.5667 | 0.541 |
| 8-Cl-ATP | Topo II | 11.2764 | −2.0944 | 11.0551 | 0.527 |
| 3t | Topo II | 10.4766 | −0.6886 | 1.7276 | 0.397 |
| Comp. 14mod | Topo II | 10.2868 | −2.5358 | 3.0286 | 0.407 |
| Comp. 14 | Topo II | 9.927 | −2.1249 | 3.0481 | 0.427 |
| AUY922 | Hsp90 | 9.8884 | −3.5617 | 3.1905 | 0.463 |
| Salvicine R | Topo II | 8.5495 | −2.5071 | 2.9333 | 0.333 |
| CCT018159 | Hsp90 | 8.0916 | −0.9934 | 2.739 | 0.382 |
| SNX0723 | Hsp90 | 8.034 | −5.2877 | 2.8872 | 0.443 |
| NSC348400 | Topo II | 7.9712 | −3.1936 | 5.9702 | 0.568 |
| Comp. 2 | Topo II | 7.9074 | −0.4726 | 2.1046 | 0.418 |
| Daurinol | Topo II | 7.564 | −1.1849 | 4.962 | 0.459 |
| Comp. 5 | Topo II | 7.4758 | −1.6766 | 2.5831 | 0.404 |
| QAP1 | Topo II | 7.3683 | −2.2725 | 2.9142 | 0.372 |
| G3130 | Hsp90 | 7.1866 | −0.3941 | 3.5543 | 0.336 |
| Comp. 1 | Topo II | 7.1634 | −1.3298 | 3.7455 | 0.235 |
| Salvicine S | Topo II | 7.0436 | −2.892 | 3.1112 | 0.461 |
| NSC35866 | Topo II | 6.9393 | −1.0221 | 2.0358 | 0.458 |
| KF58333 | Hsp90 | 6.7973 | −4.5147 | 2.4482 | 0.380 |
| CPUY201112 | Hsp90 | 6.7935 | −2.242 | 1.4817 | 0.528 |
| AT13387 | Hsp90 | 6.2906 | −9.1331 | 4.0913 | 0.415 |
| Emodin | Topo II | 6.279 | −0.6391 | 3.2784 | 0.440 |
| 2c | Topo II | 5.9724 | −5.5461 | 0.0714 | 0.342 |
| Pochonin D | Hsp90 | 5.8197 | −3.0107 | 1.6854 | 0.406 |
| o-RDC | Hsp90 | 5.743 | −6.3103 | 1.714 | 0.393 |
| Pochonin A | Hsp90 | 5.6544 | −3.0449 | 1.7182 | 0.431 |
| c-RDC | Hsp90 | 5.571 | −3.8203 | 2.7142 | 0.353 |
| Comp. 18 | Topo II | 5.2181 | −0.9817 | 2.1991 | 0.381 |
| KF25706 | Hsp90 | 4.9578 | −3.5256 | 2.2759 | 0.467 |
| KF29163 | Hsp90 | 4.8178 | −2.5315 | 2.4504 | 0.406 |
| RDC | Hsp90 | 4.2342 | −2.6456 | 1.1539 | 0.421 |
| Aigialomycin D | Hsp90 | 4.1481 | −4.3331 | 4.1964 | 0.433 |
| TSC24 | Topo II | 4.1158 | −0.8441 | 0.0514 | 0.392 |
| Gambogic acid | Topo II | 3.8992 | −7.3216 | 0.844 | 0.385 |
| D11 | Topo II | −1.4678 | −14.195 | 1.1678 | 0.361 |
| KOSN1559 | Hsp90 | −2.5462 | −14.8183 | 2.1768 | 0.428 |
| GDM | Hsp90 | −4.9652 | −18.4653 | 2.1288 | 0.358 |
| 17-AAG | Hsp90 | −6.3308 | −17.5419 | 0.7688 | 0.478 |
| 17-DMAG | Hsp90 | −8.4678 | −21.6041 | 1.4689 | 0.508 |
Total Score represents the total Surflex-Dock score expressed as –log(Kd),
Crash is the degree of inappropriate penetration by the ligand into the protein between ligand atoms that are separated by rotatable bonds. Crash scores close to 0 are favorable,
Polar values show the contribution of the polar interactions to the total score,
Similarity indicates the difference between the top scoring pose and the original ligand (AMPPNP) used as the reference.
Table 5.
Hsp90 docking results of combined inhibitors
| Name | Target | Total Score | Crash | Polar | Similarity |
|---|---|---|---|---|---|
| AUY922 | Hsp90 | 11.3281 | −2.9912 | 5.8905 | 0.566 |
| KOSN1559 | Hsp90 | 10.5386 | −4.2057 | 6.0038 | 0.497 |
| GDM | Hsp90 | 9.0405 | −4.3587 | 5.7304 | 0.506 |
| 3t | Topo II | 8.9981 | −1.9006 | 2.2646 | 0.451 |
| KF58333 | Hsp90 | 8.8035 | −2.216 | 3.7782 | 0.475 |
| PU3 | Hsp90 | 8.6779 | −1.3914 | 2.3162 | 0.465 |
| Comp. 14 | Topo II | 8.0945 | −2.9386 | 4.3295 | 0.547 |
| o-RDC | Hsp90 | 7.4652 | −3.5968 | 3.1916 | 0.421 |
| CCT018159 | Hsp90 | 7.4339 | −0.7895 | 3.2601 | 0.521 |
| SNX0723 | Hsp90 | 7.1997 | −2.4905 | 1.3399 | 0.526 |
| Salvicine S | Topo II | 7.1362 | −1.4093 | 1.6745 | 0.475 |
| AT13387 | Hsp90 | 7.1259 | −2.0725 | 1.7217 | 0.519 |
| Comp. 14mod | Topo II | 7.0102 | −2.3685 | 3.184 | 0.519 |
| G3130 | Hsp90 | 6.8551 | −0.5655 | 4.089 | 0.514 |
| QAP1 | Topo II | 6.7837 | −2.2874 | 0.9711 | 0.497 |
| CPUY201112 | Hsp90 | 6.7467 | −2.5928 | 2.7711 | 0.612 |
| NSC348400 | Topo II | 6.7386 | −1.4421 | 3.7083 | 0.511 |
| RDC | Hsp90 | 6.6884 | −2.5576 | 3.4177 | 0.666 |
| NSC35866 | Topo II | 6.649 | −1.1619 | 2.6058 | 0.486 |
| Salvicine R | Topo II | 6.4918 | −2.4288 | 2.1968 | 0.510 |
| Comp. 1 | Topo II | 6.4641 | −1.627 | 2.2857 | 0.561 |
| 2c | Topo II | 6.4036 | −3.4089 | 0.0014 | 0.329 |
| KF25706 | Hsp90 | 6.185 | −2.8315 | 3.6404 | 0.647 |
| Gambogic acid | Topo II | 6.0092 | −2.2055 | 1.3905 | 0.297 |
| Comp. 2 | Topo II | 5.952 | −1.2823 | 1.4835 | 0.524 |
| Daurinol | Topo II | 5.8292 | −0.4604 | 1.3936 | 0.556 |
| KF29163 | Hsp90 | 5.7498 | −2.3134 | 1.6213 | 0.530 |
| 17-DMAG | Hsp90 | 5.718 | −3.6373 | 1.0756 | 0.318 |
| c-RDC | Hsp90 | 5.6849 | −3.5861 | 3.0525 | 0.691 |
| Comp. 5 | Topo II | 5.6657 | −1.6574 | 1.9241 | 0.430 |
| D11 | Topo II | 5.6512 | −0.8901 | 2.5246 | 0.390 |
| Emodin | Topo II | 5.6113 | −0.429 | 1.9987 | 0.398 |
| 8-Cl-ATP | Topo II | 5.5933 | −1.2132 | 4.7994 | 0.399 |
| Pochonin D | Hsp90 | 5.5041 | −1.5103 | 2.1344 | 0.550 |
| TSC24 | Topo II | 5.477 | −1.2353 | 0.5795 | 0.418 |
| Pochonin A | Hsp90 | 5.4494 | −1.3695 | 2.6113 | 0.279 |
| 17-AAG | Hsp90 | 5.2911 | −2.6657 | 0.545 | 0.301 |
| Aigialomycin D | Hsp90 | 4.8757 | −3.236 | 3.4461 | 0.549 |
| Comp. 18 | Topo II | 4.553 | −0.4098 | 0.9114 | 0.427 |
PU3, 8-Cl-ATP and compound 3t in the docking of topo II, all showed high docking score which are all purine analogs. Fascinatingly, PU3, an Hsp90 inhibitor, scored the highest when docked to topo II. PU3 had hydrogen bonding interactions with Asn91, Asn120, Ala167 and Thr215, where they are key residues that formed hydrogen bonds with ATP (Fig. 4A). Also, PU3 had hydrophobic interactions with the residues comprising the ATP binding pocket, namely, Asn91, Asp94, Arg98, Asn120, Ile125, Ile141, Phe142, Ser149, Asn150, Thr159, Gly161, Arg162, Gly164, Ala167, Lys168 and Thr215. Compound 3t also occupied the ATP binding site and interacted with residues Asn91, Ala92, Asn95, Asn120, Pro126, Ile141, Phe142, Ser149, Gly164, Tyr165, Gly166, Ala167, Lys168, Thr215 and Ile217 (Fig. 4B). However, 3t had only one hydrogen bond interaction with residue Asn91. AUY922 is an Hsp90 inhibitor with isoxazole moiety. There are two hydroxyl substituents from the phenyl ring and amide group that can form hydrogen bonds with residues Asn95, Asn120 and Ser149. Since AUY922 is rather big molecule compared to PU3, 3t, comp. 14 and 8-Cl-ATP, larger number of residues are involved in van der Waals interaction, namely, Ile88, Asn91, Ala92, Asn95, Arg98, Ile118, Asn120, Il2125, Pro126, Ile141, Phe142, Ser149, Asn150, Gly161, Gly164, Tyr165, Gly166, Ala167, Lys168, Thr215 and Ile217. Recently from AstraZeneca, compound with benzisoxazole scaffold, ETX0914, was discovered as a novel DNA gyrase inhibitor undergoing phase II clinical trial for the treatment of uncomplicated gonorrhea (Basarab et al., 2015). There is no known topo II inhibitor reported with isoxazole scaffold to the best of our knowledge. Therefore our docking results suggest AUY922 may act as a topo II inhibitor with novel scaffold. Comp. 14 is a small compound with xanthone moiety which competed with ATP. Comp. 14 had hydrogen bond interactions with Asp94 and Thr215 and hydrophobic interactions with Asn51, Ser52, Ala55, Asp93, Ile96, Gly97, Met98, Asn106, Phe138, Val150, Thr184 and Val186.
Fig. 4.

The docking result of the selected inhibitors against topo II. The ATP binding site of topo II with inhibitors (A) PU3, (B) 3t, (C) AUY922 and (D) Comp. 14. The ligands are represented in sticks colored by atom type (magenta: carbon; red: oxygen; blue: nitrogen; orange: phosphorus) and the residues involved in hydrogen bonds are shown in dotted line colored in cyan.
In the case of docking topo II and Hsp90 inhibitors to Hsp90, the bulky Hsp90 inhibitors were high in rank as mentioned above. However, AUY922, a rather smaller isoxazole derivative compared to classical Hsp90 inhibitors such as GDM or RDC, scored highest. AUY922 also had hydrogen bond interactions with five residues in the ATP binding site of Hsp90, Asn51, Lys58, Asp93, Gly97 and Phe138. The residues involved in hydrophobic interactions are Asn51, Ala55, Lys59, Asp93, Ile96, Gly97, Met98, Asp102, Leu107, Gly135, Val136, Gly137, Phe138, Val150, Thr184 and Val186. PU3, a purine analog Hsp90 inhibitor showed good result in Hsp90 along with 3t, another purine analog topo II inhibitor.
Two purine derivatives of PU3 and 3t and two non-purine compounds of AUY922 and comp. 14 were selected for further comparison in depth since they ranked high in docking study of both topo II and Hsp90. The binding interactions of topo II and Hsp90 with compounds of PU3, 3t, AUY922 and 14 are shown in Fig. 4, 5. The hydrogen bonding residues are labeled and the bonds are displayed as light blue dashed lines. The selected common four compounds and high scoring compounds 8-Cl-ATP and KOSN1599 for topo II and Hsp90, respectively, were analyzed in detail. Table 6 summarizes the residues involved in hydrophobic and hydrogen bond interactions.
Fig. 5.

The docking result of the selected inhibitors against Hsp90. The ATP binding site of topo II with inhibitors (A) PU3, (B) 3t, (C) AUY922, and (D) Comp.14. The ligands are represented in sticks colored by atom type (yellow: carbon; red: oxygen; blue: nitrogen; orange: phosphorus) and the residues involved in hydrogen bonds are shown in dotted line colored in cyan.
Table 6.
Docking analysis of selected inhibitors
| Topo II | Hsp90 | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Name | Hydrophobic | Hydrogen bonding | Name | Hydrophobic | Hydrogen bonding |
| PU3 | Asn91, Asp94, Arg98, Asn120, Ile125, Ile141, Phe142, Ser149, Asn150, Thr159, Gly161, Arg162, Gly164, Ala167, Lys168, Thr215 | Asn91, Asn120, Ala167, Thr215 | PU3 | Asn51, Asp54, Ala55, Asp93, Ile96, Gly97, Met98, Asn106, Leu107, Gly135, Val136, Phe138, Val150, Thr184, Val186 | Asp54, Thr184 |
| 3t | Asn91, Ala92, Asn95, Asn120, Pro126, Ile141, Phe142, Ser149, Gly164, Tyr165, Gly166, Ala167, Lys168, Thr215, Ile217 | Asn91 | 3t | Asn51, Ala55, Lys58, Ile96, Gly97, Met98, Asn106, Phe138, Val150, His154, Thr184, Val186 | Lys58, Gly97 |
| Comp. 14 | Ile88, Asn91, Ala92, Asp94, Asn95, Ile118, Asn120, Ile125, Asn150, Gly161, Gly164, Tyr165, Ala167, Lys168, Thr215, Ile217 | Asp94, Thr215 | Comp. 14 | Asn51, Ser52, Ala55, Asp93, Ile96, Gly97, Met98, Asn106, Phe138, Val150, Thr184, Val186 | Asp93, Gly97, Thr184 |
| AUY922 | Ile88, Asn91, Ala92, Asn95, Arg98, Ile118, Asn120, Ile125, Pro126, Ile141, Phe142, Ser149, Asn150, Gly161, Gly164, Tyr165, Gly166, Ala167, Lys168, Thr215, Ile217 | Asn95, Asn120, Ser149 | AUY922 | Asn51, Ala55, Lys58, Asp93, Ile96, Gly97, Met98, Asp102, Leu107, Gly135, ValL136, Gly137, Phe138, Val150, Thr184, Val186 | Asn51, Lys58, Asp93, Gly97, Phe138 |
| 8-Cl-ATP | Asn91, Asp94, Asn95, Arg98, Lys123, Gly124, Ile125, Ser149, Asn150, Gly161, Arg162, Asn163, Gly164, Tyr165, Gly166, Ala167, Gln376, Lys378 | Asn91, Asp94, Asn150, Arg162, Tyr165, Gly166, Lys378 | KOSN1599 | Asn51, Ser52, Asp54, Ala55, Lys58, Asp93, Ile96, Met98, ASP102, Asn106, Leu107, Phe138, Thr184, Val186 | Ser52, Asp54, Phe138 |
Pharmacophore model analysis
Compounds PU3, 3t, AUY922 and comp. 14 were further evaluated for their dual targeting features by generating pharmacophore models. The pharmacophore model was generated using Genetic Algorithm Similarity Program (GASP) module implemented in Sybyl X-2.1.1. From the four compounds, four models were generated by GASP. The fitness score for each model ranged from 2589.82 to 2689.23 and model 2 was chosen as the best model (Table 7). Model 2 consists of two hydrophobic regions (HY, cyan), one acceptor atom (AA, green) and one donor site (DS, green) as shown in Fig. 6 with PU3 as the template. The two hydrophobic regions are about 5 Å apart and the hydrophobic region 1 and the acceptor atom is 2.5 Å apart. The pharmacophore model suggested here can be used as a template to further optimize the design of the dual inhibitor of topo II and Hsp90.
Table 7.
Results of pharmacophore hypothesis generated by GASP
| Model | Fitness | Sizea | Hitsb | Dmeanc | Featuresd |
|---|---|---|---|---|---|
| 1 | 2676.46 | 4 | 4 | 5.7693 | DS, AA, HY1, HY2 |
| 2 | 2689.23 | 4 | 4 | 3.5916 | DS, AA, HY1, HY2 |
| 3 | 2589.82 | 4 | 4 | 3.1774 | DS, AA, HY1, HY2 |
| 4 | 2663.25 | 2 | 4 | 4.5547 | HY1, HY2 |
Number of features in the model,
Number of molecules that matched during the search,
Average interpoint distance,
Pharmacophore features. DS: donor site, AA: acceptor site, HY: hydrophobic.
Fig. 6.

The pharmacophore model 2 generated from GASP. The pharmacophore features are two hydrophobic regions (HY, cyan), one acceptor atom (AA, green) and one donor site (DS, green) with PU3 as the template represented in sticks colored by atom type (gray: carbon; light blue: hydrogen; red: oxygen; blue: nitrogen; orange: phosphorus).
CONCLUSIONS
In this study, the inhibitors reported to target each ATPase domain of human topo II and Hsp90 were investigated. The structures of ATPase domains of topo II and Hsp90 were compared to evaluate how similar the environment of the receptor sites were. The topo II and Hsp90 inhibitors known to target the ATP binding site were searched and the possibility to function as a dual inhibitor was investigated in silico. All the inhibitors searched were docked to both topo II and Hsp90. Through the analysis of docking results, four candidate compounds were selected as possible dual inhibitors. These compounds were used as a template to generate pharmacophore model. This suggested pharmacophore model will be useful in developing dual inhibitor of topo II and Hsp90 by constructing 3D query for virtual screening using publically available database such as ZINC (http://zinc.docking.org/).
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF-2013R1A1A2060408), the Korean Health Technology R&D Project funded by Ministry of Health & Welfare, Republic of Korea (HI14C2469), and by the grant of the Bio & Medical Technology Development Program (NRF-2014M3A9A9073 908) of the National Research Foundation of Korea (NRF), funded by the Korean government (Ministry of Science, ICT & Future Planning).
REFERENCES
- Agatsuma T, Ogawa H, Akasaka K, Asai A, Yamashita Y, Mizukami T, Akinaga S, Saitoh Y. Halohydrin and oxime derivatives of radicicol: Synthesis and antitumor activities. Bioorg Med Chem. 2002;10:3445–3454. doi: 10.1016/S0968-0896(02)00260-2. [DOI] [PubMed] [Google Scholar]
- Barker CR, Hamlett J, Pennington SR, Burrows F, Lundgren K, Lough R, Watson AJ, Jenkins JR. The topoisomerase II-Hsp90 complex: A new chemotherapeutic target? Int. J. Cancer. 2006;118:2685–2693. doi: 10.1002/ijc.21717. [DOI] [PubMed] [Google Scholar]
- Basarab GS, Kern GH, McNulty J, Mueller JP, Lawrence K, Vishwanathan K, Alm RA, Barvian K, Doig P, Galullo V, Gardner H, Gowravaram M, Huband M, Kimzey A, Morningstar M, Kutschke A, Lahiri SD, Perros M, Singh R, Schuck VJ, Tommasi R, Walkup G, Newman JV. Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial Type II topoisomerases. Sci Rep. 2015;5:11827. doi: 10.1038/srep11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baviskar AT, Madaan C, Preet R, Mohapatra P, Jain V, Agarwal A, Guchhait SK, Kundu CN, Banerjee UC, Bharatam PV. N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIαand induce apoptosis in G1/S phase. J Med Chem. 2011;54:5013–5030. doi: 10.1021/jm200235u. [DOI] [PubMed] [Google Scholar]
- Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, Cheung KM, Collins I, Davies NG, Drysdale MJ, Dymock B, Eccles SA, Finch H, Fink A, Hayes A, Howes R, Hubbard RE, James K, Jordan AM, Lockie A, Martins V, Massey A, Matthews TP, McDonald E, Northfield CJ, Pearl LH, Prodromou C, Ray S, Raynaud FI, Roughley SD, Sharp SY, Surgenor A, Walmsley DL, Webb P, Wood M, Workman P, Wright L. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51:196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- Chene P. ATPases as drug targets: Learning from their structure. Nat Rev Drug Discov. 2002;1:665–673. doi: 10.1038/nrd894. [DOI] [PubMed] [Google Scholar]
- Chene P, Rudloff J, Schoepfer J, Furet P, Meier P, Qian Z, Schlaeppi JM, Schmitz R, Radimerski T. Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem Biol. 2009;9:1. doi: 10.1186/1472-6769-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001;8:289–299. doi: 10.1016/S1074-5521(01)00015-1. [DOI] [PubMed] [Google Scholar]
- Dutta Gupta S, Snigdha D, Mazaira GI, Galigniana MD, Subrahmanyam CV, Gowrishankar NL, Raghavendra NM. Molecular docking study, synthesis and biological evaluation of schiff bases as Hsp90 inhibitors. Biomed Pharmacother. 2014;68:369–376. doi: 10.1016/j.biopha.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. doi: 10.1016/S0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- Furet P, Schoepfer J, Radimerski T, Chène P. Discovery of a new class of catalytic topoisomerase II inhibitors targeting the atp-binding site by structure based design. Part I. Bioorg Med Chem Lett. 2009;19:4014–4017. doi: 10.1016/j.bmcl.2009.06.034. [DOI] [PubMed] [Google Scholar]
- Gui M, Shi DK, Huang M, Zhao Y, Sun QM, Zhang J, Chen Q, Feng JM, Liu CH, Li M, Li YX, Geng M, Ding J. D11, a novel glycosylated diphyllin derivative, exhibits potent anticancer activity by targeting topoisomerase IIα. Invest. New Drugs. 2011;29:800–810. doi: 10.1007/s10637-010-9425-3. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/nt. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hu CX, Zuo ZL, Xiong B, Ma JG, Geng MY, Lin LP, Jiang HL, Ding J. Salvicine functions as novel topoisomerase II poison by binding to ATP pocket. Mol Pharmacol. 2006;70:1593–1601. doi: 10.1124/mol.106.027714. [DOI] [PubMed] [Google Scholar]
- Huang H, Chen Q, Ku X, Meng L, Lin L, Wang X, Zhu C, Wang Y, Chen Z, Li M, Jiang H, Chen K, Ding J, Liu H. A series of alpha-heterocyclic carboxaldehyde thiosemicarbazones inhibit topoisomerase iialpha catalytic activity. J Med Chem. 2010;53:3048–3064. doi: 10.1021/jm9014394. [DOI] [PubMed] [Google Scholar]
- Ikuina Y, Amishiro N, Miyata M, Narumi H, Ogawa H, Akiyama T, Shiotsu Y, Akinaga S, Murakata C. Synthesis and antitumor activity of novel o-carbamoylmethyloxime derivatives of radicicol. J Med Chem. 2003;46:2534–2541. doi: 10.1021/jm030110r. [DOI] [PubMed] [Google Scholar]
- Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- Jensen LH, Liang H, Shoemaker R, Grauslund M, Sehested M, Hasinoff BB. A three-dimensional quantitative structure-activity relationship study of the inhibition of the ATPase activity and the strand passing catalytic activity of topoisomerase IIalpha by substituted purine analogs. Mol Pharmacol. 2006;70:1503–1513. doi: 10.1124/mol.106.026856. [DOI] [PubMed] [Google Scholar]
- Jensen LH, Thougaard AV, Grauslund M, Sokilde B, Carstensen EV, Dvinge HK, Scudiero DA, Jensen PB, Shoemaker RH, Sehested M. Substituted purine analogues define a novel structural class of catalytic topoisomerase II inhibitors. Cancer Res. 2005;65:7470–7477. doi: 10.1158/0008-5472.CAN-05-0707. [DOI] [PubMed] [Google Scholar]
- Jez JM, Chen JC, Rastelli G, Stroud RM, Santi DV. Crystal structure and molecular modeling of 17-DMAG in complex with human Hsp90. Chem Biol. 2003;10:361–368. doi: 10.1016/S1074-5521(03)00075-9. [DOI] [PubMed] [Google Scholar]
- Jimenez-Alonso S, Orellana HC, Estevez-Braun A, Ravelo AG, Perez-Sacau E, Machin F. Design and synthesis of a novel series of pyranonaphthoquinones as topoisomerase II catalytic inhibitors. J Med Chem. 2008;51:6761–6772. doi: 10.1021/jm800499x. [DOI] [PubMed] [Google Scholar]
- Jun KY, Lee EY, Jung MJ, Lee OH, Lee ES, Park Choo HY, Na Y, Kwon Y. Synthesis, biological evaluation, and molecular docking study of 3-(3′-heteroatom substituted-2′-hydroxy-1′-propyloxy) xanthone analogues as novel topoisomerase IIα catalytic inhibitor. Eur J Med Chem. 2011;46:1964–1971. doi: 10.1016/j.ejmech.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Kang K, Nho CW, Kim ND, Song DG, Park YG, Kim M, Pan CH, Shin D, Oh SH, Oh HS. Daurinol, a catalytic inhibitor of topoisomerase IIα, suppresses SNU-840 ovarian cancer cell proliferation through cell cycle arrest in S phase. Int J Oncol. 2014;45:558–566. doi: 10.3892/ijo.2014.2442. [DOI] [PubMed] [Google Scholar]
- Kreusch A, Han S, Brinker A, Zhou V, Choi HS, He Y, Lesley SA, Caldwell J, Gu XJ. Crystal structures of human HSP90alpha-complexed with dihydroxyphenylpyrazoles. Bioorg Med Chem Lett. 2005;15:1475–1478. doi: 10.1016/j.bmcl.2004.12.087. [DOI] [PubMed] [Google Scholar]
- Kung PP, Funk L, Meng J, Collins M, Zhou JZ, Johnson MC, Ekker A, Wang J, Mehta P, Yin MJ, Rodgers C, Davies JF, 2nd, Bayman E, Smeal T, Maegley KA, Gehring MR. Dihydroxylphenyl amides as inhibitors of the Hsp90 molecular chaperone. Bioorg Med Chem Lett. 2008;18:6273–6278. doi: 10.1016/j.bmcl.2008.09.081. [DOI] [PubMed] [Google Scholar]
- Li PH, Zeng P, Chen SB, Yao PF, Mai YW, Tan JH, Ou TM, Huang SL, Li D, Gu LQ, Huang ZS. Synthesis and mechanism studies of 1,3-benzoazolyl substituted pyrrolo[2,3-b]pyrazine derivatives as nonintercalative topoisomerase II catalytic inhibitors. J Med Chem. 2016;59:238–252. doi: 10.1021/acs.jmedchem.5b01284. [DOI] [PubMed] [Google Scholar]
- Li Y, Luan Y, Qi X, Li M, Gong L, Xue X, Wu X, Wu Y, Chen M, Xing G, Yao J, Ren J. Emodin triggers DNA double-strand breaks by stabilizing topoisomerase II-DNA cleavage complexes and by inhibiting atp hydrolysis of topoisomerase II. Toxicol Sci. 2010;118:435–443. doi: 10.1093/toxsci/kfq282. [DOI] [PubMed] [Google Scholar]
- Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Wang JG, Wang B, Li ZM. Integrating molecular docking, DFT and CoMFA/CoMSIA approaches for a series of naphthoquinone fused cyclic α-aminophosphonates that act as novel topoisomerase II inhibitors. J Mol Model. 2011;17:1899–1909. doi: 10.1007/s00894-010-0898-y. [DOI] [PubMed] [Google Scholar]
- Moulin E, Barluenga S, Winssinger N. Concise synthesis of pochonin A, an HSP90 inhibitor. Org Lett. 2005a;7:5637–5639. doi: 10.1021/ol052263+. [DOI] [PubMed] [Google Scholar]
- Moulin E, Zoete V, Barluenga S, Karplus M, Winssinger N. Design, synthesis, and biological evaluation of HSP90 inhibitors based on conformational analysis of radicicol and its analogues. J Am Chem Soc. 2005b;127:6999–7004. doi: 10.1021/ja043101w. [DOI] [PubMed] [Google Scholar]
- Murray CW, Carr MG, Callaghan O, Chessari G, Congreve M, Cowan S, Coyle JE, Downham R, Figueroa E, Frederickson M, Graham B, McMenamin R, O’Brien MA, Patel S, Phillips TR, Williams G, Woodhead AJ, Woolford AJ. Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. J Med Chem. 2010;53:5942–5955. doi: 10.1021/jm100059d. [DOI] [PubMed] [Google Scholar]
- Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–S61. doi: 10.1016/S1471-4914(02)02316-X. [DOI] [PubMed] [Google Scholar]
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009a;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer. 2009b;9:327–337. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SE, Chang IH, Jun KY, Lee E, Lee ES, Na Y, Kwon Y. 3-(3-butylamino-2-hydroxy-propoxy)-1-hydroxy-xanthen-9-one acts as a topoisomerase IIα catalytic inhibitor with low DNA damage. Eur J Med Chem. 2013;69:139–145. doi: 10.1016/j.ejmech.2013.07.048. [DOI] [PubMed] [Google Scholar]
- Patel K, Piagentini M, Rascher A, Tian ZQ, Buchanan GO, Regentin R, Hu Z, Hutchinson CR, McDaniel R. Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem Biol. 2004;11:1625–1633. doi: 10.1016/j.chembiol.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Petrelli A, Giordano S. From single- to multi-target drugs in cancer therapy: when aspecificity becomes an advantage. Curr Med Chem. 2008;15:422–432. doi: 10.2174/092986708783503212. [DOI] [PubMed] [Google Scholar]
- Pogorelcnik B, Perdih A, Solmajer T. Recent advances in the development of catalytic inhibitors of human DNA topoisomerase IIα as novel anticancer agents. Curr Med Chem. 2013;20:694–709. doi: 10.2174/092986713804999402. [DOI] [PubMed] [Google Scholar]
- Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, Hicks CD, Veal JM, Steed PM, Hyman BT, McLean PJ. Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J Pharmacol Exp Ther. 2010;332:849–857. doi: 10.1124/jpet.109.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Meng L, Hu C, Duan W, Zuo Z, Lin L, Zhang X, Ding J. Gambogic acid inhibits the catalytic activity of human topoisomerase IIα by binding to its ATPase domain. Mol Cancer Ther. 2007;6:2429–2440. doi: 10.1158/1535-7163.MCT-07-0147. [DOI] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Rowlands MG, Newbatt YM, Prodromou C, Pearl LH, Workman P, Aherne W. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal Biochem. 2004;327:176–183. doi: 10.1016/j.ab.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- Sidera K, Patsavoudi E. HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov. 2014;9:1–20. doi: 10.2174/15748928113089990031. [DOI] [PubMed] [Google Scholar]
- Soga S, Neckers LM, Schulte TW, Shiotsu Y, Akasaka K, Narumi H, Agatsuma T, Ikuina Y, Murakata C, Tamaoki T, Akinaga S. KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 1999;59:2931–2938. [PubMed] [Google Scholar]
- Soga S, Sharma SV, Shiotsu Y, Shimizu M, Tahara H, Yamaguchi K, Ikuina Y, Murakata C, Tamaoki T, Kurebayashi J, Schulte TW, Neckers LM, Akinaga S. Stereospecific antitumor activity of radicicol oxime derivatives. Cancer Chemother Pharmacol. 2001;48:435–445. doi: 10.1007/s002800100373. [DOI] [PubMed] [Google Scholar]
- Wang B, Miao ZW, Wang J, Chen RY, Zhang XD. Synthesis and biological evaluation of novel naphthoquinone fused cyclic aminoalkylphosphonates and aminoalkylphosphonic monoester. Amino Acids. 2008;35:463–468. doi: 10.1007/s00726-007-0570-8. [DOI] [PubMed] [Google Scholar]
- Wang P, Leung CH, Ma DL, Lu W, Che CM. Organoplatinum(II) complexes with nucleobase motifs as inhibitors of human topoisomerase II catalytic activity. Chem Asian J. 2010;5:2271–2280. doi: 10.1002/asia.201000451. [DOI] [PubMed] [Google Scholar]
- Wei H, Ruthenburg AJ, Bechis SK, Verdine GL. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J Biol Chem. 2005;280:37041–37047. doi: 10.1074/jbc.M506520200. [DOI] [PubMed] [Google Scholar]
- Xu XL, Bao QC, Jia JM, Liu F, Guo XK, Zhang MY, Wei JL, Lu MC, Xu LL, Zhang XJ, You QD, Sun HP. CPUY201112, a novel synthetic small-molecule compound and inhibitor of heat shock protein Hsp90, induces p53-mediated apoptosis in MCF-7 cells. Sci Rep. 2016;1900;6:4. doi: 10.1038/srep19004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Jia XZ, Feng LY, Li SY, An GS, Ni JH, Jia HT. Inhibition of topoisomerase ii by 8-chloro-adenosine triphosphate induces DNA double-stranded breaks in 8-chloro-adenosine-exposed human myelocytic leukemia K562 cells. Biochem Pharmacol. 2009;77:433–443. doi: 10.1016/j.bcp.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Yang ZQ, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. New efficient synthesis of resorcinylic macrolides via ynolides: Establishment of cycloproparadicicol as synthetically feasible preclinical anticancer agent based on Hsp90 as the target. J Am Chem Soc. 2004;126:7881–7889. doi: 10.1021/ja0484348. [DOI] [PubMed] [Google Scholar]
- Yao Q, Weigel B, Kersey J. Synergism between etoposide and 17-AAG in leukemia cells: Critical roles for Hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin Cancer Res. 2007;13:1591–1600. doi: 10.1158/1078-0432.CCR-06-1750. [DOI] [PubMed] [Google Scholar]